Chiral spherical aromatic amides: one-step synthesis and their stereochemical/chiroptical properties†
Received
7th September 2024
, Accepted 3rd November 2024
First published on 7th November 2024
Abstract
Macrocyclic aromatic amides 2, in which the meta-positions of all four benzene rings are linked by tertiary amide bonds, were successfully synthesized by a one-step condensation reaction of two monomers using dichlorotriphenylphosphorane or triphenylphosphine/hexachloroethane (dehydration-condensation reagents for carboxy and amino groups), or LiHMDS (aminolysis for esters) as coupling reagents. Single crystal X-ray analyses of the N-methyl and N-ethyl derivatives were performed and revealed that each compound adopted a spherical structure with chirality because of the fixed axis rotation and combined polarity of the amide bonds. Enantiomers of each spherical macrocycle were separated by chiral column chromatography and showed mirror-imaged CD spectra to each other.
Introduction
Molecules such as fullerenes,1 carboranes2 and adamantanes,3 which have a medium-sized rigid core structure and can be functionalized, are used widely as core structures in the design of dendritic compounds, scaffolds in functionalized 3D molecules and pharmacophores in drug discovery. Such rigid and bulky molecules form highly ordered aggregates to yield void, cavity or channel structures when crystallized.4 Moreover, these skeletons can display molecular chirality depending on the position or type of substituents and be used to design optically active materials. Therefore, developing such “block molecules” with various sizes and structures that are easily synthesized is strongly desired.
In various fields such as chemical biology, macromolecules or supermolecules amide compounds are used widely5 because of their rigidity, hydrogen bonding capacity and conformational switching between cis and trans. Recently, in drug discovery, medium-sized peptide drugs represent a new modality that mimics protein–protein interactions.5a,b Previously, aiming to discover a new functional molecule with such amide characteristics, we have also designed several macrocyclic aromatic amides6 and developed effective methods to synthesize these amides. Because of the cis-type conformational preference of tertiary aromatic amides,7 macrocyclic compounds are effectively synthesized by the amide condensation reaction using meta- or para-(alkylamino)benzoic acid as a monomer.6a,b Using 3-(ethylamino)benzoic acid as a monomer gave a cyclic trimer as the main product. This cyclic trimer displayed conformational chirality derived from the direction of the benzene rings against the amide plane and the direction of the amides and easily racemized because of the low rotation energy barrier of the benzene ring-N and benzene ring-carbonyl single bonds. For this cyclic trimer, if another benzene ring unit is linked to the compound via three amide bonds at meta-positions, that is, if the compound has four benzene rings with six amide bonds at each meta-position, the chirality of the compound will be fixed (compound 1 in Fig. 1). There are four possible structural isomers of this class of compounds because of the different combinations of amide orientations (Fig. 1, for details, see ESI†). We call these compounds ‘spherical amides’ because of the sphere-like structure formed by the four benzene rings in the core of the molecule. These compounds have fixed axial chirality and can be functionalized by substituents on amide nitrogens, which are oriented in three-dimensional directions. Three-dimensional compounds with internal voids are known as “organic cages”. Currently, various cage molecules with useful functions have been synthesized.8 For example, Lauer and coworkers developed an organic amide cage that binds nitrate efficiently.8c Tromans and coworkers introduced a cage-shaped biomimetic synthetic glucose receptor with an affinity for glucose comparable to the natural receptor.8d Thus, we also aimed to develop an efficient synthetic method of spherical amides and to present these amides as a new type of functional organic cage.
 |
| | Fig. 1 Chemical structures of structural isomers of the macrocyclic molecules (‘spherical amides’) in which each of three meta positions of four benzene rings are connected by six tertiary amide groups. Each color represents a monomer unit. Every macrocyclic structure is chiral and has its enantiomeric stereoisomer. | |
We previously synthesized one of the structural isomers of the spherical amides (isomer 1, R = CH3, CH2CH3) by a synthetic route that involves a one-step cyclization reaction of a cyclic trimer, followed by capping the cyclic trimer with one benzene unit (benzene-1,3,5-tricarboxylic acid), using dichlorotriphenylphosphorane in both steps as the condensation reagent.9 The compound formed crystals with a channel structure whose shape and size varied according to the alkyl groups on the amide nitrogen and the solvent molecules present in the crystal. However, the other structural isomers have not been synthesized and are expected to exhibit different stereochemical and chiroptical properties. Here, we report the synthesis and structural and chiroptical properties of another structural isomer (i.e., 2) composed of two sets of two different monomers.
