
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatial nano-scaled organization of heterochromatin in nuclei of endothelial cells after exposure to uremic and dialytic milieu
Maria
Bartosova Medvid
 *a,
Arslan
Saleem
b,
Myriam
Schäfer
b,
Gesine
Messerschmidt
a,
Claus Peter
Schmitt
a and
Michael
Hausmann
b
*a,
Arslan
Saleem
b,
Myriam
Schäfer
b,
Gesine
Messerschmidt
a,
Claus Peter
Schmitt
a and
Michael
Hausmann
b
aHeidelberg University, Medical Faculty Heidelberg, Centre for Pediatrics and Adolescent Medicine, Clinic 1, Pediatric Nephrology, Heidelberg, Germany. E-mail: maria.bartosovamedvid@med.uni-heidelberg.de; Tel: +49-6221-56-35267
bKirchhoff Institute for Physics, Heidelberg University, Im Neuenheimer Feld 227, 69120 Heidelberg, Germany
First published on 13th June 2025
Abstract
Cardiovascular disease is frequent in patients with chronic kidney disease (CKD); this risk increases further with peritoneal dialysis (PD), especially when high glucose degradation products (GDP) containing PD fluids (PDF) are used. We studied the spatial organization of the genome orchestrated by heterochromatin rearrangements (fluorescently labelled against H3K9me3 methylation sites) using super-resolution Single Molecule Localization Microscopy (SMLM) and novel combined, mathematical approaches. These were based for geometry and clustering on Ripley statistics, and for topology on persistent homology, persistent imaging and principal component analysis. As a model system, endothelial cells (HUVEC) were exposed to plasma from healthy and CKD individuals and to glucose-based PDF with different buffer and different GDP concentrations. H3K9me3 representing heterochromatin was clustering in endothelial cells. The clusters varied between CKD plasma donors, whereas PDF consistently increased endothelial heterochromatin relaxation and reduced H3K9me3 clustering, with significant differences based on the buffer and GDP content. Bicarbonate PDF with low-GDP concentrations induced high heterochromatin relaxation, i.e., unveiled most H3K9me3 molecules at the lowest level of relative clustering. Persistent homology varied between single and medium exposed cells, but varied less with PDF exposure. High-GDP PDF induced the highest degree of H3K9me3 disarrangement with the GDP 3,4-DGE dose-dependently increasing HC relaxation and lowering clustering to a level below controls. Structural heterochromatin (re-)organization of endothelial cells is variable in response to CKD plasma, while PDF induce major heterochromatin relaxation, depending on the buffer and GDP content, indicating additional chromatin accessibility related changes in genetic activity. Our findings provide a novel base for studying PD biocompatibility and vascular endothelial health.
Introduction
Genomic stability is an essential hallmark of organismal integrity and is therefore tightly maintained and controlled.1 This is necessary for the integrity of DNA carrying the specific genetic information for functioning of a cell. In eukaryotic cells, DNA is packed around histones creating a chromatin fibre. Chromatin as a whole forms different networks2,2a that can for instance be subdivided into the de-condensed euchromatin and the compacted (condensed) heterochromatin network.2,3 Chromatin reveals a dynamic scaffold, which responds to specific signals physically regulating the accessibility of DNA to numerous factors.4,5 This means that changes in the structure and packing density of chromatin may dynamically regulate gene expression.6 Euchromatin is rich in genes, which is why it is rather loosened-up and easily accessible for the transcriptional machinery. Heterochromatin by contrast describes the tightly packed, transcriptionally repressed genome, with fewer, mostly inactive genes or genome regions.7 Reversible changes of the spatial heterochromatin organization directly correlate with the wave-like up- and down-regulation of genes.6,8 Various pathophysiological states disrupt this structural arrangement of chromatin resulting in changes in gene function.8,9Histone H3Lys9-methylated (H3K9me3)10 heterochromatin ensures transcriptional silencing of repetitive elements and genes, and its deregulation leads to impaired cell and tissue identity, premature aging and cancer.11 Specific histone modifications, such as for instance the formation of γH2AX,12 can be induced by double strand breaks and lead to chromatin rearrangements. Damaged sites in highly condensed heterochromatin regions are transported by entropic forces towards the heterochromatin border or into the euchromatin environment, respectively.13,14 In the physiological state, double strand breaks can be perfectly repaired,15e.g., by repair mechanisms like homologous recombination16 or the rapid non-homologous end joining,17 and the heterochromatin formation is finally reconstructed.14,18 If strand breaking DNA damage exceeds repair mechanisms, or if the repair mechanisms are dysfunctional, genome stability may be severely impaired and – depending on the affected histone – result in severe consequences for the given organism.19–22
Patients with chronic kidney disease (CKD) suffer from an exceedingly high cardiovascular (CV) disease, which further increases on dialysis therapy. A 30-year-old patient on dialysis has a CV mortality corresponding to the mortality of the 80-year-old general population.23 In peritoneal dialysis (PD), glucose degradation products exert major additional endothelial toxicity.24 Accelerated endothelial aging is one of the driving forces, with children suffering from CKD5 already exhibiting premature endothelial telomere shortening.25–27
Toxins and ionizing radiation cause chromatin changes on a multiscale level, i.e. from the sub-molecular scale over the molecular nano- and meso-scale to the supra-molecular micro-scale.28 According to our best knowledge, the spatial chromatin organization in endothelial cells in response to uremic and dialytic milieu has not been studied yet.
Established methods to visualize and quantify chromatin organization are Single Molecule Localization Microscopy (SMLM) techniques,29,30 which allow studies of nano- and meso-scaled structural (re-)organization of bio-molecules.31 SMLM is a super-resolution microscopy method32 based on optical isolation of fluorescent molecules that stochastically switch between two different spectral states, e.g., an on (fluorescing) and off (non-fluorescing) state.33 These blinking events of dye molecules allow spatial separation of individual fluorescent signals which can be precisely (in the order of 10 nm) localized by calculating the intensity barycenter of the Airy disc visualized for point-like light sources (dye molecules) by the microscope objective lens used.34 SMLM data acquisition results in a matrix of point coordinates of the labelling molecules. In combination with novel image-free evaluation methods geometries of point patterns, point clustering or topologies and their degree of similarities can be calculated.35 These mathematical approaches have been successfully applied to describe chromatin networks after labelling of H3K9me3 methylation sites (heterochromatin) or ALU and L1 interspersed regions, and to differentiate cell types according to their chromatin organization.19,35 Reversible and irreversible chromatin network changes were measured during chromatin repair after DNA damaging by ionizing radiation.5 Locally the nano-organization of repair foci (γH2AX, 53BP1, MRE11 etc.) was studied along the repair process (see e.g., ref. 3, 5, 14 and 19). Also functionally correlated receptor protein clustering36 or the spatial organization of tight junction proteins37 were analysed in cell membranes.
