
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting protein protected gold and silver nanoclusters: Excited state dynamics and long-lived emission†
Shagun
Sharma
 a,
Subhadeep
Das
b,
Kush
Kaushik
a,
Abhijit
Patra
*b and
Chayan Kanti
Nandi
*a
a,
Subhadeep
Das
b,
Kush
Kaushik
a,
Abhijit
Patra
*b and
Chayan Kanti
Nandi
*a
aSchool of Chemical Sciences, Indian Institute of Technology (IIT) Mandi, H.P-175075, India. E-mail: chayan@iitmandi.ac.in
bDepartment of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, M.P.-462066, India. E-mail: abhijit@iiserb.ac.in
First published on 25th April 2025
Abstract
Owing to their intriguing photophysical properties, protein-functionalized metal nanoclusters (MNCs) have become benchmark nanomaterials in various fields, including electronics, optics, energy, sensing, and biomedicine. However, their excited-state dynamics, particularly long-lived emission, remain a subject of debate. In this work, we investigated the excited state dynamics of bovine serum albumin (BSA) functionalized Ag and Au nanoclusters (BSA-Ag NCs and BSA-Au NCs) and provided new insights into their long-lived emission. The decay dynamics of excited-state nanoclusters showed distinct photoluminescence behavior influenced by the specific metal core in the BSA-MNCs. BSA-Au NCs showed short-lived emission followed by a long-lived excited state at room temperature, originating from the core-surface state interactions. These interactions facilitate ligand-to-metal charge transfer states, which enable efficient triplet-state harvesting at room temperature. BSA-Ag NCs, on the other hand, exhibit short-lived emission originating only from the core states, resulting in fluorescence as the relaxation pathway at room temperature, which transitioned to phosphorescence with sub-millisecond lifetimes under cryogenic conditions. The time-resolved luminescence imaging microscopy of both MNCs in HeLa cells indeed confirmed the long-lived emission characteristics in BSA-Au NCs but not in BSA-Ag NCs.
Introduction
MNCs have witnessed significant progress in recent years owing to their intriguing photophysical properties such as near-infrared emission, large Stokes shift, low cytotoxicity, and long photoluminescence lifetimes.1–4 Among various ligand-protected MNCs, protein-functionalized MNCs are particularly interesting because of their facile synthesis, low toxicity, and excellent biocompatibility.5–8 This makes them highly versatile for application in electronics, optics, energy, sensing, and biomedicine.9–11 Since the discovery of the biomineralization approach for the synthesis of BSA protected gold (Au) and silver (Ag) nanoclusters, there has been a growing number of studies on these clusters.6,12–16 Although significant research has predominantly focused on their red emissive photoluminescence properties, investigations into their excited state relaxation processes remain relatively limited.One of the most intriguing features of these protein-conjugated MNCs are the nearly identical excitation spectra and red emissive luminescence spectra that originates due to the metal (M) coordination with the sulfur (S) of cysteine residue in the protein through ligand to metal charge transfer (LMCT) (Fig. 1a).17 Both static and dynamic characters of the red luminescence in the protein–Au(III) compounds were proposed. In static mechanism, BSA is considered as static cage, where Au(III) are reduced by the tyrosine residues and formed neutral Au25 gold nanoclusters.18 The second mechanism highlights the dynamic nature of BSA, where Au(III) chemisorption at a specific site resulted in the formation of a red luminophore without the reduction of Au(III) to Au(0). Overall, the studies highlight the important role of the local environment around the gold-binding site in tuning the luminescence properties.18,19 In contrast, MNCs without protein conjugation but with a different surface ligand showed dramatic change in the emission characteristics just with the incorporation of single atom in the core of the MNCs.20–22 As a result, the excited state dynamics of these MNCs remain a debatable topic, largely due to the presence of multiple emissive centers within the MNCs.23,24
 | ||
| Fig. 1 (a) Schematic representation of the possible structure and the typical lifetime decay curve of protein-conjugated MNCs. (b) The energy level diagram dictating the mechanisms for the varied lifetime range, helped in understanding the excited state dynamics. | ||
Time-resolved luminescence studies have been employed to elucidate the excited state relaxation process in these MNCs.25,26 Considering the decay characteristics, it is proposed that the nanosecond lifetime mainly originates from the metal–metal (M–M) core emission of the MNCs. A lifetime of sub-microseconds ranging from 100–200 ns is associated with the emission from the LMCT states, originating from the intramolecular interactions involving metal core and surface motifs.27 Despite the identical emission spectra of different protein-functionalized MNCs, a significant variation in lifetime occurred, potentially due to additional coordination to local residues. Most importantly, the long-lived lifetime from microseconds to milliseconds results from the thermally activated delayed fluorescence (TADF) or room temperature phosphorescence (RTP) (Fig. 1a).28–30
The Jablonski diagram presented in Fig. 1b depicts all the above relaxation processes. Both TADF and RTP mechanisms involve triplet state harvesting, with a distinct difference in the emission pathways. The thermal energy, especially singlet–triplet energy gap (ΔEst) plays a critical role in deciding the reverse intersystem crossing (rISC), which is the primary process to facilitate TADF. Typically, rISC becomes more significant at higher temperatures, while at cryogenic temperatures at around ∼77 K, rISC becomes less efficient due to insufficient thermal energy and a RTP process is favorable.31 Therefore, a characteristic feature of TADF is the redshift of emission spectra upon cooling due to the emission from lower T1 energy.32,33 A recent report on BSA-protected Au25 MNCs suggested that the red fluorescence band was attributed to the combined emission of prompt fluorescence and TADF, and the latter is more dominant.34 It has been further validated by the ΔEst estimation. However, the redshift of emission spectra upon cooling was not found in the study. In contrast, a very recent report on BSA conjugated copper nanoclusters (BSA-Cu NCs) presented the long-lived emission as RTP rather TADF.35 The observation was based on the similarity in time-resolved emission spectra at room and cryogenic temperatures (∼77 K) to support the RTP mechanism.
The above contradictory observations led us to revisit the long-lived excited state emission processes in the protein-conjugated MNCs. Thus, we aimed to investigate the intriguing relaxation mechanisms in BSA-Au NCs and BSA-Ag NCs. Interestingly, despite having the same BSA ligand, the BSA-Au NCs and BSA-Ag NCs exhibited distinct excited-state dynamics, especially the long-lived emission characteristics. While BSA-Au NCs showed a short-lived excited state followed by a long-lived emission originating from the core and surface states, the BSA-Ag NCs showed predominant short-lived emission, involving the core states only. The strong interaction between the metal core and surface motifs via the M–S bond supports LMCT states in BSA-Au NCs, whereas such interactions are weak in BSA-Ag NCs. The time-resolved emission spectra (TRES), the time-resolved area normalization emission spectra (TRANES), and the temperature-dependent luminescence measurements confirmed the RTP mechanism in BSA-Au NCs for the long-lived emission. In contrast, BSA-Ag NCs exhibit only fluorescence as the emission mechanism at room temperature (RT). However, it is transitioned to phosphorescence at 77 K which is due to the suppressed solvent and vibrational relaxations in a rigid environment. Further, the long component of BSA-Au NCs was employed for background-free intracellular imaging. The time-resolved luminescence imaging (TRLI) microscopy with the time delay is not possible with BSA-Ag NCs, confirming long-lived emission characteristics only in BSA-Au NCs.
Results and discussion
The synthesis of BSA-Ag NCs and BSA-Au NCs was carried out by the previously reported protocol.36,37 The transmission electron microscopy (TEM) data of about 100 particles suggested that BSA-Ag NCs and BSA-Au NCs have size diameters of 3.0 nm and 3.1 nm, respectively (Fig. S1d and S2d†). HR-TEM analysis of individual BSA-Ag NCs revealed an interplanar spacing of 0.338 nm, corresponding to the (012) lattice plane of monoclinic Ag2S (Fig. S1a–c†).38 Similarly, analysis of individual BSA-Au NCs showed an interplanar spacing of 0.240 nm, that corresponds to (111) lattice plane of face-centred cubic (fcc) Au (Fig. S2a–c†).39 It is important to note that TEM examines only a small portion of the sample, making it challenging to fully determine the structure and phase composition of a nanostructured material. Nevertheless, the interplanar spacings observed in the HR-TEM micrographs confirm different structural anatomies for the metal cores of BSA-Au NCs and BSA-Ag NCs. The size determined from TEM was well corroborated with the hydrodynamic diameter of the nanoclusters calculated using fluorescence correlation spectroscopy (FCS, Fig. S1e, S2e details in ESI†). The parameters obtained from the fitting analysis, namely diffusion times (τD), diffusion coefficient (Dt), number of molecules (N), hydrodynamic radius (rh), and diameter of the BSA-Ag NCs and BSA-Au NCs are presented in Tables S1 and S2,† respectively. The matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry data showed that BSA-Ag NCs consist of 30 Ag atoms (Fig. S3†). Corroborated with the literature, the number of Au atoms is assumed to be 25 in BSA-Au NCs fabricated following earlier reported method.36,40,41Furthermore, to obtain the valence states and the bonding environment in BSA-Ag NCs and BSA-Au NCs, X-ray photoelectron spectroscopy (XPS) was employed. The XPS spectra of Ag 3d provide two distinct components. The peaks with binding energy (BE) of 368.2 eV (Ag 3d5/2) and 374.2 eV (Ag 3d3/2) are designated for Ag(0), and 368.8 eV (Ag 3d5/2) and 374.8 eV (Ag 3d3/2) are assigned for Ag(I).42 The contributions of Ag(0) and Ag(I) were 67% and 33%, respectively (Fig. 2a). These results indicate that the core of the BSA-Ag NCs is stabilized both by zerovalent and univalent silver atoms. The peak at 161.4 eV in the high-resolution deconvolution data of S 2p of BSA-Ag NCs (Fig. 2b) is assigned to the metal–sulphur bond and the peak at 163.4 eV signified the unbound S of the BSA ligand.43 The XPS data of BSA-Au NCs showed two peaks at 84.8 eV and 88.4 eV, which were attributed to 4f7/2 and 4f5/2 for Au, respectively (Fig. 2c) The former peak was further deconvoluted into two distinct components at 84.6 eV and 85.4 eV, which were assigned to Au(0) (75%) and Au(I) (25%), respectively.15 The S 2p XPS spectrum of BSA-Au NCs shows two broad peaks at 161.8 eV and 164 eV, which are ascribed to sulphur bound to gold and the unbound sulphur of BSA, respectively (Fig. 2d).44
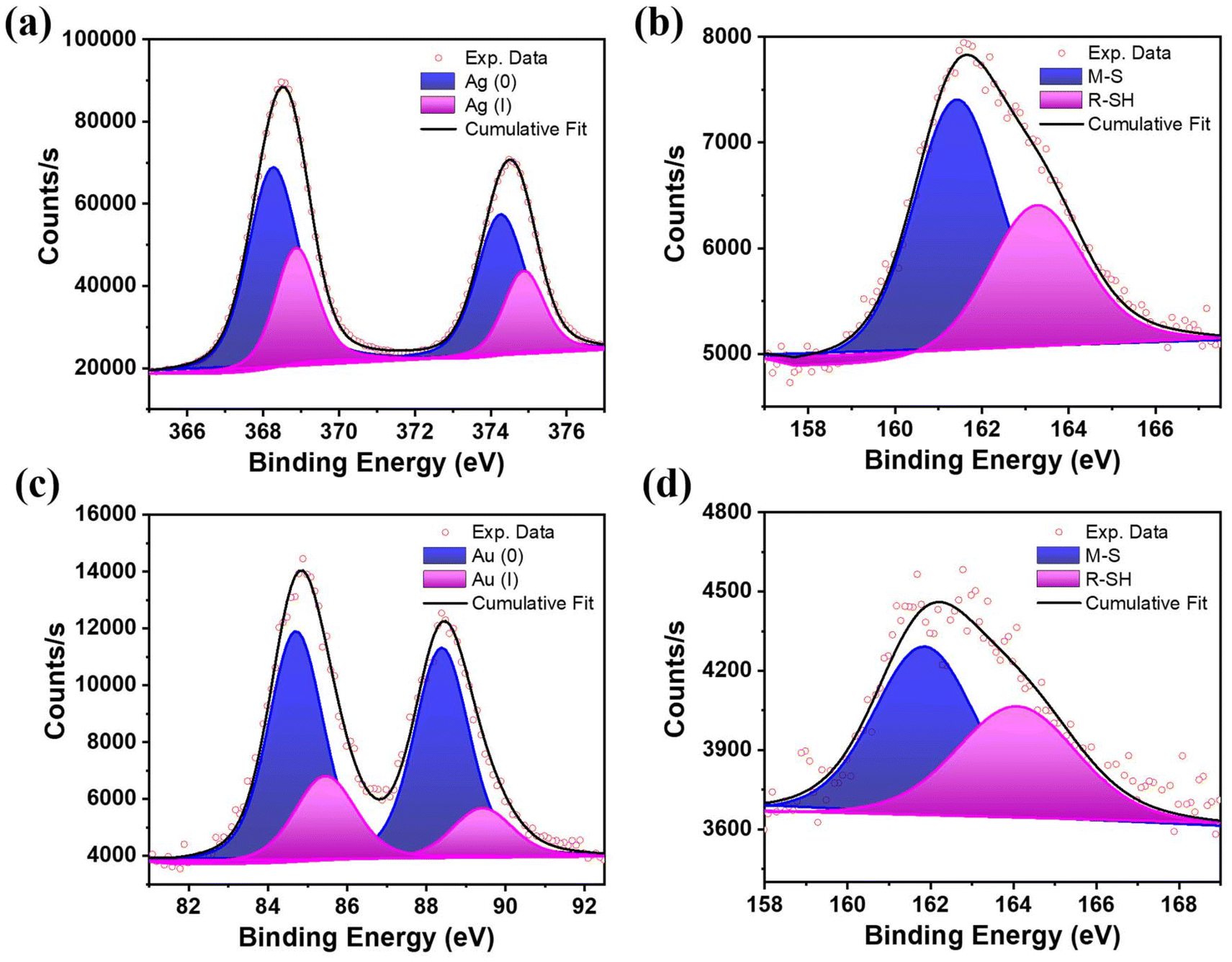 | ||
| Fig. 2 Deconvoluted X-ray photoelectron spectroscopy (XPS) profiles of (a) Ag 3d scan and (b) S 2p scan of BSA-Ag NCs. (c) Au 4f scan and (d) S 2p scan of BSA-Au NCs after etching time of 540.783 s. | ||
To understand the optical properties of BSA-Au NCs and BSA-Ag NCs, we recorded UV-vis absorption, excitation, and emission spectra, which were also shown in our previous report.45 Both the MNCs showed a broad absorption spectrum. However, a peak maximum at 280 nm corresponds to the tryptophan residues of BSA (Fig. S4a†).7,46 The emission spectra in both cases showed a red emission with a peak maxima at around 670 nm for BSA-Au NCs and 660 nm for BSA-Ag NCs when excited at 450 nm (Fig. S4b†). The photoluminescent quantum yield of BSA-Ag NCs and BSA-Au NCs were determined to be 2 ± 1% and 13 ± 1%, respectively. The excitation spectra of both the MNCs are distinctly different (Fig. S4a†). The BSA-Ag NCs show broad excitation with the excitation maximum at 400 nm, whereas the BSA-Au NCs showed progressively decreased excitation spectra with a hump at around 490 nm. This suggests that the different excited states are responsible for the emission in respective nanoclusters. The above results prompted us to understand the detailed mechanism and pathways of the excited state dynamics in both MNCs.
To investigate the excited state behaviour of red emissive MNCs, we employed luminescence lifetime measurements. BSA-Ag NCs showed the nanosecond lifetime as the dominant lifetime (inset of Fig. S4c†). The average lifetime of BSA-Ag NCs is 2.3 ns. The individual lifetime components with their amplitudes are presented in Table S3.† Additionally, a negligibly small sub-microsecond component is observed from the decay curve of BSA-Ag NCs presented in Fig. S4c.† The short and long lifetimes presented in the lifetime decay curve are tail-fitted separately (Fig. S5†) with the fitting parameters mentioned in Table S4.† On the contrary, the BSA-Au NCs showed a microsecond lifetime as the dominant one (Fig. S4c†). The average lifetime of BSA-Au NCs is found to be ∼1 μs (0.97 μs) using the triexponential function for fitting (Table S3†). The fraction of longer lifetime (A3) is more in comparison to the shorter lifetime (A1) in BSA-Au NCs.
To further understand the differences in their excited state decay profile at RT, TRES was employed to elucidate the distribution of the emissive states that evolved with time.35 For the TRES acquisition of BSA-Ag NCs, an excitation wavelength of 454 nm was used (details of the TRES acquisition are provided in ESI†). Fig. 3a displays the TRES of BSA-Ag NCs, which shows the evolution of the emission spectra over the nanosecond timescale (at certain time delays). The data showed spectral migration from 600 nm to 630 nm with the time evolution from 0.7 ns to 6.5 ns. The TRES data aligned well with the TRANES,35 showing similar spectral shifting (Fig. 3b). The spectral shifting seen in the TRES and TRANES of BSA-Ag NCs in water might be caused by the heterogeneity of the excited states arising due to intermolecular solvent relaxation.47 This might happen because the solvent molecules present in the vicinity of the nanoclusters in the excited state, induce dipole–dipole interaction, leading to a solvent-relaxed continuum of states. These states showed the observable emission detectable in the above TRES data. To get more information, TRES and TRANES were also recorded at 389 nm excitation, where no such spectral migration was seen (Fig. 3c and d).
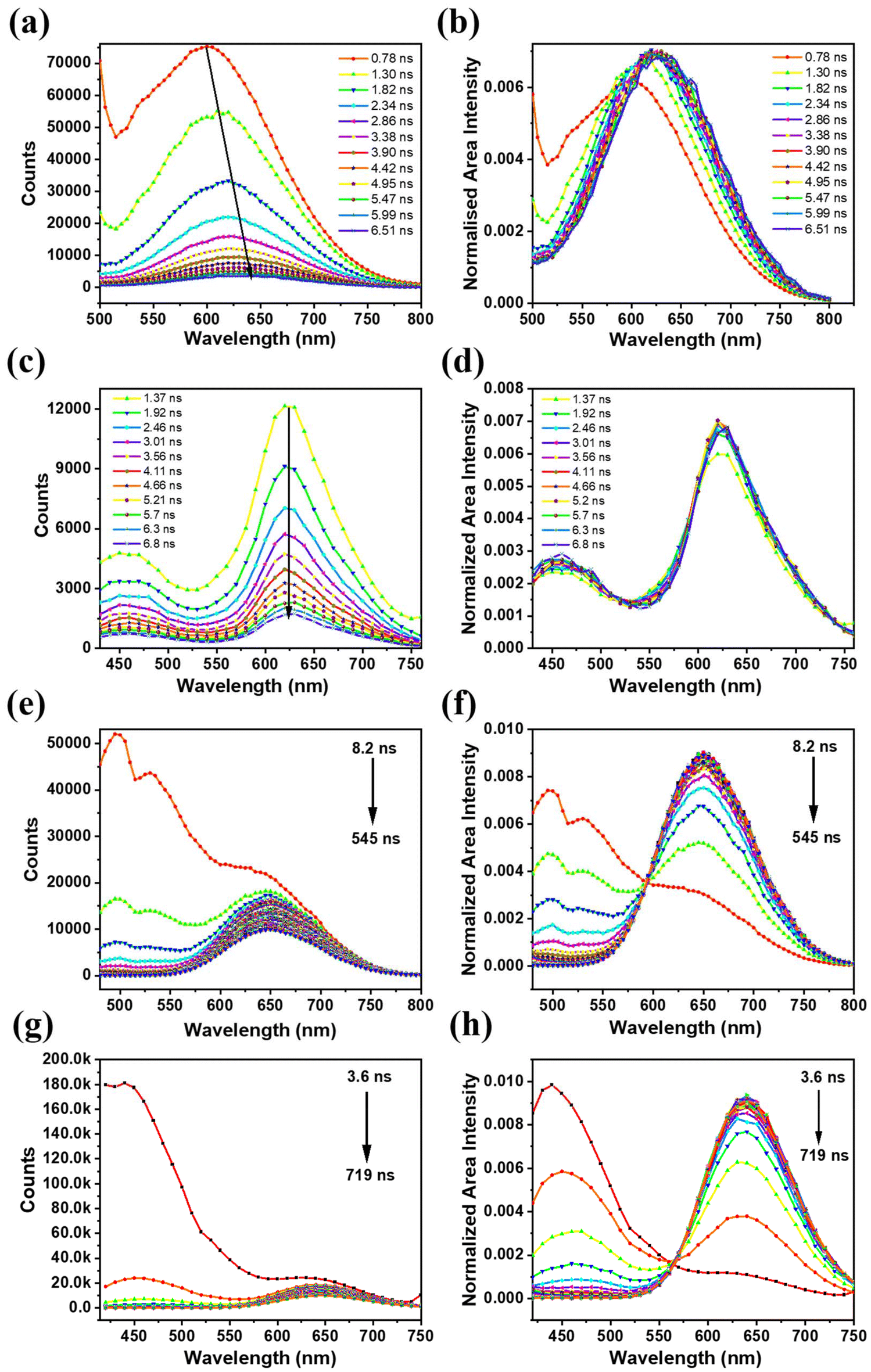 | ||
| Fig. 3 (a and c) The TRES and the (b and d) TRANES of BSA-Ag NCs [(a and b) – λex = 454 nm; (c and d) – λex = 389 nm (measurement range 100 ns)]. (e and g) The TRES and the (f and h) TRANES of BSA-Au NCs [(e and f) – λex = 454 nm; (g and h) – λex = 389 nm (measurement range 800 ns)]. | ||
Like BSA-Ag NCs, the TRES and TRANES of BSA-Au NCs were also recorded at 454 nm and 389 nm excitation. A distinct excited state dynamics was observed. The TRES data recorded at 454 nm excitation revealed the presence of two excited states responsible for emission in BSA-Au NCs. However, the spectral information of the short-lived state was inconclusive (Fig. 3e). The TRANES plotted for the TRES did not provide complete details about the short-lived state, despite the presence of an iso-emissive point shown in Fig. 3f. To fully characterize both the emissive states involved in the excited state relaxation, TRES was acquired at 389 nm excitation. The data illustrate that the short nanosecond lifetime originates from the singlet state at 450 nm, while the long-lifetime emission is associated with the triplet state at 650 nm (Fig. 3g). The short-lived emissive state exists for 54 ns, after which the long-lived state dominates (Fig. 3h).
Summarizing the above observations, both BSA-MNCs displayed heterogeneity in their excited states. However, the origin of such heterogeneity is different in both the nanoclusters. In BSA-Au NCs, excitation at 454 nm provided the emission dynamics of only the long-lived state. Connecting to the previous reports, since the long-lifetime is associated with the surface states, thus, the longer wavelength excitation (454 nm) primarily excites surface states.48 On the other side, at 389 nm excitation, both short- and long-lived emissive states in BSA-Au NCs is observed. This data indicates that the shorter wavelength excitation corresponding to 450 nm emission, is originating from core states. Also, the iso-emissive point observed in Fig. 3h signifies that core states are interacted with the surface states in BSA-Au NCs. This interaction enables the observation of surface state information along with core states at 389 nm excitation. In contrary the 454 nm excitation in BSA-Ag NCs, showed the spectral shifting which occurred exclusively at the surface motifs due to solvent interactions. In addition, unlike BSA-Au NCs, no iso-emissive point was observed in BSA-Ag NCs (Fig. 3d) when excited at 389 nm. This absence indicates that emission is coming from only the core states and the core and surface states are not interacted in BSA-Ag NCs.
The difference in the core and surface state interaction in both MNCs might arise due to the variations in the charge transfer from the surface ligands to the metal core via the sulphur–metal bond, influenced by the specific metal involved in the interaction.49 The role of charge transfer states in the emission of BSA-conjugated Au and Ag nanoclusters was demonstrated by observing the change in the emission in both clusters using a well-known electron acceptor methyl viologen (MV). To this endeavour, BSA-MNCs were incubated with the 1 mM solution of MV. To our surprise, a complete quenching of fluorescence intensity was observed from steady-state luminescence spectra of BSA-Au NCs, while the fluorescence intensity is unchanged with the addition of MV in BSA-Ag NCs (Fig. S6a and S6b†). This result indicated that MV aids the formation of the dark state by decreasing the potential energy barrier of the electron transfer process in BSA-Au NCs. Also, the charge transfer states formed due to electron donation via the S–metal bond have been quenched by the involvement of electron acceptor MV. This is due to the electron donation from the N and O-rich groups in the BSA ligand to MV.50 The complete quenching of fluorescence intensity is supposed to be due to the disruption of charge transfer states, which is responsible for the origin of emission in BSA-Au NCs. The unchanged emission intensity after MV addition confirmed the absence of LMCT states in BSA-Ag NCs.
This distinct variation in the excited state behaviour of BSA-MNCs led us to wonder if any conformational or secondary structural changes occurred in BSA during the nanocluster formation. Hence, we performed circular dichroism (CD) measurements of the BSA and BSA conjugated MNCs. Typically, BSA showed two peaks in the CD spectrum at 209 nm and 222 nm, which are the characteristics of the α-helical secondary structure of native BSA.51 The data presented in Fig. 4 showed an obvious change in the helical structure when both the BSA-MNCs were formed. We calculated the ellipticity percentage changes for BSA and BSA conjugated MNCs.52 For instance, the alpha helicity in BSA is 56.5%, while it is found to be 25.1% and 21.4% for BSA-Au NCs and BSA-Ag NCs, respectively. To further deconvolute the secondary structure of BSA on nanocluster formation, we utilized BeStSel web server,53 which estimates the secondary structure content (%) in BSA-Ag NCs and BSA-Au NCs. The method estimates alpha-helix into two forms regular (Helix1), and distorted (Helix2), where the middle part of alpha-helices is considered as regular and two residues at both ends of alpha-helices is coined as distorted. It also provides detailed information on the beta-sheets, distinguishing parallel beta-sheets and antiparallel beta-sheets with three different twists, left-hand twisted, relaxed (slightly right-hand twisted) and right-hand twisted. As shown in Table S5,† the native BSA protein consists entirely of a regular helix structure, whereas significant changes in BSA's secondary structure were observed after nanocluster formation.54 BSA-Ag NCs has more distorted helical structure than BSA-Au NCs. Also, the beta-sheet content is less than that in BSA-Au NCs. The distorted structure of BSA in BSA-Ag NCs may lead to non-radiative relaxations, hindering triplet state harvesting at room temperature.
 | ||
| Fig. 4 The CD spectra of native BSA, BSA-Ag NCs and BSA-Au NCs, all at the concentration of 0.1 mg mL−1. | ||
Another factor that may help explain the unusual results in both MNCs is the stability of BSA-MNCs, which can be assessed by monitoring changes in their chemical composition over time using XPS analysis. For that, both the as-synthesized and aged BSA-MNCs (1 week old) were subjected to XPS analysis. As evident from Fig. S7 and Table S6,† the contribution of Ag(0) and Ag(I) within the BSA-Ag NCs, has changed from the as synthesized sample to the aged sample (1 week old). Owning to the oxidation, the Ag(0) percentage decreased from 67% to 25%, whereas the binding energies remained the same. The results highlight the intrinsic instability of BSA-Ag NCs, which influences the harvesting of triplet states in the clusters. In contrast, when a similar analysis was performed on BSA-Au NCs, no significant changes in chemical composition were observed with aging; the contribution of Au(0) and Au(I) remained consistent (Fig. S7 and Table S7†).
It is now well understood that BSA-Ag NCs do not exhibit triplet state harvesting at room temperature due to their instability, distorted conformation, and the lack of core–surface state interactions, despite the presence of the electron-donor, surface ligand, BSA. For a deeper understanding of the triplet state dynamics, phosphorescence measurements of BSA-Ag NCs were done in cryogenic conditions. The phosphorescence spectrum was measured at 77 K, with a time delay of 100 μs such that all the contributions from the fluorescence and scatterings could be eliminated. Fig. 5a represents the normalized plot of fluorescence at RT and phosphorescence spectra at 77 K. It is believed that the emission is originating from the triplet state, which can be visualized from the redshift in the phosphorescence spectra of BSA-Ag NCs. Additionally, as we move from RT to 77 K, the spectral broadening decreases. This reduction may be due to rigidity induced at low temperatures, which restricts solvent-induced intermolecular interactions, which are the main cause for triplet state instability at RT. The rigidity induced at 77 K resulted in a quantum yield of 13 ± 1%, with the lifetime decay curve showing a sub-millisecond lifetime of 480 μs (Fig. 5c).
 | ||
| Fig. 5 (a) Normalized emission spectra of BSA-Ag NCs recorded under ambient conditions (T = 300 K, λex = 454 nm, no time delay) and 77 K (λex = 454 nm, delay of 100 μs), respectively. (b) Time-resolved emission spectra (TRES) profiles of BSA-Au NCs at 300 K (time delay: 1.6 μs) and 77 K (time delay: 5.0 μs). (λex = 417 nm, λem = 670 nm). (c) The lifetime decay profile of BSA-Ag NCs (τav = 0.48 ms) and BSA-Au NCs (τav = 18 μs) at 77 K; inset shows the zoomed view of the decay curves. | ||
We also unveiled the relaxation pathway of the long-lived emission in BSA-Au NCs. For that, BSA-Au NCs was analyzed using TRES, temperature-dependent steady-state measurements, and time-resolved luminescence studies, with a focus on the delayed part of emission. As the long-lived emission (microsecond) is dominating in BSA-Au NCs, a time delay of 1 μs is chosen for the temperature-dependent measurements. The emission intensity decreases with the increase in temperature from 283 K to 323 K (Fig. S8a†). As the rISC becomes prominent at high temperatures, the decrement in the intensity rules out the TADF process in BSA-Au NCs. To support this observation, the lifetime measurements were performed, which showed the obvious decrement in the lifetime values with the increment in the temperature from 283 K to 323 K (Fig. S8b†). Similarly, TRANES data, which distinguish the long-lived emissive state from the short-lived state, further rule out the possibility of a TADF pathway for emission. This suggests that RTP is likely the dominant relaxation pathway for emission in BSA-Au NCs. To further validate whether the luminescence occurred from the triplet state at RT, we compared the room-temperature and low-temperature TRES. For that, TRES are recorded at 300 K (time delay of 1.6 μs) and 77 K (time delay of 5 μs), shown in Fig. 5b. The data show an overlap with the emergence of two peaks at low temperatures. The deconvoluted peaks for 77 K are shown in Fig. S9.† The spectral overlap at two different temperatures with a microsecond time delay signifies that the dominating pathway of emission in BSA-Au NCs is RTP. Further, no lower energy spectral shift at 77 K rules out the TADF process in BSA-Au NCs. Additionally, the photoluminescence quantum yield of BSA-Au NCs at 77 K comes out to be (69 ± 2%) with an average lifetime value of 18 μs.
Based on the above results, we can comment that the two different metal cores in BSA-MNCs might be decisive in explaining the different excited state relaxation pathways involved in the BSA-Ag NCs and BSA-Au NCs. In BSA-Au NCs, the core–surface interaction-driven LMCT states cause radiative relaxation from the triplet state at RT. Meanwhile, the absence of such interactions in BSA-Ag NCs and the secondary structure distortion of BSA in BSA-Ag NCs affect its conformational stability and induces non-radiative pathways from the triplet state. This in turn, hinders the triplet state harvesting at RT.55 However, at low temperatures (77 K), the photoluminescence lifetime and quantum yield of BSA-Ag NCs are significantly enhanced due to increased structural rigidity and the suppression of thermally activated non-radiative pathways.56 Similarly, the reduced non-radiative pathways at 77 K enhance the lifetime and the quantum yield of BSA-Au NCs.57 The prominent non-radiative relaxations in BSA-Ag NCs, as compared to BSA-Au NCs at RT, resulted in a greater increase in the lifetime from RT to 77 K in BSA-Ag NCs.
The long-lived emission in BSA-MNCs prompts us to investigate the applicability of such probes in TRLI.58 The confocal laser scanning microscopy (CLSM) images of BSA-Au NCs (1 mg mL−1) stained HeLa cells depicted bright red fluorescence signals from the cytoplasmic region of the cells (Fig. S10a and S10b†). Previous studies have shown that BSA-conjugated NCs are selectively transported to lysosomes.46,59,60 Colocalization studies using LysoTracker™ Green (LTG) confirmed that BSA-Au NCs specifically localize into the lysosomes (Fig. S10b†). HeLa cells stained with BSA-Au NCs exhibited distribution of long-lived decay components in lysosomes ranging up to microsecond time domains (Fig. 6a and b). In contrast, BSA-Ag NCs exhibited only nanosecond-scale decay components under similar conditions (Fig. 6c and d). Next, we acquired TRLI images of BSA-Au NCs (1 mg mL−1) stained HeLa cells by applying different delay times (Fig. 6e). The corresponding time-gated images obtained from cells demonstrated noticeable luminescence even with a time delay of sub-microseconds (Fig. 6e, ii–iv). In contrast, upon applying a similar time delay, no noticeable luminescence was observed from HeLa cells stained with BSA-Ag NCs (1 mg mL−1), again suggesting its weak triplet harvesting nature under aqueous conditions (Fig. 6e, vi–viii).
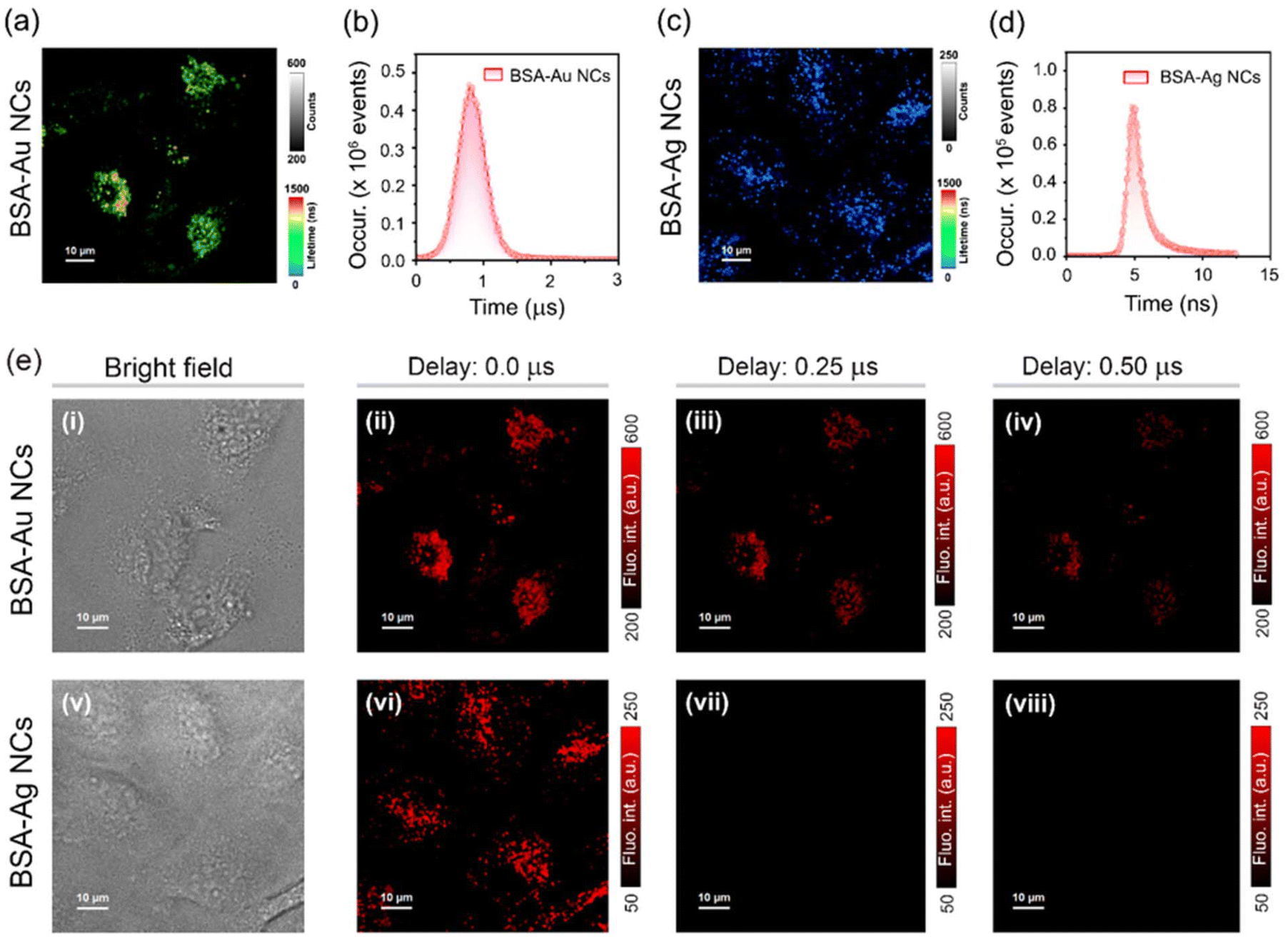 | ||
| Fig. 6 Lifetime imaging microscopy images of HeLa cells incubated with an aqueous dispersion of (a) BSA-Au NCs (λex = 470 nm, and λem = 488–800 nm) and (c) BSA-Ag NCs (λex = 470 nm, and λem = 488–800 nm); intensity and lifetime scales are also indicated; scale = 10 μm. Corresponding intracellular lifetime histograms of HeLa cells incubated with an aqueous dispersion of (b) BSA-Au NCs and (d) BSA-Ag NCs. (e) Time-resolved imaging of HeLa cells stained with an aqueous dispersion of (i–iv) BSA-Au NCs (λex = 470 nm, and λem = 488–800 nm) and (v–viii) BSA-Ag NCs (λex = 470 nm, and λem = 488–800 nm): (i and v) bright field images, (ii and vi) confocal laser scanning microscopy (CLSM) images with no time delay, time-gated images after a time delay of (iii and vii) 0.25 μs and (iv and viii) 0.50 μs; scale = 10 μm. For (ii–iv) and (vi–viii), intracellular intensity scales are also indicated. | ||
Conclusion
In summary, we have shown the distinct excited state dynamics of BSA-Au NCs and BSA-Ag NCs. It is found that the excited state relaxation pathways were significantly influenced by charge transfer states formed via M–S bonds, as well as the stability and rigidity of the protein-coated nanoclusters. From TRES and TRANES analysis, we concluded that the emission in BSA-Ag NCs comes from the core states only, while BSA-Au NCs exhibits a dual emission consisting of short- and long-lived states, originating due to core and surface state interactions. Consequently, the BSA-Ag NCs exhibit fluorescence as the emission mechanism at room temperature, transitioning to phosphorescence with sub-millisecond lifetimes under cryogenic conditions. Meanwhile, the LMCT states in BSA-Au NCs facilitate triplet-state harvesting, resulting in RTP being the dominant mechanism with a microsecond lifetime. The time-resolved luminescence imaging of both MNCs in HeLa cells ascertained the long-lived emission characteristics in BSA-Au NCs but not in BSA-Ag NCs.Author contributions
S.S. conceptualized and designed all the experiments. S.S. and S.D. conducted various steady state and time-resolved luminescence measurements and analyzed the data with the help of C.K.N. and A.P. S.D. also performed time-resolved luminescence imaging for cell-related study. K.K. wrote the MATLAB code to automate the analysis of TRANES. C.K.N. and A.P. guided the complete project thoroughly and wrote the manuscript with the help of S.S. and S.D.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI files.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Advanced Material Research Centre (AMRC) of IIT Mandi and the Indian Institute of Science Education and Research Bhopal (IISERB), India, for providing the facilities and the sophisticated instruments. The financial support from the Indo-French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA, Project No.: 6705-1) is gratefully acknowledged by A. P. C. K. N. thanks to SERB core research grant (CRG) India project number CRG/2020/000268 for the financial support. S. D. thanks, IISERB, for the fellowship. S. S. and K. K. thank the Ministry of Education, India (MoE), for research scholarships.References
- D. A. Buschmann, H. Hirai and T. Tsukuda, Inorg. Chem. Front., 2024, 11, 6694–6710 RSC.
- S. Sharma, K. Kaushik, A. Salam, R. Garg, J. Mondal, R. Lamba, M. Kaur and C. K. Nandi, ACS Appl. Nano Mater., 2024, 7, 32–60 CrossRef CAS.
- H. Lin, X. Song, O. J. H. Chai, Q. Yao, H. Yang and J. Xie, Adv. Mater., 2024, 36, 1–22 Search PubMed.
- M. Zhou and R. Jin, Annu. Rev. Phys. Chem., 2021, 72, 121–142 CrossRef CAS PubMed.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 14767 CrossRef CAS PubMed.
- L. Chen, M. Gharib, Y. Zeng, S. Roy, C. K. Nandi and I. Chakraborty, Mater. Today Chem., 2023, 29, 101460 CrossRef CAS.
- S. C. L. Tan, Z. He, G. Wang, Y. Yu and L. Yang, Molecules, 2023, 28, 5531 CrossRef CAS PubMed.
- K. Chaudhari, P. L. Xavier and T. Pradeep, ACS Nano, 2011, 5, 8816–8827 CrossRef CAS PubMed.
- I. Zare, D. M. Chevrier, A. Cifuentes-Rius, N. Moradi, Y. Xianyu, S. Ghosh, L. Trapiella-Alfonso, Y. Tian, A. Shourangiz-Haghighi, S. Mukherjee, K. Fan and M. R. Hamblin, Mater. Today, 2023, 66, 159–193 CrossRef CAS.
- E. Akyüz, F. B. Şen, M. Bener, K. S. Başkan, E. Tütem and R. Apak, ACS Omega, 2019, 4, 2455–2462 CrossRef PubMed.
- A. Aires, V. Fernández-Luna, J. Fernández-Cestau, R. D. Costa and A. L. Cortajarena, Nano Lett., 2020, 20, 2710–2716 CrossRef CAS PubMed.
- L. Yan, Y. Cai, B. Zheng, H. Yuan, Y. Guo, D. Xiao and M. M. F. Choi, J. Mater. Chem., 2012, 22, 1000–1005 RSC.
- X. Meng, I. Zare, X. Yan and K. Fan, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2020, 12, e1602 Search PubMed.
- W. Zhang, J. Ye, Y. Zhang, Q. Li, X. Dong, H. Jiang and X. Wang, RSC Adv., 2015, 5, 63821–63826 RSC.
- R. Guan, L. Tao, Y. Hu, C. Zhang, Y. Wang, M. Hong and Q. Yue, RSC Adv., 2020, 10, 33299–33306 RSC.
- W. Zhou, Y. Cao, D. Sui, W. Guan, C. Lu and J. Xie, Nanoscale, 2016, 8, 9614–9620 RSC.
- J. M. Dixon and S. Egusa, J. Phys. Chem. Lett., 2021, 12, 2865–2870 CrossRef CAS PubMed.
- D. Bain, H. Yuan, A. Pniakowska, A. Hajda, C. Bouanchaud, F. Chirot, C. Comby-Zerbino, V. Gueguen-Chaignon, V. Bonačić-Koutecký, J. Olesiak-Banska, Ž. S. Maršić and R. Antoine, Nanoscale, 2024, 16, 14953–14958 RSC.
- J. M. Dixon and S. Egusa, J. Am. Chem. Soc., 2018, 140, 2265–2271 CrossRef CAS PubMed.
- S. Maity, S. Kolay, S. Ghosh, S. Chakraborty, D. Bain and A. Patra, J. Phys. Chem. Lett., 2022, 13, 5581–5588 CrossRef CAS PubMed.
- M. Van Der Linden, A. J. Van Bunningen, L. Amidani, M. Bransen, H. Elnaggar, P. Glatzel, A. Meijerink and F. M. F. De Groot, ACS Nano, 2018, 12, 12751–12760 CrossRef CAS PubMed.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- T.-Q. Yang, B. Peng, B.-Q. Shan, Y.-X. Zong, J.-G. Jiang, P. Wu and K. Zhang, Nanomaterials, 2020, 10, 261 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- N. Basu and D. Mandal, Photochem. Photobiol. Sci., 2019, 18, 1782–1792 CrossRef CAS PubMed.
- J. Shi, J. K. Cooper, S. Lindley, O. Williams, D. S. Kliger, Y. Xu, Y. Bao and J. Z. Zhang, Chem. Phys. Lett., 2014, 610–611, 125–130 CrossRef CAS.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS PubMed.
- S. Agrawal, D. Shil, A. Gupta and S. Mukherjee, Nanoscale, 2024, 16, 20556–20569 RSC.
- H. T. Lin, K. B. Cai, H. Y. Huang, T. N. Lin, J. L. Shen, C. A. J. Lin and C. T. Yuan, J. Lumin., 2017, 187, 269–273 CrossRef CAS.
- Z. Liu, L. Luo, J. Kong, E. Kahng, M. Zhou and R. Jin, Nanoscale, 2024, 16, 7419–7426 RSC.
- F. Ni, Z. Zhu, X. Tong, M. Xie, Q. Zhao, C. Zhong, Y. Zou and C. Yang, Chem. Sci., 2018, 9, 6150–6155 RSC.
- D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 RSC.
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC.
- X. Wen, P. Yu, Y. R. Toh, A. C. Hsu, Y. C. Lee and J. Tang, J. Phys. Chem. C, 2012, 116, 19032–19038 CrossRef CAS.
- S. Sharma, S. Das, K. Kaushik, A. Yadav, A. Patra and C. K. Nandi, J. Phys. Chem. Lett., 2023, 14, 8979–8987 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- Y. Chen, T. Feng, L. Chen, Y. Gao and J. Di, Opt. Mater., 2021, 114, 111012 CrossRef CAS.
- S. Chen, I. G. Theodorou, A. E. Goode, A. Gow, S. Schwander, J. Zhang, K. F. Chung, T. D. Tetley, M. S. Shaffer, M. P. Ryan and A. E. Porter, Environ. Sci. Technol., 2013, 47, 13813–13821 CrossRef CAS PubMed.
- S. K. Sharma, R. Pasricha, J. Weston, T. Blanton and R. Jagannathan, ACS Appl. Mater. Interfaces, 2022, 14, 54992–55003 CrossRef CAS PubMed.
- X. Wen, P. Yu, Y. R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 11830–11836 CrossRef CAS.
- S. Raut, R. Chib, R. Rich, D. Shumilov, Z. Gryczynski and I. Gryczynski, Nanoscale, 2013, 5, 3441–3446 RSC.
- E. Bolli, A. Mezzi, L. Burratti, P. Prosposito, S. Casciardi and S. Kaciulis, Surf. Interface Anal., 2020, 52, 1017–1022 CrossRef CAS.
- F. Porcaro, L. Carlini, A. Ugolini, D. Visaggio, P. Visca, I. Fratoddi, I. Venditti, C. Meneghini, L. Simonelli, C. Marini, W. Olszewski, N. Ramanan, I. Luisetto and C. Battocchio, Materials, 2016, 9, 1028 CrossRef PubMed.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- S. Sharma, A. Yadav, K. Kaushik, A. Salam and C. K. Nandi, Nanoscale, 2024, 16, 16958–16966 RSC.
- A. Yadav, N. C. Verma, C. Rao, P. M. Mishra, A. Jaiswal and C. K. Nandi, J. Phys. Chem. Lett., 2020, 11, 5741–5748 CrossRef CAS PubMed.
- N. Periasamy, Chapter 2 Heterogeneity of Fluorescence Determined by the Method of Area-Normalized Time-Resolved Emission Spectroscopy, Elsevier Inc., 1st edn, 2008, vol. 450 Search PubMed.
- M. S. Devadas, J. Kim, E. Sinn, D. Lee, T. Goodson and G. Ramakrishna, J. Phys. Chem. C, 2010, 114, 22417–22423 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. Khan, N. C. Verma, A. Gupta and C. K. Nandi, Sci. Rep., 2015, 5, 11423 CrossRef CAS PubMed.
- S. Paul, N. Sepay, S. Sarkar, P. Roy, S. Dasgupta, P. S. Sardar and A. Majhi, New J. Chem., 2017, 41, 15392–15404 RSC.
- A. Baral, L. Satish, D. P. Das, H. Sahoo and M. K. Ghosh, New J. Chem., 2017, 41, 8130–8139 RSC.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y. H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- R. Barbir, I. Capjak, T. Crnković, Ž. Debeljak, D. D. Jurašin, M. Ćurlin, G. Šinko, T. Weitner and I. V. Vrček, Chem.-Biol. Interact., 2021, 335, 109364 CrossRef CAS PubMed.
- J. Dai and X. Zhang, Chem. Biomed. Imaging, 2023, 1, 796–816 CrossRef CAS PubMed.
- Q. Li, M. Zhou, W. Y. So, J. Huang, M. Li, D. R. Kauffman, M. Cotlet, T. Higaki, L. A. Peteanu, Z. Shao and R. Jin, J. Am. Chem. Soc., 2019, 141, 5314–5325 CrossRef CAS PubMed.
- M. Mitsui, J. Phys. Chem. Lett., 2024, 15, 12257–12268 CrossRef CAS PubMed.
- S. Das, A. Batra, S. Kundu, R. Sharma and A. Patra, Chem. Sci., 2024, 15, 102–112 RSC.
- R. Marwaha and M. Sharma, Bio-Protoc., 2017, 7, e2571 Search PubMed.
- A. Yadav, K. Kaushik, S. Sharma, F. Anjum and C. K. Nandi, ACS Appl. Nano Mater., 2022, 5, 9260–9265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Sections I, II and III providing the description of the materials and the synthesis procedures; Instrumentation section (IV. a–j) including brief explanation of the characterization methods, sample preparation, equations, and calculation parameters. Supplementary section (V) and (VI) containing figures and tables of material characterization, photophysical and intracellular studies. See DOI: https://doi.org/10.1039/d5nr00518c |
| This journal is © The Royal Society of Chemistry 2025 |