
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and structural coupling of pentacene on NiO(001)†
Jonah Elias
Nitschke
 a,
David Maximilian
Janas
a,
Stefano
Ponzoni
a,
Michele
Capra
b,
Elena
Molteni
c,
Andrea
Picone
b,
Alessio
Giampietri
b,
Alessandro
Ferretti
b,
Shuangying
Ma
c,
Alberto
Brambilla
b,
Giovanni
Zamborlini‡
*a,
Guido
Fratesi
*c and
Mirko
Cinchetti
a
a,
David Maximilian
Janas
a,
Stefano
Ponzoni
a,
Michele
Capra
b,
Elena
Molteni
c,
Andrea
Picone
b,
Alessio
Giampietri
b,
Alessandro
Ferretti
b,
Shuangying
Ma
c,
Alberto
Brambilla
b,
Giovanni
Zamborlini‡
*a,
Guido
Fratesi
*c and
Mirko
Cinchetti
a
aTU Dortmund University, Otto-Hahn-Straße 4, 44227 Dortmund, Germany. E-mail: giovanni.zamborlini@uni-graz.at
bDipartimento di Fisica, Politecnico di Milano, piazza Leonardo da Vinci 32, 20133 Milano, Italy
cDipartimento di Física “Aldo Pontremoli”, Università degli Studi di Milano, via Celoria 16, 20133 Milano, Italy. E-mail: guido.fratesi@unimi.it
First published on 30th June 2025
Abstract
Transition-metal oxides (TMOs) are pivotal in modern applications, with recent years seeing intensified research into their interplay with molecular layers as well as the potential of antiferromagnetic TMOs in spintronic applications. In this work we combine both approaches and investigate the adsorption of pentacene on the (001) surface of NiO. By employing a variety of methods such as scanning tunneling microscopy, low energy electron diffraction, angle-resolved photoelectron spectroscopy and density functional theory, we extract the geometrical arrangement of the molecules and their energy level-alignment. Induced by the substrate–molecule interaction, pentacene forms a self-assembled monolayer in a superstructure commensurate with the NiO substrate. Through photoemission orbital tomography, we identify the first three highest occupied molecular orbitals (HOMO, HOMO−1 and HOMO−2) in the photoemission spectra of the NiO/pentacene interface. The absence of the lowest unoccupied molecular orbital (LUMO) suggests negligible charge transfer at the interface, a finding supported by calculations. Nevertheless, we can observe an induced degeneracy of the HOMO−1 and HOMO−2 orbitals as well as an accumulation of molecular electron density toward the substrate. This preservation of the molecules free electron character of frontier orbitals points to potential applications in the optical control of THz spin dynamics in antiferromagnetic NiO, opening a promising pathway for engineering molecule-based functionalization of antiferromagnetic surfaces.
1 Introduction
Metal oxides, particularly transition metal oxides (TMOs), hold great promise in spintronics due to their adjustable composition, rich spin–orbital physics, and highly tunable electronic structure.1,2 These characteristics enable strong magnetoelectric coupling and efficient spin-charge interconversion,3 making them excellent candidates for spintronic devices. In particular, the use of antiferromagnetic (AF) insulators offers several distinct advantages.4,5 These materials allow the propagation of pure spin currents without power losses caused by Joule heating, ensuring more energy-efficient operation. The propagation of spin currents in AF oxides such as NiO6,7 and CoO8 has already been demonstrated with the observation of even μm-scales in Fe2O3.9 Furthermore, they are robust against external magnetic fields, making them suitable for a wide range of operating environments, and they can work in the terahertz (THz) range, which enables ultrafast processing speeds.10–12To fully exploit the capabilities of TMOs, recent research has shifted towards controlled structural engineering. In this context, the integration of organically functionalized solid surfaces has emerged as a powerful approach, leveraging the flexibility of organic chemistry to modify substrate properties precisely.13,14 For example, at interfaces between magnetic materials and organic molecular layers, the molecular layer can influence the spin polarization of the substrate surface.15
The synergy between TMOs and organic molecules has already been introduced to various fields such as catalysis, organic electronics, and solar cells.16 For spintronics, this combination offers significant promise, especially given recent progress in understanding and tailoring interfacial properties. Studies on the adsorption of organic molecules on TMO surfaces have revealed a universal energy level alignment, where the molecule's highest occupied orbital aligns relative to the substrate's Fermi level, governed solely by the substrate's work function and the molecule's ionization energy.16,17 As the work function of TMOs is a highly tunable parameter due to the possibility to introduce oxygen defects in a very controlled way,18 this offers an exciting opportunity to actively tailor interfacial properties to meet specific requirements.
Building on these advances, this work explores the interaction between a prototypical aromatic molecule —pentacene—and the (001) surface of the antiferromagnetic TMO nickel oxide (NiO). NiO19 is an antiferromagnetic Mott–Hubbard insulator with a high Néel temperature of 525 K and therefore allows for the study of magnetic phenomena even at room temperature. In the AF state, NiO consists of ferromagnetic (111) planes that are antiferromagnetically stacked along the [111] direction, creating rows of nickel atoms with alternating magnetization at the (001) surface. Importantly, NiO's electronic and magnetic properties can be tuned via defect engineering, enhancing its versatility for fundamental and applied studies.20 From a THz spintronics perspective, NiO is particularly attractive due to its exciton–magnon transition,12 which can be resonantly pumped to drive coherent magnons on the femtosecond timescale. Given that organic molecules host robust excitonic states, functionalizing NiO with molecules such as pentacene could provide new levers for initiating and controlling ultrafast spin dynamics in AF oxides. On the molecular side, pentacene, a polycyclic aromatic hydrocarbon (C22H14), is a benchmark p-type organic semiconductor known for its exceptionally high hole mobility.21 Its adsorption on various solid surfaces has been extensively studied, offering valuable insights into its potential role in hybrid organic–inorganic systems.22–24
In this study, we examine the crystalline, morphological, and electronic properties of pentacene adsorbed on NiO(001) grown on Ag(001) single crystals. Using low-energy electron diffraction (LEED), angle-resolved photoemission spectroscopy (ARPES) and scanning tunneling microscopy (STM), and photoemission orbital tomography (POT),25,26 we determine the structural ordering, molecular orientation, and interfacial energy-level alignment. First-principles density functional theory (DFT) calculations—including Hubbard-corrected DFT+U to treat Ni 3d electrons accurately—provide further insight into adsorption energetics and the electronic/magnetic interface properties, with a focus on the molecular contributions to the overall electronic structure.
Our combined experimental and theoretical approach reveals the formation of a well-ordered, self-assembled monolayer of pentacene on NiO(001), exhibiting a universal energy level alignment characteristic of a weak molecule–substrate interaction. Noteworthy, the pentacene molecules nonetheless adopt a commensurate registry with the NiO surface, indicating that the substrate still influences molecular ordering. Additionally, the frontier molecular orbitals largely preserve their free-molecule character, with only minimal charge redistribution toward the substrate. This retention of molecular character at the HOMO–LUMO states ensures the presence of an exciton localized at the NiO interface, which could serve as a lever for optically controlling THz spin dynamics in NiO, thereby opening new avenues for the development of hybrid spintronic architectures.
2 Methods and experimental details
2.1 Sample preparation
NiO(001) thin films were grown on an Ag(001) single crystal (from MaTeck GmbH, Germany), which was cleaned in ultra-high vacuum (UHV) by repeated cycles of sputtering with Ar+ ions at 2 keV and subsequent annealing up to 770 K for 15 min. The cleanliness of the surface was checked by means of Auger electron spectroscopy (AES) and low-energy electron diffraction (LEED). Afterwards, 10 layers of NiO (2.4 nm) were grown onto Ag(001) by molecular beam epitaxy in our preparation chamber (base pressure better than 8 × 10−11 mbar).27 The cell for the Ni deposition was pre-calibrated using a quartz micro balance and set to a deposition rate of 0.4 Å min−1. During the deposition, the sample was held at a temperature of 470 K in an oxygen atmosphere of 7 × 10−7 mbar. Afterwards, the sample was annealed for 15 min at a temperature of 770 K with an oxygen background pressure of 1 × 10−7 mbar. This post-growth annealing in oxygen atmosphere proved to be a crucial step for increasing the quality of the NiO(001) film, leading to the formation of terraces larger than 50 nm (see ESI Fig. S1† for details). Although the overall growth recipe is similar to the one employed by Das et al.,27 in our preparation we used a higher ratio of evaporation rate to oxygen partial pressure and we performed a post-annealing to improve the lateral order.Pentacene (5A) molecules were sublimated in situ via a Knudsen-type evaporator (KENTAX GmbH). At first, the deposition rate was estimated by depositing 5A on a Cu(001) surface. By stepwise evaporation and subsequent monitoring of the sharpness and intensity of the LEED spots stemming from the 5A overlayer, the optimum rate was estimated as 0.125 monolayer per min with a deposition temperature of 558 K. Afterwards, the same parameters were used to grow a saturated monolayer (ML) of 5A on the NiO(001) surface. STM measurements were performed in another UHV system, with the very same preparation procedure for the NiO layer (similarly checked by AES and LEED). In this case, molecules were sublimated in a connected UHV evaporation chamber from a home-made evaporation cell. The evaporation rate was estimated by a quartz microbalance and checked afterwards by STM.
2.2 Methods
The momentum-resolved photoemission data of the pristine Ag(001) and NiO(001)/Ag surface as well as of the molecular overlayer were acquired at room temperature using a KREIOS Photoemission Electron Microscope (PEEM), from SPECS GmbH, operated in momentum mode.28,29 The instrument allows different reciprocal space magnifications, the lowest of which results in an accessible photoelectron parallel momentum to maximum ±3 Å−1. For the reported momentum maps, we employed the second lowest k-magnification (total available parallel momentum: ±2.2 Å−1). The PEEM is coupled to a UV-light source (SPECS GmbH) which provides photons with an energy of 21.2 eV (He I α) impinging on the sample under an angle of 60° with respect to the surface normal.Real space imaging of the 5A molecular superstructure adsorbed on the NiO(001) surface was achieved by means of an Omicron Q-PLUS STM variable temperature microscope. Both large scale and molecular orbital images of pentacene were acquired at RT in constant current mode. STM measurements were performed using commercially available Q-PLUS tips (the tip material is W), purchased from Scienta Omicron.
2.3 Computational details
We performed ab initio calculations within Density Functional Theory (DFT), as implemented in the Quantum ESPRESSO distribution,30,31 with plane waves and ultrasoft pseudopotentials.32 We treat the DFT exchange–correlation (xc) term by using the vdW-DF-c09 functional,33,34 also including van der Waals molecule–surface interaction. The substrate is modelled by a slab approach, including 3- and 5 NiO(001) layers for configuration search and for the study of electronic properties, respectively. We remark that integrated quantities (such as atomic charges, adsorption energies) are weakly dependent on the slab thickness. On-site correlations at Ni atoms are approximated by the DFT+U method, with U = 4.0 eV. The chosen U value of 4.0 eV for transition metal atoms is based on the optimal match between DFT relative energy levels and many-body (GW) ones in the valence band,19 whereas fitting the bandgap would mis-reproduce the properties, e.g. of the valence band maximum.The kinetic energy cutoffs were set to 40 Ry for the plane-wave expansion and 200 Ry for the effective potential and charge density. We initialize the system magnetization so that the nonequivalent Ni sites have opposite spins, then this is optimized self-consistently without applied constraints. Equilibrium geometries were obtained by relaxing the molecule (when present) and the topmost NiO(001) surface layer only, with default thresholds (0.0001 Ry on total energies and 0.001 Ry au−1 on forces). The vacuum portion was set to about 13 Å. The surface Brillouin zone was sampled by the Γ point for adsorption structure optimization and by a (4 × 2) k-mesh for the evaluation of the electronic density of states (DOS) and for the simulated STM in the large cell with two molecules. These are evaluated within the Tersoff–Hamann scheme35 as a map of the local density of electronic states, integrated between the Fermi energy EF and EF plus the bias potential.