Here, we used SMLM in combination with the novel analysis tools to analyse the point pattern of H3K9me3 in HUVEC cells in order to elucidate the spatial and topological changes of heterochromatin in endothelial cells exposed to uremic milieu and PD fluids (PDF), respectively.
Results
Plasma from individuals with chronic kidney disease changes heterochromatin organization with high inter-individual variation
HUVEC cells, a commercially available endothelial cell model, were incubated with plasma from five healthy donors (controls) (HC1–HC5) and five CKD5 patients (U1–U5) mixed by a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 with cell medium. Visually, in endothelial cell nuclei incubated with plasma from healthy individuals (control), the typical heterochromatin ring distribution appeared more frequently, which is not obvious in the point pattern reconstructed from the fluorochrome loci. This was not observed in endothelial cells incubated with CKD5 plasma (Fig. 1A).
:4 with cell medium. Visually, in endothelial cell nuclei incubated with plasma from healthy individuals (control), the typical heterochromatin ring distribution appeared more frequently, which is not obvious in the point pattern reconstructed from the fluorochrome loci. This was not observed in endothelial cells incubated with CKD5 plasma (Fig. 1A).
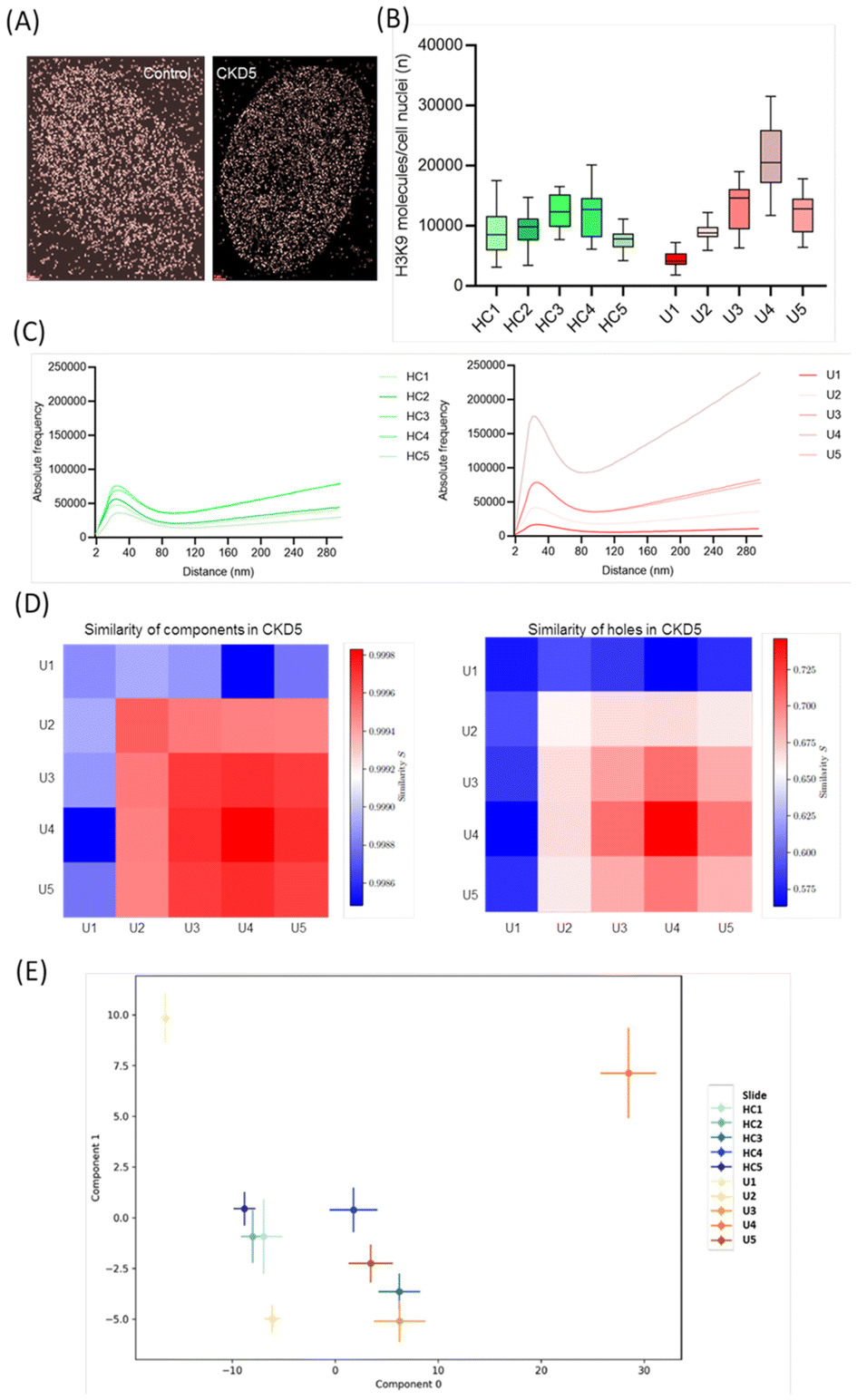 | ||
| Fig. 1 Impact of uremic plasma on heterochromatin organisation: (A) typical SMLM images of cell nuclei after anti-H3K9me3 antibody labelling. (B) Boxplots of H3K9me3 signals for cell nuclei treated with plasma of patients (U1,…,U5) and healthy controls (HC1,…,HC5). The boxplots show the median point number of the detected signals (line), the lower and upper quantile (box), and the value range within ±2 standard deviations (black line). (C) Ripley frequency histograms of pairwise point distances (left: plasma of healthy controls; right: plasma of CDK5 patients). (D) 2nd generation heatmaps obtained from persistent homology and calculation of the Jaccard indices for the patient specimens indicate a topological similarity U3, U4, U5. (E) Latent space of component 1 vs. component 0 of the specimens analysed after PCA of persistent imaging results for the holes. | ||
In Fig. 1B, the box plots of the absolute numbers of detected heterochromatin signals are shown for HUVEC cells treated with plasma from healthy individuals and CKD5 patients. Within a section of about 500 nm (typical z-section for SMLM recording) in the equatorial plane of a cell nucleus, HUVEC cells exposed to plasma from healthy individuals showed about 10092 ± 3534 signals. The fluctuations within the five data sets and each 25 recordings were comparable between the individuals. Larger fluctuations within the five data sets and each 25 recordings were observed for specimens exposed to the CDK5 plasmas. The mean number was 12091 ± 6643 considering all 125 recordings. SMLM analysis showed a higher number of event counts in 3 cases of CKD5 plasma treated cells compared to the cells treated with healthy plasma most likely representing the higher biological variation between the patients.
This stronger individual variability among the patient plasma specimens was also found for the Ripley curves in Fig. 1C representing the absolute frequency of pairwise point distances. While the peaks at small distances representing the formation of point clusters show about the same width and similar heights for the cells with plasma of healthy individuals, peak width and heights vary considerably in the patientś plasma group indicating disordered heterochromatin. This is supported for instance by the steep increase of the Ripley curve at higher distance values for U4 indicating a strong tendency to random heterochromatin organization.
Persistent homology revealed barcodes for components and holes of the networks of point patterns.38 These barcode patterns of heterochromatin labelling points for each cell were compared to each other cell by calculating the Jaccard indices.39 The Jaccard indices of each specimen pair were averaged and presented in 2nd generation heatmaps40 for components and holes. Surprisingly, U3, U4 and U5 indicate a high similarity for components and holes (Fig. 1C and D). However, the comparison of these results with the results obtained by Principal Component Analysis (PCA) after transformation of the bar codes into persistent images35 revealed a much higher variability of the patient specimens than of the control specimens in the two-dimensional latent space of the first two major components (Fig. 1E). Thus, it can be concluded from Fig. 1C–E that in the case of the specimens with patientś plasma a nearly random distribution of heterochromatin with a limited amount of clusters occurred.
Impact of peritoneal dialysis fluids on endothelial heterochromatin organization
HUVEC cells were exposed to different peritoneal dialysis fluids (PDF) and normal cell culture medium as control. These fluids contain different buffer compounds and concentrations of glucose degradation products (GDP): (a) pH neutral, bicarbonate buffered, low GDP PDF (BPDF), (b) pH neutral lactate buffered low GDP PDF (LPDF) and (c) conventional, acidic, lactate buffered high GDP PDF (CPDF).Exposure to all PDF resulted in a significantly higher amount of H3K9me3 tags molecule abundance compared to HUVEC cells exposed to cell medium only (Fig. 2A). The highest molecule counts were found in BPDF, followed by CPDF and LPDF groups which were – although statistically different – in the same range. In all cases, H3K9me3 clustering is visible as shown by the peak in the absolute and relative Ripley41 distance histograms (Fig. 2B and C). The absolute distance analysis showed distributions comparable to the molecule count histograms. BPDF has the highest peak amplitude (180000), followed by CPDF, LPDF, and the medium (100000 to 25000). The same descending order applies when considering the peak width and the gradient value at larger distances. This particular order correlates with the order of the average count numbers in the SMLM analysis, illustrating the impact of the high counts of the various treatments on the heterochromatin distribution. For BPDF, a steep increase of the curves at higher distance values indicates an increased random distribution of H3K9me3 tags.
 | ||
| Fig. 2 Impact of PDF on heterochromatin organisation: (A) and (D) box plots of the absolute numbers of heterochromatin anti-H3K9me3 tags in a ∼500 nm image section through the equatorial plane of cell nuclei exposed to different PDF in comparison to medium without PDF; (B) and (E) absolute Ripley frequency histograms of pairwise distances of H3K9me3 points for the different PDF in comparison to medium without PDF; (C) and (F) relative Ripley frequency histograms of pairwise distances of H3K9me3 points for the different PDF in comparison to medium without PDF. | ||
In the normalized distribution (equalizing the number of molecules per experimental condition) medium treated HUVEC cells have the highest peak at 40 nm with a peak-width of about 80 nm (2% of molecules were found at the distance up to 40 nm) indicating a cell typical clustering (Fig. 2C). This was also found for CPDF and LPDF. In contrast, the peak of BPDF treated cells was much lower and smaller which is compatible to a loss of heterochromatin clustering. This particular order of the Ripley curves correlates with the order of the average count numbers from the SMLM analysis, illustrating the impact of the high counts of the various treatments on the heterochromatin distribution. All relative curves show a consistent gradient after distances of approximately 80 nm (BPDF) to 110 nm (medium), depending on the peak size. Gradient values differ slightly, indicating a random point distribution at larger distances.30 Aside from the amplitudes and slight variations in peak width, the curves are generally similar.
Despite the largest differences in the H3K9me3 molecule counts, the topology of heterochromatin organisation as determined by persistent homology and 2nd generation heat maps reveals the same degree of lower similarity between medium and all PDF for components and holes. This similarity, however, is considerably higher between BPDF and CPDF, only (Fig. 3).
 | ||
| Fig. 3 2nd generation heatmaps of components (A) and holes (B) obtained from persistent homology of H3K9me3 tags of HUVEC cells under different treatments with PDF. Each heatmap pixel averages the heatmap of a given treatment. | ||
Single GDP has a dose-dependent impact on heterochromatin organisation
Next, we studied the impact of single GDP, which is present in CPDF in concentrations up to 20 μM. Endothelial HUVEC cells exposed to 10 μM and 20 μM 3,4 di-deoxy-glucosone (3,4 DGE) exhibited a dose-dependent increase in the H3K9me3 molecule counts, but the difference to the medium without PDF was smaller as compared to the effect between all PDF (Fig. 2D). Ripley absolute pairwise distance frequency analysis revealed an increase of heterochromatin clustering in a dose-dependent manner (Fig. 2E). The cluster size described by the width of the peaks in the relative pairwise distance frequency curves remained unaffected (Fig. 2F).In the cases of 2nd generation heatmaps after persistent homology, application of the higher concentration (20 μM) of 3,4 DGE showed a higher similarity to BPDF than to the medium and LPDF and CPDF.
Since the heterochromatin network has a strong impact on genome functioning, the topological analysis calculated by persistent homology and persistent imaging was especially focused on holes and meshes of the H3K9me3 pattern. While in the 2nd generation heatmaps presented for the components and holes (Fig. 3) the specimens treated with BPDF and CPDF revealed an increased similarity, the result of the topological analysis after PCA showed a completely different image (Fig. 4).
 | ||
| Fig. 4 Topological analysis of H3K9me3 tags after cell treatment with different PDF and 3,4 DGE: (A) Results of persistent imaging for component 0 in the latent spaces of PCA. Endothelial cells were incubated with peritoneal dialysis fluids containing bicarbonate (BPDF) or lactate buffer (LPDF) with content of glucose degradation products (GDP) and lactate based PD fluid with high concentration of GDP (CPDF). The pixels reveal the lifetime (length in nm) of the bars for holes in persistent homology vs. the value the bar starts (birth). The blue values are highly frequent and the content is increasing with the darkness of the pixel. The red-brown pixels are values of low (disappearing) frequency. (B) Principal component analysis of the results of persistent imaging of the holes (left: latent space of component 1 vs. component 0; right: latent space of component 2 vs. component 0). In both cases the cells incubated with BPDF were the most different compared to lactate-based PD fluids with low and high content of glucose degradation products. | ||
In Fig. 4A the results of persistent imaging are shown for the component 0, i.e. the component with the largest variance in the PCA. The data show the “lifetime” (length of the respective bar) of the holes (meshes of the heterochromatin network) vs. the birth of the holes. This gives a measure for the size of the holes at which the algorithm identifies closed holes (meshes).34 For the cells treated with medium only, the heterochromatin organization shows strongly compacted heterochromatin with meshes of 40 nm to 100 nm. Meshes of larger sizes are available but with a low frequency. For cells treated with LPDF, this strong compaction is reduced and the major mesh size is extended up to 180 nm. Also the mesh size of 250 nm to 380 nm shows an increased frequency. Cells treated with BPDF show still the majority of meshes in the range of 40 nm to 120 nm (comparable to medium only) but with a lower frequency compared to cells in medium only. Moreover, the large meshes show heterogeneity in their frequency indicating changes in the heterochromatin organization. As having shown previously, heterochromatin re-organization is correlated to changes in gene activity (up- or down-regulation).6 Thus, changes in mesh sizes of the heterochromatin network could change the accessibility of gene loci which might cause additional/other genetic activities. The situation considerably changed for cell treated with CPDF. Here the small meshes, which were the most frequent ones in cells treated with LPDF, are the ones with a disappearing frequency while the large mesh sizes become the most frequent ones. This is correlated with a complete reorganization of heterochromatin and consequently many functional changes might be induced.
In Fig. 4B, the results of PCA are presented. Concerning the three major components 0, 1 and 2, the cells treated with BPDF, are completely separated indicating a topological change in the heterochromatin organization. For comparison, the cells treated with LPDF or with 3,4 DGE (10 μM) are within the variability of the HUVEC control that was cultivated in medium without any PDF.
Discussion
Recently, it has been shown that heterochromatin organization is strongly related to gene expression.6,11 This means that heterochromatin organization may orchestrate the accessibility of the transcription machinery that impacts gene expression. So far the association of heterochromatin organization with factors associated with CKD and PD has not been investigated. In this article, we present for the first time geometrical and topological changes of heterochromatin after cell exposure to uremic milieu and different PDF.Heterochromatin usually has a condensed and tight structure, making it less accessible to proteins.5,7 In such a way heterochromatin may control molecular trafficking in cell nuclei. DNA damaging for instance by ionizing radiation, diseases or therapies, for instance cancer treatment, can lead to rearrangements of chromatin and molecular reorganization affecting genomic functioning. Here, such changes have been studied by SMLM followed by distance and topological analysis of molecular point patterns.35
We have designed a cell system based on human umbilical vein endothelial cells (HUVEC) and a heterochromatin specific antibody against H3K9me3 methylation sites11,2a which are known to exist independently of the degree of compaction of the heterochromatin.41a
The specificity of the antibodies may be considered in the context of antibody choice. The antibodies anti-H3K9me3 are known to show some but to our experience negligible cross-labelling of non-heterochromatic regions. There are other antibodies available against heterochromatin but none of them is free from cross-labelling. Detailed studies of data bases have induced the decision to use this antibody as the best compromise between labelling specificity and labelling efficiency in many studies of genome organization and radiation biophysics. Moreover, this makes the presented results compatible and comparable to many other results recently published (e.g., ref. 2, 5, 6 and 19).
The HUVEC cell system was exposed to plasma from 5 patients suffering from chronic kidney disease (CKD) and 5 healthy individuals. Patients with CKD suffer from exceedingly high risk for cardiovascular disease (CVD), the leading cause of death worldwide, with limited therapeutic options. CVD is the main complication in patients with CKD and can affect any part of the vasculature, including severe cases such as uremic arteriolopathy with varying severity depending on risk factor exposure.
Although our experiments are limited to a small number of individual patients, we studied children with CKD5 with underlying diseases limited to the kidney and urinary tract, thus largely excluding bias caused by life-style and comorbidities.41b
We did not relate heterochromatin changes induced by uremic plasma and PD fluids with gene expression. However patient 4 showed an impressive increase in heterochromatin tags compared to the controls. This might indicate a disruption of secondary and tertiary structures in chromatin.41c In another recent study42 analysing H3K9me3 in smooth muscle cells, the authors propose chromatin decompaction as a driving force for smooth muscle cell transformation into osteoclasts, a process leading to calcification of the vascular wall. The genes identified in the de-compacted heterochromatin region included members of the NF-kB inflammatory pathways and senescence associated secretory phenotype related genes, which are major drivers of vascular aging in endothelial cells as well.43 Our cell model of short-term in vitro exposure to plasma from chronically uremic patients and PD fluids has limitations, which should be addressed in in vitro repeated and longer exposure models, in respective experimental in vivo models, including the potential capacities of recovery of the heterochromatin organization and in vascular tissues from patients with CKD and on PD.
The analysis by novel mathematical approaches of H3K9me3 loci in nuclei of endothelial cells incubated with plasma from healthy individuals and children with CKD5 resulted in considerable differences in the number of tagged H3K9me3 molecules. While the mean values did not show significant differences between the groups, the variance observed among children with CKD5 was twice that of individuals with normal kidney function, most likely reflecting individual differences in the response to the uremic toxin load. Distance analysis shows cluster formations in all cells, but cell patterns of specimens treated with healthy plasma are more similar to each other than those treated with CKD5 plasma. The part in the distance frequency histograms related to random point distribution is up to three times higher in CDK5 plasma treated cells compared to healthy plasma treated cells, indicating relaxation of the heterochromatin structures. The analysis showed more organized and homogeneous clusters in all endothelial cells treated with plasma from healthy individuals, while in endothelial cells incubated with plasma from CKD5 children the clusters showed irregularities and were less pronounced. Higher numbers of H3K9me3 molecules coupled with a reduced number of clusters imply a relaxation of HC in the uremic milieu, suggesting higher transcriptional activity due to better accessibility of the chromatin. Topological analysis35 employed to describe the chromatin architecture confirms this observation of high variations among individual CKD5 donors.
Although only standardized tools of data evaluation were used, which have been applied for several applications of SMLM, the cells in many cases showed a broad variability in the quantitative values. Using standard box plot graphics allowed for the description of the major differences between the cell treatments. Calculation of p-values gave hints about the quality of significance of differences.
Nevertheless, it might be apposite to ask for other reasons of this variability than the treatment. At a first glimpse one might think about preparation effects that on one hand can never be excluded completely. However, the specimens were prepared by well skilled persons without visible differences in quality. So, on the other hand, real biological reasons have to be included into the consideration. Recently, Alekseenko et al.44 have pointed out reasons for fundamentally low reproducibility of quantitative results between individual cells. For each cell that could be subjected to single cell experiments slightly different conditions in the microenvironment might lead to small functional differences in the individual cells although they all contain to the same entity. This might be negligible in bulk experiments but in single cell experiments as presented here, it could become more prominent. Or with other words, the more precisely single cell measurements are performed, the more variations between individual cells may become obvious.
In the experiments applying plasma of uremic individuals, additional variability is introduced since the composition of the plasma is influenced by various factors, such as the duration and degree of chronic kidney disease, nutrition, medication and the intestinal microbiome.41c
Heterochromatin in HUVEC also responded to various PD fluids and their related glucose degradation products (GDP), particularly 3,4 DGE, which was applied in two concentrations. GDP are known to be very highly reactive and form advanced glycation end-products, which are deposited in the endothelium.24 These trigger tight junction disintegration and consequently cell death, well known mechanisms associated with vascular disease development.24 Although PD solutions with low amount of GDP are available, these still induce major vascular changes, predominantly due glucose-induced immune response and angiogenesis.25 The effects of these compounds on heterochromatin organization caused a pronounced increase in visible H3K9me3 tag molecules compared to cells treated with medium only. This indicates not only heterochromatin changes but also heterochromatin relaxation. The bicarbonate buffer leads to almost the double numbers of registered molecules compared to the other PD fluids, indicating a de-compaction of heterochromatin and thus a better accessibility for the anti-H3K9me3 antibodies for heterochromatin labelling.
Between the lactate buffered PDF the higher GDP containing CPDF results in higher count numbers compared to LPDF. Examining the highly toxic, well characterized GDP, the 3,4 DGE, an increase in concentration resulted in a higher number of detected molecules, which also corresponds to the observations made between the lactate buffered PDF. Distance analysis showed an increased random distribution at greater distances. Lactate buffered PDF show more clustering and less random distribution, hence less relaxation of the examined H3K9me3, compared to BPDF. The whole distribution in the topological analysis indicates that the high GDP containing CPDF has the highest number of similarly sized hole structures on high mesh sizes. On the other hand, cells treated with BPDF show a completely different chromatin organization than cells exposed to the other PDF.
The biocompatibility of low GDP-PDF is widely debated. Despite increasing clinical and experimental evidence supporting the benefits of low GDP-PDF, many countries still use high GDP-PDF. GDPs are rapidly absorbed from the peritoneal cavity and increase the concentration of advanced systemic glycation end products. Multiple randomized studies have shown the advantages of fluids with low GDP in contrast to fluids with high GDP.45–47
In conclusion, the results for spatial heterochromatin organization obtained by SMLM and advanced mathematical tools demonstrated that different PDF with different impact on cells caused heterochromatin reorganization on different levels of compaction and de-compaction. Future systematic studies will further improve the knowledge of PDF impact on chromatin organization and genome functioning.
Experimental
Cell culture and immunostaining
Polarized human umbilical vein endothelial cells (HUVEC) were purchased commercially (PromoCell, Heidelberg, Germany) and cultured as recommended by the manufacturer (for further details see ref. 37). They had been grown on transwells (Greiner, Heidelberg, Germany) and were exposed to plasma from healthy donors (n = 5) and CKD5 patients (n = 5) mixed 1:4 with cell medium (plasma samples collected within the International Peritoneal Biobank25) and to different peritoneal dialysis fluids (PDF). The five CKD5 patients suffered from congenital abnormalities of kidney and urinary tract.
PDF (pH neutral, bicarbonate buffered, low GDP-PDF (BPDF), pH neutral lactate buffered low GDP-PDF (LPDF) and conventional, acidic, lactate buffered high GDP-PDF (CPDF)) were mixed 1:1 with cell medium. Glucose degradation product (GDP) 3,4 di-deoxy-glucosone (3,4-DGE) was used in concentrations found in CPDF.
Anti-H3K9me3 (ab8898, 1:1000, Abcam, Cambridge, UK) was diluted in blocking buffer containing 5% fetal bovine serum.37 Cell nuclei were stained with DAPI, the filters were cut out and fixed with ProlongGold until imaging.