Results and discussion
Synthesis and reaction optimization
Initially, 5-(alkylamino)isophthalic acid (7a–d) and 3,5-bis(alkylamino)benzoic acid (11a–d) were synthesized (a: methyl; b: ethyl; c: propyl; d: isobutyl) as two monomers required for synthesizing structural isomer 2. Compounds 6a and 10a (R = methyl) were synthesized according to Scheme 1 by the N-alkylation method using dimethyl carbonate with NaY zeolite as the catalyst,10 followed by hydrolysis using aqueous NaOH (4 M) to give compounds 7a and 11a. Dimethyl 5-(alkylamino)isophthalates (6b–d) were obtained from dimethyl 5-aminoisophthalate (5) by N-alkylation using a reductive amination reaction with 2-picoline borane,11 and were hydrolyzed to give compounds 7b–d (Scheme 2, top). Ethyl 3,5-diaminobenzoate (9) was synthesized from 3,5-diaminobenzoic acid (8) by the Fischer esterification reaction,12 followed by N-alkylation11 and hydrolysis to give compounds 11b–d (Scheme 2, bottom).
 |
| | Scheme 1 Synthetic routes of 7a and 11a. | |
 |
| | Scheme 2 Synthetic routes of 7b–d and 11b–d. | |
Next, a condensation reaction to a mixture of 7c and 11c at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was performed in 1,1,2,2-tetrachloroethane (0.05 M) using dichlorotriphenylphosphorane (7.2 eq.) as the coupling reagent at 120 °C (Scheme 3; entry 1 in Table 1).6 The obtained crude product was purified by silica gel column chromatography followed by gel permeation chromatography, resulting in the desired product in which two sets of monomers were condensed to give a 13.6% yield. The reaction using triphenylphosphine and hexachloroethane that generate dichlorotriphenylphosphorane in the reaction system13 gave the target compounds with similar yields (13.0%, entry 2 in Table 1). Then, the reaction conditions (reagent amount, solvent, temperature) were optimized in this system (entries 3–10). The best amount of coupling reagent was found to be 7.2 eq., which is 2.4 eq. per one amide bond formation (entries 3 and 4). Lower and higher temperatures gave lower yields (entries 5 and 6). Other solvents (toluene, chloroform, pyridine or DMF) gave remarkably low yields (entries 7–10). Moreover, when SiCl4, an amide coupling reagent used for the synthesis of a macrocyclic amide in previous studies,7b,14 was used (entry 11), polymeric compounds were obtained instead of the target product, although the monomers were consumed. Another synthetic approach used LiHMDS as a coupling reagent for corresponding esters 6 and 10 as monomers (Scheme 3(b); entries 12–16).15 The reactions were carried out under a combination of reaction conditions with different amounts of reagents and reaction concentrations, and although the target compounds were obtained, yields were very low (1.40–2.80%), and separation of impurities was difficult.
:1 ratio was performed in 1,1,2,2-tetrachloroethane (0.05 M) using dichlorotriphenylphosphorane (7.2 eq.) as the coupling reagent at 120 °C (Scheme 3; entry 1 in Table 1).6 The obtained crude product was purified by silica gel column chromatography followed by gel permeation chromatography, resulting in the desired product in which two sets of monomers were condensed to give a 13.6% yield. The reaction using triphenylphosphine and hexachloroethane that generate dichlorotriphenylphosphorane in the reaction system13 gave the target compounds with similar yields (13.0%, entry 2 in Table 1). Then, the reaction conditions (reagent amount, solvent, temperature) were optimized in this system (entries 3–10). The best amount of coupling reagent was found to be 7.2 eq., which is 2.4 eq. per one amide bond formation (entries 3 and 4). Lower and higher temperatures gave lower yields (entries 5 and 6). Other solvents (toluene, chloroform, pyridine or DMF) gave remarkably low yields (entries 7–10). Moreover, when SiCl4, an amide coupling reagent used for the synthesis of a macrocyclic amide in previous studies,7b,14 was used (entry 11), polymeric compounds were obtained instead of the target product, although the monomers were consumed. Another synthetic approach used LiHMDS as a coupling reagent for corresponding esters 6 and 10 as monomers (Scheme 3(b); entries 12–16).15 The reactions were carried out under a combination of reaction conditions with different amounts of reagents and reaction concentrations, and although the target compounds were obtained, yields were very low (1.40–2.80%), and separation of impurities was difficult.
 |
| | Scheme 3 One-pot condensation reaction using (a) Ph3PCl2 or Ph3P/(CCl3)2; (b) LiHMDS. | |
Table 1 Optimization of the reaction conditions for synthesizing 2c
| Entry |
Reagent |
Eq. |
Solvent |
Conc. (M) |
Temp. (°C) |
Yield (%) |
| Reaction conditions: entries 1–10: 7c (0.2 mmol), 11c (0.2 mmol), for 2 h under argon bubbling; entry 11: 7c (0.5 mmol), 11c (0.5 mmol), for 24 h under argon atmosphere; entries 12–16: 6c (0.2 mmol), 10c (0.2 mmol), for 2 h under argon atmosphere. Containing an impurity. |
| 1 |
Ph3PCl2 |
7.2 |
(CHCl2)2 |
0.05 |
120 |
13.6 |
| 2 |
Ph3P/(CCl3)2 |
7.2 |
(CHCl2)2 |
0.05 |
120 |
13.0 |
| 3 |
Ph3P/(CCl3)2 |
4.8 |
(CHCl2)2 |
0.05 |
120 |
7.9 |
| 4 |
Ph3P/(CCl3)2 |
3.0 |
(CHCl2)2 |
0.05 |
120 |
7.3 |
| 5 |
Ph3P/(CCl3)2 |
7.2 |
(CHCl2)2 |
0.05 |
80 |
10.9 |
| 6 |
Ph3P/(CCl3)2 |
7.2 |
(CHCl2)2 |
0.05 |
150 |
7.3 |
| 7 |
Ph3P/(CCl3)2 |
7.2 |
Toluene |
0.05 |
Reflux |
3.3 |
| 8 |
Ph3P/(CCl3)2 |
7.2 |
CHCl3 |
0.05 |
Reflux |
7.5 |
| 9 |
Ph3P/(CCl3)2 |
7.2 |
Pyridine |
0.05 |
Reflux |
4.8 |
| 10 |
Ph3P/(CCl3)2 |
7.2 |
DMF |
0.05 |
120 |
— |
| 11 |
SiCl4 |
9.0 |
Pyridine |
0.025 |
90 |
— |
| 12 |
LiHMDS |
3.0 |
THF |
0.1 |
rt |
2.2a |
| 13 |
LiHMDS |
6.0 |
THF |
0.1 |
rt |
2.8a |
| 14 |
LiHMDS |
9.0 |
THF |
0.1 |
rt |
2.5a |
| 15 |
LiHMDS |
6.0 |
THF |
0.025 |
rt |
1.4a |
| 16 |
LiHMDS |
6.0 |
THF |
0.1 |
−20 |
— |
Characterization of spherical amides 2
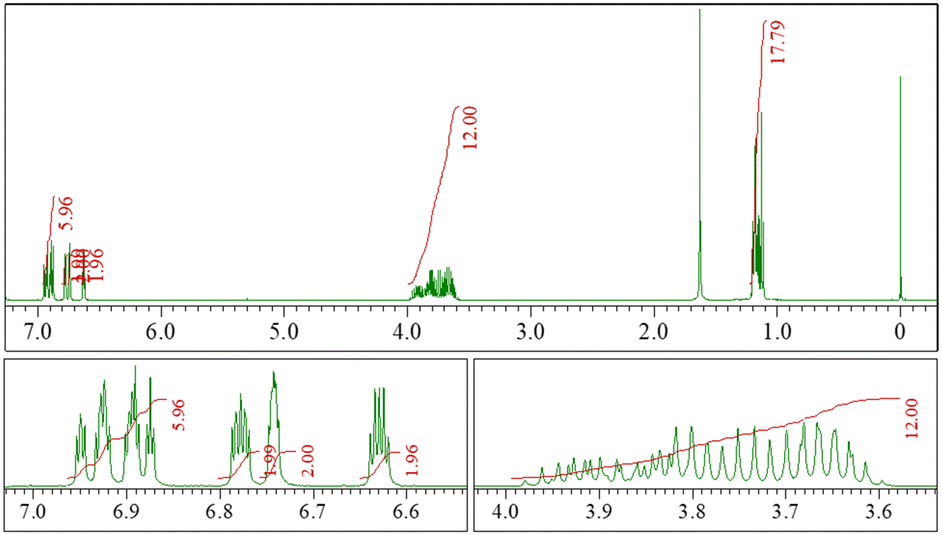
Compound 2 was characterized by 1H NMR (see ESI† for details) and X-ray crystallographic analysis. 1H NMR spectra of 2b (Fig. 2) showed that all the aromatic protons on the four benzene rings of the core structure appeared to be non-equivalent in the high magnetic region (6.5–7.0 ppm) because of the symmetry of its structure, i.e., the point group of the structure is C1. Furthermore, the prochiral methylene protons on the amide nitrogens (3.5–4.1 ppm) were observed non-equivalently, and these complex sets of signals were also observed in the spectra of 2c and 2d. These results revealed that these molecules had cyclic and fixed chiral structures. In comparing the spectra of 2a–d, signals of aromatic protons were similar, which suggested that the rigidity of the core structure was kept independent from alkyl substituents.
 |
| | Fig. 2 (top) 1H NMR spectra of 2b (400 MHz, in CDCl3): (bottom left) signals representing aromatic protons; (bottom right) signals representing N-C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2. 2. | |
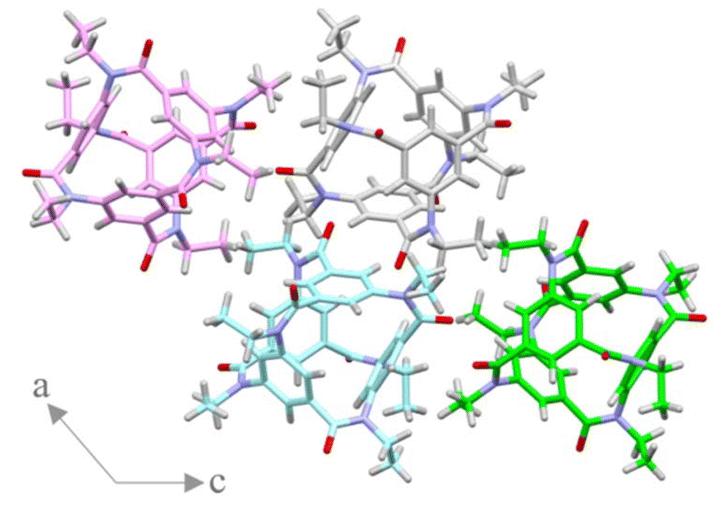
Next, X-ray crystallographic analyses were performed on single crystals of 2a (N-methyl, racemate) and 2b (N-ethyl, racemate). Crystals were obtained by slow evaporation from a chloroform/methanol solution of 2a and a chloroform/tetrahydrofuran solution of 2b. The structures of 2a and 2b in the crystal are shown in Fig. 3. The crystal of 2a belonged to the space group R3c, and that of 2b belonged to the space group P21/c. Each molecule of both compounds had a spherical shape surrounded by four benzene rings, and the radius of the inner space were about 1 Å estimated by voids calculation using Mercury (ver. 2024.1.0, CCDC), which is too small to include an organic molecule. In the crystals of 2b, two pairs of enantiomers existed in the unit cell (Fig. 4). The molecules were packed in the unit cells without the channel-forming arrangement that was previously observed in the crystals of derivatives of 1.9
 |
| | Fig. 3 Single molecular structures of 2a and 2b. C: gray; N: blue; O: red; H: white. | |
 |
| | Fig. 4 Packing structure in a racemic crystal of 2b (viewed along the b-axis). A pair of pink/blue and grey/green colored molecules have the same structure, and the other pairs (e.g., pink/grey) are enantiomers. | |
Chiral separation and circular dichroism (CD) spectra
Chiral separations of the racemic mixture 2a–d were carried out by chiral HPLC, and all separations were successful using a DAICEL CHIRALPAK IC column. In separating each compound, two eluted fractions were obtained in a 1:1 area ratio. A CD spectra of each compound showed a mirror image in acetonitrile (Fig. 5 and S5.2†). Furthermore, the spectra of 2a–d were similar, especially in the 230–310 nm range (Fig. S5.2†), which also proved the rigidity of the core structure.
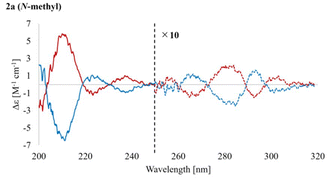 |
| | Fig. 5 CD spectra of 2a (N-methyl) in acetonitrile. Red line: 1st eluent. Blue line: 2nd eluent. The dashed line (250–320 nm) is displayed at 10× for each eluent. The sign of 2a [(+) or (−)] is based on the respective sign of specific rotation in a solution of MeOH:CHCl3 = 1:1. | |
DFT calculations
We performed computational calculations to estimate the Cotton effects in CD spectra for spherical amide 2a. Initially, for the molecular structure of one enantiomer (−)-2a, a conformational calculation based on MD calculations was performed on a Macromodel (worked on Maestro 13.9, Schrödinger, Inc.) using coordinate data of racemic molecules obtained by X-ray crystallographic analysis as initial structure (for details, see ESI†). Next, for the most stable conformer, density-functional theory (DFT) calculations were performed to optimize the structure at the B3LYP/6-31G+(d) level with the addition of the acetonitrile solvent effect utilizing the polarizable continuum model (Fig. 6), followed by time-dependance DFT (TD-DFT) calculations were performed to simulate CD spectra. One hundred and twenty states were required to simulate the calculated spectra in the 200–320 nm region. Fig. 7 shows the simulated and experimental UV/CD spectra. An intense negative Cotton effect was observed in the calculated spectra at ∼210 nm, corresponding to one in the experimental spectra (blue line: 2nd eluent of the chiral HPLC analysis). At wavelengths greater than 230 nm, peaks arising from the Cotton effect did not completely match when comparing the simulated and experimental spectra. These results were used to estimate the absolute structure of (−)-2a.
 |
| | Fig. 6 Optimized structure of (−)-2a by the DFT calculation. | |
 |
| | Fig. 7 Simulated (dashed line) and experimental (solid line) UV/CD spectra. | |
Possible reaction pathways in cyclization
In the condensation reaction (Scheme 3), the target compounds were obtained as the main product without byproducts, except for polymeric compounds; nevertheless, multiple reaction points exist (amino and carboxy groups). We propose the reaction pathway of the present reaction as follows (Fig. 8) and based on the substituent effect and the selectivity of the condensation in the polymer chain reaction reported by Yokozawa et al.16 At the beginning of the reaction, two monomers A and B exist in equal amounts. The nucleophilicity of amino group a of A with two electron-withdrawing carboxy groups is weaker than that of the amino group a′ of B with one carboxy group because of the lower electron density of the benzene ring. In contrast, the electrophilicity of carboxy group b of A is stronger than that of carboxy group b′ of B because B has two electron-donating amino groups. Therefore, the fastest reaction is a nucleophilic attack of a′ of B to b of A, which gave dimer C. Although, as the reaction proceeds, the amount of dimer C increases, the nucleophilicity of amino group a′ in dimer C is weaker than that of B because the electron-donating ability of the other amino group is weakened by being connected with the carboxyl group in its amide bond. Then, almost the whole amount of A and B is converted to dimer C. In dimer C, amino group a′, which is stronger than a in nucleophilicity, attacks carboxyl group b, which is stronger than b′ in electrophilicity, to give tetramer D. Next, a cyclization reaction that gives a cyclized product containing a cyclic trimer moiety proceeds faster than the formation of a cyclic tetramer because the reaction point to form a cyclic trimer is preferable than that for the cyclic tetramer.6b Finally, the cyclic trimer moiety is capped by a pendant monomeric part to give the spherical amide 2 and its enantiomer.
 |
| | Fig. 8 Proposed synthesis mechanism of the spherical amide 2. | |
Conclusions
In this report, we synthesized enantiomeric pairs of stereoisomers of spherical macrocyclic aromatic amides by a short-step synthesis, including reductive amination and the one-step amide coupling reaction of two sets of two components. Covalent bonds fixed the chirality of the spherical amides, and the chiral separations of these compounds were successful. The enantiomeric pairs of these stereoisomers had mirror-image CD spectra. The core structure of the spherical amides was bulky and rigid, and the directions of the six N–C bonds (from nitrogen of amide to carbon of substituent) were fixed. This short-step synthetic method opens the way to readily designing spherical amides as various three-dimensional scaffolds. Additionally, this scaffold may afford possible chiral building blocks for pharmaceuticals, such as drugs, or optical materials that display circularly polarized luminescence by introducing fluorescent structures.
Experimental
See ESI† for details.
Author contributions
D. Koike: writing – original draft, data curation, formal analysis, investigation; H. Masu: writing – review & editing, investigation; S. Kikkawa: writing – review & editing, investigation; A. Chiba: investigation; K. Kamohara: investigation; A. Okuda: investigation; H. Hikawa: writing – review & editing; I. Azumaya: supervision, conceptualization, writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2a and 2b have been deposited at the CCDC under 2377216 and 2382135,† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank Dr Katsuyoshi Mitsunaga (Faculty of Pharmaceutical Sciences, Toho University) for assistance with mass spectrometry. We also thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
References
-
(a) J. F. Nierengarten, N. Armaroli, G. Accorsi, Y. Rio and J. F. Eckert, Chem. – Eur. J., 2003, 9, 36–41 CrossRef CAS PubMed;
(b) F. Diederich and C. Thilgen, Science, 1996, 271, 317–323 CrossRef CAS;
(c) M. Abellán-Flos, M. Tanç, C. T. Supuran and S. P. Vincent, Org. Biomol. Chem., 2015, 13, 7445–7451 RSC;
(d) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed;
(e) E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC;
(f) C. Fu, S. Gong, L. Lin, Y. Bao, L. Li and Q. Chen, Biomed. Pharmacother., 2024, 176, 116828 CrossRef CAS PubMed.
-
(a) M. C. Parrott, E. B. Marchington, J. F. Valliant and A. Adronov, J. Am. Chem. Soc., 2005, 127, 12081–12089 CrossRef CAS PubMed;
(b) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS;
(c) P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC;
(d) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. De Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef;
(e) X. Li, Q. Zhou, M. Zhu, W. Chen, B. Wang, Y. Sha and H. Yan, New J. Chem., 2021, 45, 7496–7500 RSC.
-
(a) H. Akiyama, K. Miyashita, Y. Hari, S. Obika and T. Imanishi, Tetrahedron, 2013, 69, 6810–6820 CrossRef CAS;
(b) D. Ranganathan and S. Kurur, Tetrahedron Lett., 1997, 38, 1265–1268 CrossRef CAS;
(c) K. Nasr, N. Pannier, J. V. Frangioni and W. Maison, J. Org. Chem., 2008, 73, 1056–1060 CrossRef CAS PubMed;
(d) A. A. Spasov, T. V. Khamidova, L. I. Bugaeva and I. S. Morozov, Pharm. Chem. J., 2000, 34, 1–7 CrossRef CAS;
(e) L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed;
(f) A. Ragshaniya, V. Kumer, R. K. Tittal and K. Lal, Arch. Pharm., 2024, 357, e2300595 CrossRef PubMed.
-
(a) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed;
(b) M. Okayasu, T. Sunakawa, M. Ikeda, T. Namikawa, R. Hagura, S. Kikkawa, H. Hikawa and I. Azumaya, ChemistrySelect, 2021, 6, 13336–13341 CrossRef CAS;
(c) S. J. Dalgarno, G. W. V. Cave and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 45, 570–574 CrossRef CAS PubMed;
(d) M. J. Bojdys, M. E. Briggs, J. T. A. Jones, D. J. Adams, S. Y. Chong, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 16566–16571 CrossRef CAS PubMed.
-
(a) W. Wang, X. Dong and G. Li, Signal Transduction Targeted Ther., 2024, 9, 81 CrossRef CAS;
(b) O. N. Pattelli, E. M. Valdivia, M. S. Beyersdorf, C. S. Regen, M. Rivas, K. A. Hebert, S. D. Merajver, T. Cierpicki and A. K. Mapp, Angew. Chem., Int. Ed., 2024, 63, e202400781 CrossRef CAS PubMed;
(c) Y. Sawatari, Y. Shimomura, M. Takeuchi, R. Iwai, T. Tanaka, E. Tsurumaki, M. Tokita, J. Watanabe and G. Konishi, Aggregate, 2024, 5, e507 CrossRef CAS;
(d) S. Kimura, F. Takeda, A. Ikeda, A. Tanimoto, K. Katagiri, M. Kawahata, Y. Okada, N. Kobayashi, H. Kagechika and A. Tanatani, Bull. Chem. Soc. Jpn., 2024, 97, uoae094 CrossRef.
-
(a) I. Azumaya, T. Okamoto, F. Imabeppu and H. Takayanagi, Tetrahedron, 2003, 59, 2325–2331 CrossRef CAS;
(b) F. Imabeppu, K. Katagiri, H. Masu, T. Kato, M. Tominaga, B. Therrien, H. Takayanagi, E. Kaji, K. Yamaguchi, H. Kagechika and I. Azumaya, Tetrahedron Lett., 2006, 47, 413–416 CrossRef CAS;
(c) Y. Saito, M. Satake, R. Mori, M. Okayasu, H. Masu, M. Tominaga, K. Katagiri, K. Yamaguchi, S. Kikkawa, H. Hikawa and I. Azumaya, Org. Biomol. Chem., 2020, 18, 230–236 RSC;
(d) A. Yokoyama, A. Ishii, T. Ohishi, S. Kikkawa and I. Azumaya, Tetrahedron Lett., 2021, 62, 152704 CrossRef CAS.
-
(a) A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177–6180 CrossRef CAS;
(b) I. Azumaya, H. Kagechika, K. Yamaguchi and K. Shudo, Tetrahedron Lett., 1996, 37, 5003–5006 CrossRef CAS.
-
(a) C. Zhai, C. Xu, Y. Cui, L. Wojtas and W. Liu, Chem. – Eur. J., 2023, 29, e202300524 CrossRef CAS PubMed;
(b) K. G. Andrews, P. N. Horton and S. J. Coles, Chem. Sci., 2024, 15, 6536–6543 RSC;
(c) J. C. Lauer, A. S. Bhat, C. Barwig, N. Fritz, T. Kirschbaum, F. Rominger and M. Mastalerz, Chem. – Eur. J., 2022, 28, e202201527 CrossRef CAS PubMed;
(d) R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed.
- H. Masu, K. Katagiri, T. Kato, H. Kagechika, M. Tominaga and I. Azumaya, J. Org. Chem., 2008, 73, 5143–5146 CrossRef CAS PubMed.
-
(a) M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2003, 68, 7374–7378 CrossRef CAS PubMed;
(b) M. Selva, P. Tundo and T. Foccardi, J. Org. Chem., 2005, 70, 2476–2485 CrossRef CAS PubMed.
- S. Sato, T. Sakamoto, E. Miyazawa and Y. Kikugawa, Tetrahedron, 2004, 60, 7899–7906 CrossRef CAS.
- R. R. Perrotta, A. H. Winter and D. E. Falvey, Org. Lett., 2011, 13, 212–215 CrossRef CAS PubMed.
-
(a) G.-c. Wu, H. Tanaka, K. Sanui and N. Ogata, Polym. J., 1982, 14, 797–801 CrossRef CAS;
(b) K. Takagi, D. Miyamoto, H. Yamaguchi and I. Azumaya, Bull. Chem. Soc. Jpn., 2022, 95, 47–51 CrossRef CAS.
- R. Yamakado, S. Matsuoka, M. Suzuki, D. Takeuchi, H. Masu, I. Azumaya and K. Takagi, Chem. Commun., 2015, 51, 5710–5713 RSC.
-
(a) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165–4172 CrossRef PubMed;
(b) A. Yokoyama, M. Karasawa, M. Taniguchi and T. Yokozawa, Chem. Lett., 2013, 42, 641–642 CrossRef CAS.
-
(a) Y. Ohta, Y. Kamijyo, S. Fujii, A. Yokoyama and T. Yokozawa, Macromolecules, 2011, 44, 5112–5122 CrossRef CAS;
(b) T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Ayako
Chiba
a,
Kaho
Kamohara
a,
Arisa
Okuda
a,
Hidemasa
Hikawa
a,
Ayako
Chiba
a,
Kaho
Kamohara
a,
Arisa
Okuda
a,
Hidemasa
Hikawa