3 Results and discussion
3.1 Structural characterization
First, we characterize the geometric properties of a self-assembled monolayer of pentacene atop NiO(001) by means of STM. The technique allows to determine both the azimuthal orientation of the molecule and the resulting unit-cell upon self-assembly. A large scale STM image (Fig. 1a) shows that 5A grows in a well-ordered fashion across different NiO terraces. Higher-resolution images obtained by zooming in on the boundary between two terraces (Fig. 1b) enables the identification of single pentacene molecules within the organic overlayer, as well as their alignment with respect to the high symmetry directions of the substrate surface. Our STM images reveal that the molecules, within a single rotational domain, align slightly off with respect to the [110] direction of NiO, by 3.5° ± 2° (also confirmed by the momentum resolved images, which will be discussed later). Knowing the orientation and size of the substrate unit cell, it is also possible, from the high-resolution STM images, to determine the unit cell of the molecular overlayer and provide the corresponding epitaxial matrices. For 5A/NiO(001), we obtained the following matrix: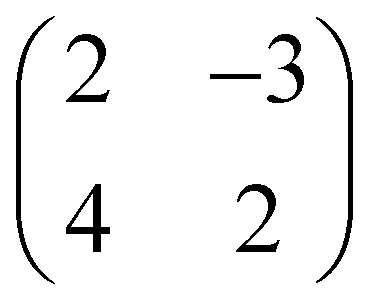 , to which we can associate the corresponding symmetry-equivalent domains
, to which we can associate the corresponding symmetry-equivalent domains  , and
, and  .
.
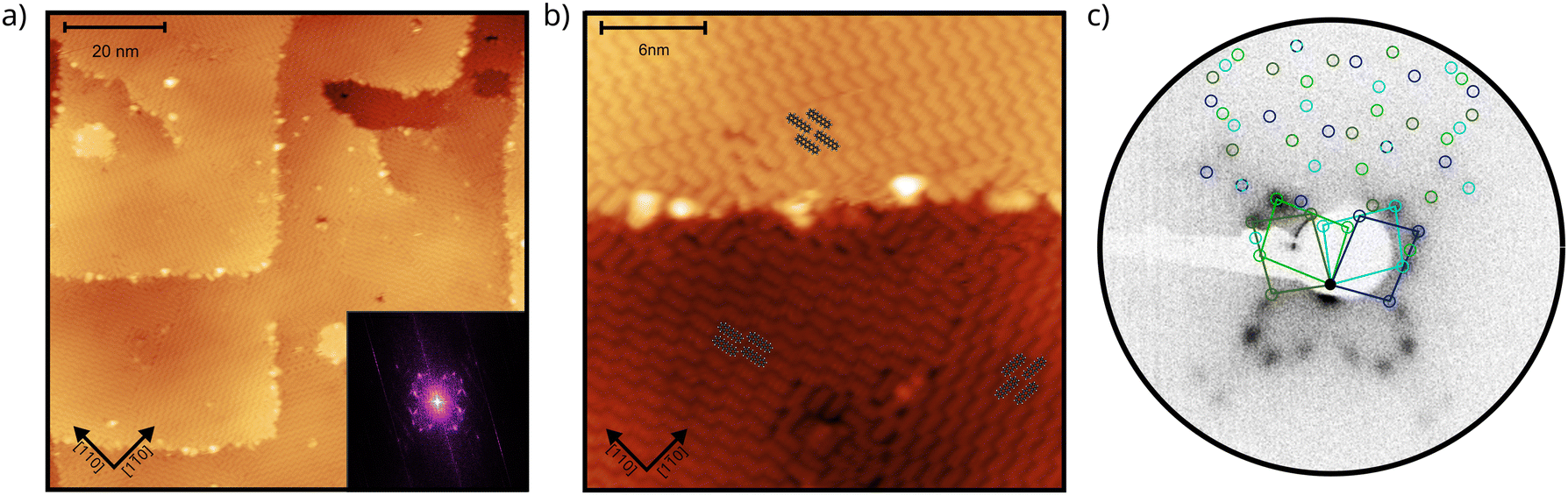 | ||
| Fig. 1 (a) STM image with a size of 100 × 100 nm2, showing the different terraces of the pentacene-covered NiO(001). The inset shows the 2D FFT of the image, which is well consistent with the pattern observed in LEED (see c). Setpoint: ΔV = −2.2 V, I = 0.1 nA. (b) STM closeup with a size of 26 × 26 nm2, showing the different orientations of the pentacene molecules overlayed with ball and stick models. Setpoint ΔV = −2.2 V, I = 0.1 nA. (c) LEED pattern of 1 ML pentacene on NiO(001), taken at 20 eV. The diffraction spots due to the molecular superstructure are indicated by the blue and green circles and the corresponding unit cell, referring to the different domains on the substrate, is also reported. | ||
The observed unit cell and overlayer matrix are also confirmed by LEED measurements on the molecular layer, shown in Fig. 1c. As all the matrix elements are integer numbers, the molecular overlayer grows indeed commensurate with respect to the NiO(001) surface. Starting from the matrices determined above, we can simulate the expected LEED pattern and compare it to the measured data. The excellent agreement between the two (see Fig. 1c, where the colored circles are simulated LEED spot positions marked in different tones of blue and green to distinguish the 4 symmetry-equivalent domains) confirms the STM findings, also indicating the existence of a long-range order across the surface. It is also worth noticing that the obtained LEED pattern matches the 2D Fast Fourier Transform (FFT) (Fig. 1a, inset) obtained from the large scale STM image.
We thus observe that, although the molecule–substrate interaction does not involve an appreciable redistribution of the electron density (as we will be discussing in section 3.3), it appears to be enough to make the 5A molecules arrange in register with the substrate structure underneath (consistently with the computed corrugation of the potential energy surface discussed in section 3.2).
3.2 DFT structural analysis
Although from STM and LEED is possible to determine the azimuthal orientation of pentacene with respect to the surface and the extension of the unit cell, our experimental data cannot alone provide the complete adsorption configuration, as information on the adsorption site is still missing. In this regard, DFT can give additional insights. To explore different possible adsorption configurations of pentacene on the NiO(001) surface, we initially considered a rather diluted overlayer of 5A molecules, i.e., having 24 surface Ni atoms per molecule in either a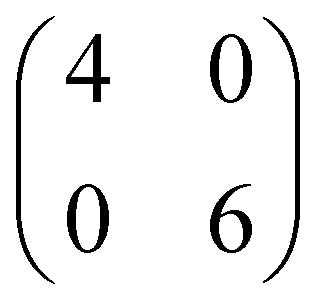 or
or  periodicity (relative to the Ni square lattice with unit length 2.94 Å). We investigated three different in-plane orientations of the molecules (always lying planar on the surface), namely with their main axis along rows of Ni atoms with the same magnetization ([
periodicity (relative to the Ni square lattice with unit length 2.94 Å). We investigated three different in-plane orientations of the molecules (always lying planar on the surface), namely with their main axis along rows of Ni atoms with the same magnetization ([![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10], i.e., at 135° from [100]), perpendicular to them ([110], at +45° from [100]), or in an intermediate orientation ([010], i.e., equivalent to [100]) (more details in ESI section S2†). For each of these orientations we looked at four different adsorption sites, taking the position of the central ring as a reference: above a Ni atom (“Ni”); above O (“O”); and above a Ni–Ni bridge site – in this case either between Ni atoms of the same spin (“b11” and the equivalent “b22”) or between atoms of opposite spin (“b12”). Among these cases, the most favorable configurations (nearly isoenergetic) are b12–135° and b11–45° (see Table 1).
10], i.e., at 135° from [100]), perpendicular to them ([110], at +45° from [100]), or in an intermediate orientation ([010], i.e., equivalent to [100]) (more details in ESI section S2†). For each of these orientations we looked at four different adsorption sites, taking the position of the central ring as a reference: above a Ni atom (“Ni”); above O (“O”); and above a Ni–Ni bridge site – in this case either between Ni atoms of the same spin (“b11” and the equivalent “b22”) or between atoms of opposite spin (“b12”). Among these cases, the most favorable configurations (nearly isoenergetic) are b12–135° and b11–45° (see Table 1).
| Unit cell | Site | Angle | E ads/5A (eV) | E ads/area (eV Å−2) |
|---|---|---|---|---|
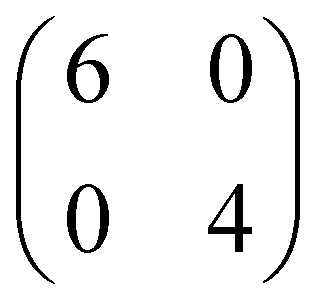
|
b11 | 45° | −3.432 | −0.0165 |

|
b12 | 135° | −3.443 | −0.0165 |

|
b12 | 135° | −3.410 | −0.0245 |

|
b11 | 45° | −3.588 | −0.0258 |

|
b12, b21 | 135° | −3.605 | −0.0259 |

|
b11, b22 | 45° | −3.389 | −0.0244 |
They are stabilized by the matching of the two central C atoms with the two Ni atoms of the bridge site (ESI Fig. S2b†). Conversely, other orientations/sites of the molecule are found at higher energy (from 0.6 eV to 1.3 eV, see Table S1†), justifying the adsorption of molecules in registry with the substrate lattice. We then investigated various possibilities for the overlayer periodicity. All investigated sites and orientations are summarized in Fig. 2 and Fig. S2a† with energies reported in Table S1.† Among them, a full overlayer of pentacene molecules on NiO(001) (Fig. 2), with surface unit cell and molecule orientation suggested by LEED measurements and by STM and ARPES measurements (see below in section 3.3), resulting in the epitaxy matrix  depicted in Fig. 2a, that can accommodate one flat-lying pentacene molecule. An inequivalent domain results by rotating the overlayer lattice by 90° or by rotating the rows of Ni atoms having the same magnetization from ([10] to [110]), leading to a
depicted in Fig. 2a, that can accommodate one flat-lying pentacene molecule. An inequivalent domain results by rotating the overlayer lattice by 90° or by rotating the rows of Ni atoms having the same magnetization from ([10] to [110]), leading to a  matrix (dashed line in Fig. 2a). However, this does not match the magnetic NiO(001) unit cell, that is
matrix (dashed line in Fig. 2a). However, this does not match the magnetic NiO(001) unit cell, that is  considering the alternating spin polarizations along [110]. Hence, simulations require a doubled overlayer cell,
considering the alternating spin polarizations along [110]. Hence, simulations require a doubled overlayer cell,  , containing two molecules in different sites. We restrict this investigation to bridge sites which we had found to be preferred in our study of diluted configurations and obtain the four cases shown in Fig. 2b–e. Therein, models shown in Fig. 2c and d are the most stable, with a mild energy difference of 0.02 eV per molecule. We hence take these as the models for the two rotated domains of 5A/NiO(001).
, containing two molecules in different sites. We restrict this investigation to bridge sites which we had found to be preferred in our study of diluted configurations and obtain the four cases shown in Fig. 2b–e. Therein, models shown in Fig. 2c and d are the most stable, with a mild energy difference of 0.02 eV per molecule. We hence take these as the models for the two rotated domains of 5A/NiO(001).
 | ||
Fig. 2 (a) Scheme of the NiO(001) square lattice highlighted in different colors according to the Ni magnetization direction (spin up/down in light blue/dark orange). A ball-stick model, with O shown as red circles, is partially over imposed. Light and dark yellow circles mark the epitaxy matrix suggested by experiments, 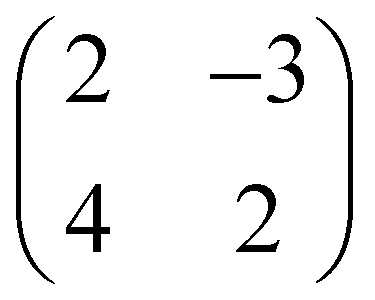 in terms of the square lattice vectors a1 and a2, and its 90°-rotation in terms of the square lattice vectors a1 and a2, and its 90°-rotation  . The latter being doubled, to respect the surface magnetization. (b) and (c): adsorption models for pentacene on NiO(001) on bridge sites within a . The latter being doubled, to respect the surface magnetization. (b) and (c): adsorption models for pentacene on NiO(001) on bridge sites within a  lattice. (d) and (e): similarly, for lattice. (d) and (e): similarly, for  lattice. lattice. | ||
In full agreement with the experimental findings, the models constructed to satisfy the LEED and STM observations represent the most stable configurations among all the considered ones, both for diluted molecules and for other possible overlayers. This stability is reflected in terms of adsorption energy per molecule (≈−3.6 eV) and per unit surface area (−26 meV Å−2, see Table 1 and ESI Table S1†). The molecule–surface interaction results in little structural modifications that we quantify by the vertical displacement from the planar position; its unsigned average amounts to 0.01 Å for 5A and to 0.02 Å for the topmost NiO layer.
3.3 Electronic properties
We now focus on the electronic structure of the 5A/NiO(001) interface, where our main interest lies in the frontier orbitals of pentacene. Fig. 3a shows the momentum-integrated data (over a range of ±1.7 Å−1) of the clean Ag(001) substrate (blue line), of the 10 layer-thick NiO film (red line) and of 1 ML 5A on NiO (yellow line). Please note that, in the following, we will set our zero of the Binding Energy (BE) scale at the Fermi level EF of the Ag(001) substrate. As we use an ultraviolet light source with a fixed photon energy (21.2 eV) and keep the same pass energy for all measurements, the same energy scaling obtained for the clean Ag surface can be applied to the NiO/Ag system. | ||
| Fig. 3 (a) Valence band structure of 1 ML of pentacene on top of NiO, in comparison to the substrate. The top curve (green line) shows a fit of the difference between the signal with and without molecules based on multiple Gaussian contributions. The different features are marked A–C. The momentum resolved maps for the Gaussian contribution to the molecular feature A (b) and B (c) were integrated over the corresponding energy range shown in (a). A comparison of the experimentally obtained intensity distribution (top part, contrast enhanced) with the simulated momentum maps (bottom part) enables us to assign feature A to the HOMO of the pentacene (in gas phase) and feature B to the degenerate HOMO−1/HOMO−2 levels. The calculated maps were generated by aligning the molecules azimuthally according to the geometrical structure extracted from the LEED and STM measurements (for more details, see Fig. S5†). | ||
The Ag(001) spectrum close to EF shows a rather featureless plateau, originating from the 5s states, up to 3.7 eV of binding energy. At this BE, the spectrum suddenly rises due to the 4d band. Upon deposition of 10 ML of NiO, a peak appears at a BE of 2.45 eV, while the higher BE region is dominated by a broader resonance having the maximum intensity at 5.75 eV and a shoulder-like feature at around 3.8 eV. The origin of these features is known from previous measurements:27 the two at lower BE originate from the Ni 3d states, whereas the peak at 5.75 eV from the oxygen 2p states.
Upon deposition of a saturated monolayer of pentacene, we observe no changes in the energy region very close to EF (in the [0;1] eV interval range), while the peak at 2.45 eV witnesses a noticeable shift of 0.3 eV to lower binding energies with respect to the bare NiO substrate. At the same time, a new peak rises at around 3.8 eV. To better highlight the photoemission features originating from the organic film, the difference between the spectra of the 5A/NiO(001) and of the bare NiO(001) is shown in the upper part of Fig. 3a. This allows us to identify three main features (A–C) that can be associated with the 5A overlayer.
For the purpose of identifying the 5A molecular orbitals that contribute to these resonances in the valence band spectrum, we can measure the momentum distribution of the photoemitted electrons at these specific binding energies and compare them to simulated orbital patterns based on DFT calculations within the Photoemission Orbital Tomography (POT) approach.36–38 Within this approximation, the final state of the photoemitted electron is approximated by a plane wave. Thus, the angular dependence of the photoemission intensity can be related to the modulus square of the Fourier Transform (FT) of the respective initial state wave function.39 Because PEEM is a spatially – averaging technique that integrates over a large area (hundreds of μm2) comprising all the symmetry-equivalent domains, the different possible azimuthal orientations of the 5A molecule must be considered. This has been done by taking as an input the alignment inferred from the STM measurements and corroborated by the DFT structural analysis (section 3.2, more details in ESI Fig. S3†). Fig. 3b and c show the measured photoemission intensity distribution for the two identified peaks A and B (upper half) in comparison to theoretically calculated momentum maps26,40 (lower half). For both peaks (i.e., A and B), the molecular features are located at high k-values (> ± 1.0 Å−1), while the features in the center of the image (< ± 0.5 Å−1) stem from the NiO substrate (more details in ESI Fig. S4†). We can clearly ascribe the momentum pattern originating from peak A at 2.05 eV to the highest molecular orbital (HOMO) of the pentacene molecule in the gas-phase. In contrast, the momentum feature at 3.8 eV (B) unveils signatures of the degenerate HOMO−1/HOMO−2 orbitals. This is unexpected: we would assume that, because of absence of appreciable charge transfer between the molecule and the oxide, the orbital order would resemble the one of the gas-phase, where these two states are separated in energy by 0.585 eV (DFT value). However, as suggested by DFT investigations (see later discussion of Fig. 4), the energy overlaps of such and other states should occur because of surface hybridization. The good agreement between the measured and simulated data confirms the alignment of the molecular long axis almost parallel (3.5° off) with the [110] direction of NiO(001) (see ESI Fig. S5†), as determined in the previous sections.
 | ||
| Fig. 4 Theory data. (a) DOS of gas phase pentacene (grey), adsorbed pentacene (green), and the topmost NiO(001) layer (red). The Fermi energy EF of the pentacene/NiO(001) system is set to 0, and the LUMO of gas phase pentacene is then aligned with the bottom conduction state of pentacene/NiO(001). Values in states per eV per cell, summed over spin. (b) Projected DOS for various 5A orbitals ranging from HOMO−5 up to LUMO+1 (MOPDOS). | ||
Fig. 4 reports the computed density of electronic states (DOS) of the most energetically stable adsorption configuration of pentacene on NiO(001), namely the “b12–b21” in the  cell, see Fig. 2c. The spin unbalance is very small, so we report spin-integrated results. We show in Fig. 4a the DOS of the adsorbed system projected on the topmost NiO(001) layer in the NiO(001) slab (red) and on the 5A molecules (green curve); the latter one is compared to the DOS of the gas phase 5A (grey). Looking at the occupied states of 5A, we find that the sharp peaks corresponding to molecular orbitals of free 5A hybridize with substrate ones and spread in energy. One observes three structures, at −0.8 eV, −2.3 eV, and −4.5 eV (we remark that the energy scale used in the theoretical computation is referred to the Fermi level of the full system, arbitrarily placed within its band gap), whose energy spacing appears to correspond to the A, B, and C features observed in Fig. 3a, yet with significant differences such as a B–C separation larger than in the experiments. To understand the molecular contributions to such features, we report in Fig. 4b the 5A DOS now resolved in its molecular orbitals (MOPDOS41). While the first peak can be assigned to the HOMO, at lower energies we can see that many molecular orbitals hybridize so strongly to spread over wide energy ranges. Hence, several orbitals are found to contribute at the same energy, even though no degenerate orbitals are expected in the gas phase (see ESI Fig. S6†). In the unoccupied states, we first observe the LUMO of 5A, lying within the bulk NiO energy gap, as can be identified in the MOPDOS. Löwdin partitioning of the electronic density points to a charge displacement of less than 0.5e/5A from molecule to substrate. However, inspection of the charge density upon adsorption (see Fig. 5) reveals that charge accumulates in the region between the molecule and the substrate, without localization on specific parts of the molecule. This result, obtained for the two most stable adsorption configurations, is consistent with a polarization of the molecular electron density towards the substrate, without any change in orbital occupation.
cell, see Fig. 2c. The spin unbalance is very small, so we report spin-integrated results. We show in Fig. 4a the DOS of the adsorbed system projected on the topmost NiO(001) layer in the NiO(001) slab (red) and on the 5A molecules (green curve); the latter one is compared to the DOS of the gas phase 5A (grey). Looking at the occupied states of 5A, we find that the sharp peaks corresponding to molecular orbitals of free 5A hybridize with substrate ones and spread in energy. One observes three structures, at −0.8 eV, −2.3 eV, and −4.5 eV (we remark that the energy scale used in the theoretical computation is referred to the Fermi level of the full system, arbitrarily placed within its band gap), whose energy spacing appears to correspond to the A, B, and C features observed in Fig. 3a, yet with significant differences such as a B–C separation larger than in the experiments. To understand the molecular contributions to such features, we report in Fig. 4b the 5A DOS now resolved in its molecular orbitals (MOPDOS41). While the first peak can be assigned to the HOMO, at lower energies we can see that many molecular orbitals hybridize so strongly to spread over wide energy ranges. Hence, several orbitals are found to contribute at the same energy, even though no degenerate orbitals are expected in the gas phase (see ESI Fig. S6†). In the unoccupied states, we first observe the LUMO of 5A, lying within the bulk NiO energy gap, as can be identified in the MOPDOS. Löwdin partitioning of the electronic density points to a charge displacement of less than 0.5e/5A from molecule to substrate. However, inspection of the charge density upon adsorption (see Fig. 5) reveals that charge accumulates in the region between the molecule and the substrate, without localization on specific parts of the molecule. This result, obtained for the two most stable adsorption configurations, is consistent with a polarization of the molecular electron density towards the substrate, without any change in orbital occupation.
 | ||
| Fig. 5 Electron density displacement upon 5A adsorption on Ni(001) for the two most stable configurations (panels (a) and (b) here refer to Fig. 2c and d, respectively). Yellow/cyan isosurfaces delimit regions whose electron density increases/decreases by 0.01 e Å−3. | ||
The analysis of the visible molecular orbitals and the comparison with DFT+U calculations indicate that negligible charge transfer occurs between the substrate and the molecules. Consequently, the position of the molecular HOMO relative to the Fermi level should be determined solely by the surface potential of the substrate.16 This allows us to compare the observed HOMO binding energy with predictions based on the universal energy level alignment model for transition metal oxides proposed by Greiner et al.17,20 According to their model, the energy level alignment of molecules on metal oxide surfaces follows a universal trend where the HOMO position relative to the Fermi level (ΔEH) is governed by the oxide's work function (ϕ) and the ionization energy of the adsorbed molecule (IEorg). While for the case of ϕ > IEorg a more or less constant ΔEH of 0.3 eV is expected, for the case of ϕ < IEorg the approximation is defined by:
| ΔEH ≈ (IEorg − ϕ) + 0.3 eV. |
For pentacene, the ionization energy in gas phase is reported to be around 6.61 eV.42 The observed work function for our in situ grown NiO is around 5.1 eV, consistent with previous studies.18,27,43 Using the model above, the expected energy difference for the 5A HOMO with respect to the detector's Fermi level is ΔEH = 1.81 eV, which agrees well with the experimentally observed value of 1.9 eV. Remarkably, the model holds despite the hybridization between molecular and substrate orbitals causing the former to broaden in energy, but no appreciable charge transfer is measured.
The nature of the molecular contributions to the electronic features of the 5A/NiO(001) system can be discussed also by looking at STM images with molecular resolution, reported in Fig. 6a for a negative bias ΔV = −2 V. There, the observed image of a single molecule can be superimposed very well to the electron density in real space for the HOMO of pentacene in the gas phase, depicted in Fig. 6b. A very similar electron density can be seen in the simulated STM at negative bias in Fig. 6c. Both in the experimental and in the simulated image, the longitudinal nodal plane is evident. Similarly, a simulated image including empty states shown in Fig. 6d testifies the contribution of the LUMO. Unfortunately, no high quality high-resolution STM images could be obtained for positive biases.
 | ||
| Fig. 6 (a) STM image with a size of 9 × 9 nm2 showing the single molecules oriented along the [110] direction of the NiO(001) surface, overlayed by three molecules with the real space electron distribution of the pentacene HOMO. Setpoint ΔV = −2 V, I = 0.1 nA. (b) Calculated electron density in real space for the gas phase HOMO of pentacene. (c and d) Calculated STM images for the most stable pentacene/NiO(001) configuration, obtained by imaging (c) filled states (ΔV = −1 V) and (d) empty states (ΔV = +1 V). | ||
By DFT, we further investigated the interface magnetic structure by evaluating the modification of magnetic moments at the NiO surface and the occurrence of magnetic moments induced on pentacene molecules upon adsorption. In all the cases (diluted molecules as of Fig. S2† and the two most stable overlayers as of Fig. 2c and d), we overall observe small variations in magnetic moments. In particular, the magnetic moments of surface Ni atoms lying below pentacene molecules slightly reduce their absolute value (by up to 34 and 50 mμB in the configurations of Fig. 2c and d, respectively, see Fig. S7a† for details). Simultaneously, carbon atoms attain a nonzero magnetic moment which amounts to less than 20 mμB in the configuration of Fig. 2c, but that can reach up to 100 mμB for the central C atoms when 5A molecules align parallel to rows of nickel atoms of the same magnetization as in Fig. 2d. Such enhanced effect is found regardless of the specific intermolecular arrangement within the overlayer, as can be seen in Fig. S7b.†
4 Conclusion
We report the successful growth of a self-assembled monolayer of pentacene molecules on a 2.4 nm thick NiO(001) film grown on an Ag(001) substrate. Using LEED, STM and POT, we determined the superstructure and molecular orientation relative to the high symmetry axes of the substrate and identified the momentum dependent signatures of several molecular orbitals. No charge transfer occurs and the LUMOs remain unpopulated while the HOMOs remain populated. This confirms the general behavior observed on non-magnetic transition metal oxide surfaces, where the highest occupied molecular orbital (HOMO) aligns with the substrate's Fermi level based solely on the substrate's work function and the molecule's ionization energy.Despite the negligible charge transfer, pentacene molecules adopt an ordered arrangement in registry with the NiO(001) surface, indicating a non-negligible influence of the substrate on molecular growth by providing a preferential adsorption site where the central C atoms match with two surface Ni atoms. This is consistent with our observation that POT, supported by DFT calculations, suggests a degeneracy of the HOMO−1 and HOMO−2 orbitals in the adsorbed phase, in contrast to the gas-phase situation. Furthermore, DFT reveals an accumulation of molecular electron density toward the substrate. Although this does not induce a pronounced spin imbalance, the preservation of strong free-molecule character in the HOMO and LUMO orbitals points to potential applications in the optical control of THz spin dynamics in NiO functionalized with pentacene. These findings open a promising route for engineering molecule-based functionalities on antiferromagnetic surfaces for next-generation spintronic devices.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
The raw data that support the findings of this study are available in the ZENODO database. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.Acknowledgements
This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under grant agreement no. 964396 FET-Open SINFONIA (Selectively activated INFOrmation technology by hybrid Organic Interfaces).We acknowledge the CINECA award under the ISCRA initiative (grants IscrC-PentaNiO-HP10C1B51Q, IscrB-ORGAFINT-HP10BC1AM9), for the availability of high-performance computing resources and support. The momentum microscope was financed by the DFG through the project INST 212/409 and by the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen.
References
- R. Ramesh and D. G. Schlom, Creating emergent phenomena in oxide superlattices, Nat. Rev. Mater., 2019, 4, 257–268 CrossRef.
- Q. Wang, Y. Gu, C. Chen, F. Pan and C. Song, Oxide Spintronics as a Knot of Physics and Chemistry: Recent Progress and Opportunities, J. Phys. Chem. Lett., 2022, 13, 10065–10075 CrossRef CAS PubMed.
- Y. Han, et al., Transition metal oxides: a new frontier in spintronics driven by novel quantum states and efficient charge-spin interconversion, Front. Mater., 2024, 11, 1444769 CrossRef.
- S. Ramazanov, et al., Surface Modification and Enhancement of Ferromagnetism in BiFeO3 Nanofilms Deposited on HOPG, Nanomaterials, 2020, 10, 1990 CrossRef CAS PubMed.
- T. Kosub, et al., Purely antiferromagnetic magnetoelectric random access memory, Nat. Commun., 2017, 8, 13985 CrossRef CAS PubMed.
- C. Hahn, et al., Conduction of spin currents through insulating antiferromagnetic oxides, Europhys. Lett., 2014, 108, 57005 CrossRef.
- H. Wang, C. Du, P. C. Hammel and F. Yang, Antiferromagnonic Spin Transport from Y3Fe5O12 into NiO, Phys. Rev. Lett., 2014, 113, 097202 CrossRef CAS PubMed.
- W. Lin, K. Chen, S. Zhang and C. L. Chien, Enhancement of Thermally Injected Spin Current through an Antiferromagnetic Insulator, Phys. Rev. Lett., 2016, 116, 186601 CrossRef PubMed.
- R. Lebrun, et al., Tunable long-distance spin transport in a crystalline antiferromagnetic iron oxide, Nature, 2018, 561, 222–225 CrossRef CAS PubMed.
- R. Cheng, M. W. Daniels, J.-G. Zhu and D. Xiao, Ultrafast switching of antiferromagnets via spin-transfer torque, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 91, 064423 CrossRef.
- R. Khymyn, I. Lisenkov, V. Tiberkevich, B. A. Ivanov and A. Slavin, Antiferromagnetic THz-frequency Josephson-like Oscillator Driven by Spin Current, Sci. Rep., 2017, 7, 43705 CrossRef PubMed.
- D. Bossini, et al., Ultrafast Amplification and Nonlinear Magnetoelastic Coupling of Coherent Magnon Modes in an Antiferromagnet, Phys. Rev. Lett., 2021, 127, 077202 CrossRef CAS PubMed.
- I. Žutić, J. Fabian and S. D. Sarma, Spintronics: Fundamentals and applications, Rev. Mod. Phys., 2004, 76, 323–410 CrossRef.
- W. J. M. Naber, S. Faez and W. G. Wiel, van der. Organic spintronics, J. Phys. D: Appl. Phys., 2007, 40, R205 CrossRef CAS.
- X. Sun, M. Jibran, A. Pratt, B. Wang and Y. Yamauchi, Spin Polarization Enhancement of an Fe3O4(100) Surface by Coadsorption of Atomic Hydrogen and Molecular Nitric Oxide, J. Phys. Chem. C, 2019, 123, 12813–12817 CrossRef CAS.
- M. T. Greiner and Z.-H. Lu, Thin-film metal oxides in organic semiconductor devices: their electronic structures, work functions and interfaces, NPG Asia Mater., 2013, 5, e55–e55 CrossRef CAS.
- M. T. Greiner, et al., Universal energy-level alignment of molecules on metal oxides, Nat. Mater., 2012, 11, 76–81 CrossRef CAS PubMed.
- M. T. Greiner, L. Chai, M. G. Helander, W. Tang and Z. Lu, Transition Metal Oxide Work Functions: The Influence of Cation Oxidation State and Oxygen Vacancies, Adv. Funct. Mater., 2012, 22, 4557–4568 CrossRef CAS.
- C. Rödl, F. Fuchs, J. Furthmüller and F. Bechstedt, Quasiparticle band structures of the antiferromagnetic transition-metal oxides MnO, FeO, CoO, and NiO, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79, 235114 CrossRef.
- M. T. Greiner, M. G. Helander, Z.-B. Wang, W.-M. Tang and Z.-H. Lu, Effects of Processing Conditions on the Work Function and Energy-Level Alignment of NiO Thin Films, J. Phys. Chem. C, 2010, 114, 19777–19781 CrossRef CAS.
- A. Facchetti, Semiconductors for organic transistors, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- P. Hurdax, et al., Controlling the Charge Transfer across Thin Dielectric Interlayers, Adv. Mater. Interfaces, 2020, 7, 2000592 CrossRef CAS.
- A. Baby, H. Lin, G. P. Brivio, L. Floreano and G. Fratesi, Core-level spectra and molecular deformation in adsorption: V-shaped pentacene on Al(001), Beilstein J. Nanotechnol., 2015, 6, 2242–2251 CrossRef CAS PubMed.
- A. Baby, et al., Anchoring and Bending of Pentacene on Aluminum (001), J. Phys. Chem. C, 2015, 119, 3624–3633 CrossRef CAS.
- S. Weiß, et al., Exploring three-dimensional orbital imaging with energy-dependent photoemission tomography, Nat. Commun., 2015, 6, 8287 CrossRef PubMed.
- D. Brandstetter, X. Yang, D. Lüftner, F. S. Tautz and P. Puschnig, kMap.py: A Python program for simulation and data analysis in photoemission tomography, Comput. Phys. Commun., 2021, 263, 107905 CrossRef CAS.
- J. Das and K. S. R. Menon, A revisit to ultrathin NiO(001) film: LEED and valence band photoemission studies, J. Electron Spectrosc. Relat. Phenom., 2015, 203, 71–74 CrossRef CAS.
- S. Ponzoni, et al., Dirac Bands in the Topological Insulator Bi2Se3 Mapped by Time–Resolved Momentum Microscopy, Adv. Phys. Res., 2023, 2, 2200016 CrossRef.
- J. E. Nitschke, et al., Valence band electronic structure of the van der Waals antiferromagnet FePS3, Mater. Today Electron., 2023, 6, 100061 CrossRef.
- P. Giannozzi, et al., QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, et al., Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Pseudopotentials for high-throughput DFT calculations, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
- V. R. Cooper, Van der Waals density functional: An appropriate exchange functional, Phys. Rev. B:Condens. Matter Mater. Phys., 2010, 81, 161104 CrossRef.
- K. Lee, É.D Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Higher-accuracy van der Waals density functional, Phys. Rev. B:Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- J. Tersoff and D. R. Hamann, Theory of the scanning tunneling microscope, Phys. Rev. B:Condens. Matter Mater. Phys., 1985, 31, 805–813 CrossRef CAS PubMed.
- G. Zamborlini, et al., Multi-orbital charge transfer at highly oriented organic/metal interfaces, Nat. Commun., 2017, 8, 335 CrossRef PubMed.
- H. M. Sturmeit, et al., Molecular anchoring stabilizes low valence Ni(i)TPP on copper against thermally induced chemical changes, J. Mater. Chem. C, 2020, 7, 105–111 Search PubMed.
- D. M. Janas, et al., Metalloporphyrins on oxygen-passivated iron: Conformation and order beyond the first layer, Inorg. Chim. Acta, 2023, 557, 121705 CrossRef CAS.
- P. Puschnig, et al., Reconstruction of Molecular Orbital Densities from Photoemission Data, Science, 2009, 326, 702–706 CrossRef CAS PubMed.
- P. Puschnig, Molecular Orbital Database - Search. https://physikmdb.uni-graz.at:5001/.
- A. Ravikumar, A. Baby, H. Lin, G. P. Brivio and G. Fratesi, Femtomagnetism in graphene induced by core level excitation of organic adsorbates, Sci. Rep., 2016, 6, 24603 CrossRef CAS PubMed.
- T. M. Halasinski, D. M. Hudgins, F. Salama, L. J. Allamandola and T. Bally, Electronic Absorption Spectra of Neutral Pentacene (C22H14) and Its Positive and Negative Ions in Ne, Ar, and Kr Matrices, J. Phys. Chem. A, 2000, 104, 7484–7491 CrossRef CAS.
- K. Marre and H. Neddermeyer, Growth of ordered thin films of NiO on Ag(100) and Au(111), Surf. Sci., 1993, 287, 995–999 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00700c |
| ‡ Present Address: Institute of Physics, NAWI Graz, University of Graz, Universitätsplatz 5, 8010 Graz, Austria. |
| This journal is © The Royal Society of Chemistry 2025 |