Single molecule localization microscopy (SMLM)
SMLM is an advanced technique of super-resolution fluorescence microscopy29 and makes use of stochastic blinking of dye molecules for precise localization in the ten-nanometre range of single molecules. The dye molecules stochastically switch between two different spectral states, on- (fluorescing) and off- (non-fluorescing).33,34 This “blinking” allows spatial separation of individual fluorescent molecules.30 The SMLM setup described in14,48 has an oil-immersion objective lens (100×/NA 1.46) and four different lasers for illumination with the wavelengths 405 nm/491 nm/561 nm/642 nm with maximal laser powers of 120 mW/200 mW/200 mW/140 mW, respectively. The illumination laser light path is equipped with a LightHub—laser combiner (Omicron Laserprodukte GmbH, Rodgau-Dudenhofen, Germany) and a polychromatic AOTF (AA Opto Electronic, Orsay Cedex, France) for switching between the laser wavelengths. The shapes of the laser beams are formed by a variable beam expander 10BE03-2-8 (Standa Ltd, Vilnius, Lithuania) and a Flat-Top-Profile forming optics—PiShaper (AdlOptica GmbH, Berlin, Germany). To separate illumination and fluorescence, two quadband interference filter glasses F73-410 and F72-866 (AHF Analysentechnik AG, Tübingen, Germany) are used. Fluorescence images with the blinking events are recorded by an Andor Ultra EMCCD (iXonUltra 897, Andor Technology, Belfast, Northern Ireland). Data are from five independent sets of experiments from each treatment 25 cells were recorded. The individual cell nuclei were selected by uniform shape based on DAPI staining (using the 405 nm laser). Up to 2000 image frames were acquired at each illumination wavelength used with an exposure time of 100 ms per image. No background was recorded showing high specificity of the H3K9me3 immunostaining.Software and Statistics
Using an in-house software package,35 super-resolution signal coordinates were calculated according to an algorithm subtracting the brightness values of two successive image frames. Dark states over more than two successive frames were registered, the barycentre calculated,49 the loci coordinates of all molecules registered in the ‘orte-matrix’. Signals with intensities below the threshold were sorted out.In Fig. 5, the further evaluation process35 is schematically visualized: Ripley statistics41,50 was applied to the coordinate values of the points and point-to-point distances were calculated and normalized. For topological analysis, persistent homology,38,51 persistent imaging52 and principal component analysis53 were applied as described in detail in ref. 35. These analyses depend on the extraction of major structures of a pointillist pattern. Mathematically, this means a transformation in the topological space spanned by these structures. We calculated two parameters: the number of components (number of points with which the analysis starts) and holes (closed configurations like meshes of a net occurring and disappearing during the mathematical evaluation process). For this calculation, each point (“component”) registered in the orte-matrix is virtually surrounded by a circle which radius is increasing. Each component is represented by a bar starting at 0 (i.e., the given point) and ending at the radius value where two circles attach. At this moment the two components merge into one and one of the two bars stops further growing. During this process enlarged components can arise, forming a closed structure and showing free space inside not covered by the increasing virtual circles (“hole”). With the birth of a hole another bar starts. When the hole is completely covered by the increasing virtual circles, also this bar ends. In this way, the measured point pattern is transferred to a bar code pattern of components and holes.
 | ||
| Fig. 5 Schematic representation of the different evaluation steps of SMLM datasets. After acquisition of a time series of image frames, the coordinates and other values of all the blinking events of a cell nucleus were integrated into a matrix, the so-called “orte-matrix” (top-left). In the one-channel analysis, the coordinate values of the points were subjected to Ripley statistics. For the representative graph (top right), the point-to-point distances were calculated and represented in a normalized distance frequency histogram. In the lower part, the processes of persistent homology, persistent imaging, and principal component analysis are shown. The point pattern is transferred into a bar code description of components and holes. The lengths of the bars (difference of the α values of the end and the beginning of a particular bar) provide the lifetime; the beginning provides the birth in the one-point cloud. The one-point clouds for all cell nuclei that are considered for evaluation are transferred in pixel images (persistent imaging). Each pixel is compared for all images. The results of these comparisons span an n-dimensional orthogonal vector space. The variations of the pixel values determine the components of the principal component analysis. Finally, the outcome for the components (orthogonal vector values) with the largest variation and the second largest variation determines the latent space (graph bottom right) (for further details, see text in Material and Methods). Note: this figure was originally published under CC-BY licence in ref. 5. | ||
To compare the bar codes of individual cells with each other, we calculated the Jaccard index,39 a standard similarity measure of two given bar codes. The Jaccard index is normalized and results in values between 0 (no overlap of two bar codes) and 1 (two identical bar codes). The Jaccard indices of the individual cells of one individual are represented in a 1st generation heatmap, to compare the individuals with each other the heatmap values of one individual are averaged and the mean values of all individuals are compared with each other by the representation in a 2nd generation heatmap.40
The bar-codes of the holes are transferred into a diagram of “bar life-time” vs. “bar birth”, overlaid by a pixel scan (persistent image). The intensity of each pixel correlates to the number of points in the given pixel. Persistent images are obtained for each cell. Finally, the persistent images are subjected to principal component analysis (PCA).35 The values of the 1st, the 2nd, the 3rd, the 4th, …, the nth pixel of all individual persistent images are separately compared and build an n-dimensional orthogonal vector space. The dimension with the larges variances forms the component 0 in the final diagram. Component 1 is a dimension perpendicular to component 0 and shows the second large variance of values. In this way, the results of a complex point pattern can be reduced to a latent space of two dimensions representing the largest changes in topology. This means that only two principal components describe the main features and their changes of the labelled molecules while small variations (“biological noise”) are neglected.
Conclusions
By means of SMLM we measured the spatial organization of heterochromatin methylation points (H3K9me3) and subjected the results to mathematical procedures that gave us geometrical and topological information so that feature parameters could be extracted and compared. It was shown that the spatial heterochromatin organization of endothelial cells is variable in response to treatment with CKD plasma. PDF induce major heterochromatin relaxation, depending on the buffer and GDP content. This indicates that additional chromatin accessibility could be opened and genetic activity may change. Our findings provide a novel base for measuring and improving PD biocompatibility which may show the vascular endothelial health during PD.Author contributions
Conceptualization, M. B. and A. S.; methodology, M. B., G. M. and M. H.; software, A. S. and M. S.; validation, M. B., A. S., and M. S.; formal analysis, M. B., A. S., G. M. and M. S.; investigation, M. B., C. S. and M. H.; resources, C. S. and M. H.; data curation, A. S. and M. S.; writing—original draft preparation, M. B. and M. H.; writing—review and editing, M. B., A. S., M. S., G. M., C. S. and M. H.; visualization, A. S. and M. S.; supervision, C. P. and M. H.; project administration, M. B., C. S. and M. H.; funding acquisition, M. B. and C. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
Authors declare no conflict of interest.Data availability
Data and software are available upon request from corresponding author.Acknowledgements
This work has been part of the IMPROVE-PD project, that has received funding from the European Union's Horizon 2020 Research and Innovation Programs under the Marie Sklodowska-Curie Grant Agreement Number 812699 (C. P. S.). M. B. was funded by the Olympia Morata Fellowship from Heidelberg University and acknowledges Baden-Württemberg Stiftung for the financial support by the “Eliteprogramme für Postdocs”. C. P. S. has obtained funding from European Nephrology and Dialysis Institute (ENDI). The authors are indebted to Felix Bestvater, German Cancer Research Centre (DKFZ), Heidelberg, for his continuous instrumental support.References
- J. X. Feng and N. C. Riddle, Mamm. Genome, 2020, 31, 181–195 CrossRef PubMed.
- (a) J. Erenpreisa, J. Krigerts, K. Salmina, B. I. Gerashchenko, T. Freivalds, R. Kurg, M. Krufczik, R. Winter, P. Zayakin, M. Hausmann and A. Giuliani, Cells, 2021, 10, 1582 CrossRef CAS PubMed; (b) D. Nicetto and K. S. Zareth, Curr. Opin. Genet. Dev., 2019, 55, 1–10 CrossRef CAS PubMed.
- M. Hausmann, G. Hildenbrand and G. Pilarczyk, in Nuclear, Chromosomal, and Genomic Architecture in Biology and Medicine, ed. M. Kloc and J. Z. Kubiak, Results and Problems in Cell Differentiation, 2022, vol. 70, pp. 3–34 Search PubMed.
- S. L. Klemm, Z. Shipony and W. J. Greenleaf, Nat. Rev. Genet., 2019, 20, 207–220 CrossRef CAS PubMed.
- M. Schäfer, G. Hildenbrand and M. Hausmann, Int. J. Mol. Sci., 2024, 25, 12843 CrossRef PubMed.
- J. Krigerts, K. Salmina, T. Freivalds, P. Zayakin, F. Rumnieks, I. Inashkina, A. Giuliani, M. Hausmann and J. Erenpreisa, Biophys. J., 2021, 120, 711–724 CrossRef CAS PubMed.
- M. Falk, Y. Feodorova, N. Naumova, M. Imakaev, B. R. Lajoie, H. Leonhardt, B. Joffe, J. Dekker, G. Fudenberg, I. Solovei and L. A. Mirny, Nature, 2019, 570, 395–399 CrossRef CAS PubMed.
- M. Tsuchiya, P. Brazhnik, M. Bizzarri and A. Guiliani, Int. J. Mol. Sci., 2023, 18, 24 Search PubMed.
- G. Zimatore, M. Tsuchiya, M. Hashimoto, A. Kasperski and A. Giuliani, Biophys. Rev., 2021, 2, 031303 CrossRef CAS PubMed.
- G. Millán-Zambrano, A. Burton, A. J. Bannister and R. Schneider, Nat. Rev. Genet., 2022, 23, 563–580 CrossRef PubMed.
- C. Y. Ho, M.-Y. Wu, J. Thammaphet, S. Ahmad, J. Ho C. S., L. Draganova, G. Anderson, U. S. Jonnalagadda, R. Hayward, R. Shroff, W. Tan Lek Wen, A. Verhulst, R. Foo and C. M. Shanahan, Circ. Res., 2025, 136, xx CrossRef PubMed.
- E. P. Rogakou, C. Boon, C. Redon and W. M. Bonner, J. Cell Biol., 1999, 146, 905–916 CrossRef CAS PubMed.
- B. Jakob, J. Splinter, S. Conrad, K.-O. Voss, D. Zink, M. Durante, M. Löbrich and G. Taucher-Scholz, Nucleic Acids Res., 2011, 39, 6489–6499 CrossRef CAS PubMed.
- M. Hausmann, E. Wagner, J.-H. Lee, G. Schrock, W. Schaufler, M. Krufczik, F. Papenfuß, M. Port, F. Bestvater and H. Scherthan, Nanoscale, 2018, 10, 4320–4331 RSC.
- J. Stadler and H. Richly, Int. J. Mol. Sci., 2017, 18, 1715 CrossRef PubMed.
- W. D. Wright, S. S. Shah and W.-D. Heyer, J. Biol. Chem., 2018, 293, 10524–10535 CrossRef CAS PubMed.
- H. H. Y. Chang, N. R. Pannunzio, N. Adachi and M. R. Lieber, Nat. Rev. Mol. Cell Biol., 2017, 18, 495–506 CrossRef CAS PubMed.
- Y. Zhang, G. Máté, P. Müller, S. Hillebrandt, M. Krufczik, M. Bach, R. Kaufmann, M. Hausmann and D. W. Heermann, PLoS One, 2015, 10, e0128555 CrossRef PubMed.
- J. Erenpreisa, A. Giuliani, K. Yoshikawa, M. Falk, G. Hildenbrand, K. Salmina, T. Freivalds, N. Vainshelbaum, J. Weidner, A. Sievers, G. Pilarczyk and M. Hausmann, Int. J. Mol. Sci., 2023, 24, 2658 CrossRef CAS PubMed.
- B. I. Gerashchenko, K. Salmina, J. Eglitis, A. Huna, V. Grjunberga and J. Erenpreisa, Histochem. Cell Biol., 2016, 145, 497–508 CrossRef CAS PubMed.
- J. Erenpreisa, A. Giuliani and M. S. Cragg, Int. J. Mol. Sci., 2023, 24, 14567 CrossRef PubMed.
- L. Mosteiro, C. Pantoja, A. de Martino and M. Serrano, Aging Cell, 2018, 17, e12711 CrossRef PubMed.
- R. N. Foley, P. S. Parfrey and M. J. Sarnak, Am. J. Kidney Dis., 1998, 32(5 Suppl 3), S112–S119 CrossRef CAS PubMed.
- M. Bartosova, C. Zhang, B. Schaefer, R. Herzog, D. Ridinger, I. Damgov, E. Levai, I. Marinovic, C. Eckert, P. Romero, P. Sallay, A. Ujszaszi, M. Unterwurzacher, A. Wagner, G. Hildenbrand, B. A. Warady, F. Schaefer, S. G. Zarogiannis, K. Kratochwill and C. P. Schmitt, Circ. Res., 2021, 129, e102–e118 CrossRef CAS PubMed.
- B. Schaefer, M. Bartosova, S. Macher-Goeppinger, P. Sallay, P. Vörös, B. Ranchin, K. Vondrak, G. Ariceta, A. Zaloszyc, A. K. Bayazit, U. Querfeld and R. Cerkauskiene, et al. , Kidney Int., 2018, 94, 419–429 CrossRef CAS PubMed.
- T. Levstek and K. Trebušak Podkrajšek, Antioxidants, 2023, 12, 579 CrossRef CAS PubMed.
- M. Bartosova, S. G. Zarogiannis and C. P. Schmitt, Members of the International Pediatric Biobank, Mol. Cell. Pediatr., 2022, 9, 9 CrossRef PubMed.
- A. V. Solov'yov, A. V. Verkhovtsev, N. J. Mason, R. A. Amos, I. Bald, G. Baldacchino, B. Dromey, M. Falk, J. Fedor and L. Gerhards, et al. , Chem. Rev., 2024, 124, 8014–8129 CrossRef PubMed.
- M. Lelec, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffie, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev. Methods Primers, 2021, 1, 39 CrossRef PubMed Erratum in: M. Lelec, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffie, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev. Methods Primers, 2022, 2, 70 CrossRef.
- M. Hausmann, N. Ilić, G. Pilarczyk, J.-H. Lee, A. Logeswaran, A. P. Borroni, M. Krufczik, F. Theda, N. Waltrich and F. Bestvater, et al. , Int. J. Mol. Sci., 2017, 18, 2066 CrossRef PubMed.
- M. Hausmann, C. Neitzel, H. Hahn, R. Winter, I. Falkova, D. W. Heermann, G. Pilarczyk, G. Hildenbrand, H. Scherthan and M. Falk, Med. Sci. Forum, 2021, 3, 15 Search PubMed.
- I. M. Khater, I. R. Nabi and G. Hamarneh, Patterns, 2020, 1, 100038 CrossRef CAS PubMed.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS PubMed.
- H. Deschout, F. Cella Zanacchi, M. Mlodzianoski, A. Diaspro, J. Bewersdorf, S. T. Hess and K. Braeckmans, Nat. Methods, 2014, 11, 253–266 CrossRef CAS PubMed.
- J. Weidner, C. Neitzel, M. Gote, J. Deck, K. Küntzelmann, G. Pilarczyk, M. Falk and M. Hausmann, Comput. Struct. Biotechnol. J., 2023, 21, 2018–2034 CrossRef CAS PubMed.
- K. Jahnke, N. Struve, D. Hofmann, M. J. Gote, M. Bach, M. Kriegs and M. Hausmann, Nanoscale, 2024, 16, 15240–15255 RSC.
- M. Bartosova, R. Herzog, D. Ridinger, E. Levai, H. Jenei, C. Zhang, G. T. González Mateo, I. Marinovic, T. Hackert, F. Bestvater, M. Hausmann, M. López Cabrera, K. Kratochwill, S. G. Zarogiannis and C. P. Schmitt, Biomolecules, 2020, 10, 1178 CrossRef CAS PubMed.
- A. Hofmann, M. Krufczik, D. W. Heermann and M. Hausmann, Int. J. Mol. Sci., 2018, 19, 2263 CrossRef PubMed.
- P. Jaccard, Bull. Soc. Vaudoise Sci. Nat., 1901, 37, 547–579 Search PubMed.
- H. Hahn, C. Neitzel, O. Kopecná, D. W. Heermann, M. Falk and M. Hausmann, Cancers, 2021, 13, 5561 CrossRef CAS PubMed.
- (a) B. D. Ripley, J. R. Stat. Soc. Series B, 1977, 39, 172–192 CrossRef; (b) F. Natale, A. Rapp, W. Yu, A. Maiser, H. Harz, A. Scholl, S. Grulich, T. Anton, D. Hörl, W. Chen, M. Durante, G. Taucher-Scholz, H. Leonhardt and M. C. Cardoso, Nat. Commun., 2017, 8, 15760 CrossRef CAS PubMed; (c) A. Martin, J. MacDonald and J. Moore, Anaesth. Intensive Care Med., 2015, 16, 267–274 CrossRef; (d) S. Raghunathan, T. Jaganade and U. D. Priyakumar, Biophys. Rev., 2020, 12, 65–84 CrossRef CAS PubMed.
- C. Y. Ho, M. Y. Wu, J. Thammaphet, S. Ahmad, C. S. J. Ho, L. Draganova, G. Anderson, U. S. Jonnalagadda, R. Hayward, R. Shroff, W. T. L. Wen, A. Verhulst, R. S. Foo and C. M. Shanahan, Circ. Res., 2025, 4, 379–399 CrossRef PubMed.
- S. Costantino, F. Paneni and F. Cosentino, J. Physiol., 2016, 8, 2061–2073 CrossRef PubMed.
- I. Alekseenko, L. Kondratyeva, I. Chernov and E. Sverdlov, Int. J. Mol. Sci., 2023, 24, 2796 CrossRef CAS PubMed.
- M. Zeier, V. Schwenger, R. Deppisch, U. Haug, K. Weigel, U. Bahner, C. Wanner, H. Schneider, T. Henle and E. Ritz, Kidney Int., 2003, 1, 298–305 CrossRef PubMed.
- C. P. Schmitt, D. von Heyl, S. Rieger, K. Arbeiter, K. E. Bonzel, M. Fischbach, J. Misselwitz, A. K. Pieper and F. Schaefer, Nephrol. Dial. Transplant., 2007, 7, 2038–2044 CrossRef PubMed.
- D. W. Johnson, F. G. Brown, M. Clarke, N. Boudville, T. J. Elias, M. W. Foo, B. Jones, H. Kulkarni, R. Langham, D. Ranganathan, J. Schollum, M. Suranyi, S. H. Tan and D. Voss, J. Am. Soc. Nephrol., 2012, 23(6), 1097–1107 CrossRef PubMed.
- M. Eryilmaz, E. Schmitt, M. Krufczik, F. Theda, J.-H. Lee, C. Cremer, F. Bestvater, W. Schaufler, M. Hausmann and G. Hildenbrand, Cancers, 2028, 10, 25 CrossRef PubMed.
- F. Grüll, M. Kirchgessner, R. Kaufmann, M. Hausmann and U. Kebschull, Proc. 2011 Int. Conf. Field Programmable Logic and Applications (FPL), Chania, Greece, September 5th–11th, 2011, 1–5.
- G. Máté and D. W. Heermann, Front. Phys., 2014, 2, 20 Search PubMed.
- R. Ghrist, Bull. Am. Math. Soc., 2008, 45, 61–75 CrossRef.
- H. Adams, S. Chepushtanova, T. Emerson, E. Hanson, M. Kirby, F. Motta and L. Ziegelmeier, J. Mach. Learn. Res., 2017, 18, 1–35 Search PubMed.
- K. Pearson, London, Edinburgh Dublin Philos. Mag. J. Sci., 1901, 2, 559–557 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |