
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBottlebrush polymer patches template heterometal growth on a gold nanoparticle surface†
Minjun
Kim‡
,
Jiyun
Nam‡
,
Jiseok
Kim
,
Hyunsik
Hwang
,
Myungeun
Seo
 * and
Hyunjoon
Song
*
* and
Hyunjoon
Song
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: seomyungeun@kaist.ac.kr; hsong@kaist.ac.kr
First published on 24th April 2025
Abstract
We report a new bottlebrush copolymer (BBCP) ligand design as robust patches for gold nanoparticles (Au NPs) to construct a rigid template guiding heterometal deposition on the surface. Given the spatial congestion of the side chains, the BBCP rapidly forms dense and stationary patches on Au NPs and effectively blocks additional metal deposition. Reducing solvent quality varies the phase segregation of the BBCP and subsequently restricts metal deposition to specific locations, fabricating diverse bimetallic heterostructures. The resulting morphology exhibits a unique orientation-dependent scattering property that thermodynamic configuration cannot achieve.
Introduction
Anisotropically functionalizing nanoparticles to form “patches” is an appealing route for breaking the symmetry on the surface and guiding the growth of asymmetric hetero-nanostructures and programmed self-assembly.1–3 Polymeric ligands have been proven particularly useful for patch formation. As the entropic gain for a polymer upon mixing decreases exponentially with its chain length (i.e., the degree of polymerization), strong incompatibility of the polymeric ligands with other molecular or polymeric co-ligands enables selective surface decoration driven by segregation.4–7 The incompatibility with the medium can also be exploited to collapse polymeric ligands and expose uncovered surfaces between the patches with controlling patch numbers and spatial distribution.3,8,9 Intriguing self-assembly behaviors suggest the potential of patchy metal nanoparticles toward constructing customized self-assemblies and superlattices.5–7 Regioselective deposition onto the exposed surface has produced blocky nanorods and Janus heterodimers.9–11While diverse surface morphologies composed of multiple polymer patches have been reported, translating the template into complex hetero-nanostructures through a second deposition is challenging. In particular, linear polymers, such as thiol-terminated polystyrene, have been widely used for nanoparticle surface modification; however, they often require prolonged annealing processes to promote the thermodynamic segregation and collapse of the polymeric ligands into distinct patches with the designated number.8,10,11 Moreover, linear polymers are usually sensitive to poor reaction media, making it hard to control the surface morphology during the deposition.3 Even if the multi-patch structure persists during the deposition reaction, the thin layer cannot entirely prevent the second element from diffusing into the underlying surface, leading to uneven and irregular deposition.
In this work, we propose utilizing bottlebrush polymers for rapid, stable, and robust polymer patch fabrication to overcome the limitations of linear polymeric ligands. Bottlebrush polymers contain densely grafted polymer chains for every repeating unit of the backbone and exhibit distinct physical properties compared to their linear analogs. The backbone adopts a stretched conformation because of steric congestion between the side chains.12,13 While ultrahigh molecular weights can be readily attained, fewer entanglements between stiffer bottlebrush polymers yield relatively fast chain stabilization.14,15 We exploit the bottlebrush architecture to rapidly collapse the polymer ligands in a selective solvent and form robust patches on a Au nanoparticle (Au NP) surface (Scheme 1a).
 | ||
| Scheme 1 Synthesis of Au–Ag hetero-nanostructures using patches formed through the segregation of the BBCP by solvent quality control. | ||
Introducing a nonsolvent to reduce the solvent quality pins the bottlebrush block and quickly forms discrete patches. The number of patches strongly depends on the composition of the medium, which allows us to access various morphologies. Notably, the morphology persists during the second deposition, utilizing the uncovered surface as a seed to template an inverse structure on the Au NP surface. The deposition of surface plasmon-active Ag, as the second element, induces unique surface plasmon resonance scattering depending on the orientation of Ag islands (Scheme 1b). The strategy can be extended to fabricating Au–Pt heterostructures with morphological control, demonstrating the versatility of the bottlebrush approach.
Results and discussion
To ensure strong binding between Au NPs and the whole bottlebrush polymer, we designed the polymer with a lengthy linear polymer block carrying numerous sulfur-rich trithiocarbonate (TTC) moieties.16 As schematically shown in Scheme 1a, we aim to prepare a bottlebrush copolymer (BBCP)–Au NP hybrid (BBCP–Au) that possesses a Au NP in the core tightly wrapped with the TTC-abundant linear blocks and a rigid bottlebrush corona radiating from the core.First, we copolymerized a TTC-tethered norbornene monomer (Fig. S1†) with bare norbornene as a spacer to adjust the TTC density to 50 mol% in the chain. Subsequent chain extension with a norbornene-based polystyrene (PS) macromonomer (Fig. S2†) produced the target BBCP (Scheme S1 and Fig. S3†). Size exclusion chromatography (SEC) analysis shows a complete shift of the trace to higher molar mass after the chain extension, supporting the successful synthesis of the BBCP (Fig. S4†). Based on the conversion, we estimate that the BBCP contains approximately 320 norbornene repeating units per backbone chain with nearly symmetric lengths of the TTC-containing linear and PS-containing bottlebrush blocks. The linear block containing 80 TTC groups corresponds to the number-average molar mass (Mn) of 59 kg mol−1. As the PS macromonomer with an Mn value of 2.7 kg mol−1 includes approximately 25 styrene repeating units, the bottlebrush block comprising 160 PS side chains equals an Mn value of 432 kg mol−1 with 4000 styrene units, reflecting its massive nature (Table S1†). The 1H NMR spectrum of the BBCP supports the predicted composition and indicates that the TTC groups are retained during polymerization (Fig. S5†).
In a typical synthesis of BBCP–Au, we used citrate-capped Au NPs with an average diameter of 48 ± 5 nm (Fig. S6a†). We performed ligand exchange in dimethylformamide (DMF), where Au NPs and BBCP were dispersed well. While the Au NP morphology and dispersity were unchanged, transmission electron microscopy (TEM) images revealed the presence of PS shells after the BBCP conjugation, which appeared light grey on the Au NP surface by negative staining (Fig. S6b†). Elemental mapping with scanning transmission electron microscopy-energy dispersive spectroscopy (STEM-EDS) shows a uniform distribution of sulfur over the Au surface, corroborating the binding of the TTC-rich linear block with Au (Fig. S6c†) and confirming the previously reported strong binding of TTC-containing ligands to gold nanoparticles.16 The BBCP–Au conjugate is sufficiently stable even to run preparative SEC using chloroform as an eluent (Fig. S7†). A clear shift to higher molar mass compared to that of the BBCP indicates that BBCP–Au behaves as an ultrahigh-molar mass macromolecule with increased hydrodynamic size as the PS bottlebrush corona fully stabilizes the Au NP core, and there is no free BBCP residue in the conjugate. This result exemplifies the utility of SEC for the effective purification of conjugated nanoparticles.17,18 Dynamic light scattering (DLS) measurement also supports the increase of the hydrodynamic diameter of the Au NPs from 49 to 112 nm after the BBCP conjugation (Fig. S6†). A change in zeta potential from −19.81 to −3.93 mV after the conjugation is consistent with the ligand exchange of citrate anions to the BBCP (Table S2†). The 3.5% weight loss above 400 °C in thermogravimetric analysis (TGA) corresponds to the decomposition of the BBCP (Fig. S8†). Roughly, we estimate that 50 BBCPs are bound to each Au NP on average. Considering the surface area of the Au NP core, the grafting density of BBCPs on Au NPs is calculated to be 0.007 polymers per nm2. BBCP–Au was stable over three months in aqueous media (Fig. S9†).
We induced the collapse of the PS bottlebrush by decreasing the solvent quality, i.e., adding water to the BBCP–Au dispersion in DMF. We adjusted the water volume fraction from 0 to 50% to control the patch morphology. The patches were produced entirely by 20 min of stirring (Fig. S9†). Fig. 1a shows negatively stained TEM images of BBCP–Au at different water percentages in DMF. The morphology gradually changes from an even shell (0 vol%) to an uneven shell (10 vol%), multiple patches (30 vol%), and an asymmetric single patch (50 vol%) with increasing the water percentage, consistent with reducing the surface area exposed to the more incompatible medium. Injecting an Ag precursor into the dispersion in the presence of hydrazine at room temperature and stirring vigorously for 5 min produced Au–Ag heterostructures via the seed-mediated growth mechanism. The spatial location of Ag domains on the Au core was complementary to that of the polymer patches; smooth core–shell, multi-domain core–shell, multiple-island, and Janus-type heterodimers were obtained by varying the water percentage (Fig. 1b). While most BBCP domains (bright) remain on the Au surface, the TEM image suggests that a small fraction migrates and stabilizes the Ag domains (gray) during the reaction. STEM-EDS mapping corroborates the Ag growth on the Au core as separate domains without alloying (Fig. 1c). The low-resolution TEM images clearly distinguish between the Au–Ag heterostructures with different solvent qualities (Fig. 1d).
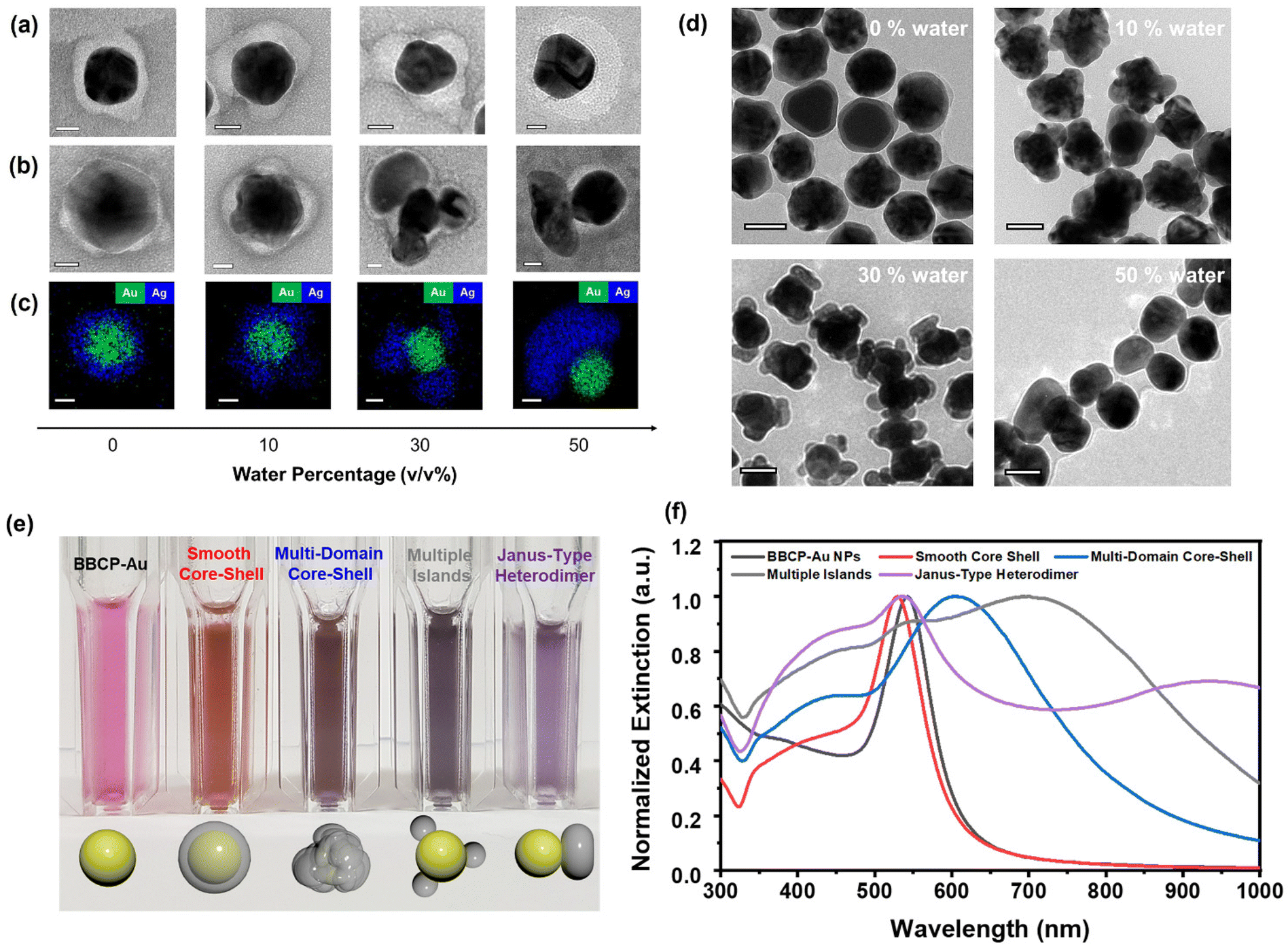 | ||
| Fig. 1 Polymer patch formation and selective Ag deposition on Au NPs. TEM images and negative staining of (a) BBCP–Au and (b) Au–Ag heterostructures with decreasing the solvent quality of the dispersion medium. (c) STEM-EDS mapping of the corresponding Au–Ag heterostructures. The scale bars represent 20 nm. (d) Wide-view TEM images of Au–Ag heterostructures. The scale bars represent 50 nm. (e) Colors and (f) UV-Vis-NIR spectra of Au–Ag heterostructures dispersed in chloroform. | ||
The inverse replication of the PS bottlebrush corona structure with Ag is key to Au–Ag heterostructure formation with the desired morphology. In pure DMF with a 0% water content, Ag species seem to diffuse through the solvent-swollen PS corona to reach the Au surface and form the smooth Ag shell. Upon water addition, the PS bottlebrush starts to collapse locally, increasing the resistance to Ag diffusion. The undulating PS corona templates the uneven Ag shell with multiple domains. Increasing the water fraction to 30 vol% drives PS segregation and forms several discrete PS domains on the Au NP surface while exposing the rest. As the dense PS domain effectively blocks the Ag diffusion, the seed-mediated growth occurs from the exposed area to generate multiple Ag islands (Fig. S10 and S11†). At 50 vol% water, the PS bottlebrushes coagulate into a single crescent-like domain and promote Ag domain growth from the opposite side—the Janus heterodimer forms.
The bottlebrush architecture constructed with hydrophobic PS side chains is necessary to rapidly produce thick, robust, surface-protecting polymer patches. When the brush backbone containing PS chains is extended twice to 320 norbornene repeating units, the Au–Ag morphology change is similar to that using the original BBCP, from core–shell nanoparticles to heterodimers; however, the patch formation requires less water content for assembly (Fig. S12†). As a control experiment, thiol-terminated linear PS (Mn = 104.5 kg mol−1) as the Au NP ligand only produced Au–Ag core–shell structures regardless of the solvent quality, indicating that the collapsed linear chains are permeable to Ag diffusion under our reaction conditions (Fig. S13†). The linear PS is known to produce a diverse patch morphology but requires precise solvent control with a long annealing process over 24 h.3 Linear PS carrying the TTC group at the chain end (Mn = 25 kg mol−1) resulted in the irregular growth of the Ag structure (Fig. S14†). We suggest that the conjugation mediated by the single TTC group may not be stable under the Ag deposition conditions. Hydrophilic poly(vinyl pyrrolidone) also yielded concentric Au–Ag core–shell structures, indicating the importance of grafted polymer chains (Fig. S15†).
Au and Ag exhibit strong surface plasmon resonance scattering in the visible wavelength range.19 The Ag deposition on BBCP–Au significantly changes dispersion color in chloroform depending on the Ag domain morphology, from red to orange (smooth core–shell), brown (multi-domain core–shell), gray (multiple-island), and purple (Janus) (Fig. 1e), although the amount of deposited Ag is similar (see the Experimental). In the UV-Vis-NIR spectra, the characteristic localized surface plasmon resonance (LSPR) peak of BBCP–Au at 543 nm shows a blue shift by 13 nm upon forming a smooth Ag shell (Fig. 1f).20,21 Multiple broad, red-shifted peaks appear from other morphologies, suggesting changes in the LSPR modes because of their structural complexity and roughness.22,23
By varying the Ag precursor amount, we examined the morphological evolution during Ag deposition for the multiple-island Au–Ag heterostructure. The entire growth was rapidly completed under the reaction conditions within 1 min; therefore, we decreased the Ag precursor concentration to (i) 5% to (ii) 10%, (iii) 25%, and (iv) 50% of the original growth condition, and Ag was fully reduced by hydrazine. After the rapid reaction, a bathochromic shift in the UV-Vis-NIR spectra occurs, signaling the Ag nucleation and growth on the Au surface (Fig. 2a). The TEM images indicate that each Ag island directly nucleates at specific positions of the Au surface and gradually grows, accompanied by further red shifts in the UV-Vis-NIR spectra upon increasing the Ag concentration. The increasing extinction at ∼400 nm indicates the development of Ag LSPR dipoles, and the redshift and splitting of the long-wavelength peaks with pronounced broadening are due to the development of asymmetric structures from the spherical gold cores.
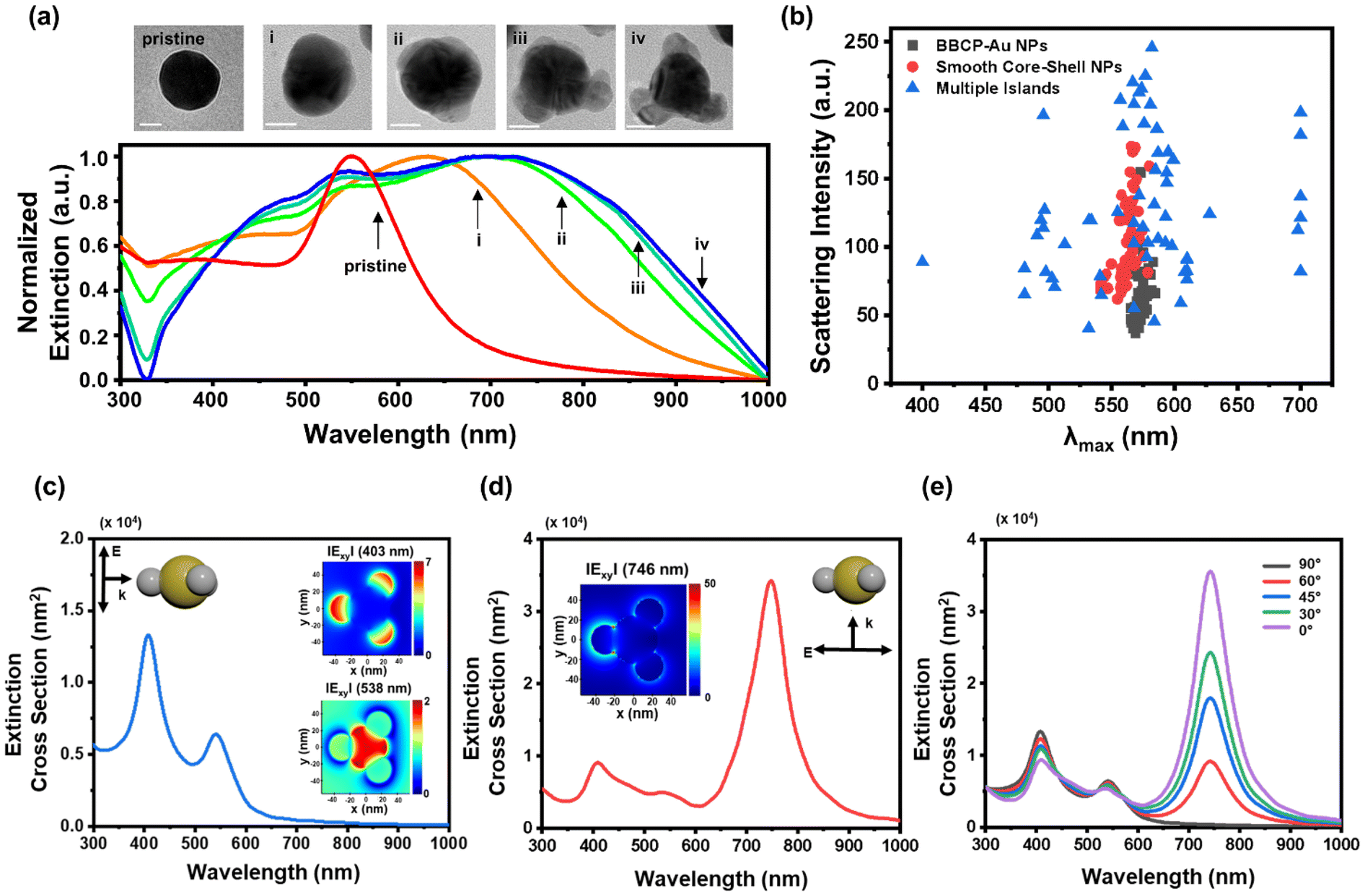 | ||
| Fig. 2 (a) TEM images and extinction spectra of Ag multiple islands (i–iv) grown on Au nanospheres using different Ag precursor concentrations. The scale bars represent 20 nm. (b) A scatter plot showing peak wavelength distribution from single-particle scattering measurements of BBCP–Au nanospheres (gray), smooth core–shell (red), and multiple islands (blue). Extinction cross-section spectra resulting from polarized light incident (c) parallel to and (d) normal to the symmetry axis of Ag multiple islands on Au spheres simulated by FDTD. (insets) The embedded electric field mappings are simulation plots at (c, top) 403 nm, (c, bottom) 538 nm, and (d) 746 nm. (e) Spectral changes of the Ag multiple islands on Au spheres to the incident light angle. | ||
We performed single-particle scattering imaging using transmission dark-field microscopy to investigate the unique optical properties of Au–Ag heterostructures (Fig. S16†).24 Nanoparticles were dispersed onto ITO-coated slide glass, and each particle was positioned on the substrate with three-dimensional random orientations, assisted by the polymer capping agent (Fig. S17†). Scattering peak wavelength and intensity of individual particles were extracted from scattered images through the RGB-to-wavelength (RTW) process using custom MATLAB code (see page S14†).25,26
The RTW process was applied to 60 particles at each structure, including BBCP–Au, smooth core–shell, and multiple-island, to generate a plot based on their scattering light information (Fig. 2b). Concentrically symmetric structures, such as BBCP–Au and smooth core–shell structures, exhibited uniform scattering signals at 560–570 nm. In contrast, the multiple-island Au–Ag structure showed a notably wide distribution from 400 nm to 700 nm. The highly sensitive nature of the scattering will help track the orientation of individual particles.27 The scattering peak wavelengths can be roughly categorized into 500, 580, and 700 nm.
To analyze the optical changes observed in ensemble and single-particle analyses, we used the finite-difference time-domain (FDTD) method to simulate the extinction cross-sections of model structures. The simulation of a multiple-island Au–Ag heterostructure involved an idealized particle model with three Ag islands attached to the Au core (Fig. S18†). We found that the extinction cross-section significantly depends on the direction of incident polarized light (Fig. S19†). When light is irradiated parallel to the symmetry axis, two intense scattering peaks appear at 403 and 538 nm, attributed to out-of-plane dipolar excitation of the Ag islands and the Au core, respectively (Fig. 2c and insets). On the other hand, perpendicular irradiation generates the most intense signals at 746 nm, corresponding to the in-plane excitation of the longitudinal Au–Ag (Fig. 2d and inset).28 As a result, the total extinction pattern is primarily changed by the incident beam angle to the symmetry axis of the nanoparticle (Fig. 2e). Considering the random orientation of the Au–Ag heterostructure in the dispersion, the sum of the simulated extinction cross-section spectra matches the three distinguishable absorption maxima, which confirms the patchy morphology as the origin of the spectral change (Fig. 2c–e and S18, S19†).
In contrast, two isotropic model structures, Au sphere and concentric Au–Ag core–shell structure, exhibit strong Au LSPR peaks at 542 nm and 529 nm in simulation (Fig. S20†), consistent with the nearly invariable scattering peaks in single-particle measurements shown in Fig. 2b.
Experimental details
Preparation of citrate-capped Au NPs
Citrate-stabilized Au NPs with an average diameter of 45 nm were synthesized according to the literature.29 For Au seeds (10 nm), a mixture of 150 mL of freshly prepared sodium citrate aqueous solution (SC, 2.2 mM) was heated in a 100 mL two-neck round-bottom flask under vigorous stirring. When the temperature reached boiling, 1.0 mL of gold chloride hydrate (HAuCl4, 25 mM) was injected. The solution color changed rapidly to black-gray and then to wine-red in 2–3 min. The solution was kept for 5 min to reduce the gold precursor completely. Immediately after the Au seed formation, the reaction was cooled until the solution temperature reached 90 °C. Then, the sample was diluted by extracting 55 mL and adding 55 mL of the SC solution (2.2 mM). When the temperature reached 90 °C, 1.0 mL of HAuCl4 (25 mM) was twice injected at a time interval of 30 min. This growing step was repeated until the particles reached 45 nm.Synthesis of the linear block-bottlebrush copolymer (BBCP)
The BBCP was synthesized by sequential polymerization of trithiocarbonate-norbornene (TTC-N), norbornene (N), and polystyrene macromonomers (PS-MM). TTC-N and PS-MM were prepared according to the literature (Fig. S1 and S2; see Table S1† for characterization details).30,31N and TTC-N were polymerized by using G3 with a feed ratio of [N]0 : [TTC-N]0 : [Ru]0 of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80:1 in toluene ([M]0 = 0.2 M), respectively. When the consumption of N and TTC-N was completed, PS-MM was sequentially polymerized with the molar ratio of [PS-MM]0 : [Ru]0 of 160:1 in toluene ([MM]0 = 0.3 M) (see Table S1† for the characterization details). Next, we describe the detailed conditions of ring-opening metathesis polymerization. N (0.0053 g, 0.056 mmol) and TTC-N (0.036 g, 0.056 mmol) were added and dissolved in toluene (0.56 mL) in a 5 mL reaction vial equipped with a stir bar. A stock solution of G3 (150 μL, 0.00069 mmol) was injected into the solution to initiate the polymerization. After 6 min, the consumption of the N and TTC-N was completed. PS-MM (0.30 g, 0.11 mmol) was dissolved in toluene (0.37 mL) and added to the mixture. The reaction mixture was further stirred for 2 h at room temperature. Then, several drops of ethyl vinyl ether were added to quench the polymerization. Small aliquots were taken out for SEC analysis. A trace amount of residual MM was removed by preparative SEC. The remaining polymerization mixture was precipitated with methanol, and the collected BBCP was dried in a vacuum oven overnight.
:80:1 in toluene ([M]0 = 0.2 M), respectively. When the consumption of N and TTC-N was completed, PS-MM was sequentially polymerized with the molar ratio of [PS-MM]0 : [Ru]0 of 160:1 in toluene ([MM]0 = 0.3 M) (see Table S1† for the characterization details). Next, we describe the detailed conditions of ring-opening metathesis polymerization. N (0.0053 g, 0.056 mmol) and TTC-N (0.036 g, 0.056 mmol) were added and dissolved in toluene (0.56 mL) in a 5 mL reaction vial equipped with a stir bar. A stock solution of G3 (150 μL, 0.00069 mmol) was injected into the solution to initiate the polymerization. After 6 min, the consumption of the N and TTC-N was completed. PS-MM (0.30 g, 0.11 mmol) was dissolved in toluene (0.37 mL) and added to the mixture. The reaction mixture was further stirred for 2 h at room temperature. Then, several drops of ethyl vinyl ether were added to quench the polymerization. Small aliquots were taken out for SEC analysis. A trace amount of residual MM was removed by preparative SEC. The remaining polymerization mixture was precipitated with methanol, and the collected BBCP was dried in a vacuum oven overnight.
Preparation of BBCP–Au
The surface functionalization of Au NPs was carried out in DMF, where the solvent was compatible with both the BBCP and Au NPs. The dispersion of the as-synthesized 45 nm-diameter Au NPs was concentrated from 50 mL to approximately 100 μL using centrifugation (5000 rpm, 30 min, 25 °C) and subsequent supernatant removal. The resulting dispersion was diluted with 5.0 mL of DMF with sonication. The BBCP solution (0.10 mg mL−1) was typically prepared in DMF. Then, the NP dispersion was added to the BBCP solution drop by drop. The resulting solution was incubated at room temperature overnight. After that, the resulting BBCP–Au was washed by six cycles of centrifugation/redispersion to eliminate excess polymer residues. TTC-PS, HS-PS, and PVP-capped Au NPs were also prepared using an identical protocol by substituting the BBCP with these ligands.Seed-mediated growth of Ag on BBCP–Au
Au–Ag hetero-nanostructures were synthesized by seed-mediated growth of Ag on the exposed surfaces of BBCP–Au. In a typical experiment, water was added to the BBCP–Au dispersion in a desired ratio, and the mixture was incubated for 1 h at room temperature. 3.0 mL of segregated BBCP–Au was mixed with PVP (10 wt%, 0.30 mL) and hydrazine (0.25 M, 0.30 mL). Then, 30 μL of AgNO3 (6.8 mg mL−1) was added and vigorously stirred for 5 min at room temperature. After the reaction, the NPs were collected by centrifugation and washed with DMF and chloroform twice. For the intermediate analysis, the precursors with the original Ag amounts of 5%, 10%, 25%, and 50% were added under identical reaction conditions. TTC-PS, HS-PS, and PVP-capped Au NPs were used instead of BBCP–Au for control experiments.Negative staining method
For transmission electron microscopy (TEM) analysis, the polymer domains within BBCP–Au and Au–Ag heterostructures were visualized using a negative staining technique with uranyl acetate. A staining solution was prepared by dissolving 30 mg of uranyl acetate in 1.0 mL of deionized water. A 15 μL droplet of this solution was dispensed onto a hydrophobic parafilm surface, and the TEM grid bearing the sample was placed face-down onto the droplet for 2 min at room temperature. Following incubation, the excess staining solution was gently removed using filter paper, and the grid was air-dried prior to imaging.Monitoring single-particle scattering imaging
A conductive ITO-coated glass slide (10 ohms, 76 × 26 × 1 mm, Thermo Scientific) used for SEM imaging was sonicated in acetone, ethanol, and deionized water for 30 min, and then dried with nitrogen gas. 5.0 μL of diluted colloidal solution was added dropwise to achieve single-particle level dispersion of Au–Ag nanoparticles on the substrate.Dark-field imaging was performed using optical microscopy (Carl Zeiss, Scope A1). High-angle incident white light from a halogen lamp (12 V, 100 W) was directed through a dark-field condenser (N.A. 1.2–1.4). The scattered light from the nanoparticles was selectively detected using a 40× object lens (EC Plan-Neofluar, N.A. 0.5). The scattering images were captured with a 2584 × 1936-pixel color video camera (Axiocam MRc 5).
FDTD calculation
Far-field scattering spectra and extinction cross-section were calculated using Mie theory. The modeling of Au–Ag heterostructures was constructed using FDTD solution (Ansys Lumerical). (n,k) values of gold and silver for FDTD calculation were obtained from the reference of Palik for small-size noble metal particles and Jonson and Christy (Fig. S16†).32,33Conclusions
We demonstrated that the PS bottlebrushes can serve as robust and tunable surface-blocking patches of Au NPs, templating a second metal deposition. We developed the PS bottlebrush-containing macromolecular BBCP ligand by combining the bottlebrush block with a TTC-rich linear chain. While the multiple TTC groups firmly bound to the Au surface, the massive PS bottlebrush segment effectively stabilized Au NPs, even enabling chromatographic experiments. In the BBCP–Au conjugate dispersed in DMF, we found that the PS bottlebrush corona configuration can be controlled by adding water as a nonsolvent to PS. The collapse of the PS bottlebrush to minimize its contact with water rapidly formed dense PS domains and voids on the Au NP surface. By increasing the water percentage in DMF, we controlled the PS corona morphology from an isotropic shell to an undulating shell, multiple patches, and a crescent-like single patch. As the dense PS domain was impermeable to Ag diffusion, Ag nucleation occurred selectively on the exposed Au surface. It produced Au–Ag heterostructures with inversely replicated corona morphology, including smooth core–shell, multi-domain core–shell, multiple-island, and Janus Au–Ag heterodimers. We further analyzed unique orientation-dependent LSPR scattering features of the multiple-island Au–Ag by FDTD simulation and single-particle scatterometry. We suggest that bottlebrush macromolecular ligands hold promise for fabricating more complex anisotropic heterometal nanostructures, given the vast synthetic freedom in the bottlebrush parameter space combined with applicability to other metals. The resulting heterostructures offer significant potential for diverse applications.34 Their tunable surface morphologies and heterometal interfaces make them promising candidates for catalytic processes, such as oxygen and carbon dioxide reduction reactions, where precise control of active sites is critical. Additionally, the unique structural features and plasmonic properties of these materials enable their use in advanced optical applications, including surface plasmonic and surface-enhanced Raman scattering sensors for highly sensitive molecular detection. Asymmetric structures enhance effective interactions with biological systems, providing potential bioimaging and drug delivery applications.35 These versatile functionalities highlight the broad applicability of our heterostructures in both environmental and analytical technologies.Author contributions
M. K. and J. N. contributed equally to this work. M. K. and H. S. designed and planned the experiments. M. K., J. N. and J. K. carried out all the experiments. H. H. performed the single-particle study and FDTD simulation. All authors contributed to writing the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KAIST Grand Challenge 30 project (KC30) funded by the MSIT and KAIST, Korea (1711100600/N11190146). This work was also supported by the National Research Foundation of Korea (NRF), funded by the Korean Government (MSIT) (NRF-2018R1A5A1025208, 2023R1A2C2005705, RS-2023-00208239, and RS-2024-00405261).References
- J. Du and R. K. O'Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416 RSC.
- Y. Cui, J. Wang, J. Liang and H. Qiu, Small, 2023, 19, e2207609 CrossRef PubMed.
- R. M. Choueiri, E. Galati, H. Thérien-Aubin, A. Klinkova, E. M. Larin, A. Querejeta-Fernández, L. Han, H. L. Xin, O. Gang, E. B. Zhulina, M. Rubinstein and E. Kumacheva, Nature, 2016, 538, 79–83 CrossRef CAS PubMed.
- A. M. Percebom, J. J. Giner-Casares, N. Claes, S. Bals, W. Loh and L. M. Liz-Marzán, Chem. Commun., 2016, 52, 4278–4281 RSC.
- A. Kim, S. Zhou, L. Yao, S. Ni, B. Luo, C. E. Sing and Q. Chen, J. Am. Chem. Soc., 2019, 141, 11796–11800 CrossRef CAS PubMed.
- M. Lattuada and T. A. Hatton, J. Am. Chem. Soc., 2007, 129, 12878–12889 CrossRef CAS PubMed.
- T. Chen, M. Yang, X. Wang, L. H. Tan and H. Chen, J. Am. Chem. Soc., 2008, 130, 11858–11859 CrossRef CAS PubMed.
- Y. Yang, C. Yi, X. Duan, Q. Wu, Y. Zhang, J. Tao, W. Dong and Z. Nie, J. Am. Chem. Soc., 2021, 143, 5060–5070 CrossRef CAS PubMed.
- Z. Wang, B. He, G. Xu, G. Wang, J. Wang, Y. Feng, D. Su, B. Chen, H. Li, Z. Wu, H. Zhang, L. Shao and H. Chen, Nat. Commun., 2018, 9, 563 CrossRef PubMed.
- H. Duan, T. Malesky, J. Wang, C.-H. Liu, H. Tan, M.-P. Nieh, Y. Lin and J. He, Nanoscale, 2022, 14, 7364–7371 RSC.
- B. Liu, S. Thanneeru, A. Lopes, L. Jin, M. McCabe and J. He, Small, 2017, 13, 1700091 CrossRef PubMed.
- J. Paturej, S. S. Sheiko, S. Panyukov and M. Rubinstein, Sci. Adv., 2016, 2, e1601478 CrossRef PubMed.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Nat. Mater., 2016, 15, 183–189 CrossRef CAS PubMed.
- W. Gu, J. Huh, S. W. Hong, B. R. Sveinbjornsson, C. Park, R. H. Grubbs and T. P. Russell, ACS Nano, 2013, 7, 2551–2558 CrossRef CAS PubMed.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef CAS.
- L. Gromotka, M. J. Uttinger, C. Schlumberger, M. Thommes and W. Peukert, Nanoscale, 2022, 14, 17354–17364 RSC.
- L. Pitkanen and A. M. Striegel, Trends Anal. Chem., 2016, 80, 311–320 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- C. Zhang, B.-Q. Chen, Z.-Y. Li, Y. Xia and Y.-G. Chen, J. Phys. Chem. C, 2015, 119, 16836–16845 CrossRef CAS.
- D.-K. Lim, I.-J. Kim and J.-M. Nam, Chem. Commun., 2008, 5312–5314 RSC.
- Z. Feng, Y. Jia and H. Cui, J. Colloid Interface Sci., 2024, 672, 1–11 CrossRef CAS PubMed.
- M. Sovizi and M. Aliannezhadi, Optik, 2022, 252, 168518 CrossRef CAS.
- H. Hwang and H. Song, Chem. Phys. Rev., 2022, 3, 031301 CrossRef CAS.
- H. Hwang, H. Oh and H. Song, Small, 2023, 19, e2301241 CrossRef PubMed.
- C. Jing, Z. Gu, Y.-L. Ying, D.-W. Li, L. Zhang and Y.-T. Long, Anal. Chem., 2012, 84, 4284–4291 CrossRef CAS PubMed.
- J. Hu, T. Liu, P. Choo, S. Wang, T. Reese, A. D. Sample and T. W. Odom, ACS Cent. Sci., 2020, 6, 2339–2346 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, X. Song, S. Xing and H. Chen, Chem. Sci., 2017, 8, 430–436 RSC.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS PubMed.
- M. Alaboalirat, L. Qi, K. J. Arrington, S. Qian, J. K. Keum, H. Mei, K. C. Littrell, B. G. Sumpter, J.-M. Y. Carrillo, R. Verduzco and J. B. Matson, Macromolecules, 2019, 52, 465–476 CrossRef CAS.
- E. D. Palik and G. Ghosh, Handbook of Optical Constants of Solids, Academic Press, 1998 Search PubMed.
- P. B. Johnson and R. W. Christy, Phys. Rev., 1972, 6, 4370–4379 CAS.
- Q. N. Nguyen, C. Wang, Y. Shang, A. Janssen and Y. Xia, Chem. Rev., 2023, 123, 3693–3760 CrossRef CAS PubMed.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753–10758 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr01001b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |