
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free graphitic carbon nitride nanosheet for dual mode fluorescence and electrochemical detection of para-nitrophenol†
Ankush Kumar
Singh‡
 a,
Aayoosh
Singh‡
b,
Mithilesh
Patel
a,
Vinod P.
Singh
b and
Rosy
*a
a,
Aayoosh
Singh‡
b,
Mithilesh
Patel
a,
Vinod P.
Singh
b and
Rosy
*a
aDepartment of Chemistry, IIT(BHU), Varanasi, India
bDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi, India. E-mail: rosy.chy@iitbhu.ac.in
First published on 7th May 2025
Abstract
para-Nitrophenol (p-NP) contamination poses significant risks to both environmental and human health, highlighting the urgent need for sensitive and selective methods for its detection. In this study, a graphitic carbon nitride sheet (g-CNS) synthesized via a one-step hydrothermal method is proposed as a bi-functional probe for p-NP sensing. The fluorescence activity of the g-CNS was first optimized, and its quenching on the addition of p-NP was used for the fluorometric detection of p-NP. A broad linear response to p-NP concentrations ranging from 1 to 100 μM was observed, with a detection limit of 36.76 nM. The sensor exhibited excellent performance in the presence of potential interferences and was successfully applied to real sample analysis. To enhance on-site detection applicability, a g-CNS modified voltammetric sensor was developed. The g-CNS was electrodeposited on a glassy carbon electrode (GCE) using cyclic voltammetry and characterized using a range of techniques to confirm the successful modification. When applied to p-NP detection, the modified GCE demonstrated high sensitivity, with a limit of detection (LOD) of 218 nM. Furthermore, the stability, reusability, and reproducibility of the modified electrode were thoroughly evaluated, confirming its reliability for long-term use in electrochemical sensing applications.
1. Introduction
para-Nitrophenol (p-NP), an important aromatic nitro chemical, is a vital raw material and additive in a variety of sectors, including explosives, insecticides, dyes, medicines, and plastics.1,2 Furthermore, it is a hydrolysis product of several organophosphorus pesticides. Water contamination has been exacerbated by the discharge of wastewater from industrial, agricultural, and municipal sources.3p-NP is a persistent pollutant that has been detected not only in industrial effluents but also in freshwater and marine environments.4 Owing to its stability in the natural environment, p-NP can migrate over long distances and accumulate in organisms, leading to significant ecological harm and potential long-term impacts on aquatic and terrestrial lives. The US Environmental Protection Agency (EPA) has classified p-NP and its derivatives as possible environmental pollutants because of their high toxicity, limited biodegradability, and prolonged chemical stability.5,6 The EPA has set a maximum permissible value of 60 mg L−1 (0.43 μM) for p-NP in drinking water owing to their environmental persistence and possible health hazards. Throughout the food chain, an increase in p-NP levels in the environment presents major health hazards, resulting in diseases including hypoxia, brain and lung edema, dehydration, and extensive vascular damage.6 Therefore, it is essential to accurately identify p-NP in water samples to guarantee the safety of the environment and human health. For its precise identification and quantification, a number of analytical methods have been investigated, such as gas chromatography-mass spectrometry,7,8 capillary electrophoresis,9,10 high-performance liquid chromatography,11 liquid chromatography-mass spectrometry,12 and micelle electrokinetic capillary chromatography.13 Although these analytical techniques are quite effective in detecting p-NP, they mostly require highly expensive and sophisticated instruments operated specifically by trained personnel. The actual detection procedure takes a long time and necessitates cumbersome sample preparations. Their use is further restricted by high operating costs and requirement for frequent maintenance, especially for large-scale environmental evaluations and real-time monitoring. As a result, there is an increasing need for p-NP detection alternatives that are more effective, affordable, and easy to use.Compared to conventional analytical techniques, fluorescence and electrochemical methods offer significant advantages, including rapid response, ease of operation, and possible miniaturization.5,14–17 Several studies have reported the successful application of these two approaches for the detection of p-NP. Fluorescence-based detection relies on materials exhibiting strong and stable fluorescence properties, enabling sensitive and selective analyte recognition. In contrast, electrochemical detection depends on the presence of an effective surface modifier/recognition element that enhances the electrocatalytic activity, thereby improving the sensitivity and accuracy of p-NP quantification. Several research groups have reported that metal nanoparticles are effective fluorescent probes and/or recognition elements which show considerable interaction with p-NP.18–20 Despite their efficiency, metal-based nanoparticles are typically costly and harmful. Furthermore, the disposal and management of waste resulting from these materials provide substantial environmental concerns. These challenges have encouraged research into metal-free, carbon-based nanomaterials, which provide a viable alternative due to their cheaper cost, lower toxicity, and more controlled environmental effects.
Metal-free quantum dots have become a focus point in sensing research during the last decade due to their non-toxic and ecologically friendly features, which differ significantly from those of standard semiconductor metal quantum dots.21,22 Notably, graphitic carbon nitride dots have sparked widespread attention due to their bright fluorescence, remarkable stability, excellent water solubility, intrinsic biocompatibility, and non-toxicity.23–25 However, the electrocatalytic activity of quantum dots (QDs) is typically very low, and they generally exhibit insulating behaviour when employed as electrochemical surface modifiers. To overcome this limitation and enable dual-mode detection of p-NP via fluorescence and electrochemical methods, we have hydrothermally synthesized a liquid solution of graphitic carbon nitride sheet (g-CNS). In fluorescence detection we directly used the g-CNS; however, for electrochemical detection, we electrodeposited the synthesized g-CNS to form a conductive polymerized film on the electrode surface. This as-synthesized g-CNS showcased bi-functional roles as a fluorescent probe and as an electrode modifier for p-NP detection. To the best of our knowledge, this manuscript represents the first report of dual-mode p-NP detection utilizing a single g-CNS nanomaterial.
2. Chemicals and experimental procedures
Refer to the ESI.†3. Results and discussion
3.1 Material characterization: examining the g-CNS formation
The XRD pattern of synthesized g-CNS depicted two prominent peaks: one at 2θ ∼ 10° and a second broad peak near 2θ ∼ 23.7° corresponding to the (100) and (002) planes, respectively, as shown in Fig. 1(a). The appearance of a broad (002) plane at an angle lower than 2θ = 27° clearly hinted that g-CNS has both sp2 and sp3 characters.26 The increased broadness, decreased intensity and shift to lower 2θ values compared to bulk 2D g-CN manifest the formation of thin g-CN sheets.27 IR spectrum was investigated to obtain detailed insights into the functional group of the synthesized material. Fig. 1(b) shows various bands of g-CNS, starting with the characteristic N–H stretching vibrations near 3430 cm−1 and the N–H deformation band near 1630 cm−1. The presence of C–H sp2 and C–H sp3 stretching bands is also detected above and below 3000 cm−1. The band near 2070 cm−1 is due to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N characteristic stretching vibration. Several stretching bands appear due to C–O, C–N, and C–C. The characteristic out-of-plane bending mode28 of the g-CNS triazine ring also appears at a higher wavenumber near 830 cm−1. The shift to high wavenumbers or lower wavelengths further indicates weak interaction between the layers, resulting in the formation of thin individual g-CN nanosheets. The detailed characterization of XRD and IR confirms the successful synthesis of g-CNS. Next, the surface functionalities and chemical state of synthesized g-CNS was investigated using X-ray photoelectron spectroscopy (XPS).
N characteristic stretching vibration. Several stretching bands appear due to C–O, C–N, and C–C. The characteristic out-of-plane bending mode28 of the g-CNS triazine ring also appears at a higher wavenumber near 830 cm−1. The shift to high wavenumbers or lower wavelengths further indicates weak interaction between the layers, resulting in the formation of thin individual g-CN nanosheets. The detailed characterization of XRD and IR confirms the successful synthesis of g-CNS. Next, the surface functionalities and chemical state of synthesized g-CNS was investigated using X-ray photoelectron spectroscopy (XPS).
 | ||
| Fig. 1 (a) XRD; (b) FT-IR; and XPS spectra for (c) C 1s and (d) N 1s; (e) HR-SEM and (f) HR-TEM images of the g-CNS synthesized at 160 °C with 180 minutes reaction time. | ||
Fig. S1† shows the full survey spectra of g-CNS, showing three peaks originating near 284 eV, 399 eV, and 532 eV due to the presence of C 1s, N 1s, and O 1s, respectively. The deconvoluted C 1s spectra shown in Fig. 1(c) depicted three prominent peaks at 284.6 eV, 286.5 eV, and 288.6 eV binding energy.29,30 The peak at 284.6 eV is attributed to the surface adventitious carbon resulting from C–C coordination. The weak peak observed at 286.5 eV is plausibly attributable to nitrogen-related defects that are present in the g-CNS system and are associated with the N–C(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonding within the system. The peak at 288.6 eV is attributed to sp2 C atoms in the aromatic ring connected to the primary and secondary amines (carbon atoms coupled with three N neighbours, NC–(N)–NH, NC–(N)–NH2). Similarly, the N 1s spectra of the material showed three signals from distinct nitrogen environments in the triazine structural unit,30–32 with binding energies of 399.5 eV, 400.5 eV, and 401.5 eV, as shown in Fig. 1(d). The peak at 399.5 eV represents nitrogen atoms bound to three sp2 carbon atoms in the C–N network [N–(C)3]. The peak at 400.5 eV is attributed to N–H groups located at the open ends of the g-CN network. Quaternary nitrogen, which are oxidized nitrogen species, are detected in the N 1s spectra at a binding energy of 401.5 eV. The deconvoluted O 1s spectra in Fig. S2† show two peaks at binding energies of 531.6 eV and 532.8 eV. The peak at 531.6 eV is due to N–C–O functionality, while the peak at 532.8 eV is a characteristic peak of absorbed moisture (H2O).33 The morphological properties of g-CNS were also investigated using HR-SEM and HR-TEM. The HR-SEM image of g-CNS drop-cast onto a glass slide shows the existence of nanoscale plate-like structures, as shown in Fig. 1(e). To get a clearer picture of the thin sheet-like g-CN, HR-TEM was used, and the image is shown in Fig. 1(f). The HR-TEM image showcased the thin edges of the sheets, further validating the material's sheet-like structure. Dynamic light scattering (DLS) was used to determine the particle size of the as-synthesized material. Fig. S3† shows the distribution of particle size. DLS spectrum demonstrated two peaks centered at 51 and 220 nm. The area under the curve illustrated that ∼93% of the particles were included in the peak residing at 220 nm, indicating particle distribution in the 100–350 nm range.
N bonding within the system. The peak at 288.6 eV is attributed to sp2 C atoms in the aromatic ring connected to the primary and secondary amines (carbon atoms coupled with three N neighbours, NC–(N)–NH, NC–(N)–NH2). Similarly, the N 1s spectra of the material showed three signals from distinct nitrogen environments in the triazine structural unit,30–32 with binding energies of 399.5 eV, 400.5 eV, and 401.5 eV, as shown in Fig. 1(d). The peak at 399.5 eV represents nitrogen atoms bound to three sp2 carbon atoms in the C–N network [N–(C)3]. The peak at 400.5 eV is attributed to N–H groups located at the open ends of the g-CN network. Quaternary nitrogen, which are oxidized nitrogen species, are detected in the N 1s spectra at a binding energy of 401.5 eV. The deconvoluted O 1s spectra in Fig. S2† show two peaks at binding energies of 531.6 eV and 532.8 eV. The peak at 531.6 eV is due to N–C–O functionality, while the peak at 532.8 eV is a characteristic peak of absorbed moisture (H2O).33 The morphological properties of g-CNS were also investigated using HR-SEM and HR-TEM. The HR-SEM image of g-CNS drop-cast onto a glass slide shows the existence of nanoscale plate-like structures, as shown in Fig. 1(e). To get a clearer picture of the thin sheet-like g-CN, HR-TEM was used, and the image is shown in Fig. 1(f). The HR-TEM image showcased the thin edges of the sheets, further validating the material's sheet-like structure. Dynamic light scattering (DLS) was used to determine the particle size of the as-synthesized material. Fig. S3† shows the distribution of particle size. DLS spectrum demonstrated two peaks centered at 51 and 220 nm. The area under the curve illustrated that ∼93% of the particles were included in the peak residing at 220 nm, indicating particle distribution in the 100–350 nm range.
3.2 Investigating the fluorescence detection of para-nitrophenol by g-CNS
N or conjugated CN hybridized groups. These absorption features correspond to the π → π* electronic transitions typically observed in s-triazine rings of graphitic carbon, as well as the n → π* transitions associated with CN bonds in the graphitic carbon nitride structure.34 The carbon nitride sheet also demonstrated strong photoluminescence, exhibiting intense emissions across a broad range of excitation wavelengths. The fluorescence emission properties of g-CNS synthesized under various hydrothermal conditions (different temperatures and reaction times) were systematically investigated using an excitation wavelength of 330 nm and a laser slit width of 1 nm. As illustrated in Fig. S4(a),† an increase in hydrothermal temperature enhances the fluorescence intensity of the g-CNS. However, when the hydrothermal duration is extended, the fluorescence initially increases but subsequently declines beyond a certain threshold. Notably, three specific samples synthesized at 160 °C for 180 minutes, 160 °C for 360 minutes, and 180 °C for 180 minutes exhibited comparable fluorescence intensities. Based on these observations, the g-CNS synthesized at 160 °C for 180 minutes was selected for further optimization, as it was obtained under a relatively milder temperature and shorter reaction time. Subsequently, the optimization of different slit widths was investigated, as shown in Fig. S4(b).† The results clearly indicate that a laser with a slit width of 2 nm yields the highest fluorescence intensity, suggesting that it is the optimal condition for further studies. Furthermore, the excitation wavelength was systematically optimized for hydrothermally synthesized g-CNS (160 °C, 180 minutes) while maintaining a slit width of 2 nm. The excitation wavelength was varied between 300 and 360 nm, and the corresponding fluorescence emission intensities were analyzed. As depicted in Fig. S4(c),† the fluorescence intensity increases with excitation wavelength, reaching a maximum at 330 nm, beyond which a gradual decline is observed. Notably, the emission wavelength remains unchanged at higher excitation wavelengths, indicating an excitation-independent fluorescence behaviour. Typically, excitation-dependent fluorescence arises from nanoparticle size variations, distinct emissive traps, or surface defects associated with oxygen-rich functional groups. The findings suggest that excitation at 330 nm yields the most optimized emission with a 2 nm slit width and confirms the uniform size distribution of the synthesized g-CNS, with minimal structural defects. Moreover, these g-CNS exhibit a light brown appearance under visible light and appear bright blue upon exposure to UV light, as illustrated in Fig. S5.† The enhanced photoluminescence (PL) intensity is primarily attributed to electron-withdrawing nitrogen- and oxygen-rich edge functional groups originating from precursor materials. The fluorescence intensity of the as-synthesized material is known to be affected by the pH of the solution. Therefore, the effect of pH on the fluorescence intensity of g-CNS in the presence and absence of 1 mM p-NP solution was investigated in the pH range of 2 to 10. As shown in Fig. S6,† the fluorescence intensity of g-CNS increases with the rise in pH up to pH 8; after that, a sharp decline in intensity was observed. The fluorescence intensity of g-CNS was fully quenched by adding 1 mM p-NP solution at all pH values. p-NP is generally found in water and the optimal pH of water bodies is near 7. Consequently, subsequent fluorometric investigations were carried out at pH 7.
 | ||
| Fig. 2 (a) UV-Vis spectrum of the synthesized g-CNS; (b) p-NP detection in a linear range of 1–1000 μM concentration; (c) linear regression plot between fluorescence intensity vs. the concentration of p-NP (1–100 μM); (d) interference study of different analytes with g-CNS (Al3+, Co2+, Cr3+, Cu2+, Fe3+, Hg2+, Na+, K+, Mn2+, Ni2+, Pb2+, Zn2+, F−, Cl−, I−, OH−, SO42−, HSO4−, CO32−, CH3COO−, CN−, 2-aminophenol, 4-aminophenol, 2,4-dichlorophenol (DCP), 2,4-dinitrophenol, 2-nitrophenol, phenol, 2,4,6-trichlorophenol, and p-NP); (e) Stern–Volmer plot for the fluorescence quenching of g-CNS in the presence of p-NP; and (f) time-resolved fluorescence decay profile of g-CNS alone and g-CNS with p-NP. | ||
The fluorescence intensity was plotted against the p-NP concentration, as shown in Fig. 2(c). A linear correlation was observed within the concentration range of 1.0 to 100 μM, with an excellent coefficient of determination R2 = 0.9923. The limit of detection (LOD) was determined to be 36.76 nM. The proposed fluorescence sensing approach demonstrates superior or comparable analytical performance for p-NP detection relative to previously reported methods (Table S1†).
Most of the previously reported material given in the Table is synthesized by a multistep process, either by compositing with two materials or by doping one element to another. Furthermore, the reported material mostly relied on expensive metals (Au, Ag) and hazardous heavy metals like Cd. In contrast, our work reports a fairly simple, metal-free g-CNS synthesized via a single-step hydrothermal process as the fluorescent probe. The proposed method using a g-CNS fluorescent probe demonstrated superior sensitivity, as reflected by the lowest LOD and wide linear dynamic range.
The fluorescence intensity at 330 nm excitation was recorded in the absence and presence of potential interferents. Fig. 2(d) and Fig. S7† demonstrate that only p-NP effectively quenches the fluorescence of g-CNS among the other interferents, leading to approximately 50% decrease in fluorescence intensity with 100 μM p-NP. In addition to metal cations and anions, the other phenol molecules and nitrophenol isomers exhibited negligible fluorescence quenching effects, confirming the sensor's ability to distinguish p-NP from its structural analogues. Moreover, none of the tested anions, metal ions, or phenolic molecules significantly affected the fluorescence response of the g-CNS, further demonstrating the high specificity of the developed sensor.
Additionally, competitive selectivity studies were conducted by analyzing the fluorescence response of g-CNS in the presence of 100 μM p-NP along with the same concentrations of all the above-mentioned potential interferents (Fig. S8†). The results indicate that the presence of these interferents did not affect the fluorescence quenching behaviour of p-NP, reaffirming the exceptional selectivity of this sensing platform for p-NP detection. It is worth mentioning that the synthesized material shows interference-free results in the presence of 2-aminophenol and 4-aminophenol, which is very similar to p-NP. This is mainly because synthesized g-CNS already has –NH2 groups in its open-end structure, which plays a key role in the interaction between p-NP (–NO2 group) and synthesized material (–NH2 group) through hydrogen bonding. However, hydrogen bonding is not possible with molecules such as 2-aminophenol and 4-aminophenol. These findings highlight the robustness and specificity of the g-CNS based fluorescence sensor, making it a promising candidate for reliable p-NP detection in complex environmental matrices.
To further investigate the type of quenching, the time-resolved photoluminescence (TRPL) and quantum yield (Φ) measurements37 were performed with g-CNS (10 μL mL−1) alone and with p-NP (100 μM) (Table S2†). Fig. 2(f) shows the fluorescence decay curves of the g-CNS before and after the addition of p-NP. The bi-exponential fitted fluorescence decay profiles of the g-CNS is almost unchanged with or without p-NP. The fluorescence decay of g-CNS alone and with p-NP is almost unaltered, with decay constant values of 0.049 ns and 0.048 ns, respectively. However, quantum yield (Φ) values were found to decrease (Φ = 1.69 × 10−3 and 0.40 × 10−3 for g-CNS and g-CNS with p-NP, respectively), confirming substantial quenching upon the addition of p-NP. The decrease in the radiative (Kr) decay rate constant (34.42 × 106 s−1) and slight increase in the non-radiative (Knr) rate constant (2.03 × 1010 s−1) for g-CNS, in contrast to g-CNS with p-NP (Kr = 8.35 × 106 s−1 and Knr = 2.08 × 1010 s−1), indicates deactivation via non-radiative decay paths.38
The fitting of the Stern–Volmer equation, decreased quantum yield value, constant lifetime of the quenched fluorescence, and the modulation of UV-Vis absorbance suggested the presence of static quenching caused by the formation of a stable non-fluorescent interaction between g-CNS and p-NP.
 | ||
| Fig. 3 (a) Schematic illustration for the preparation of cotton swab test kit. (b) Fluorometric response of g-CNS adsorbed cotton swab test kits with the introduction of 100 μM of the following analytes: (1) Al3+, (2) Co2+, (3) Cr3+, (4) Cu2+, (5) Fe3+, (6) Hg2+, (7) Na+, (8) K+, (9) Mn2+, (10) Ni2+, (11) Pb2+, (12) Zn2+, (13) F−, (14) Cl−, (15) I−, (16) p-NP, (17) OH−, (18) SO42−, (19) HSO4−, (20) CO32−, (21) CH3COO−, (22) CN−, (23) 2-aminophenol, (24) 4-aminophenol, (25) 2,4-dichlorophenol (DCP), (26) 2,4-dinitrophenol, (27) 2-nitrophenol, (28) phenol, and (29) 2,4,6-trichlorophenol. Photographs were taken under UV light. | ||
3.3 Investigating the applicability of g-CNS for the electrochemical detection of para-Nitrophenol
Electrochemical sensors pose several advantages over fluorescence sensing, including the possibility of on-site detection, cost-effectiveness, and miniaturization. After using g-CNS as a fluorescence probe for indirect fluorometric sensing of p-NP, its applicability was investigated for direct electrochemical sensing of p-NP. The ability of g-CNS to specifically interact with p-NP was exploited by using it as a recognition element for developing the surface-modified sensors. | ||
| Fig. 4 (a) Electrodeposition cyclic voltammogram of g-CNS on GCE. Comparative SWV current responses of 1 mM K3[Fe(CN)6] solution recorded using g-CNS modified with (b) varying concentrations of g-CNS suspension and (c) different numbers of CV cycles. (d) HR-SEM image. (e) C 1s and (f) N 1s XPS spectra of the electrodeposited g-CNS|GCE interface. | ||
The thickness of the deposited g-CNS layer has a major impact on the electrode's electrocatalytic activity. Different g-CNS suspension concentrations were electrodeposited onto the electrode surface to establish the optimum thickness. Square wave voltammetry (SWV) was used to assess the electrocatalytic activity of the modified electrodes in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) solution of 1 mM K3[Fe(CN)6] and 1 M KCl.
:1 (v/v) solution of 1 mM K3[Fe(CN)6] and 1 M KCl.
Fig. 4(b) shows that electrodes treated with 100 μL of g-CNS solution showed lower current responses after 15 CV cycles, whereas electrodes treated with 250 μL, 500 μL, and 1000 μL of g-CNS solution showed comparable current responses. These results led to 250 μL being chosen as the optimal solution volume for additional electrodeposition optimization. Then, the influence of varying numbers of CV cycles on the electrocatalytic activity was examined, and the associated current responses were measured in the prepared K3[Fe(CN)6] in KCl solution. The current response began to increase as the number of CV cycles increased, peaking at 15 cycles, as seen in Fig. 4(c). After this, the current responses for cycles 25 and 30 were constant, suggesting that the electrocatalytic interface was saturated. Thus, 250 μL of g-CNS solution and 15 CV cycles were considered as the optimal electrodeposition conditions, offering the best possible compromise between film thickness and electrocatalytic activity.
The electrocatalytic performance of the electrodeposited g-CNS interface was systematically compared with the conventional drop-casting technique. The electrochemical responses of the bare GCE, drop-casted g-CNS modified GCE, and electrodeposited g-CNS modified GCE were analysed. As depicted in Fig. S9,† the electrodeposited interface demonstrated a markedly higher current response, indicating improved electron transfer kinetics and superior electrocatalytic activity. Conversely, the drop-casted g-CNS film exhibited even lower current responses than bare GCE, which can be attributed to material agglomeration, non-uniform deposition, and inadequate interfacial contact, together impeding the charge transfer.40 In contrast, electrodeposition provides precise control over the film thickness through adjustable deposition cycles and suspension concentration, resulting in a uniform surface growth of the modifier.41,42 Due to its enhanced electrochemical performance and structural homogeneity, the electrodeposited g-CNS coated scaffold (g-CNS|GCE) was selected for further characterization to investigate its compositional, morphological, and topographical attributes.
C), which indicates the presence of graphitic structures. The signal at 287.1 eV represents the distinctive triazine ring arrangement in graphitic carbon nitride, specifically the double-bonded carbon (N–CHN) associated with g-CNS. Furthermore, the peak at 288.8 eV is ascribed to sp2-hybridized carbon atoms inside the aromatic ring that are bound to both primary and secondary amines, notably carbon atoms coordinated with three nitrogen neighbours (NC–(N)–NH, NC–(N)–NH2). Fig. 4(f) shows the high-resolution XPS spectra of the N 1s region, which indicates three distinct peaks with binding energies of 399.6 eV, 400.6 eV, and 401.8 eV.43 The 399.6 eV peak indicates nitrogen atoms trigonally bound to three sp2-hybridized carbon atoms in the C–N network (N–(C)3). The peak at 400.6 eV can be attributed to the open-ended –NH2 functional group in the g-CNS structure. Furthermore, the signal at 401.8 eV is attributed to oxidised nitrogen species or quaternary nitrogen. The O 1s spectra show three peaks at binding energies of 531.5 eV, 532.9 eV and 536.5 eV, as shown in Fig. S11.† The first two peaks are similar to synthesized material peaks, while the third peak is mainly due to the strong binding of oxygen with the glassy carbon electrode. The combined examination of the C 1s and N 1s XPS spectra reveals that g-CNS is uniformly decorated on the GCE surface.
The HR-SEM image and the electrochemical response indicate a significant increase in the number of electroactive sites on the electro-functionalized substrate. This enhancement is expected to contribute to an increased electrochemical surface area and improved charge transfer kinetics. To quantitatively assess the electroactive surface area, a comparative evaluation was conducted for both the g-CNS|GCE and the bare GCE.
Cyclic voltammetry (CV) measurements were recorded by varying the scan rate from 5 to 100 mV s−1 in a 1:1 (v/v) mixture of 1 mM K3[Fe(CN)6] and 1 M KCl. The corresponding voltammograms depicted in Fig. S12(a and b)† exhibited two well-defined redox peaks, indicative of the reversible oxidation and reduction of the Fe2+/Fe3+ couple. The absence of a significant potential shift at higher scan rates suggests that the redox process is diffusion-controlled and follows a reversible electron transfer mechanism.
The Randles–Ševčík equation was applied at a controlled temperature of 25 °C to further quantify the electrochemically active surface area:26
| Ip = 2.69 × 105n3/2AD1/2Cν1/2 |
By evaluating the slope of the linear plot of Ipversus ν1/2, the electroactive surface area of the bare GCE was calculated to be 0.0431 cm2, while the electrodeposited g-CNS|GCE exhibited an increased electroactive surface area of 0.0533 cm2, approximately 1.24 times larger than that of the unmodified electrode. This enhancement in electroactive surface area directly contributes to the observed increase in current response following electrodeposition. The increase in available electroactive sites facilitates improved charge transfer kinetics, thereby enhancing the overall electrocatalytic efficiency of the modified electrode.
Cyclic voltammetry (CV) was used to compare the electrocatalytic behaviour of the g-CNS coated surface to the bare GCE for detecting p-NP. Fig. 5(a) shows the electrochemical reactions of 100 μM p-NP recorded using bare and g-CNS modified GCE. Both electrodes showed three distinct peaks:46–49R1 (∼−0.76 V), O1 (∼0.14 V), and R2 (∼0.08 V), which are consistent with earlier studies. In particular, the O1 and R2 peaks had reversible behavior, whereas the R1 peak was irreversible. The R1 signal is related to the reduction of p-NP to hydroxyaminophenol by the four electron (4e−) and four proton (4H+) transfer (Scheme 1). Alternatively, the O1 and R2 peaks constitute a redox pair that represents the reversible conversion of hydroxylaminophenol to nitrosophenol. During the forward potential sweep, the hydroxylamine group (–NHOH) is oxidized (O1) by the transfer of 2e− and 2H+, forming the nitroso group (–NO). During the reverse sweep, the nitroso group is reduced to hydroxylaminophenol (–NHOH) at peak R2via the identical 2e− and 2H+ transfers. The g-CNS modified GCE exhibits noticeably higher catalytic activity than the bare GCE. This is evidenced by increased current responses at all three peaks (R1, O1 and R2), indicating more efficient electron transfer and faster redox processes. Several factors contribute to the increased catalytic activity of the g-CNS coated electrode, including π–π stacking interactions between the aromatic rings of g-CNS and p-NP, hydrogen bonding between the –NH2 groups of g-CNS and the –NO2 groups of p-NP, electrostatic interactions, and larger surface area. These parameters aid in the effective diffusion of p-NP to the coated interface, increasing electron and proton transfer processes and overall electrochemical performance.
 | ||
| Fig. 5 (a) Comparative CV of 100 μM p-NP recorded using bare GCE and g-CNS|GCE. (b) CV of 100 μM p-NP as a function of varying scan rates ranging from 0.02 V s−1 to 0.2 V s−1 recorded using g-CNS|GCE. (c) Plot of logIpvs. logν for the R1 peak. (d) Comparative SWV graph of 100 μM p-NP recorded using bare GCE and g-CNS|GCE. (e) SWV peaks corresponding to increasing concentrations of p-NP ranging from 1 μM to 100 μM recorded using g-CNS|GCE. (f) Linear regression plot between peak current and concentration of p-NP. All the measurements were carried out using phosphate buffer of pH 7 as supporting electrolyte. | ||
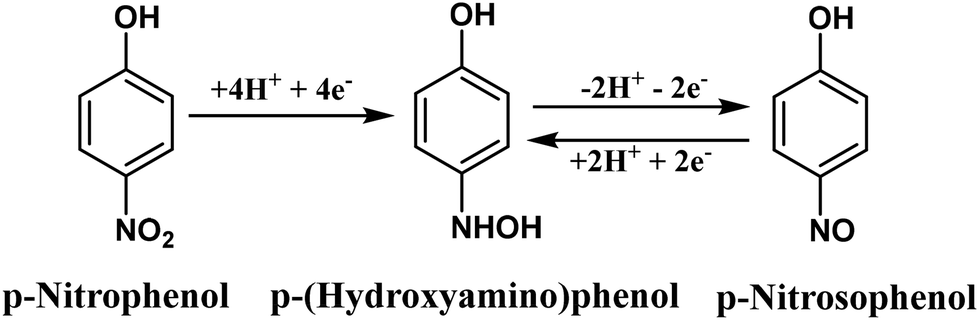 | ||
| Scheme 1 Electrochemical reduction mechanism of p-NP. | ||
The effect of the pH of the electrolyte on the reduction peak of 45 μM p-NP was investigated. As the pH increases, the reduction peak shifts towards a more negative potential and gives a maximum peak current at pH 7, as shown in Fig. S13(a).† On plotting peak potential as a function of pH (Fig. S13(b)†), we obtained a linear plot with a linear regression equation as given below:
| Ep = −0.047 pH − 0.35 (R2 = 0.99) |
The obtained slope is in good agreement with the Nernst equation at 25 °C. For the equal number of electron and proton transfers, the theoretical slope is predicted as −0.059 V/pH. Hence, the pH study is in good agreement with the previously reported 4e− and 4H+ transfer mechanism for the conversion of the –NO2 group to the –NHOH group.50
A scan rate analysis was carried out for a 100 μM p-NP solution in phosphate buffer (pH 7) with scan rates ranging from 0.02 V s−1 to 0.2 V s−1 to obtain additional understanding of the underlying mechanisms and processes. Fig. 5(b) shows that the current for all three p-NP peaks increases as the scan rate increases. For all three peaks, a linear relationship between the peak current (Ip) and the square root of the scan rate (ν1/2) was found, suggesting that the electrochemical process is diffusion-controlled (Fig. S14(a–c)†). To further comprehend the behaviour occurring at the g-CNS coated interface, a scan rate study was recorded again in the range of the R1 peak (Fig. S14(d)†), and a plot of logIpversus logν was constructed as shown in Fig. 5(c). According to theoretical models,51 the slopes of the logIpvs. logν plots for pure diffusion-controlled and pure adsorption-controlled processes are expected to be 0.5 and 1, respectively. The process is classified as pure adsorption-controlled if the slope is between 0.75 and 1.0, mixed adsorption-diffusion-controlled if the slope is between 0.5 and 1, diffusion-controlled if the slope is between 0.2 and 0.5, and mixed diffusion-adsorption if the slope is between 0.6 and 0.75. The slope found in the present study is 0.24, indicating that the reduction process occurring at peak R1 is diffusion-controlled. To calculate the number of electron transfers during the irreversible reduction process R1, a graph is plotted between ER1 and the natural logarithm of the scan rate (Fig. S15†). The linear regression equation between ER1 and ln(ν) was obtained as follows:
| ER1(p-NP)/V = −0.0301ln(ν) − 0.76 (R2 = 0.99) |
The above linear equation was compared with Nicholson's equation,52,53 which is generally used for the irreversible reaction and is given below:
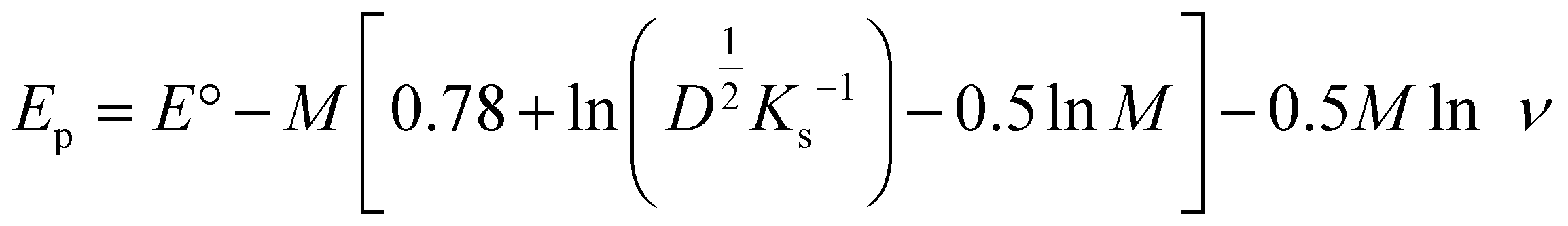
For the oxidation process:
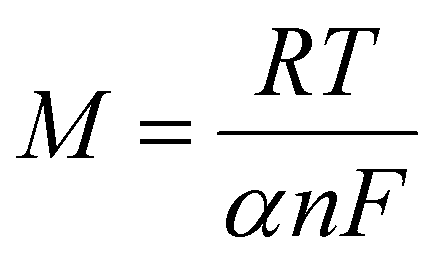
For the reduction process:

After comparing the slope value for the reduction process R1 and substituting the R, T, and F values for 25 °C, we obtained the value of (1 − α)n, which equates to 0.43. For an irreversible process, the value of α can be calculated with the help of the following given formula:54

Using the CV measurement conducted at 0.2 V s−1, the values of Ep and Ep/2 are obtained as −0.7180 and −0.6647, respectively. Inserting these two values in the above equation, we obtain the value of α = 0.894. Inserting this value of α in (1−α)n, which is 0.43, results in the 4 electron transfers. From the pH study, it is already clear that the number of electron transfers is equal to the number of proton transfers.
For reversible processes (O1 and R2), the values of |Ep − Ep/2| are independent of the scan rate, and the value of n can be determined using the following equation:51
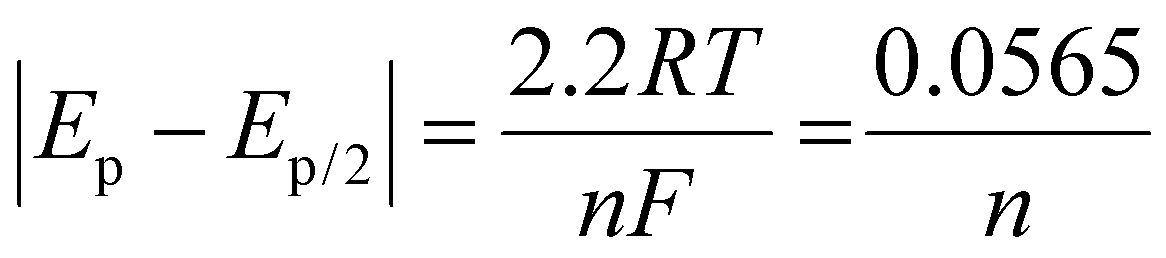
Taking the CV peak of the 0.2 V s−1 scan rate from Fig. 5(b) into consideration, the value of EP and Ep/2 for O1 is obtained as 0.143 and 0.110, respectively and that for R2 is obtained as 0.99 and 0.132, respectively. Putting these values in the above equation, the number of electron transfers for both the oxidation and reduction processes equals 1.71. The number of electron transfers will always be an integer. Hence, we can conclude that the number of electron transfers is 2 for both the O1 and R2 processes. The pH study and scan rate study confirms the p-NP reduction using the above-proposed mechanism.
SWV was used for the quantitative p-NP detection. Fig. 5(d) shows the SW voltammograms for a 100 μM p-NP solution using both bare and modified GCE. The comparative analysis of the square wave voltammograms revealed that the current response for the g-CNS|GCE was approximately 1.7 times higher than that for the bare GCE. Moreover, the g-CNS|GCE exhibited a p-NP peak at −0.66 V, compared to −0.74 V for the bare GCE. This shift of approximately 0.08 V underscores the accelerated redox process, thereby demonstrating the electrocatalytic effect of g-CNS. The enhanced electroactive surface area of the electrode, coupled with the unique π–π interactions between the g-CNS and the aromatic ring of the p-NP molecule, and the hydrogen bonding contribute to the significant increase in current and potential shift. For a more detailed quantitative analysis, a calibration curve was constructed by examining the correlation between the p-NP concentration and the corresponding current responses for the bare and modified GCE electrodes. The results revealed a linear increase in the peak current with rising p-NP concentrations for g-CNS|GCE electrodes, as illustrated in Fig. 5(e and f). The relationship between peak current (Ip) and p-NP concentration (C) was found to follow the linear regression equations provided below.

The results revealed that the g-CNS|GCE demonstrated a significantly lower LOD of 218 nM. To further assess the advantages of the newly developed sensor, its performance was compared with several previously reported p-NP electrochemical sensors, which utilized g-CNS or other similar surface modifications. As detailed in Table S4,† the proposed sensor exhibited superior attributes in terms of ease of fabrication and sensitivity. Additionally, while all methodologies reported in the literature involve the separate synthesis of various components followed by their combination and drop-casting onto the electrode, the proposed approach enhances repeatability, practicality, and scalability by employing a single-step electrodeposition technique, allowing precise control over the layer thickness.
:1 (v:v) solution of 1 mM K3[Fe(CN)6] and 1 M KCl. The peak current responses obtained from the first 40 SWV cycles using the same g-CNS|GCE are presented in Fig. 6(b), confirming the stability of the electrode during repeated measurements. The stability studies were also carried out in the presence of p-NP. Fig. S18(a)† presents the 15 consecutive SWV of 45 μM p-NP in pH 7 phosphate buffer using g-CNS|GCE. There was no substantial decay in the current or shift in the potential, indicating the stability of g-CNS|GCE for multiple scans.
 | ||
| Fig. 6 (a) Long cycle CV stability of g-CNS|GCE electrode. (b) Repetitive SWV current response. (c) Reproducibility study at different GCE's after modification with g-CNS in a 1:1 (v:v) solution of 1 mM K3[Fe(CN)6] and 1 M KCl. | ||
To verify the reproducibility of the electrodeposition method, SWV measurements were performed on three separate GCE electrodes simultaneously modified with g-CNS using the same electrodeposition protocol. The electrochemical responses of the modified GCE were recorded using 1 mM K3[Fe(CN)6] solution with 1 M KCl and a 45 μM p-NP with a phosphate buffer solution of pH 7, respectively. The current responses obtained using three different electrodes are shown in Fig. 6(c) and Fig. S18(b).† The SWV demonstrates exceptional reproducibility, validating the reliability of the proposed electrodeposition protocol for consistent and uniform modification of the GCE.
4. Conclusions
This study presents the synthesis and dual-mode application of a graphitic carbon nitride sheet (g-CNS) for the detection of p-nitrophenol (p-NP). The synthesized g-CNS material was extensively characterized using various analytical techniques to confirm its properties. The manuscript is organized into two primary sections: fluorescence-based and electrochemical-based p-NP detection.For fluorescence detection, the fluorescence properties of g-CNS were first optimized by adjusting key experimental parameters: excitation wavelength, slit width, and pH. The g-CNS exhibited the most intense fluorescence at an excitation wavelength of 330 nm and a slit width of 2 nm. The fluorescence intensity of g-CNS was found to decay in the presence of p-NP, leading to its indirect fluorometric sensing, demonstrating a limit of detection (LOD) of 36.76 nM. The detection mechanism, interference studies, and analysis of real samples were comprehensively discussed.
In the electrochemical detection approach, a step-by-step, optimized electrodeposition protocol was employed to modify the working electrode, offering an advantage over the conventional drop-casting method. The surface, chemical composition, and electrochemical behavior of the modified electrode revealed successful electrodeposition of g-CNS exhibiting enhanced electrochemical characteristics. The p-NP reduction peak at −0.66 V in SWV enabled direct voltammetric sensing of p-NP using g-CNS modified GCE. The proposed method resulted in a significantly lower LOD of 218 nM in a linear dynamic range of 1–100 μM. Furthermore, the modified electrode was also tested for p-NP sensing in real water samples and in the presence of 25 interfering species. The proposed electrodeposition protocol demonstrated superior stability, reusability, and reproducibility, making it a promising candidate for reliable and consistent detection of p-NP.
In conclusion, the g-CNS based sensors offer a practical and sensitive platform for p-NP detection, combining fluorescence and electrochemical techniques with superior performance, high reproducibility, and practical applicability in real-world samples.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. Singh is thankful to UGC for awarding the SRF. A. Singh is thankful to the University Grants Commission, New Delhi, for the UGC Research Fellowship (R/Dev./IX-Sch. (BHU Res. Sch.)2021-22/37605). M. Patel is thankful to the UGC for providing the JRF fellowship. V. P. Singh is thankful to Banaras Hindu University for a grant under the IoE Scheme (Dev. Scheme No. 6031). Rosy is thankful to IIT (BHU) Varanasi for providing the seed funding to carry out this project.We are also thankful to the Central Instrumentation Facility (CIF), IIT (BHU) Varanasi, for providing access to the sophisticated instrumental facilities.
References
- A. O. Cardoso Juarez, E. Ivan Ocampo Lopez, M. K. Kesarla and N. K. R. Bogireddy, ACS Omega, 2024, 9, 33335–33350 Search PubMed.
- M. S. Qatan, F. Arshad, M. Miskam and G. A. Naikoo, Int. J. Environ. Sci. Technol., 2024, 21, 5247–5268 Search PubMed.
- M. Ahmaruzzaman, S. R. Mishra, V. Gadore, G. Yadav, S. Roy, B. Bhattacharjee, A. Bhuyan, B. Hazarika, J. Darabdhara and K. Kumari, J. Environ. Chem. Eng., 2024, 12, 112964 Search PubMed.
- P. Deng, Z. Xu, Y. Feng and J. Li, Sens. Actuators, B, 2012, 168, 381–389 Search PubMed.
- N. Xiao, S. G. Liu, S. Mo, N. Li, Y. J. Ju, Y. Ling, N. B. Li and H. Q. Luo, Talanta, 2018, 184, 184–192 Search PubMed.
- B. B. Xing, Y. S. Wang, T. Zhang, J. Y. Liu, H. Jiao and L. Xu, Mater. Adv., 2025, 6, 756–765 Search PubMed.
- D. B. Barr, W. E. Turner, E. DiPietro, P. C. McClure, S. E. Baker, J. R. Barr, K. Gehle, R. E. Grissom, R. Bravo, W. J. Driskell, D. G. Patterson, R. H. Hill, L. L. Needham, J. L. Pirkle and E. J. Sampson, Environ. Health Perspect., 2002, 110, 1085–1091 Search PubMed.
- M. K. Trivedi, A. Branton, D. Trivedi, G. Nayak, K. K. Sethi and S. Jana, Am. J. Chem. Eng., 2016, 4, 68–77 Search PubMed.
- X. Guo, Z. Wang and S. Zhou, Talanta, 2004, 64, 135–139 Search PubMed.
- J. Fischer, J. Barek and J. Wang, Electroanalysis, 2006, 18, 195–199 Search PubMed.
- A. Brega, P. Prandini, C. Amaglio and E. Pafumi, J. Chromatogr. A, 1990, 535, 311–316 Search PubMed.
- Z. Kitanovski, I. Grgić, R. Vermeylen, M. Claeys and W. Maenhaut, J. Chromatogr. A, 2012, 1268, 35–43 Search PubMed.
- F. Huo, H. Yuan, X. Yang, D. Xiao and M. M. F. Choi, Anal. Sci., 2011, 27, 879–879 Search PubMed.
- X. Peng, Y. Wang, Z. Luo, B. Zhang, X. Mei and X. Yang, Microchem. J., 2021, 170, 106735 Search PubMed.
- Y. Li, Y. Ma, E. Lichtfouse, J. Song, R. Gong, J. Zhang, S. Wang and L. Xiao, J. Hazard. Mater., 2022, 421, 126718 Search PubMed.
- S. D. Lawaniya, G. Pandey, Y. Yu and K. Awasthi, Nanoscale, 2024, 16, 13915–13924 Search PubMed.
- S. Gond, P. Yadav, A. Singh, S. Garai, A. Shekher, S. C. Gupta and V. P. Singh, Org. Biomol. Chem., 2023, 21, 4482–4490 Search PubMed.
- A. Das and S. Biswas, Sens. Actuators, B, 2017, 250, 121–131 Search PubMed.
- J. Li, Y. Liang, S. Wu, Y. Zhang, M. Zhu and E. Gao, Inorg. Chem. Commun., 2022, 143, 109724 Search PubMed.
- J. Fu, S. Zhou, P. Zhao, X. Wu, S. Tang, S. Chen, Z. Yang and Z. Zhang, Biosens. Bioelectron., 2022, 198, 113848 Search PubMed.
- W. Zhang, H. Zhong, P. Zhao, A. Shen, H. Li and X. Liu, Food Control, 2022, 133, 108591 Search PubMed.
- M. J. Deka, D. Chowdhury and B. K. Nath, Carbon Lett. 2022 325, 2022, 32, 1131–1149 Search PubMed.
- X. Lu, L. Liao, Y. Cui, L. Cheng, M. Zhang, F. Lv, C. Zhu, J. Liu, W. Kong, G. Yu, F. Liu, Y. Yang and W. Li, J. Lumin., 2024, 269, 120491 Search PubMed.
- S. Pattnayak, U. Sahoo, S. Choudhury, P. Aparajita and G. Hota, J. Photochem. Photobiol. A Chem., 2023, 443, 114819 Search PubMed.
- X. Guo, J. Yang, Y. Wei, L. Wang and J. Pi, ACS Appl. Nano Mater., 2022, 5, 4230–4240 Search PubMed.
- A. K. Singh, A. Singh and Rosy, Microchem. J., 2024, 204, 110998 Search PubMed.
- R. Banavath, A. Abhinav, S. S. Nemala, R. Srivastava and P. Bhargava, Mater. Adv., 2022, 3, 9019–9029 Search PubMed.
- A. K. Singh, P. Keshari, A. Saroj, V. Ramanathan and Rosy, Surf. Interfaces, 2023, 42, 103316 Search PubMed.
- E. Alwin, W. Nowicki, R. Wojcieszak, M. Zieliński and M. Pietrowski, Dalton Trans., 2020, 49, 12805–12813 Search PubMed.
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol., A, 2021, 39, 13204 Search PubMed.
- B. Choudhury, K. K. Paul, D. Sanyal, A. Hazarika and P. K. Giri, J. Phys. Chem. C, 2018, 122, 9209–9219 Search PubMed.
- A. K. Singh, R. Yadav, A. Singh and Rosy, Nano-Struct. Nano-Objects, 2024, 39, 101284 Search PubMed.
- S. Wang, F. Wang, Z. Su, X. Wang, Y. Han, L. Zhang, J. Xiang, W. Du and N. Tang, Catalysts, 2019, 9, 439 Search PubMed.
- M. M. Xavier, N. N. Adarsh, P. R. Nair and S. Mathew, ACS Omega, 2021, 6, 22840–22847 Search PubMed.
- F. Wang, X. Fu, X. Chai, Q. Han, H. Wang and Q. Hao, Microchem. J., 2021, 168, 106389 Search PubMed.
- E. Ciotta, P. Prosposito and R. Pizzoferrato, J. Lumin., 2019, 206, 518–522 Search PubMed.
- P. Yadav, A. Singh, G. Kumar, S. Singh and V. P. Singh, Spectrochim. Acta, Part A, 2025, 329, 125557 Search PubMed.
- A. Singh, P. Yadav, S. Singh, P. Kumar, S. Srikrishna and V. P. Singh, J. Mater. Chem. C, 2023, 11, 13056–13066 Search PubMed.
- A. K. Singh, Tarul, A. P. S. Bais and Rosy, Microchem. J., 2024, 203, 110833 Search PubMed.
- A. K. S. Kumar, Y. Zhang, D. Li and R. G. Compton, Electrochem. Commun., 2020, 121, 106867 Search PubMed.
- A. K. Singh, R. Yadav, A. Singh and Rosy, Mater. Chem. Phys., 2024, 328, 129941 Search PubMed.
- A. K. Singh, Tarul, M. Patel and Rosy, J. Environ. Chem. Eng., 2025, 13, 115476 Search PubMed.
- N. Hellgren, R. T. Haasch, S. Schmidt, L. Hultman and I. Petrov, Carbon, 2016, 108, 242–252 Search PubMed.
- C. Li, X. Yang, B. Yang, Y. Yan and Y. Qian, Mater. Chem. Phys., 2007, 103, 427–432 Search PubMed.
- X. Li, J. Zhang, L. Shen, Y. Ma, W. Lei, Q. Cui and G. Zou, Appl. Phys. A, 2009, 94, 387–392 Search PubMed.
- Renu, Komal, R. Kaur, J. Kaur, Jyoti, V. Kumar, K. B. Tikoo, S. Rana, A. Kaushik and S. Singhal, J. Electroanal. Chem., 2021, 887, 115161 Search PubMed.
- Q. Y. Wang, H. R. Li, Z. Y. Zheng, H. Xu, J. M. Tao and S. S. Li, Microchem. J., 2025, 208, 112359 Search PubMed.
- H. R. Li, H. Xu, Q. Y. Wang and S. S. Li, Talanta, 2025, 285, 127360 Search PubMed.
- G. B. P. Ngassa, I. K. Tonlé and E. Ngameni, Talanta, 2016, 147, 547–555 Search PubMed.
- T. Yu, K. Herdman, P. R. Kasturi and C. B. Breslin, Microchem. J., 2023, 195, 109453 Search PubMed.
- N. Jaiswal, I. Tiwari, C. W. Foster and C. E. Banks, Electrochim. Acta, 2017, 227, 255–266 Search PubMed.
- X.-L. Cheng, X. Xia, Q.-Q. Xu, J. Wang, J.-C. Sun, Y. Zhang and S.-S. Li, Sens. Actuators, B, 2021, 348, 130692 Search PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 Search PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Methods: Fundamentals and Applications, Wiley, 2022, 3rd edn, ISBN: 978-1-119-33405-7 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00874c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |