
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic Fe(OH)x@Co0.8Fe0.2-MOF/NF composites as effective electrocatalysts for the production of 2,5-furandicarboxylic acid from 5-hydroxymethylfurfural†
Mehran
Nozari-Asbemarz
 a,
Simin
Arshi
a,
Behnam
Babaei
b,
Italo
Pisano
a,
Edmond
Magner
*a and
James J.
Leahy
*a
a,
Simin
Arshi
a,
Behnam
Babaei
b,
Italo
Pisano
a,
Edmond
Magner
*a and
James J.
Leahy
*a
aDepartment of Chemical Sciences, Bernal Institute, University of Limerick, V94 T9PX, Ireland. E-mail: edmond.magner@ul.ie
bDepartment of Chemistry, Faculty of Basic Science, University of Mohaghegh Ardabili, Ardabil, Iran
First published on 11th March 2025
Abstract
A wide range of catalytic techniques have been explored for the use of biomass components. For example, the electrocatalytic oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) can be performed with excellent energy efficiency under safe operating conditions and with fine control of the production parameters. Metal–organic framework (MOF) catalysts with active metal centres have been prepared as electrocatalysts for the oxidation of HMF to FDCA. A Fe(OH)x@Co0.8Fe0.2-MOF/nickel foam (NF) was made via two steps: Co0.8Fe0.2-MOF/NF was synthesized by in situ solvothermal methods followed by the electrodeposition of Fe(OH)x. X-ray photoelectron spectroscopy (XPS) analysis confirmed the successful electrodeposition of Fe(OH)x on Co0.8Fe0.2-MOF/Ni. Fe(OH)x@Co0.8Fe0.2-MOF/NF demonstrated enhanced electrocatalytic activity for the oxidation of HMF in 1M KOH, requiring an overpotential of 236 mV and 263 mV versus RHE to achieve current densities of 50 and 100 mA cm−2, respectively, with an apparent Tafel slope of 92 mV. The electrochemically active surface area of the catalysts showed that Fe(OH)x incorporated samples possessed a higher number of active sites compared to Co0.8Fe0.2-MOF/Ni, enhancing efficiency and improving the yield of 5−Hydroxymethylfurfural oxidation reaction (HMFOR).
Introduction
Utilizing the possibilities offered by renewable energy sources, such as solar, wind, and other energy sources, is vital for future energy supply systems. Hydrogen (H2) can be produced from renewable electricity using electrochemical processes, such as water electrolysis. This technology not only produces high-purity H2 but also helps to address the temporal and spatial intermittency issues related to renewable electricity.1–3 Water splitting enables efficient storage and conversion of renewable energy, contributing to the development of carbon-neutral economies.4,5 The slow kinetics of the oxygen evolution reaction (OER) negatively impact the energy efficiency of water electrocatalytic systems. Biomass electrooxidation has become increasingly investigated as an emerging technology for effectively transforming HMF into FDCA. This interest is attributed to its advantages, including low energy consumption and environmentally friendly operating conditions. Notably, the HMFOR can be readily integrated with the hydrogen evolution reaction (HER), allowing for the simultaneous synthesis of hydrogen producing value-added products at both electrodes, enhancing the energy conversion efficiency.6–9 Upgrading biomass-derived chemicals10,11 such as HMF12,13 to value-added products such as 2-hydroxymethyl-5-(methylamino methyl)furan (HMMAMF) and FDCA using renewable resources is gaining significant attention.14,15HMF, synthesized by dehydrating hexose-based biomass such as fructose and glucose, is an important renewable platform chemical.16 It is a versatile substrate with functional groups that include aldehyde (–CHO), hydroxyl (–OH), and furan rings that enable conversion into platform chemicals such as 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 2,5-diformylfuran (DFF), 5-formyl-2-furancarboxylic acid (FFCA), and FDCA. FDCA is a precursor for the synthesis of polyethylene furan-dicarboxylate (PEF),17,18 a bio-based polymer that can act as a substitute for petroleum-based plastics such as polyethylene terephthalate (PET).19–22
The incorporation of metal catalysts on MOF supports can serve as electrocatalysts for the HMFOR reaction.4,23 Fine-tuning of the energy levels of the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) at the interface can be accomplished through deliberate adjustments in the electronic structure, composition, and ligands in MOFs. In ligand field theory, ligands, such as carboxylates, which possess both σ-donor and π-donor/acceptor properties, can significantly impact the energy levels of the d orbitals of metals in MOFs.24,25 In particular, both the σ and π contributions from the ligands can influence the energy gap between the T2g and eg* energy levels in an octahedral environment (Fig. 1).
 | ||
| Fig. 1 The synergistic effect of ligand field on the pseudo-octahedral coordination of transition metal ions in the MOF, illustrating crystal field splitting and the stabilisation of T2g orbitals through interaction with the π-manifold of the linker. | ||
The pKa value of the linker can be correlated with the electronic properties of the ligand. A linear association between the pKa value and the expected band energy in the MOF has been reported,24 indicating that the pKa value can help determine whether if the linker behaves as a π acceptor or donor.24
The observation that fewer acidic linkers typically exhibit π-donor behavior suggests that they can raise the energy levels of the metal's d orbitals. In metal–organic frameworks (MOFs), the metal centre's d orbitals split into different energy levels due to the coordination environment. In an octahedral geometry, the d orbitals split into two sets: the lower-energy T2g orbitals (dxy, dxz, dyz) and the higher-energy eg* orbitals (dx2–y2, dz2). By donating electron density, π-donor ligands raise the energy of the T2g and eg* orbitals while reducing the metal's ability to accept further electron density. The shift in the d-orbital energy levels directly affects the MOF's overall electronic structure, subsequently affecting its conductive and catalytic characteristics. In contrast, metals with a complete d10 electron configuration in tetrahedral systems (like Zn or Cu(I)) do not participate in d-orbital bonding in the same manner. Instead, they affect the valence band energy levels by contributing to ligand-to-metal charge transfer processes, rather than splitting the d orbitals into T2g and eg* levels. The electron conduction pathway between the donor (ligand) and acceptor (metal center) in MOFs is essential for modifying the energy levels of the HOMO and the LUMO. This process promotes electron transfer and influences the electronic properties of the MOF, affecting its functioning, including conductivity and electrocatalysis. Understanding and controlling these electrical interactions in order to create MOFs with specialized properties for certain applications. For instance, by adjusting the metal nodes and ligands, MOFs’ electrocatalytic characteristics can be modified. This will change the electronic structure by adjusting the interactions between the metal and the ligand.24,26–28
Nevertheless, growing 2D MOF nanoarrays in situ on conductive scaffolds offers a solution to the issue of poor mass and electron transport typically observed in 3D MOF structures. By reducing transport resistance, decreasing diffusion pathways, and enhancing activity, these 2D nanoarrays improve the overall performance of the electrocatalyst. Ge et al.29 reported that the 3D structure of MOFs can hinder mass and electron transport due to longer diffusion distances and increased resistance. Additionally, the color change of the 2D MOF-Fe/Co nanosheet from orange to black following cyclic voltammetry cycling is significant, as it indicates a change in the material's oxidation state and electronic properties—often correlated with enhanced catalytic performance. X-ray photoelectron spectroscopy (XPS) revealed that the binding energies of Fe3+ 2p3/2 and Co2+ 2p3/2 shifted to higher values, indicating firm contact and partial charge transfer from Co2+ to Fe3+ through oxygen ligands. This ‘firm contact’ refers to the strong electronic interaction between the metal centers (Co2+ and Fe2+), facilitated by oxygen, which stabilizes the structure and enhances charge transfer efficiency, ultimately improving the catalytic activity of the electrocatalysts.29–33 Enhancing the concentration of active sites and the adsorption capacity in MOFs can be achieved by adjusting the electron density by adding electron-rich or electron-poor groups. Charge transfer within the MOF can be facilitated by forming heterojunctions with materials that possess different electron densities, thereby increasing catalytic activity. Furthermore, controlled defects can be introduced by creating vacancies or structural distortions within the MOF, which not only generates more active sites but also alters their electronic properties to improve overall performance. Additionally, advancements in synthesis techniques, such as thermochemical processing, heteroatom incorporation,34,35 plasma treatment,36 or doping with noble metals can boost catalytic activity.37–40 Developing an ultrathin oxyhydroxide layer with improved durability and catalytic activity is crucial. Transition metal-based compounds, including cobalt (Co), iron (Fe), nickel (Ni), molybdenum (Mo), and other transition metals, have demonstrated significant effectiveness in both the OER and HMFOR.5,41–43 Oxyhydroxides, particularly those derived from transition metals, are pivotal in electrocatalysis as active species. Achieving precise control over the thickness of the oxyhydroxide layer is essential for maximizing electrocatalytic performance.44–47
This study describes an electrocatalyst prepared by synthesizing Co0.8Fe0.2-MOF on an NF substrate using an in situ solvothermal method. Subsequently, the Co0.8Fe0.2-MOF/NF catalyst underwent activation via electrodeposition process of Fe(OH)x at ambient temperature, forming a hybrid crystalline-amorphous heterostructure, Fe(OH)x@Co0.8Fe0.2-MOF/NF. In an H-type electrochemical cell, Fe(OH)x@Co0.8Fe0.2-MOF/NF was used as the electrocatalyst at the anode for the HMF oxidation reaction. The Co- and Fe-based nanocomposite electrocatalyst demonstrated excellent stability, an acceptable Tafel slope, and lower overpotentials during the electrochemical HMFOR process.
Experimental
Materials
Trimesic acid (C6H3(CO2H)3), cobalt(II) nitrate hexahydrate (Co(NO3)3·6 H2O), iron(III) nitrate nonahydrate (Fe(NO3)3·9 H2O), iron(II) sulfate heptahydrate (FeSO4·7H2O), potassium hydroxide (KOH), hydrochloric acid (HCl), ethanol (C2H5OH), methanol (CH3OH) and N,N-dimethylformamide of analytical reagent grade were sourced from Merck. HMF, FDCA, DFF, FFCA, HMFCA, and ammonium formats were purchased from Sigma-Aldrich. All solutions were prepared using double-distilled water. NF measuring 1.5 × 1.0 cm2 and having a thickness of 0.9 mm, a bulk density of 0.62 g cm−3, a porosity of 93%, and an average of 20 pores per centimeter was procured from Goodfellow (Hamburg, Germany).Synthesis of Co0.8Fe0.2-MOF/NF, Co0.5Fe0.5-MOF/NF, Co-MOF/NF, and Fe-MOF/NF
The Co0.8Fe0.2-MOF/NF composite was synthesized using a one-step solvothermal approach on an NF substrate. In this method, a solution was prepared by mixing (Co(NO3)2·6H2O), Fe(NO3)3·9H2O, and trimesic acid (organic ligand) in a molar ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:5, along with 35 mL of N,N-dimethylformamide and 10 mL of methanol. Magnetic stirring (500 rpm) was employed for 1 hour to ensure the mixture was uniformly mixed. Subsequently, a pre-cleaned NF substrate measuring 1 × 1.5 cm2 was introduced into a 50 mL Teflon™-lined stainless-steel autoclave containing the prepared solution. Before this step, the NF substrate was thoroughly cleaned through sequential 30 minute sonication in 3 M HCl, ethanol, and deionized water solutions.
:2:5, along with 35 mL of N,N-dimethylformamide and 10 mL of methanol. Magnetic stirring (500 rpm) was employed for 1 hour to ensure the mixture was uniformly mixed. Subsequently, a pre-cleaned NF substrate measuring 1 × 1.5 cm2 was introduced into a 50 mL Teflon™-lined stainless-steel autoclave containing the prepared solution. Before this step, the NF substrate was thoroughly cleaned through sequential 30 minute sonication in 3 M HCl, ethanol, and deionized water solutions.
The solvothermal reaction was initiated by sealing the autoclave and heating it at 150 °C for 18 hours. After the designated reaction time, the autoclave was allowed to cool to room temperature, and the solid was then carefully retrieved. The material was washed thoroughly with deionized water to remove any remaining impurities and dried overnight in an oven at 80 °C to remove moisture, resulting in the final Co0.8Fe0.2-MOF/NF composite.
Syntheses were carried out similarly to produce Co-MOF, Fe-MOF, and Co0.5Fe0.5-MOF/NF separately. The variations in Fe and Co were introduced to emphasize their effects on catalytic activity.
Synthesis of Fe(OH)x@Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF
A three-electrode system was employed to prepare the electrocatalyst, consisting of Co0.8Fe0.2-MOF/NF, Pt, and a saturated Ag/AgCl electrode as the working, counter, and reference electrodes. The electrodeposition process was initiated by applying an aqueous solution containing 0.01 M Fe(NO3)3·9 H2O and 0.01 M FeSO4·7H2O onto the Co0.8Fe0.2-MOF/NF working electrode. During the electrodeposition step, a consistent potential of 1.2 V versus RHE was applied under continuous stirring at 500 rpm for 600 seconds at room temperature. During the electrodeposition process, Fe(OH)x species were formed and deposited onto the surface of the Co0.8Fe0.2-MOF/NF electrode. After the electrodeposition step, the Co0.8Fe0.2-MOF/NF electrode with the deposited Fe(OH)x material was carefully rinsed with deionized water to remove residual impurities. Following the rinsing step, the electrode was dried at 80 °C overnight to eliminate any remaining moisture.Fe(OH)x/NF was fabricated independently from Co0.8Fe0.2-MOF/NF using a similar method. The synthesis utilized either Fe(NO3)3·9H2O, FeSO3·7H2O or a combination of both exclusively on an NF substrate. As a result, the Fe(OH)x species were electrodeposited onto the NF surface.
Materials characterization
The microstructure morphology of the materials was analyzed using a ThermoFisher Helios G4 CX dual beam focused ion beam scanning electron microscope (FIB-SEM). Energy-dispersive X-ray (EDX) analysis, with supplementary data collected at a 20 kV accelerating voltage using an Oxford Instruments X-Max detector. X-ray diffraction (XRD) patterns were obtained using a PANalytical Empyrean X-ray diffractometer with Cu K radiation (λ = 0.15406 nm) and a scanning rate of 0.04° s−1 over a 2θ range from 10° to 80°. X-ray photoelectron spectroscopy (XPS) was conducted using a Kratos AXIS ULTRA spectrometer equipped with a monochromatic Al K X-ray gun (energy of 1486.58 eV). Calibration was performed using the C 1s line at 284.8 eV, and peak fitting and data analysis were carried out using Casa XPS software. A high-performance liquid chromatography (HPLC) system was used to evaluate and identify the products, utilizing an Agilent 1260 Infinity Series instrument (Agilent Technologies, Palo Alto, USA) with a Beckman Coulter Ultrasphere C18 (5 m × 4.6 mm × 25 cm) column.Electrochemical measurements
The measurements of electrochemical activity were performed on CHI630A potentiostat (CH Instruments, Austin, Texas) at ambient temperature. The experimental setup involved a three-electrode system comprising a self-supporting nickel foam electrode with an effective geometric area as the working electrode, a platinum (Pt) counter electrode, and a saturated Ag/AgCl reference electrode. The electrolyte solution used for OER tests was 1.0 M KOH. For the HMFOR, the electrolyte solution contained 1.0 M KOH and 10.0 mM HMF (pH 13.76). The KOH electrolyte underwent a 30 minute N2 bubbling treatment before OER testing. Polarization curves were obtained using linear sweep voltammetry (LSV) at a scan rate of 10.0 mV s−1. For HMFOR and OER, measurements were performed in the potential range of 1.0 to 1.8 V versus RHE. All potentials were reported versus the reversible hydrogen electrode (RHE): | (1) |
The effects of iR drop were addressed using iR compensation (90%), with a resistance (R) value of 1.83 Ω, determined from electrochemical EIS measurements at high frequency. The overpotential (ηj) at a particular current density (j) was calculated using (2):
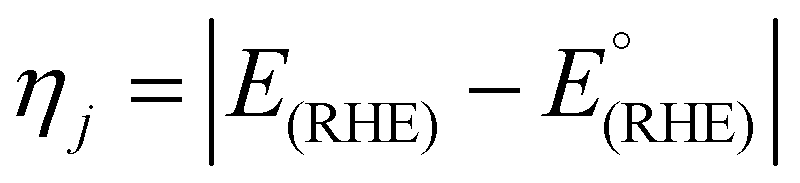 | (2) |
The standard electrode potential E° for the OER is 1.23 V versus the RHE. Controlled potential electrolysis (CPE) experiments were conducted using an H-type cell, which consists of two compartments separated by a Nafion™ 117 membrane. The working electrode, made of modified NF, and the reference electrode (Ag/AgCl) were immersed in 18 mL of electrolyte solution, while the counter electrode was placed in a separate compartment containing another 18 mL of electrolyte solution. A stirring rate of 500 rpm was maintained throughout the experiment. Tafel plots were utilized to evaluate the kinetics of the electrocatalytic process at the electrode surface. In order to conduct electrochemical impedance spectroscopy (EIS) investigations, an alternating current (AC) voltage amplitude of 5 mV was applied over a frequency range of 100 kHz to 0.1 Hz. The acquired Nyquist plots were fitted using a condensed Randles circuit to analyze the electrochemical system and determine the charge transfer resistance (Rct). To determine the electrochemically active surface area (ECSA), cyclic voltammetry (CV) was performed in a non-faradaic potential region at scan rates ranging from 20 to 100 mV s−1. The ECSA was estimated by calculating the double-layer capacitance (Cdl) using eqn (3):
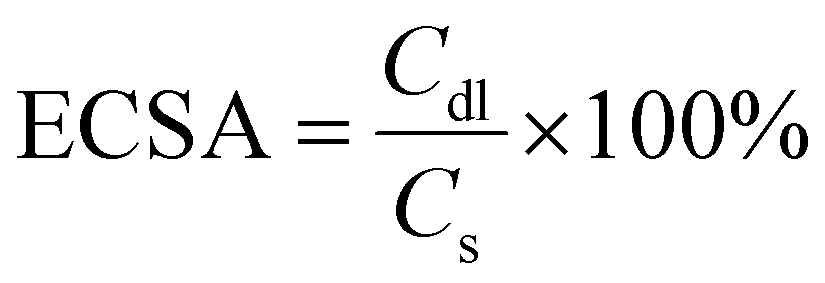 | (3) |
The Cdl was calculated by dividing the difference in charging current density (Δj) by ν, where Δj = (ja − jc) and ν represents the scanning rate. The specific capacitance (Cs), commonly reported as 0.040 mF cm−2 (ref. 48 and 49) in 1 M KOH, was used in estimating the ECSA using eqn (3).
High-performance liquid chromatography (HPLC) analysis
High-performance liquid chromatography (HPLC) was used to analyze HMF, DFF, FFCA, FDCA, and HMFCA with a C18 column at detection wavelengths of 265 nm and 285 nm. The mobile phase consisted of methanol (A) and 5 mM ammonium formate (B). Separation was performed using gradient elution, involving an increase in mobile phase composition from 25% A to 30% A and 75% to 70% B during 0 to 4 minutes, followed by an increase to 35% A and 65% B during 4 to 5 minutes then kept for 3 minutes constant at a flow rate of 0.7 ml min−1. The column oven temperature was kept at 30 °C. In order to prepare HPLC samples, 100 μL of the reaction mixture was diluted with 900 μL of distilled water. Standard calibration curves were prepared from the stock solution to quantify the concentration of HMF and FDCA. DFF, FFCA, and HMFCA were detected on the basis of retention time but not quantified. Fig. S1† exhibits the standard chromatograms of HMF and DFF, Fig. S2† displays the standard chromatograms of FDCA, Fig. S3† illustrates standard chromatograms of FFCA, and Fig. S4† depicts HMFCA standard chromatograms. The amount of conversion of HMF, yield of FDCA, and the faradaic efficiency (FE) were calculated using eqn (4)–(6): | (4) |
 | (5) |
 | (6) |
Results and discussion
Characterization
The Co0.8Fe0.2-MOF nanorods exhibited a typical perpendicular alignment on the NF substrate, demonstrating a highly consistent unidirectional orientation (Fig. 2A). This unique structural morphology can be attributed to the ordered alignment of the nanorods on the substrate. The Co0.8Fe0.2-MOF nanorods underwent significant transformation following a subsequent electrochemical deposition reaction utilizing a solution enriched with Fe3+ and Fe2+ (Fig. 2B). A distinct layer can be seen adorning the surface of the nanorods, indicating successful modification. Energy-dispersive X-ray EDX analysis (Fig. 2C–F) reveals a consistent and homogeneous distribution of cobalt (Co), iron (Fe), nickel (Ni), carbon (C), and oxygen (O) on the surface. The observed dispersion of Co, Fe, Ni, C, and O throughout the electrode structure via EDX mapping analysis indicates the successful integration of these elements into the MOF substrate. | ||
| Fig. 2 SEM images of (A) Co0.8Fe0.2-MOF/NF; (B) Fe(OH)x@Co0.8Fe0.2-MOF/NF; (C–H) elemental mapping of Co0.8Fe0.2-MOF/NF. | ||
XPS was utilized to examine the chemical composition and valence state of the surface of the Co0.8Fe0.2-MOF/NF electrode before and after deposition of Fe(OH)x This investigation focused on analyzing the Co 2p and O 1s spectra to gain insight into the electrocatalytic behavior of the material. The high-resolution Co 2p spectra revealed distinct changes following the electrodeposition of Fe(OH)x. Specifically, the Co0.8Fe0.2-MOF/NF spectra exhibited two satellite peaks at 788.99 eV and 803.39 eV, accompanied by two broad primary peaks corresponding to the 2p3/2 and 2p1/2 spin–orbit lines at 781.89 and 797.74 eV, respectively (Fig. 3A).50–53
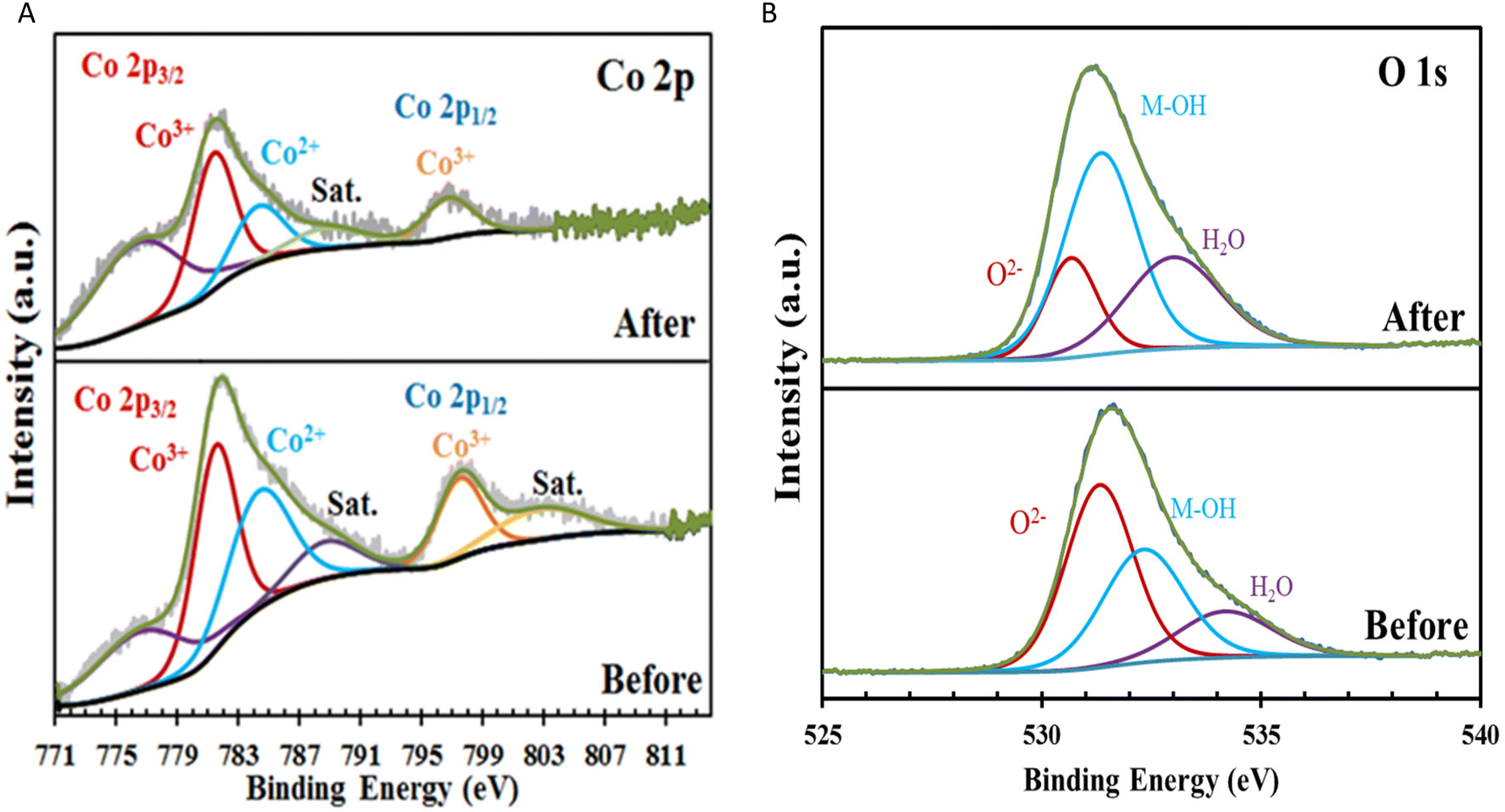 | ||
| Fig. 3 (A) Co 2p XPS spectra of Co0.8Fe0.2-MOF/NF before and after electrodeposition of (Fe3+, Fe2+); (B) O 1s XPS spectrum of Co0.8Fe0.2-MOF/NF before and after electrodeposition of (Fe3+, Fe2+). | ||
As shown in Table S1,† when compared to Co0.8Fe0.2-MOF, the binding energy peaks associated with the Co 2p levels in Fe(OH)x@Co0.8Fe0.2-MOF decreased by 0.331 and 0.881 eV for the 2p3/2, 2p1/2 levels, respectively.
This shift in binding energy is attributed to electron donation from Fe(OH)x to the Co 2p orbitals in Fe(OH)x@Co0.8Fe0.2-MOF.54 This phenomenon can be explained by the lower electronegativity of Fe (1.83) compared to Co (1.88), along with the higher electronegativity of O (3.44). These factors contribute to a decrease in the electron density of the Fe species, resulting in a shift of the Co 2p3/2 peak toward lower binding energy levels.55 The O 1s spectrum of the electrode surface was analyzed before and after electrodeposition (Fig. 3B). The XPS analysis revealed distinct oxygen species, including hydroxide ions (OH−) or surface-adsorbed oxygen associated with CoO (Co–OH) at 530.709 eV. This assignment is corroborated by Co 2p spectra, which exhibit characteristic satellite peaks at 789.14 eV and 803.39 eV, confirming the presence of CoO and its contribution to the observed oxygen chemistry. Bridging oxygen species (Co–O–Fe) were identified at 531.359 eV, attributed to a shift in binding energy induced by the electronic influence of Fe, distinguishing it from Co–O–Co bonds. Surface-bound water molecules were observed at 533.009 eV, completing the spectrum of oxygen species in the system.56 The Fe(OH)x@Co0.8Fe0.2-MOF/NF composite exhibited a higher concentration of bound OH groups (44.25%) when compared with Co0.8Fe0.2-MOF/NF (35.59%), indicating that the electrodeposition of Fe(OH)x increased the concentration of M-OH species within the Fe(OH)x@Co0.8Fe0.2-MOF/NF composite (Table S2†).57 XRD patterns were acquired at different stages of electrode preparation (Fig. S5A†). The bare Ni foam substrate initially exhibited two distinct peaks at 44.5° and 51.8° (JCPDS 04-0850) (Fig. S5A(d)†). In addition to the peaks corresponding to the Ni foam substrate at 44.5° and 51.8°, a number of distinct diffraction peaks were observed over the range of 10° to 70° that were indicative of the presence of Co0.8Fe0.2-MOF (Fig. S5A(b)†). Diffraction patterns corresponding to Fe(OH)x (JCPDS 38-0032) (Fig. S5A(c)†) were observed in the Fe(OH)x/NF sample, confirming that Fe(OH)x nanoparticles were electrodeposited onto Co0.8Fe0.2-MOF/NF, resulting in the formation of the composite material Fe(OH)x@Co0.8Fe0.2-MOF/NF (Fig. S5A(a)†). In Fe(OH)x@Co0.8Fe0.2-MOF/NF (Fig. S5B†), an XPS survey scan confirmed the presence of Co, Fe, Ni, and C, confirming the results obtained by elemental analysis. The peaks observed at 715.8 eV and 725.6 eV correspond to Fe 2p3/2 and Fe 2p1/2, respectively (Fig. S5C†).
Electrocatalytic HMFOR performance
The electrocatalytic performance of Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)2/NF, and bare NF for the oxidation of HMF in a solution containing 1 M KOH and 10 mM HMF (pH 13.76) was evaluated using the LSV method. The kinetics and efficiency of the HMFOR processes in concentrated alkaline HMF solutions are primarily governed by electrode kinetics (i.e., reaction rates at the electrode surface).58 However, additional factors such as electrode structure and gas diffusion can also affect their activity. In some cases, polarization curves with uncompensated or partially compensated iR drop were used for Tafel analysis, which ideally requires steady-state data that is fully compensated for iR effects.59,60 Employing a lower scan rate helps minimize the effects of the catalyst metal oxidation peak and double-layer charging, as the delays caused by resistance–capacitance (RC) coupling in series are reduced, thereby lowering the onset overpotential. As low scan rates minimize interference.61,62 To address potential interference from the oxidation peaks related to the formation of active sites (Co3+, Fe3+) and HMFOR onset currents, a slow scan rate of 10 mV s−1 and 90% iR drop correction were employed.Based on the polarization curves (Fig. 4A), it is evident that the Fe(OH)x@Co0.8Fe0.2-MOF/NF configuration displayed the highest current density for the HMFOR. At a current density of 50 mA cm−2, the overpotentials (calculated relative to the OER standard potential) for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)2/NF, and bare NF were 236 mV, 317 mV, 336 mV, and 452 mV, respectively (Fig. 4B). At 100 mA cm−2, these overpotentials increased to 263 mV, 344 mV, 373 mV, and 545 mV, respectively.
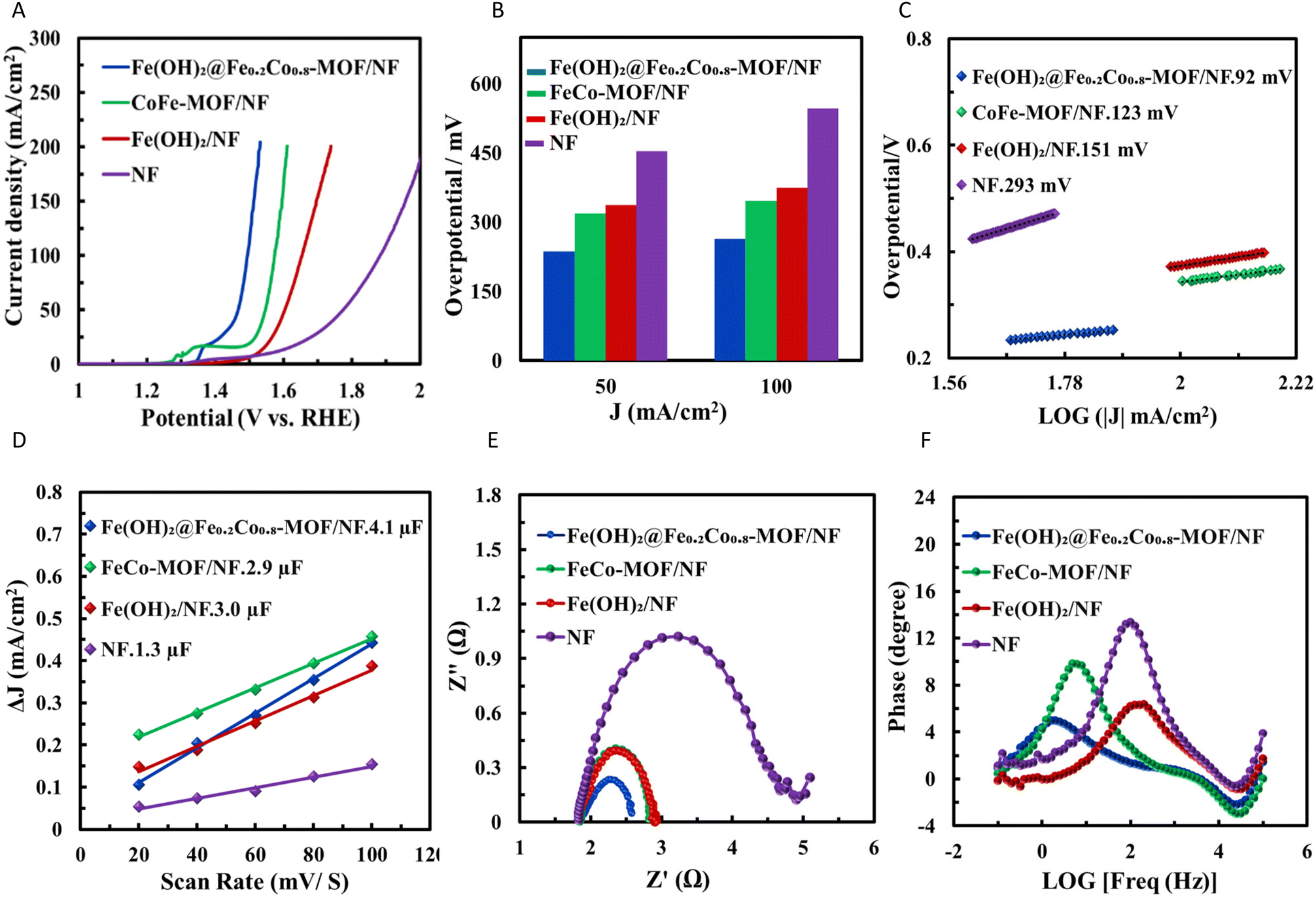 | ||
| Fig. 4 (A) Polarization curves (HMFOR) for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF in a KOH 1.0 M and HMF 10 mM electrolyte, with a scan rate of 10 mV s−1; (B) overpotential at current densities of 50 and 100 mA cm−2; (C) Tafel plots; (D) Double-layer capacitance plots; (E) Nyquist plots and; (F) Bode plots at a potential of 273 mV. | ||
The overpotentials of Fe(OH)x@Co0.8Fe0.2-MOF/NF were significantly lower than those of Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF, indicating that Fe and Co components collaborate for enhanced catalytic activity. The incorporation of Fe into the Co-MOF boosts catalytic efficiency primarily due to these synergistic effects. Fig. S6† illustrates the impact of increasing Fe content on electrocatalytic activity. The most favorable performance for HMFOR was observed with Co0.8Fe0.2-MOF/NF, where the metal oxidation peak decreased by 34 mV to a lower onset potential of 1.12 V versus RHE compared to Co-MOF/NF, indicative of a decrease in the overpotential for the oxidation process. The comparison of metal oxidation peaks for Fe-MOF/NF, Co-MOF/NF, Co0.5Fe0.5-MOF/NF, and Co0.8Fe0.2-MOF/NF (Fig. S6†) shows that Co0.8Fe0.2-MOF/NF exhibits a visibly higher charge, and although precise charge calculation is challenging due to potential inaccuracies in integrating current density over potential, the visual trend in Fig. S6† clearly highlights its superior performance, indicating a greater metal concentration.
This study suggests doping with iron in a particular ratio can alter the electrocatalytic performance of cobalt-based materials. The anodic shift in the oxidation potential of the Co3+/Co2+ redox couple is attributed to electronic interactions that modify the catalyst's electronic structure. Voltammetry shows a strong dependence of the Co3+/Co2+ redox potential on the Fe(OH)x content, indicative of strong electronic coupling between Fe and Co in the solid matrix. This supports the hypothesis that the cobalt-based material, with its conductive, chemically stable, and intrinsically porous structure, provides a suitable environment for Fe incorporation. The Fe ions substitute for Co in the lattice, acting as the most active sites for HMFOR catalysis (Fig. S6†).63–65 In addition, Fe3+ is frequently employed in organic transformations, particularly in aromatic electrophilic substitution reactions involving aryl rings. It is widely regarded as one of the most robust and versatile transition metal-based Lewis acids due to its ability to maintain catalytic activity under a wide range of reaction conditions, including variations in temperature, pH, and competing ligands or reagents.66,67 The high electrophilicity of Fe3+ significantly influences the electronic properties of other cations within the host material. According to Li et al.,68 Fe3+ ions, with a pKa of 2.2, enhance the acidity of the hydroxyl proton coordinated to the Co site. This increase in acidity facilitates proton dissociation, creating a more favourable environment for the oxidation of Co2+ to Co3+. By stabilising the transition state and reducing the energy barrier required for this oxidation process, Fe3+ effectively lowers the activation threshold. This mechanism promotes enhanced HMFOR activity by accelerating the generation of catalytically active Co3+ species. The Lewis acidic nature of Fe3+ also results in an anodic shift in the potential for the formation of the oxyhydroxide species in Co-based HMFORs. This potential shift occurs because the increased covalent character of the Co2+–O bonds requires a higher potential for oxidation. Consequently, HMFOR activity is greatly influenced by this difference in electronic states. These results agree with previous reports that attributed the enhanced activity to the robust electronic interactions between Fe and Co.60 While the exact mechanism behind this enhancement is not yet fully understood, ongoing investigations highlight the potential of bimetallic structures in electrocatalytic applications. Results from the analysis of a range of iron compounds, including Fe3O4, Fe2O3, Fe(III) hydroxide, Fe(II) hydroxide, and a mixture of Fe(III)/Fe(II) hydroxide (Fig. S7†) suggest that maintaining a 1:1 ratio of Fe(III) to Fe(II) in Fe(OH)X significantly boosts catalytic HMFOR activity with Fe(III) and Fe(II) responsible for this increase. This enhanced activity is attributed to the combined roles of Fe(III) and Fe(II), which facilitate key steps in the reaction mechanism. However, this study also reveals that the synergistic interaction between Fe and Co in Fe–Co mixed oxide catalysts is crucial, indicating that Fe alone may not always be the active site for HMFOR catalysis. Instead, the interplay between Fe and Co is essential for achieving optimal catalytic performance.
Conversely, HMFOR intermediates exhibit considerably weaker adsorption interactions with Co and Ni than Fe. This difference adsorption arises from the electronic properties of Co and Ni, which favour the formation of more covalent M–O bonds rather than strong ionic interactions. As a result, the intermediates bind less firmly to the surface, facilitating their desorption and accelerating turnover during the catalytic cycle. This behavior enhances the catalytic efficiency of Co and Ni by promoting faster reaction kinetics and reducing the risk of surface poisoning by strongly adsorbed intermediates. Consequently, this property is reflected in the high selectivity and nearly 100% faradaic efficiency observed for FDCA production, as previously reported.69 Additionally, studies have shown that the kinetic isotope effect indicates a proton-independent electron transfer process as the rate-determining step in HMF oxidation, further supporting the high charge transfer efficiency of these catalysts.69 The kinetic parameters of HMFOR catalysis were evaluated using the Tafel slope for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF electrodes (Fig. 4C). The Tafel slope provides insights into the charge transfer kinetics and the rate-limiting step of the reaction.
Additionally, the exchange current density (j0) was determined to characterize the inherent activity of the electrocatalysts under equilibrium conditions. The transfer coefficient, which reflects how the surface potential influences the free energy barrier and electron exchange, was also considered in evaluating the electrocatalytic process. The apparent Tafel slopes obtained for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF and bare NF electrodes were 92 mV, 123 mV, 151 mV and 293 mV, respectively. The lower apparent Tafel slopes for HMFOR at Fe(OH)x@Co0.8Fe0.2-MOF/NF indicated a higher charge transfer rate compared to the other modified electrodes. The intercept of the current density axis (log10(j)) at (η = 0) (Fig. S8†) yields the value of j0, which characterizes the inherent activity of an electrocatalyst under equilibrium conditions. The calculated j0 value for Fe(OH)x@Co0.8Fe0.2-MOF/NF was 6.41 mA cm−2, which is higher than the values for Co0.8Fe0.2-MOF/NF (4.57 mA cm−2), Fe(OH)x/NF (2.39 mA cm−2), and bare NF (1.49 mA cm−2). The exchange current density of Fe(OH)x@Co0.8Fe0.2-MOF/NF increased by a factor of 1.40, 2.68, 4.31 compared to Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF respectively, significantly enhancing the HMFOR rate.
The electrooxidation of HMF involves the transfer of 6 electrons at a temperature of 293.15 K. Determining the values of transfer coefficients (α) in electrochemical reactions such as the oxidation of HMF enables a deeper understanding of the mechanism of electron transfer at the electrode–electrolyte interface. Transfer coefficients of 0.105, 0.078, 0.064, and 0.033 were obtained for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF electrodes, respectively. The high transfer coefficient (α) of the Fe(OH)x@Co0.8Fe0.2-MOF/NFelectrode indicates its outstanding electrocatalytic capability. This improvement results from combining three factors: integrating Fe(OH)x enhances the catalyst's electronic characteristics, the increased surface area provides abundant active sites, and the well-designed morphology promotes efficient mass and electron transfer. In the electrocatalytic conversion of HMF to FDCA, a higher α value denotes a more efficient electron transfer mechanism. The exchange current density and Tafel slope lend credence to the system's lowered energy barriers and quicker reaction kinetics. Fe(OH)x@Co0.8Fe0.2-MOF/NF is a very efficient electrode for HMF electrooxidation because of these combined benefits.
The increased catalytic activity of Fe(OH)x@Co0.8Fe0.2-MOF/NF in HMFOR (Table 1) was examined in detail. The ECSA was calculated from the Cdl (from CV curves in the non-faradaic region (Fig. S9†)) and used to estimate the catalytic active sites’ performance during electrochemical processes. The slope representing the Cdl values is shown in Fig. 4D. Fe(OH)x@Co0.8Fe0.2-MOF/NF exhibits the highest value of Cdl compared to the other configurations, indicating that Fe(OH)x@Co0.8Fe0.2-MOF/NF provides the most electrocatalytic surface area. It is critical to compare intrinsic activities based on active site density to make meaningful activity comparisons and understand their fundamental origins. McCrory et al. proposed a standard protocol for assessing ECSA by measuring double-layer capacitance in a potential region with no faradaic response.70 Fig. S10† presents the LSV curves for Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF electrodes after normalization concerning their ECSA. The EIS technique provides valuable insights into the kinetics associated with the HMFOR. EIS analysis was performed under uniform conditions to investigate the factors influencing the catalytic activities of Fe(OH)x@Co0.8Fe0.2-MOF/NF, Co0.8Fe0.2-MOF/NF, Fe(OH)x/NF, and bare NF. Fig. 4E and F display representative EIS plots, including Nyquist and Bode (Phase angle vs. logarithm of frequency) diagrams. These measurements were taken under an applied overpotential of 273 mV across a frequency range from 100 kHz to 0.1 Hz. The Randles circuit was used to model the electrodes’ electrical properties, incorporating solution resistance (Rs), double-layer capacitance (Cdl), and charge transfer resistance (Rct). The Rct values were determined: Fe(OH)x@Co0.8Fe0.2-MOF/NF (0.75 Ω), Co0.8Fe0.2-MOF/NF (0.98 Ω), Fe(OH)x/NF (1.06 Ω), and bare NF (3.07 Ω). Fe(OH)x@Co0.8Fe0.2-MOF/NF exhibited the lowest Rct, indicating superior conductivity. Furthermore, electron–hole recombination data obtained from the Bode plot is shown in Fig. 4F, which provides information on the material's charge transfer processes. The Bode Fig. 4F shows the system's frequency response, and the phase angle reflects how well the charge separation and recombination processes work. A small phase angle at low frequencies indicates better charge transfer and a greater chance of effective catalytic activity, predicting less electron–hole recombination. Since effective charge separation is essential for improving reaction kinetics and overall efficiency, this analysis is essential for comprehending the material's performance in electrocatalytic applications. The electron lifetime (τr) was estimated using the frequency (fp) at the mid-frequency peak (1–100 Hz) according to the eqn (7):
 | (7) |
| Electrocatalyst | Electrode | Electrolyte | HMF conc. (mM) | Applied potential (V vs. RHE) | FDCA yield (%) | FE of FDCA (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Carbon fibre paper. b Carbon cloth. c Glassy carbon. d Carbon paper. | |||||||
| NiCoFe-LDHs | CFPa | 1 M NaOH | 10 | 1.54 | ∼82 | — | 73 |
| NiCo2O4 | NF | 1 M KOH | 5 | 1.50 | 72 | 80 | 74 |
| CuCo2O4 | NF | 1 M KOH | 10 | 1.45 | 93.7 | 94 | 42 |
| NiCo2O4 | NF | 1 M KOH | 10 | 1.55 | 90 | 100 | 21 |
| 3%Co-NiO | CCb | 1 M KOH | 10 | 1.47 | 94.8 | 94.6 | 75 |
| NiCo2O4 | NF | 1 M KOH | 10 | 1.45 | 99 | 99 | 76 |
| Ni0.5Co2.5O4 | GCc | 1 M KOH | 10 | 1.5 | 92.4 | 90.3 | 77 |
| NiCo2O4 | CPd | 1 M KOH | 10 | 1.45 | 97 | 96.5 | 78 |
| Co@NiCo-LDH | NF | 1 M KOH | 10 | 1.4 | 100 | 95.2 | 79 |
| Fe(OH)x@Co0.8Fe0.2-MOF | NF | 1 M KOH | 10 | 1.42 | 95.83 | 96.26 | This work |
The Bode plot indicates that the Fe(OH)x@Co0.8Fe0.2-MOF/NF electrode exhibited a longer electron lifetime compared to the other electrodes due to its lower fp, suggesting enhanced electron lifetime and improved HMFOR activity.71,72 The conclusions drawn from the EIS analyses affirm prior discoveries from XPS analysis regarding the Co 2p binding energies shift towards lower binding energies in the Fe(OH)x@Co0.8Fe0.2-MOF system. This aligns with LSV observations, Tafel analysis, and Cdl evaluations, confirming the superior performance of Fe(OH)x@Co0.8Fe0.2-MOF/NF as an HMFOR electrocatalyst (Table 1).
Product analysis and mechanism study
The study explored using HMFOR instead of OER in water electrolysis, highlighting its overpotential for significantly reducing energy consumption. The effectiveness of Fe(OH)x@Co0.8Fe0.2-MOF/NF as an electrocatalyst for HMFOR was investigated in two different electrolyte solutions: 1.0 M KOH with and without the addition of 10 mM HMF. Fig. 5A presents the iR-corrected LSV curves, revealing significant findings. The data indicates a lower potential is required to achieve the same current density when HMF is present. Specifically, at a current density of 100 mA cm−2, HMFOR requires an overpotential that is 60 mV lower than that of OER, and at 200 mA cm−2, it requires a 90 mV lower overpotential. Additionally, HMFOR facilitates the generation of valuable chemicals at the anode.80,81 The electrocatalytic efficiency of Fe(OH)x@Co0.8Fe0.2-MOF/NF for the oxidation of HMF was assessed under 1.42 VRHE applied potential using separate cells. Each compartment was filled with 18 mL of an electrolyte solution. The cathode side contained 1 M KOH solution, while the anode side was filled with 1 M KOH and 10 mM HMF at a pH of 13.76. The electrocatalytic oxidation process of HMF to FDCA primarily consists of a two-step oxidation sequence involving an alcohol group and an aldehyde group transitioning into a carboxyl group (Fig. 5B). This reaction pathway encompasses two feasible oxidation routes.82 The first pathway entails the oxidation of the alcohol group, leading to the formation of DFF. Subsequently, the aldehyde group undergoes oxidation, resulting in the formation of FFCA. Finally, the second oxidation of the aldehyde group leads to the formation of FDCA. In contrast, the second pathway involves the initial oxidation of the aldehyde group, yielding an intermediate compound known as HMFCA. After this step, the alcohol group undergoes oxidation, generating FFCA. Ultimately, the second oxidation of the aldehyde group leads to the formation of FDCA (Fig. 5D).83–85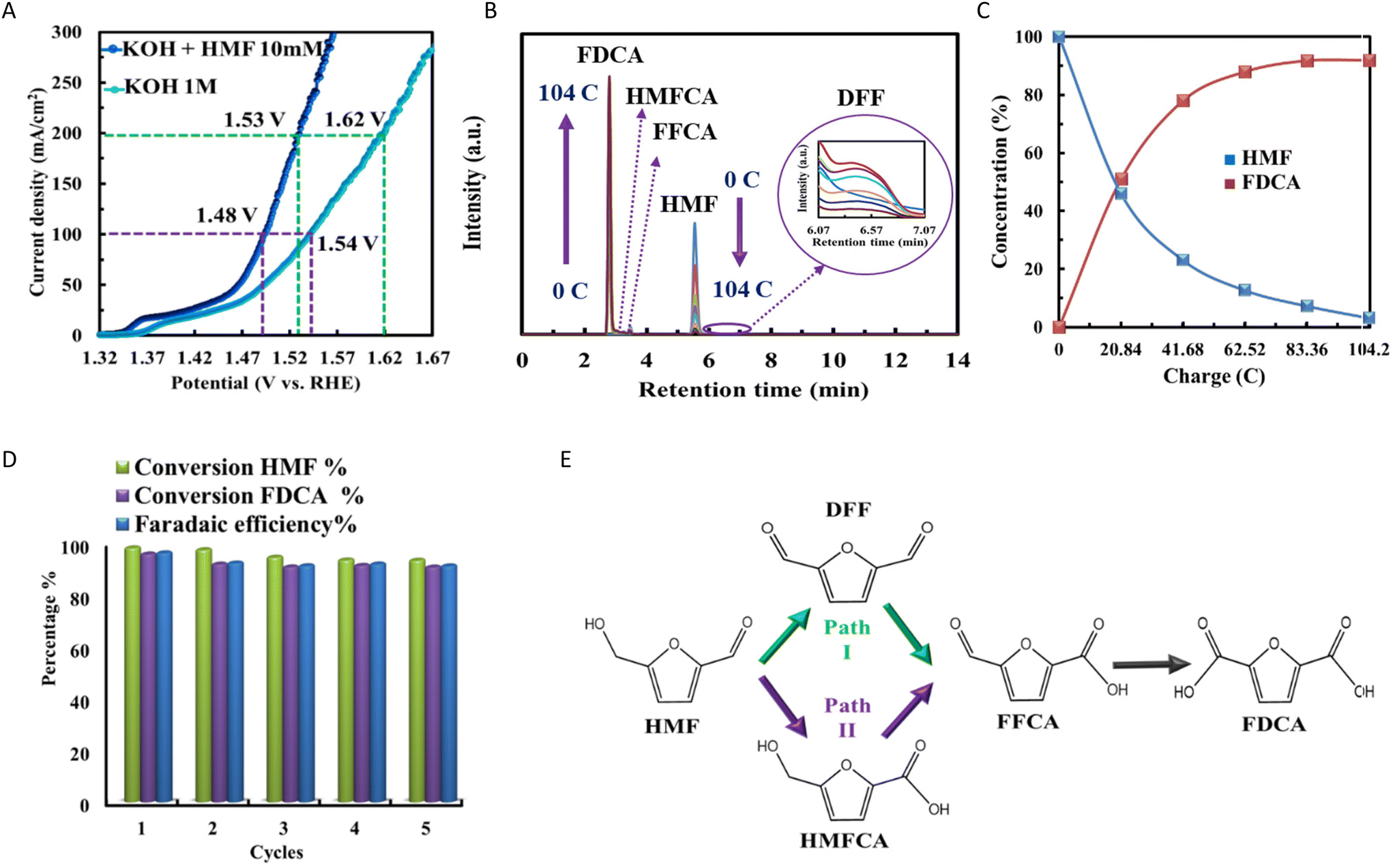 | ||
| Fig. 5 (A) LSV curves with and without 10 mM HMF for the Fe(OH)x@Co0.8Fe0.2-MOF/NF electrocatalyst in a 1 M KOH electrolyte; (B) HPLC profiles showing HMF electrolysis at 1.42 V versus RHE in 1 M KOH containing 10 mM HMF; (C) changes in the concentrations of FDCA and HMF during electrolysis with the amount of transmitted charge; (D) two proposed HMF oxidation mechanisms have been discussed in the literature; (E) performance of the effectiveness and efficiency of Fe(OH)x@Co0.8Fe0.2-MOF/NF in converting 10 mM HMF to FDCA through electrooxidation over five consecutive cycles at 1.42 V versus RHE. The assessment is conducted using HPLC during the electrochemical process. | ||
HPLC was utilized to characterize the reaction pathway of HMF electrooxidation, as depicted in Fig. 5B. The conversion of HMF and the yield of oxidation products with the charges used by the Fe(OH)x@Co0.8Fe0.2-MOF/NF electrocatalyst is displayed in Fig. 5C. The standard curves for HMF and FDCA are in Fig. S11 and S12,† respectively. The signal related to HMF appeared at a retention time of 5.5 minutes and consistently diminished, whereas the signal corresponding to FDCA, detected at 2.8 minutes, shows a significant increase. The intermediates HMFCA, FFCA, and DFF were detected at retention times of 3.1, 3.4, and 6.7 minutes, respectively, demonstrating that the oxidation of HMF occurs through both of the outlined pathways. Interestingly, Path 1 is the predominant route, highlighting its primary role in the reaction process (Fig. 5B–D).
The catalytic stability was examined. At a constant current of 15 mA cm−2, each 7200 s period can be regarded as a single electrolysis cycle. The LSV curves before and after 10 hours of electrolysis show no significant difference (Fig. S13†), while the selectivity and faradaic efficiency (FE) for the production of FDCA remained stable, demonstrating the stability of Fe(OH)x@Co0.8Fe0.2-MOF/NF (Fig. 5E). The average selectivity and FE for FDCA were 92.16% and 92.61% over five testing cycles.
Conclusion
In summary, the incorporation of Fe(OH)x significantly modified the electrochemical properties of Co0.8Fe0.2-MOF/NF, enhancing the transformation of biomass derived HMF. The findings from this work, along with existing literature, underscore the importance of Fe(OH)x in increasing the number of active sites, which in turn improves the adsorption capacity of aldehyde functional groups. Moreover, the increased catalytic activity in Fe(OH)x@Co0.8Fe0.2-MOF can be attributed to the synergistic effect of these groups. Notably, the Fe(OH)x@Co0.8Fe0.2-MOF/NF catalyst demonstrated excellent performance in the HMFOR, achieving an E100 of 1.49 V versus RHE, and in the OER, with an E100 of 1.54 V versus RHE. HMF oxidation occurs via two distinct pathways: Path 1 (HMF → HMFCA → FFCA → FDCA) and Path 2 (HMF → DFF → FFCA → FDCA), with Path 1 being the dominant route, as confirmed by the detection of key intermediates. The conversion of HMFCA to FFCA is identified as the rate-determining step, highlighting a critical stage in the catalytic process. Importantly, after five cycles at 1.42 V versus RHE, the HMFOR activity consistently maintained high conversion rates for both HMF and FDCA, as well as faradaic efficiency.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the Interreg NWE BioWILL project (NWE964) and funding from the Science Foundation Ireland Research Centre for Pharmaceuticals (SSPC) under Grant Number 12/RC/2275.References
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS.
- B. Dasgupta, J. N. Hausmann, R. Beltrán-Suito, S. Kalra, K. Laun, I. Zebger, M. Driess and P. W. Menezes, Small, 2023, 2301258 CrossRef CAS PubMed.
- M. Nozari-asbmarz, M. Amiri, A. Bezaatpour and S. Arshi, Appl. Chem., 2021, 16, 137–148 Search PubMed.
- M. Nozari-Asbemarz, M. Amiri, H. Imanzadeh, A. Bezaatpour, S. Nouhi, P. Hosseini, M. Wark and D. Seifzadeh, Int. J. Hydrogen Energy, 2022, 47, 5250–5259 CrossRef CAS.
- T. Wu, Z. Xu, X. Wang, M. Luo, Y. Xia, X. Zhang, J. Li, J. Liu, J. Wang and H.-L. Wang, Appl. Catal., B, 2023, 323, 122126 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- O. Simoska, Z. Rhodes, S. Weliwatte, J. R. Cabrera-Pardo, E. M. Gaffney, K. Lim and S. D. Minteer, ChemSusChem, 2021, 14, 1674–1686 CrossRef CAS PubMed.
- Y. Feng, K. Yang, R. L. Smith and X. Qi, J. Mater. Chem. A, 2023, 11, 6375–6383 RSC.
- D. D. Liu and E. Y.-X. Chen, Green Chem., 2014, 16, 964–981 RSC.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- B. Kuster, Starch – Stärke, 1990, 42, 314–321 CrossRef CAS.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- Y. Xiao, C. Shen, Z. Xiong, Y. Ding, L. Liu, W. Zhang and Y. A. Wu, Mater. Today Phys., 2023, 101122 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- X. Jiang, W. Li, Y. Liu, L. Zhao, Z. Chen, L. Zhang, Y. Zhang and S. Yun, SusMat, 2023, 3, 21–43 CrossRef CAS.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS PubMed.
- D. A. Giannakoudakis, J. C. Colmenares, D. Tsiplakides and K. S. Triantafyllidis, ACS Sustainable Chem. Eng., 2021, 9, 1970–1993 CrossRef CAS.
- W. Li, L. Zhao, X. Jiang, Z. Chen, Y. Zhang and S. Wang, Adv. Funct. Mater., 2022, 32, 2207727 CrossRef CAS.
- W. Zhang, B. Zhang, Y. Li, E. Zhang, Y. Zhang, Q. Wang and Y. Cong, Int. J. Hydrogen Energy, 2022, 47, 1633–1643 CrossRef CAS.
- B. G. Diamond, L. I. Payne and C. H. Hendon, Commun. Chem., 2023, 6, 67 CrossRef CAS PubMed.
- C. Qi, S. Han, J. Lin, J. Cheng, K. Du, Y. Hu and Y. Chen, Catalysts, 2022, 12, 671 CrossRef CAS.
- J. Jiang, X. L. Zhou, H. G. Lv, H. Q. Yu and Y. Yu, Adv. Funct. Mater., 2023, 33, 2212160 CrossRef CAS.
- L. Shen, J. Ying, K. I. Ozoemena, C. Janiak and X. Y. Yang, Adv. Funct. Mater., 2022, 32, 2110851 CrossRef CAS.
- D. Guo, S. Wang, J. Xu, W. Zheng and D. Wang, J. Energy Chem., 2022, 65, 448–468 CrossRef CAS.
- K. Ge, S. Sun, Y. Zhao, K. Yang, S. Wang, Z. Zhang, J. Cao, Y. Yang, Y. Zhang and M. Pan, Angew. Chem., Int. Ed., 2021, 60, 12097–12102 CrossRef CAS PubMed.
- C. A. Downes and S. C. Marinescu, ChemSusChem, 2017, 10, 4374–4392 CrossRef CAS PubMed.
- X. Liu, W. Zang, C. Guan, L. Zhang, Y. Qian, A. M. Elshahawy, D. Zhao, S. J. Pennycook and J. Wang, ACS Energy Lett., 2018, 3, 2462–2469 CrossRef CAS.
- M. Cai, Y. Zhang, Y. Zhao, Q. Liu, Y. Li and G. Li, J. Mater. Chem. A, 2020, 8, 20386–20392 RSC.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S.-Z. Qiao, Chem. Commun., 2017, 53, 10906–10909 RSC.
- R. Luo, Y. Li, L. Xing, N. Wang, R. Zhong, Z. Qian, C. Du, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 CrossRef CAS.
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS.
- K. Ye, K. Li, Y. Lu, Z. Guo, N. Ni, H. Liu, Y. Huang, H. Ji and P. Wang, TrAC, Trends Anal. Chem., 2019, 116, 102–108 CrossRef CAS.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- B. Zhou, Y. Li, Y. Zou, W. Chen, W. Zhou, M. Song, Y. Wu, Y. Lu, J. Liu and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 22908–22914 CrossRef CAS PubMed.
- J. Wu, Z. Kong, Y. Li, Y. Lu, P. Zhou, H. Wang, L. Xu, S. Wang and Y. Zou, ACS Nano, 2022, 16, 21518–21526 CrossRef CAS PubMed.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- X. Ren, C. Wei, Y. Sun, X. Liu, F. Meng, X. Meng, S. Sun, S. Xi, Y. Du and Z. Bi, Adv. Mater., 2020, 32, 2001292 CrossRef CAS PubMed.
- B. Jiang, Z. Wan, Y. Kang, Y. Guo, J. Henzie, J. Na, H. Li, S. Wang, Y. Bando and Y. Sakka, Nano Energy, 2021, 81, 105644 CrossRef CAS.
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang and Q. Liao, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS.
- N. Han, F. Zhao and Y. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 RSC.
- P. Chen, T. Zhou, S. Wang, N. Zhang, Y. Tong, H. Ju, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 15471–15475 CrossRef CAS PubMed.
- M.-H. Tsai, Y. Juang, C.-C. Hu, L.-C. Hua, B. K. Mahata and C. Huang, Electrochim. Acta, 2023, 446, 142130 CrossRef CAS.
- Z. Zhao, H. Zhao, X. Du and X. Zhang, Int. J. Hydrogen Energy, 2024, 49, 811–822 CrossRef CAS.
- F. Shahbazi Farahani, M. S. Rahmanifar, A. Noori, M. F. El-Kady, N. Hassani, M. Neek-Amal, R. B. Kaner and M. F. Mousavi, J. Am. Chem. Soc., 2022, 144, 3411–3428 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Z. Yang, S. Yan, C. Zhang and S. Liu, ACS Sustainable Chem. Eng., 2019, 8, 1004–1014 CrossRef.
- C. Yan, H. Shan, H. Yang, W. Zhang, S. Wang, Y. Zhang, J. Qin, W. Li, W. Xiao and Z. Zhou, ChemElectroChem, 2021, 8, 4745–4749 CrossRef CAS.
- X. Li, Z. Ao, J. Liu, H. Sun, A. I. Rykov and J. Wang, ACS Nano, 2016, 10, 11532–11540 CrossRef CAS PubMed.
- Y. Xu, X. Li, J. Wang, Q. Yu, X. Qian, L. Chen and Y. Dan, Chem. – Eur. J., 2020, 26, 1298–1305 CrossRef CAS PubMed.
- G. Mu, J. Zhou, B. Ye, T. Zhang, C. Li and Q. Zhao, J. Electron. Mater., 2023, 52, 4951–4958 CrossRef CAS.
- H. Zhang, J. Zhang, Y. Li, H. Jiang, H. Jiang and C. Li, J. Mater. Chem. A, 2019, 7, 13506–13510 RSC.
- C. Xiong and C. Cai, Nanomaterials, 2022, 12, 1788 CrossRef CAS PubMed.
- J. Bockris, Modern Electrochemistry 2A: Fundamentals of Electrodics, 2002, pp. 1042–1055 Search PubMed.
- J. M. Bockris, A. Reddy, M. Gamboa-Aldeco and L. Peter, Platinum Met. Rev., 2002, 46, 15–17 CrossRef.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- Q. Wang, H. Guesmi, S. Tingry, D. Cornu, Y. Holade and S. D. Minteer, ACS Energy Lett., 2022, 7, 952–957 CrossRef CAS.
- M. Nozari-Asbemarz, M. Amiri, A. Khodayari, A. Bezaatpour, S. Nouhi, P. Hosseini, M. Wark, R. Boukherroub and S. Szunerits, ACS Appl. Energy Mater., 2021, 4, 2951–2959 CrossRef CAS.
- Y. Zhang, T. Gao, Z. Jin, X. Chen and D. Xiao, J. Mater. Chem. A, 2016, 4, 15888–15895 RSC.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- P. Arunkumar, S. Gayathri, A. Rajasekar, S. S. Kumar, S. K. Kamaraj and J. H. Han, J. Colloid Interface Sci., 2024, 656, 424–439 CrossRef CAS PubMed.
- X. X. Peng, S. Gai, Z. Liu, K. Cheng and F. Yang, ChemSusChem, 2024, 17, e202301227 CrossRef CAS PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. Von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- M. Zhang, Z. Zheng, X. Zhang, Z. Jiang, X. Yong, K. Li, X. Tu and K. Yan, JACS Au, 2025, 5, 937–947 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Razavi, M. Sookhakian, B. T. Goh, H. Bahron, E. Mahmoud and Y. Alias, Nanoscale Res. Lett., 2022, 17, 1–10 CrossRef PubMed.
- C.-Y. Ho and H.-W. Wang, Appl. Surf. Sci., 2015, 357, 147–154 CrossRef CAS.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- Y. Yang, D. Xu, B. Zhang, Z. Xue and T. Mu, Chem. Eng. J., 2022, 433, 133842 CrossRef CAS.
- Z. Zhou, Y.-n. Xie, L. Sun, Z. Wang, W. Wang, L. Jiang, X. Tao, L. Li, X.-H. Li and G. Zhao, Appl. Catal., B, 2022, 305, 121072 CrossRef CAS.
- Y. Lu, T. Liu, Y.-C. Huang, L. Zhou, Y. Li, W. Chen, L. Yang, B. Zhou, Y. Wu and Z. Kong, ACS Catal., 2022, 12, 4242–4251 CrossRef CAS.
- S. Yang, X. Xiang, Z. He, W. Zhong, C. Jia, Z. Gong, N. Zhang, S. Zhao and Y. Chen, Chem. Eng. J., 2023, 457, 141344 CrossRef CAS.
- S. Fan, B. Zhu, Y. Zhong, S. Shi, J. Zhang and C. Chen, Chem. Eng. J., 2023, 474, 145905 CrossRef CAS.
- Y. Song, Z. Li, K. Fan, Z. Ren, W. Xie, Y. Yang, M. Shao and M. Wei, Appl. Catal., B, 2021, 299, 120669 CrossRef CAS.
- C. Yang, C. Wang, L. Zhou, W. Duan, Y. Song, F. Zhang, Y. Zhen, J. Zhang, W. Bao and Y. Lu, Chem. Eng. J., 2021, 422, 130125 CrossRef CAS.
- X. Pang, H. Bai, H. Zhao, W. Fan and W. Shi, ACS Catal., 2022, 12, 1545–1557 CrossRef CAS.
- P. Zhou, G. Hai, G. Zhao, R. Li, X. Huang, Y. Lu and G. Wang, Appl. Catal., B, 2023, 325, 122364 CrossRef CAS.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li and X. Zhong, Adv. Funct. Mater., 2023, 2214488 CrossRef CAS.
- S. Liu, S. Dou, J. Meng, Y. Liu, Y. Liu and H. Yu, Appl. Catal., B, 2023, 331, 122709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00346f |
| This journal is © The Royal Society of Chemistry 2025 |