DOI:
10.1039/D5NR00910C
(Paper)
Nanoscale, 2025,
17, 14271-14277
Functionalization with polymer ligands enhances the catalytic activity of surfactant-stabilized gold nanoparticles†
Received
1st March 2025
, Accepted 17th May 2025
First published on 23rd May 2025
Abstract
Plasmonic nanoparticles are increasingly explored as catalytically active entities to affect (photo-)catalytic transformations. Synthesis methods are established for accessing such nanoparticles with defined dimension and shape, thereby controlling their plasmonic behavior, including the possibility of resonance engineering and the control of chiral plasmonic properties of single plasmonic nanoparticles that can be used to drive asymmetric photocatalytic transformations. However, the most productive nanoparticle synthesis procedures employ surfactants that lead to comparably dense surface layers and may limit surface accessibility. In this work, we demonstrate that functionalization of such surfactant-stabilized plasmonic gold nanoparticles with a brush-type polymer-ligand layer results in both significantly increased catalytic activity in the colloidal solution state (by a factor of ∼4) and excellent colloidal stability, enabling several catalytic cycles. Heterogeneous catalysis experiments performed after surface deposition allowed the comparison between polymer ligand-grafted and ligand-free gold nanoparticles, which show comparably weak differences in catalytic rate (a factor of ∼1.8). These results pave the way for efficient (photo-)catalysis driven by well-defined plasmonic nanoparticles in colloidal solution.
Introduction
Catalysis by gold nanoparticles (AuNPs) has first gained interest when it was discovered that – opposed to bulk gold – nanoparticulate gold is active in reactions such as selective CO and H2 oxidation, CO2 hydrogenation or methanol oxidation.1–3 Later, AuNPs were successfully used in a variety of organic reduction reactions, such as reductions by borohydride, reduction of carbonyls and N-alkylation of amines.2 When the reaction progress is coupled to a change of solution color, as is the case e.g. for the reduction of nitrophenols, structure–activity relationships can be established in a convenient manner.4–6 AuNP catalysis is still a vibrant domain of materials science, which recently gained additional momentum because the optical properties of gold also enable plasmonic catalysis, i.e. plasmon-mediated chemical reactions driven by light of visible frequencies.7 Three main mechanistic modalities of plasmonic catalysis can be discriminated: (localized) plasmonic heating,8 local field enhancement,9,10 and direct hot-carrier transfer of electrons.11–13 Appropriate chemical design of the catalytically active entity allows to isolate such plasmonic effects on catalytic activity from non-plasmonic pathways.14 Recently, there has been abiding research effort dedicated to the mechanistic understanding of reactions catalyzed or promoted by AuNPs, especially in the domain of heterogeneous catalysis.15–17 Starting from tailored particles using surfactants, ligand exchange and subsequent deposition can achieve heterogeneous catalysts with a complexity and homogeneity that is unattainable with top-down methods.18
In comparison to the mentioned heterogeneous catalytic systems AuNP-mediated catalysis in colloidal solution requires AuNP stabilization. The most common stabilizing agents that derive from AuNP synthesis procedures are citrate19 and quaternary ammonium surfactants such as cetyltrimethylammonium bromide (CTAB)20 or cetyltrimethylammonium chloride (CTAC).21 Both citrate and surfactant capping agents only provide colloidal stability in a limited solvent/ionic-strength parameter space and are easily displaced by other competitive small-molecule ligands, which is why often times alternative surface coatings are applied after AuNP synthesis. Such surface coatings may provide additional opportunities for controlling catalytic activity but may also impair it. Opportunities can emerge from additional control over the reaction that can be exerted by distinct coatings. For example, additional chemisorption of poly(vinyl pyrrolidone) onto silica-supported AuNPs was demonstrated to greatly enhance the selectivity for benzaldehyde in the aerobic oxidation of benzyl alcohol.22 Polymer functionalization also enables the on-demand switching of reaction rate in response to external stimuli that are processed by the polymer coating in different ways, leading to e.g. volume phase transitions23,24 or triggered de-sorption from the surface.25 Moreover, catalyst recycling can be facilitated by special solvency behavior of a polymer coating.26 However, concomitantly with surface functionalization arises the question of surface accessibility.27,28 Especially citrate-capped AuNPs are catalytically active but limited in terms of colloidal stability. Table S1† summarizes publications where catalysis in colloidal solution from gold nanoparticles is studied, and the catalytic activity before and after functionalization with polymer molecule ligands via the grafting-to method is compared: upon such surface functionalization, a reduction of catalytic performance is experimentally observed, with the exception only of grafted polymers carrying N-heterocyclic carbene end groups that act as gold anchoring moieties.27,29–34 It was demonstrated that catalytic activity is directly correlated with the grafting density of adsorbed (polymeric) ligands (in units of adsorbed molecules per surface area).30 In the case of CTAB/CTAC-stabilized nanoparticles, on the other hand, huge part of the Au surface is blocked by the dense surfactant bilayer,35 while molecular dynamics simulations also predicted the formation of channels that give rise to some limited accessibility of the surface.36 However, many recent advancements in design and synthesis of plasmonic nanoparticles were enabled by using quaternary ammonium surfactants: complex shapes such as near-perfect spheres,37 cubes,38 rods,39 triangles,40 stars,41 nanorattles,42 and even chiral nanoparticles43 have been synthesized, as well as bimetallic particles of well-defined shape and composition.39,44 These structures feature unique plasmonic properties that in turn enable otherwise not attainable catalytic possibilities, such as asymmetric organic catalysis in which the handedness of circularly polarized light translates into the chiral properties of the reaction products.45 Moreover, when polymer ligands enable catalysis in colloidal solution the available surface is intrinsically increased as the NPs are in contact with the reaction medium from all sides.27 Concomitantly, the advantages of heterogeneous and homogenous catalysis can be combined, such as simple catalyst removal in a classic batch reactor process.27 Thus, leveraging the catalytic performance of surfactant-stabilized AuNPs by improving surface access is an important goal toward efficient solar-to-chemical energy conversion.
The purpose of this work was twofold: (i) We investigated the effect of polymer functionalization on the catalytic behavior of surfactant-stabilized gold nanoparticles; and (ii) compared the catalytic activity with substrate-deposited systems after ligand removal. We employed polymer ligands synthesized by the Reversible Addition–Fragmentation chain Transfer (RAFT)46 method. Polymer molecules synthesized by that method can be readily anchored to gold surfaces via their trithiocarbonate end groups.47–50 As a model reaction we chose the oxygen-assisted decomposition of hydrogen peroxide and subsequent decolorization of methylene blue (MB), which occurs in the dark but is further accelerated by illumination,51 which allows us to investigate the catalytic effect of AuNPs on both reaction pathways. Our results demonstrate colloidal stability of the polymer-NP hybrids under reaction conditions and significantly increased reaction rate when high molar-mass polymer ligands replaced the initial CTAC surfactant, indicating enhanced surface access. These polymer-grafted gold nanoparticles thereby approach the catalytic activity of the ligand-free Au surface as demonstrated by experiments after deposition on a substrate.
Experimental section
Ascorbic acid (AA, >99%), hydrogen tetrachloroaurate (HAuCl4, >99.9%), sodium borohydride (NaBH4, 99%), dioxane, 2-(dodecyl-thiocarbonothioylthio)-2-methylpropionic acid (DDMAT, 98%), azobis(isobutyronitril) (AIBN) and N,N-dimethylacrylamide (DMAM, 99%) were obtained from SigmaAldrich. Hexadecyl-trimethylammonium bromide (CTAB, 99%) and hexadecyltrimethylammonium chloride (CTAC, >99%) were obtained from Molekula. AIBN was recrystallized from toluene/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and DMAM purified through an aluminumoxide column to remove the inhibitor. All other chemicals and solvents were used as received. Fresh purified water (Milli-Q grade, 18.2 MΩ cm at 25 °C) was used in all experiments.
:3) and DMAM purified through an aluminumoxide column to remove the inhibitor. All other chemicals and solvents were used as received. Fresh purified water (Milli-Q grade, 18.2 MΩ cm at 25 °C) was used in all experiments.
Seeded-growth synthesis of AuNPs
All glassware and stir bars used in the synthesis were thoroughly cleaned with aqua regia. The synthesis of AuNPs using surfactants followed a protocol from Schletz et al.:37 A solution of 4.7 mL CTAB (100 mM) and 1.25 μmol HAuCl4 was heated at 32 °C for 10 minutes. Then, 300 μL of a freshly prepared 10 mM NaBH4 solution was rapidly injected while stirring vigorously, continuing for 30 seconds, followed by aging for 30 minutes. 40 mL of CTAC (200 mM), 5 mL of AA (1 M), and 1 mL of the Wulff-seed solution were mixed, 40 mL of a CTAC (200 mM) and HAuCl4 (1 mM) solution added under mild stirring, and after 15 minutes purified by twofold centrifugation-redispersion in 10 mL of 10 mM CTAC. To obtain larger particles, 200 mL of CTAC (60 mM) and AA (3.9 mM) were mixed with varying amounts of the seed solution, larger amounts resulting in smaller particle diameter (see volumes and concentrations listed in Table S2.†) Then, 360 mL of a 1.5 mM HAuCl4 solution in CTAC was added at 1 mL min−1 with a syringe pump and ECOFLO® Scalp vein sets 21 G green. Afterward, 40 mL of the same solution were added to etch the particles into spheres and after 30 minutes the solution was purified by twofold centrifugation-redispersion in 10 mM CTAC.
Synthesis of poly(N,N-dimethylacrylamide)
Linear polymer linker (a) PDMAM4.5 and (b) PDMAM49 was synthesized via reversible addition–fragmentation chain transfer polymerization.46 DDMAT ((a) 40 mg, 0.106 mmol, (b) 4 mg, 0.011 mmol), AIBN ((a) 5.8 mg, 0.0356 mmol, (b) 0.58 mg, 0.0036 mmol), and DMAM (0.441 mL, 4.277 mmol) were dissolved in 1.8 mL dioxane, purged with Ar for 10 min, and polymerized at 60 °C for 5 hours. The polymer was purified by precipitation from Et2O and dissolution in DCM which was repeated four times, then dried under vacuum at 40 °C for 12 hours. The molecular weight was determined from size exclusion chromatography (ESI, Fig. 1†) as (a) MGPCn = 4500 g mol−1, Đ = 1.06 and (b) MGPCn = 49000 g mol−1, Đ = 1.21.
Ligand exchange from CTAC-capped AuNPs
To facilitate the targeted ligand exchange, in a first step excess surfactant needs to be removed. Here, we used a comparatively simple approach: a small quantity of sample was centrifuged and re-dispersed in pure water after supernatant removal, repeated n times until aggregation visibly occurred. For the functionalization with polymer ligands, n − 1 centrifugation-redispersion cycles were used prior to polymer grafting (see details in Table S3†). This method achieved reproducible results and NP dispersions that showed no aggregation. The particles could be centrifuged and redispersed multiple times. Less than n − 1 centrifugation/re-dispersion cycles prior to polymer functionalization on the other hand resulted in poor colloidal stability. This was associated with incomplete re-dispersion after centrifugation and can be attributed to an insufficient polymer grafting density. Schulz et al. proposed 2-phase extraction as a method to remove excess CTAC.52 For comparison, the 2-phase method was tried and no further improvement regarding colloidal stability and catalytic activity could be achieved compared with the approach used in this work (ESI, Fig. S2†). An ultrasonic bath was used to promote the polymer-grafting, which is an activated barrier-controlled process.53 Comparison between extinction spectra of PDMAM@AuNPs and CTAC-functionalized particles reveals no changes in the linewidth of the plasmon peak indicating absence of aggregation and colloidal stability (ESI, Fig. S3†). Electrophoretic measurements reveal successful polymer grafting by a sign change in zeta-potential value. Since the grafted polymer is electroneutral, this is explained by the cationic surfactant bilayer being removed during the polymer functionalization and chemisorbed chloride ions being left at the gold surface, which changes the zeta potential from strongly positive to a negative value (Fig. S3†), as previously demonstrated.54,55 Further proof for functionalization with polymer ligands and the stabilizing role of the polymeric ligand is obtained in control experiments (Fig. S4a and b†), TEM images and DLS measurements (Fig. S4c and d†).
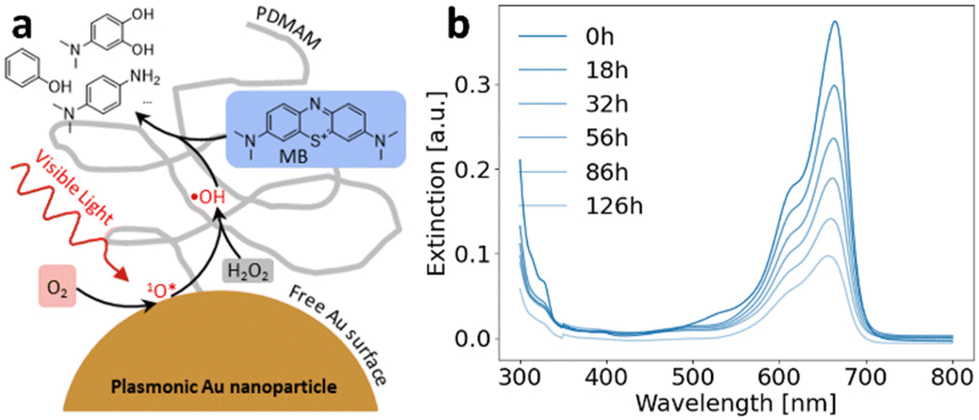
Model reaction for catalytic activity
A dye decomposition reaction was chosen as it can be easily followed by UV-vis spectroscopy (Fig. 1b). More specifically, the decolorization of methylene blue by oxygen-assisted hydrogen peroxide decomposition on AuNPs was studied (proposed mechanism see Fig. 1a). It was proposed by Wen et al. that in this reaction oxygen is first adsorbed on the gold surface.51 Strongly promoted by the localized surface plasmon resonance (LSPR), an active oxygen intermediate that resembles singlet oxygen is created and subsequently reacts with hydrogen peroxide to create reactive hydroxyl radicals.51 The decomposition of MB by this radical species has been thoroughly investigated for wastewater treatment.56 According to these investigations, in a first step the OH radical attaches to the MB molecule in one of the available positions. In the following, the intermediates undergo a second reaction with radical OH that is followed in most cases by full decomposition.56 For illumination a Cree XHP50 SMD-LED (6500 K) was used and the distance varied for different light intensities. The conditions for catalytic experiments (Table S4†), calculation of total Au surface and method for the kinetic fit are explained in section 6 of the ESI.†
 |
| | Fig. 1 (a) Proposed mechanism for the hydrogen peroxide-mediated decomposition of methylene blue by reactive oxygen species generated on free plasmonic nanoparticle surface enabled through PDMAM ligand stabilization. (b) loss of MB absorption peak over time. | |
Sample preparation for heterogeneous catalysis
Glass substrates (25 × 25 × 1.1 mm, redox.me) were washed using sonication (5 min, 25 °C, 37 kHz) in acetone and isopropanol, consecutively. The substrates were then treated with oxygen plasma for 5 minutes. The PDMAM49@Au43 nanoparticle solution was transferred to MeOH by 3× centrifugation – redispersion to give a highly concentrated dispersion (39.7 mM). 20 μL of this solution were used per sample in the spin coating process. To ensure a good quality of the layer, extinction spectra of the layer were recorded and the best samples selected (clear plasmon absorption signal, minimal aggregation visible). Of those samples, one was used in the catalysis as prepared. The others were treated with oxygen plasma (if not further specified for 10 min). During this treatment, the polymer was removed, and agglomeration occurred to some extent. Half of the plasma treated samples were then re-functionalized with polymer to allow direct comparison with the ligand-free surface without influence of the formed agglomerates.
Results and discussion
Stability during the reaction
While the polymer-stabilized particles are fully stable in water and other good solvents for PDMAM over long periods of time, keeping them fully dispersed during the catalytic reaction proved to be dependent on the solvent used. In pure water, it was observed that particles assembled at the water/glass and water/air interfaces during the catalyzed reaction. Although the characteristic red color was maintained and particles can be easily redispersed using ultrasonic treatment, attempts to prevent the segregation process by stirring or shaking during the reaction were unsuccessful. Changing the solvent to MeOH provided better colloidal stability, but the reaction rate decreased significantly. A 1:1 mixture of THF and water however gives the same reaction rate as pure water together with complete colloidal dispersion throughout the reaction: full methylene blue decomposition can be achieved without any loss in characteristic AuNP absorption (Fig. 2a). Because of the excellent colloidal stability, catalyst recycling could be successfully demonstrated with again no loss of gold absorption during the catalyzed reaction (Fig. 2b). The biggest challenge regarding catalyst re-use here is associated with the handling during the step of centrifugation – redispersion after the reaction took place. Here, some agglomeration was observed and after the second cycle not all colloidal gold could be retrieved – in consequence the half-life time of the reaction increases. To ensure full catalyst recycling, more optimization of this centrifugation/re-dispersion step or alternative methods like filtering may prove useful. Fig. 2c shows the strong visible difference between the stable (solvent mixture) and non-stabilized (pure water) system. Zeta potential measurements revealed that during the reaction negative charges accumulate on the particle surface (Fig. 2d). In MeOH the surface becomes positively charged, which, together with the strongly reduced reaction speed, indicates that the catalytic reaction is impaired. It is shown that in THF/H2O, the most successful system, negative charges also accumulate, indicating that the reaction mechanism is not strongly affected. The net values of zeta potential are smaller compared to pure water; however, this is also the case before the reaction (empty bars) and presumably results from more effective separation of ion pairs in pure water.
 |
| | Fig. 2 (a) Extinction spectra at different points of the reaction. (b) Reaction cycles with NP centrifugation in-between. (c) Images from MB decomposition reaction in water and H2O/water. (d) Zeta potentials before (empty bars) and after (filled bars) the reaction in different solvents. | |
Catalysis depending on gold-nanoparticle dimension and chain-length of surface ligands
The catalytic activity of differently sized AuNPs with (a) CTAC surfactant, (b) PDMAM 4.5 kDa and (c) PDMAM 49 kDa were studied and referenced against the reaction without catalyst present. The absorption peak of methylene blue was observed by recording extinction spectra of the reaction solution at different points of time during the reaction. The reaction rate coefficients from first order fit to the obtained MB absorption maxima over time are displayed in Fig. 3. The lowest reaction rate coefficients among the catalyzed cases are observed for CTAC@AuNPs. Interestingly, a relatively strong size dependency is observed: larger particles cause only slight enhancement compared to the non-catalyzed reaction and a maximum at 28 nm is found. This observation of a “magic-size effect” is consistent with findings from Fenger et al. and Piella et al. who observed very similar behavior with a maximum around 20 nm when studying nitrophenol reduction with CTAB57 and citrate29 stabilized particles, respectively. Upon functionalization with PDMAM Mn = 4.5 kDa, we observe a significant increase in reaction rate coefficients, which becomes even stronger when PDMAM ligands with Mn = 49 kDa are used. Upon functionalization with these higher Mn PDMAM ligands, an enhancement up to factor 4 for the larger particles is achieved. This demonstrates that high-Mn polymer ligands can stabilize particles and at the same time provide access to the catalytic surface, by forming a polymer layer that is not dense in a good solvent: the grafting-to approach leads to smaller grafting densities when polymer ligands with higher Mn are employed in the grafting process.58 Therefore, small molecules can easily penetrate into the brush layer and diffuse to and from the catalyst surface.
 |
| | Fig. 3 Reaction rates for MB decomposition from first order fit using different ligands: CTAC, 4.5 kDa PDMAM and 49 kDa PDMAM. Vertical error bars from repeated experiments to ensure reproducibility. Horizontal error bars from deviation in particle size as found in TEM images. | |
Comparison to ligand-free gold surface
To put the experiments in colloidal solution into perspective, the catalytic activity was compared to that of a ligand-free gold surface. To make that comparison, the NPs were deposited on a substrate and used in a heterogeneous catalysis setup. After spin coating PDMAM49@Au43 from MeOH onto cleaned glass substrates, the samples were treated with oxygen plasma to remove the polymer ligands. To ensure complete removal of the polymer ligand as well as the quality of colloidal gold after plasma treatment, transmission spectra were recorded between the processing steps (Fig. 4). After plasma treatment (orange), a vertical shift across the whole spectral range is observed, which is reverted upon re-grafting polymer ligands (highlighted by black arrows, Fig. 4 upper panel). We attribute this observation to light scattering resulting from the refractive-index mismatch between the additional interfaces (air/dry PDMAM and PDMAM/Au), which are absent in the ligand-free gold system after plasma treatment. Additionally, after the plasma treatment an additional peak appears at the low-energy shoulder of the plasmon peak, which indicates some agglomeration. Because refractive-index sensitivity generally is proportional to resonance wavelength,59 upon re-functionalization the change in refractive index has the biggest effect on the low-energy region of the spectrum, red-shifting the resonance position and increasing extinction values.60 Fig. S5† shows findings on the removal of polymer as well as aggregate formation during plasma treatment. Because polymer re-grafting is unlikely to affect the agglomeration state of deposited nanoparticles, it is reasonable to assume a comparable degree of agglomeration for both samples after the plasma treatment (orange and green trace in Fig. 4, panel c). We therefore compare their catalytic performance for MB decomposition (additionally, the performance of the sample before plasma treatment (blue) is given in Fig. S6†). The results are shown in Fig. 4, panel c. Compared to a blank experiment, PDMAM49@Au and Au on glass samples both show distinct catalytic activity. The reaction rate coefficient acquired from catalysis with PDMAM49@Au samples was found to be 55% of the reaction rate coefficient from pure AuNP catalysis, i.e. a factor of 1.8. Thus, while the catalytic activity of the ligand-free reference system is higher compared with the grafted system PDMAM49@Au, the relative difference in activity is comparably modest.
 |
| | Fig. 4 (a) Extinction spectra corresponding to different substrate-deposited NP samples for comparison in heterogeneous catalysis. (b) Schematic representation of the corresponding fabrication steps (same colours to match with spectra in a). (c) MB decomposition from heterogeneous catalysis with reaction rates shown in the inlet. Filled/empty markers indicate separate measurements to ensure reproducibility. | |
Light response
We chose the model reaction such that we can investigate the dark reaction alongside potential plasmon-assisted acceleration, since a colloidal gold catalyst in principle provides both possibilities. To assess LSPR contribution the reaction was tested at different light intensities (Fig. 5a) and compared to the dark reaction at different temperatures (Fig. 5c). At all tested light intensities, the reaction followed first order kinetics and the obtained reaction rate coefficients were fitted using an adapted Mitscherlich equation (Fig. 5b, ESI†), which is typically used for light response curves.61 It is observed that, although a quite pronounced dark reaction is present, the slope at low intensities is steeper compared to the non-catalyzed reaction. At the highest recorded light intensities, the rate is already approaching light saturation. In consequence, the biggest enhancement is observed at medium light intensity. Since the reaction shows a very strong temperature dependence (Fig. 5c and d) with an apparent activation energy of only 63 kJ mol−1 according to an Arrhenius fit, plasmonic heating could in principle affect reaction acceleration. However, we designed our experiments such that plasmonic heating can be ruled out as a cause for rate enhancement. We used an unfocussed light source, which resulted in very small power density and therefore no significant heating effect (order of 10–6 K, calculation see ESI†). Additionally, it was observed that the reactions at elevated temperatures follow zero order reaction kinetics opposed to the light-enhanced reactions that clearly show first order reaction kinetic, which supports the assumption that thermal enhancement in this case results in a different reaction mechanism and is not contributing in the purely light-driven experiments. Likewise, Wen et al. report that the LSPR is responsible for the creation of the active oxygen species that results in MB decomposition and could exclude a purely thermal pathway.51 Therefore, we attribute the observed enhancement to a non-thermal light-related effect.
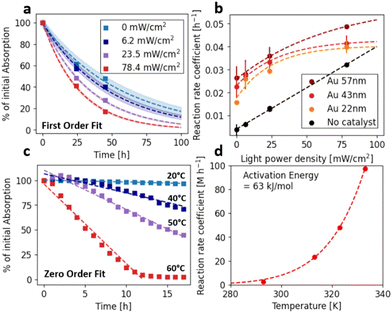 |
| | Fig. 5 (a) Loss in MB decomposition over time with PDMAM@Au43nm as catalyst and different light intensities. (b) Reaction rate coefficients for different sizes of NPs and without NPs vs. light intensity. (c) like (a) but with variation in temperature. (d) Reaction rate coefficient from zero order fit of data (c) vs. temperature with Arrhenius fit. | |
Summary
We demonstrated that functionalization of surfactant-coated gold nanoparticles with polymer-molecule ligands results in increased catalytic activity. This enhancement is more pronounced for high-Mn polymer-molecule ligands, which under good solvency conditions form a more dilute layer covering the gold nanoparticles. This polymeric coating also provides colloidal stability, enabling catalyst recovery by centrifugation and repeated re-use. Compared with the situation where nanoparticle ligands were removed after surface deposition and plasma treatment, the polymer-grafted system displays only moderately reduced catalytic activity. Polymer-grafted gold nanoparticles can therefore be applied as both highly active and colloidally stable catalysts in solution. We demonstrated two possibilities for accelerating/controlling reactivity via distinct mechanisms: (i) thermal activation by heating of the bulk solution; and (ii) light-driven reaction. While our work concentrated on spherical CTAC/CTAB-surfactant stabilized gold nanoparticles, it can clearly be expanded to other nanoparticle shapes, thereby leveraging the variety of gold colloids for specific (photo-)catalytic transformations.
Data availability
The data supporting this article have been included as part of the ESI.† Specific data files (such as instrument files or data spreadsheets) related to this study are available upon request from the corresponding author.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under grant 466182523. The authors acknowledge support by GRK 2767 – Projektnummer 451785257 (DFG, German Research Foundation). Olga Kuharenko is acknowledged for the preparation of RAFT polymers, Lukas Mielke for help with substrate-deposited samples and Gabriele Carelli for recording AFM images. Emiliano Cortés is acknowledged for fruitful discussions. CR acknowledges receipt of a Liebig fellowship (Fonds der Chemischen Industrie).
References
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- P. Zhao, X. Feng, D. Huang, G. Yang and D. Astruc, Coord. Chem. Rev., 2015, 287, 114–136 CrossRef CAS.
- C. Zhan, J. Yi, S. Hu, X.-G. Zhang, D.-Y. Wu and Z.-Q. Tian, Nat. Rev. Methods Primers, 2023, 3, 1–21 CrossRef.
- M. Zakia and S. I. Yoo, Soft Matter, 2020, 16, 10252–10259 RSC.
- T. Torimoto, H. Horibe, T. Kameyama, K. Okazaki, S. Ikeda, M. Matsumura, A. Ishikawa and H. Ishihara, J. Phys. Chem. Lett., 2011, 2, 2057–2062 CrossRef CAS.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Nano Lett., 2017, 17, 3710–3717 CrossRef CAS PubMed.
- E. Cortés, W. Xie, J. Cambiasso, A. S. Jermyn, R. Sundararaman, P. Narang, S. Schlücker and S. A. Maier, Nat. Commun., 2017, 8, 14880 CrossRef PubMed.
- R. Schürmann, A. Dutta, K. Ebel, K. Tapio, A. R. Milosavljević and I. Bald, J. Chem. Phys., 2022, 157, 084708 CrossRef PubMed.
- R. Schürmann and I. Bald, Nanoscale, 2017, 9, 1951–1955 RSC.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS PubMed.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 1–16 CrossRef PubMed.
- M. Herran, S. Juergensen, M. Kessens, D. Hoeing, A. Köppen, A. Sousa-Castillo, W. J. Parak, H. Lange, S. Reich, F. Schulz and E. Cortés, Nat. Catal., 2023, 6, 1205–1214 CrossRef CAS.
- Y. Kang, S. M. João, R. Lin, K. Liu, L. Zhu, J. Fu, W.-C. (Max) Cheong, S. Lee, K. Frank, B. Nickel, M. Liu, J. Lischner and E. Cortés, Nat. Commun., 2024, 15, 3923 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- Y. Zheng, Y. Ma, J. Zeng, X. Zhong, M. Jin, Z.-Y. Li and Y. Xia, Chem. – Asian J., 2013, 8, 792–799 CrossRef CAS PubMed.
- K. Chen, H. Wu, Q. Hua, S. Chang and W. Huang, Phys. Chem. Chem. Phys., 2013, 15, 2273–2277 RSC.
- R. Roa, S. Angioletti-Uberti, Y. Lu, J. Dzubiella, F. Piazza and M. Ballauff, Z. Phys. Chem., 2018, 232, 773–803 CrossRef CAS.
- S. Wu, L. Lei, Y. Xia, S. Oliver, X. Chen, C. Boyer, Z. Nie and S. Shi, Polym. Chem., 2021, 12, 6903–6913 RSC.
- S. Wu, T. Wang and H. Xu, ACS Macro Lett., 2020, 9, 1192–1197 CrossRef CAS PubMed.
- S. M. Ansar, B. Fellows, P. Mispireta, O. T. Mefford and C. L. Kitchens, Langmuir, 2017, 33, 7642–7648 CrossRef CAS PubMed.
- S. Chakraborty and C. L. Kitchens, J. Phys. Chem. C, 2019, 123, 26450–26459 CrossRef CAS.
- B. Liu, H. Yao, W. Song, L. Jin, I. M. Mosa, J. F. Rusling, S. L. Suib and J. He, J. Am. Chem. Soc., 2016, 138, 4718–4721 CrossRef CAS PubMed.
- J. Piella, F. Merkoçi, A. Genç, J. Arbiol, N. G. Bastús and V. Puntes, J. Mater. Chem. A, 2017, 5, 11917–11929 RSC.
- S. M. Ansar and C. L. Kitchens, ACS Catal., 2016, 6, 5553–5560 CrossRef CAS.
- L. Zhang, Z. Wei, M. Meng, G. Ung and J. He, J. Mater. Chem. A, 2020, 8, 15900–15908 RSC.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- R. Ciganda, N. Li, C. Deraedt, S. Gatard, P. Zhao, L. Salmon, R. Hernández, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 10126–10129 RSC.
- M. S. Álvarez Cerimedo, L. G. Baronio, C. E. Hoppe and M. A. Ayude, ChemistrySelect, 2019, 4, 608–616 CrossRef.
- A. Rogolino, N. Claes, J. Cizaurre, A. Marauri, A. Jumbo-Nogales, Z. Lawera, J. Kruse, M. Sanromán-Iglesias, I. Zarketa, U. Calvo, E. Jimenez-Izal, Y. P. Rakovich, S. Bals, J. M. Matxain and M. Grzelczak, J. Phys. Chem. Lett., 2022, 13, 2264–2272 CrossRef CAS PubMed.
- S. K. Meena and M. Sulpizi, Langmuir, 2013, 29, 14954–14961 CrossRef CAS PubMed.
- D. Schletz, J. Schultz, P. L. Potapov, A. M. Steiner, J. Krehl, T. A. F. König, M. Mayer, A. Lubk and A. Fery, Adv. Opt. Mater., 2021, 9, 2001983 CrossRef CAS.
- A. M. Steiner, M. Mayer, D. Schletz, D. Wolf, P. Formanek, R. Hübner, M. Dulle, S. Förster, T. A. F. König and A. Fery, Chem. Mater., 2019, 31, 2822–2827 CrossRef CAS.
- M. Tebbe, C. Kuttner, M. Mayer, M. Maennel, N. Pazos-Perez, T. A. F. König and A. Fery, J. Phys. Chem. C, 2015, 119, 9513–9523 CrossRef CAS PubMed.
- C. Kuttner, M. Mayer, M. Dulle, A. Moscoso, J. M. López-Romero, S. Förster, A. Fery, J. Pérez-Juste and R. Contreras-Cáceres, ACS Appl. Mater. Interfaces, 2018, 10, 11152–11163 CrossRef CAS PubMed.
- A. Guerrero-Martínez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzán, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef.
- M. J. Schnepf, M. Mayer, C. Kuttner, M. Tebbe, D. Wolf, M. Dulle, T. Altantzis, P. Formanek, S. Förster, S. Bals, T. A. F. König and A. Fery, Nanoscale, 2017, 9, 9376–9385 RSC.
- B. Ni, M. Mychinko, S. Gómez-Graña, J. Morales-Vidal, M. Obelleiro-Liz, W. Heyvaert, D. Vila-Liarte, X. Zhuo, W. Albrecht, G. Zheng, G. González-Rubio, J. M. Taboada, F. Obelleiro, N. López, J. Pérez-Juste, I. Pastoriza-Santos, H. Cölfen, S. Bals and L. M. Liz-Marzán, Adv. Mater., 2023, 35, 2208299 CrossRef CAS PubMed.
- M. Hoffmann, D. Schletz, A. M. Steiner, D. Wolf, M. Mayer and A. Fery, J. Phys. Chem. C, 2022, 126, 2475–2481 CrossRef CAS.
- P. Bainova, J.-P. Joly, M. Urbanova, D. Votkina, M. Erzina, B. Vokata, A. Trelin, P. Fitl, G. Audran, N. Vanthuyne, J. Vinklarek, V. Svorcik, P. Postnikov, S. R. A. Marque and O. Lyutakov, ACS Catal., 2023, 13, 12859–12867 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. L. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef CAS.
- C. Rossner, B. Ebeling and P. Vana, ACS Macro Lett., 2013, 2, 1073–1076 CrossRef CAS PubMed.
- S. Slavin, A. H. Soeriyadi, L. Voorhaar, M. R. Whittaker, C. R. Becer, C. Boyer, T. P. Davis and D. M. Haddleton, Soft Matter, 2011, 8, 118–128 RSC.
- C. Rossner, V. Roddatis, S. Lopatin and P. Vana, Macromol. Rapid Commun., 2016, 37, 1742–1747 CrossRef CAS PubMed.
- T. Wen, H. Zhang, Y. Chong, W. G. Wamer, J.-J. Yin and X. Wu, Nano Res., 2016, 9, 1663–1673 CrossRef CAS.
- F. Schulz, W. Friedrich, K. Hoppe, T. Vossmeyer, H. Weller and H. Lange, Nanoscale, 2016, 8, 7296–7308 RSC.
- Q. Tang, C. Rossner, P. Vana and M. Müller, Biomacromolecules, 2020, 21, 5008–5020 CrossRef CAS PubMed.
- S. Vazirieh Lenjani, M. Mayer, R. Wang, Y. Dong, A. Fery, J.-U. Sommer and C. Rossner, J. Phys. Chem. C, 2022, 126, 14017–14025 CrossRef CAS.
- H. Zhu, E. Prince, P. Narayanan, K. Liu, Z. Nie and E. Kumacheva, Chem. Commun., 2020, 56, 8131–8134 RSC.
- Q. V. Vo, L. T. T. Thao, T. D. Manh, M. V. Bay, B.-T. Truong-Le, N. T. Hoa and A. Mechler, RSC Adv., 2024, 14, 27265–27273 RSC.
- R. Fenger, E. Fertitta, H. Kirmse, A. F. Thünemann and K. Rademann, Phys. Chem. Chem. Phys., 2012, 14, 9343–9349 RSC.
- H. Yockell-Lelièvre, J. Desbiens and A. M. Ritcey, Langmuir, 2007, 23, 2843–2850 CrossRef PubMed.
- E. Martinsson, M. M. Shahjamali, N. Large, N. Zaraee, Y. Zhou, G. C. Schatz, C. A. Mirkin and D. Aili, Small, 2016, 12, 330–342 CrossRef CAS PubMed.
- C. Rossner, T. A. F. König and A. Fery, Adv. Opt. Mater., 2021, 9, 2001869 CrossRef CAS.
- Q. Liu, W. Jia and F. Li, Sci. Rep., 2020, 10, 11664 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab and
Christian
Rossner
ab and
Christian
Rossner