
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe efficacy of oleic acid treatment in passivating MAPbI3 films†
Ghada
Abdelmageed
 a,
Rashad F.
Kahwagi
a,
Joelle
Korkomaz
a,
Anthony
El-Halaby
a,
Adam F. G.
Leontowich
bc,
Sean
Hinds
a and
Ghada I.
Koleilat
*a
a,
Rashad F.
Kahwagi
a,
Joelle
Korkomaz
a,
Anthony
El-Halaby
a,
Adam F. G.
Leontowich
bc,
Sean
Hinds
a and
Ghada I.
Koleilat
*a
aDepartment of Process Engineering and Applied Science, Dalhousie University, 1360 Barrington Street, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: ghada.koleilat@dal.ca
bCanadian Light Source Inc., 44 Innovation Boulevard, Saskatoon, SK S7N 2 V3, Canada
cDepartment of Chemistry, University of Saskatchewan, Saskatoon, SK S7N 5C9, Canada
First published on 11th April 2025
Abstract
Reliability, scalability, and excellent film properties with large crystals and low grain boundaries are essential for successfully commercializing perovskites in optoelectronic applications. Our previous reports introduced meniscus-guided blade coating, or shearing, which is referred to as one-step blade coating in the present study, as a promising method for depositing scalable perovskite films with millimetre-sized crystals, fulfilling two of the essential criteria. As a subsequent study, we investigated the stability of the films in response to humidity by employing a readily accessible hydrophobic molecule, oleic acid (OA), through surface passivation. We compared the quality of the surface treatment on films produced via one-step and two-step deposition methods utilizing spin and blade coating techniques while subjecting them to continuous exposure to high humidity levels. Initially, we applied OA to the films using spin-coating, which is the standard method for surface passivation. Our results prove that the film properties resulting from the deposition technique determine the effectiveness of the passivation process. A quick surface treatment using OA via spin coating can be highly effective for perovskite films with smooth surfaces and smaller grain sizes, in contrast to textured films with larger crystal sizes. By tailoring the surface treatment method from spin coating to dip coating, we demonstrated that OA can prolong the stability of perovskites for months under continuous high-humidity exposure.
1. Introduction
Metal halide perovskites (MHPs) with their various compositions have received enormous attention for their highly promising performance in solar cells.1 The rapid progress of their reported power conversion efficiency (PCE) from 3.8% in 2009 to 27.0% in 2025 has been unprecedented, as evident in the National Renewable Energy Laboratory (NREL) efficiency chart.2 However, the stability of the metal halide perovskites is highly dependent on their compositions and has been a major obstacle to the potential commercialization of perovskite solar cells.3–6 Humidity, in particular, is a primary concern as it rapidly induces degradation in MHPs due to the high ionic nature of the material.7,8 Many studies have focused on deciphering humidity-induced degradation pathways as a first step to tackle and solve this issue.9–11 Methylammonium lead iodide (CH3NH3PbI3, or MAPbI3), arguably the most unstable perovskite composition, has been proposed to decompose into the hydrate intermediate compound according to eqn (1) as humidity permeates through the thin film grain boundaries.12–16| 4CH3NH3PbI3 + 2H2O → (CH3NH3)4PbI6·2H2O + 3PbI2 | (1) |
In this equation, isolated octahedra hydrated compound (PbI64−) forms and subsequently PbI2 separates and crystalizes, while volatile gases (e.g., HI and CH3NH2 (MA)) form are released. In a detailed study, Song et al. have elucidated that humidity-induced degradation occurs by the forming of a low-dimensional, intermediate hydrated perovskite layer due to the dissolution of the Pb–I framework superficial layer and subsequent interaction with the organic species.17 The intermediate hydrated layer may act as a protective barrier at the beginning of the exposure process, shielding the bulk from water molecules and suppressing the generation of free electrons caused by water doping. This could explain why controlled humidity can be beneficial.18 Nonetheless, prolonged exposure to high levels of humidity is sure to promote an irreversible decomposition of perovskite into a solid-state degradation byproduct (PbI2) and volatile gaseous byproducts.14 Alternatively, the degradation pathway develops through direct deprotonation of the organic cation, leading the perovskite to decompose directly into the degradation byproduct, bypassing the formation of an intermediate phase, as described by Lu et al.19 They concluded that the termination of the MAPbI3 surface and grain boundaries dictates the mechanism of the degradation pathway, whether through hydration or deprotonation, as illustrated in Fig. 1. Such termination can result from temperature preparation conditions where high temperatures may promote the breakage of the weak bonding of MA+, leading to an increased presence of Pb-rich sites at the film's surface. MAI-terminated MAPbI3 films, as those produced with low-temperature preparation conditions, were shown to degrade by allowing water molecules to penetrate and form hydrated compounds, similar to the interaction outlined in eqn (1). The reaction of the hydrated compound can be reversed with limited exposure to humidity. However, with prolonged exposure, the structure breaks down permanently into HI, CH3NH2, and PbI2. The PbI2-terminated MAPbI3 films degrade directly into PbI2 through an irreversible interaction of water molecules with iodine, producing volatile HI and OH− ions that subsequently deprotonate MA+ ions, as demonstrated in Fig. S1b† and detailed in eqn (2) and (3).
| CH3NH3PbI3 + H2O → CH3NH3·PbI2 + OH− + HI↑ | (2) |
| CH3NH3·PbI2 + OH− → H2O↑ + PbI2 + CH3NH2↑ | (3) |
 | ||
| Fig. 1 Schematic illustrations of humidity-induced degradation pathways of MAPbI3 surface layer with (a) MAI termination, (b) PbI2 termination. Schematic illustrations of MAPbI3 films preparation methods: (c) SC-1, and SC-2, (d) BC-1, and BC-2. SEM images (5k magnification, bar denotes 10 μm) of MAPbI3 films prepared via different deposition methods: (e) SC-1, (f) SC-2, (g) BC-1, (h) BC-2. (i) Schematic illustrations of oleic acid molecules anchoring with the surface of MAPbI3 films with different terminations. | ||
Numerous potential solutions have been explored, including encapsulation and the implementation of chemical treatments, such as employing additives in solution and film surface passivation strategies.20–24 Hydrophobic molecules, such as polyethylene terephthalate (PET), oleic acid, and polystyrene (PS), have been utilized to reinforce the long-term stability of perovskite.22,25 Oleic acid (C18H34O2) has been reported as an effective facile surface passivation ligand for perovskite and other nanocrystals as it prevents aggregation, improves chemical stability, and enhances optoelectrical properties.26,27 Additionally, as a surface passivation treatment, oleic acid was reported to enhance perovskite solar cell efficiency by substantially reducing recombination.25,28 Owing to its high hydrophobicity, the long-chained fatty acid theoretically should form a barrier to repel water molecules from the perovskite films surface thus suppressing that mechanism of degradation. A previous study reported improved stability for two-step spin-coated perovskite films and devices over 4 weeks of exposure to humidity.25 While spin coating remains highly adopted in lab settings, blade coating, enables large-scale production with customizable surface coverage and is compatible with roll-to-roll (R2R) manufacturing essential for a wide range of applications, a feature that spin coating lacks. Furthermore, the deposition technique and its controls play a crucial role in the crystal growth process and the resulting thin film-forming properties. These properties, including morphology, crystal size, grain boundaries, and surface termination, significantly impact stability and morphological characteristics of the resulting films as demonstrated in our previous work.29,30 Recently, we produced highly oriented millimetre-sized crystals by meniscus-guided blade coating.29 We demonstrated that the pristine blade coated MAPbI3 films with their macroscale crystal grains are inherently more stable that spin coated films when stored in nitrogen or ambient air over a one week period. Beyond that, we have not closely analyzed the properties of our films: to our knowledge no surface passivation has been reported to prolong the stability of the films.
Herein, we theorize that such vast changes in the film morphology and crystal formation process inherently alter the efficacy of widely reported, exclusively on spin-coated films, surface passivation treatments such as the facile oleic acid surface treatment when films are exposed to severe continuous high humidity conditions and thus, require an in-depth evaluation and mapping of the films decomposition pathways. We initially hypothesized that films with large highly oriented crystals should in principle be vastly more stable than films with smaller polycrystalline regions: the results show that crystal packing, grain boundary depths and surface terminations directly impact the lifetime of the films exposed to severe humidity.
We used spin coating, the one-step method as well as the two-step method, both widely reported in the literature,31 as comparative baseline methods of MAPbI3 deposition techniques to be evaluated in conjunction with one-step and two-step blade coated MAPbI3 films. Here the two-step refers to depositing PbI2 first followed by MAI while the one-step refers to depositing both precursors simultaneously.32 We assess the impact of the instantaneous oleic acid treatment on the various surfaces by analyzing degradation-induced changes in the perovskite films using various spectroscopic techniques, such as UV-Vis absorbance spectroscopy, photoluminescence (PL) spectroscopy, X-ray diffraction (XRD), and Fourier transform infrared (FT-IR) spectroscopy. We also rely on X-ray photoelectron spectroscopy (XPS) to elucidate the film's surface composition and deduce the degradation mechanics involved.
2. Experimental
2.1. Materials
All chemicals were used without further purification, including lead iodide (PbI2, 99%, Sigma-Aldrich), methylammonium iodide (MAI, ≥99% anhydrous, Sigma-Aldrich), N,N-dimethylformamide (DMF, spectroscopic grade, Sigma-Aldrich), dimethyl sulfoxide (DMSO, anhydrous ≥99.9%, Sigma-Aldrich), diethyl ether (≥99.9%, Sigma Aldrich), 2-propanol (99.8%, Sigma-Aldrich), oleic acid (analytical grade, Supelco Inc., Millipore Sigma), and toluene (99.8%, Sigma-Aldrich). Borosilicate glass slides were used as substrates, and the dry aging conditions were achieved employing Drierite™ indicating absorbents (W.A. Hammond).2.2. Preparation methods
Borosilicate glass slides were cut into 25 mm × 25 mm pieces, then cleaned by sonication in Triton solution, de-ionized water, acetone, and 2-propanol respectively for 20 minutes, and then dried using compressed N2 gas. The dried slides were treated with ultraviolet ozone (UVO) for 20 minutes right before the perovskite deposition to enhance the wettability of the slides. The perovskite samples were prepared using spin and blade coating techniques (Fig. 1c and d) with the following details:1 One-step spin coating (SC-1) deposition: the perovskite solution was prepared as follows: a 645 mg of PbI2 and a 222 mg of MAI were dissolved in 1 mL DMF by stirring on a hotplate at 100 °C for 1 hour. Before deposition, the solution was filtered using a syringe filter (PVDF, 22 μm pore size). 100 μL of the perovskite solution was spin coated in air on cleaned glass slides with speed of 3000 rpm for 25 seconds. 250 μL of chlorobenzene was dropped on the spinning slides in the final 10 seconds. Then, the films were transferred immediately to anneal on a hotplate (100 °C) for 10 minutes.
2 Two-steps spin coating (SC-2) deposition: PbI2 (461 mg) was dissolved in DMF (1 mL) by stirring on a hotplate (80 °C, 400 rpm) for 30 minutes. The PbI2 solution (50 μL) was spin-coated on the glass slide in air at 6000 rpm for 5 seconds, followed by an annealing step (100 °C) for 30 minutes in air. Then, the lead iodide films were dipped in the MAI solution for 5 minutes, as illustrated in Fig. 1b, converting them to MAPbI3. The resultant perovskite films were rinsed with 2-propanol and dried on a hotplate (85 °C) for 2 minutes in air.
3 One-step blade coating (BC-1) deposition: 0.4 M of MAPbI3 precursors solutions were prepared by dissolving PbI2 (184 mg) and MAI (64 mg) in anhydrous DMSO (1 mL) in a N2 filled glovebox. The solutions were stirred in the glovebox under dark conditions on a hotplate at 70 °C, 400 rpm, for 7 hours. Subsequently, the perovskite solutions were filtered (PVDF syringe filter, 0.22 μm). The deposition was performed in ambient atmosphere using a custom-made blade coating station that allow a heat assisted deposition. For the film formation, a 70 μL of the perovskite solution was blade coated on a clean glass slide at 160 °C, with a speed of 0.1 mm s−1, and blade gap of 300 μm. The films were annealed for 20 minutes at 100 °C in inert atmosphere.
4 Two-step blade coating (BC-2) deposition: in this method, the PbI2 and MAI solutions were prepared like in two-step spin coating. For the deposition, a 50 μL of PbI2 solution was blade coated at 100 °C, with a speed of 1 mm s−1, and blade gap of 300 μm and subsequently annealed for 30 minutes at 100 °C. Next, the as prepared PbI2 films were dipped in the MAI solution for 5 minutes, and the resultant perovskite films were rinsed and dried in air in the same details as two step spin coating (SC-2).
2.3. Surface passivation treatment and aging procedure
Equal amounts of toluene and oleic acid were stirred for 10 minutes in air, then 50 μL of the prepared solution was spin-coated on MAPbI3 films for 10 seconds at 0 rpm (loading time) then at 25 seconds at 6000 rpm in air, followed by drying on a hotplate for 5 minutes at 70 °C in air. For dip coating, the films were dipped in the oleic acid solution for 10 minutes. Then the films were rinsed briefly with toluene to remove excess oleic acid molecules, dried on the hotplate for 5 minutes at 70 °C, and stored overnight under vacuum to ensure the complete dryness. To test the effectiveness of the surface passivation treatment against high humidity levels, we followed a previously reported method in which the untreated and treated samples were placed inside an airtight container with saturated sodium chloride solutions in dark conditions to avoid any photoinduced degradation. In such a method, water evaporates from the salt solution inside the enclosure to reach equilibrium at relative humidity levels of around 76% at room temperature.33,34 The salt mixtures were monitored throughout the experiment and replenished when needed to ensure the steadiness of the humidity levels. Duplicate control and treated samples were stored in dark, dry conditions using indicating Drierite™ to monitor any parasitic effect of oleic acid on the perovskite stability.2.4. Characterization techniques
The humidity-induced structural changes of the perovskite films were examined using UV-Vis absorbance spectroscopy (Agilent Cary 60 Spectrophotometer), and photoluminescence (PL) spectra were measured using a Cary Eclipse fluorescence spectrometer. The excitation wavelength used was 532 nm, with a penetration length of 80 nm at the MAPbI3 surface, and the films were measured from the film side.35 X-ray diffraction (XRD, Bruker D8 Advance X-ray diffractometer, Cu Kα source, λ = 1.5406 Å), and Fourier Transform Infrared (FT-IR) spectroscopy (Agilent Cary 630 FTIR). Grazing-incidence wide-angle X-ray Scattering (GIWAXS) was conducted on perovskite films at the Brockhouse X-ray Diffraction and Scattering Sector Wiggler Low Energy (BXDS-WLE) beamline of the Canadian Light Source (CLS) synchrotron, using a photon energy of 15.1 keV (λ = 0.81931 Å).36In addition, the morphology and surface properties of the samples were analyzed using scanning electron microscopy (SEM, Hitachi S-4700, cold field emission), and X-ray photoelectron spectroscopy (XPS, Thermofisher Multilab 2000). The XPS instrument had a magnesium anode (1253.6 eV) with the sample chamber under pressure range from 6 × 10−10 to 2 × 10−9 Torr. The samples’ exposure to the X-rays was limited to less than 20 minutes. All measurements were conducted in dark conditions to prevent any additional photoinduced degradation mechanism. The X-ray penetration depth into the film was calculated to be <10 nm. All XPS data analysis was performed using CasaXPS software (ver. 2.3.23).37 Background subtraction was performed using a Shirley background and peak fitting uses symmetric Gaussian/Lorentzian peak shapes. The elemental atomic concentrations of the perovskite surface were computed from ratios of XPS peaks intensities after appropriate normalization via atomic sensitivity factors.38,39
3. Results and discussion
We prepared the perovskite film samples through two different deposition techniques, namely blade coating and spin coating, either via one-step or two-step methods as illustrated in Fig. 1c and d. Each preparation method resulted in different film-forming properties with different grain sizes, revealed in scanning electron micrographs in Fig. 1e–h and S4.† The blade coating method produced sizably larger crystal sizes compared to spin coating, with one-step blade coating, in particular, producing the biggest grain size of all. The large crystals created by one-step blade coating agree with previous studies that proved that meniscus-guided blade deposition can yield millimeter-sized crystal grains.29,30,40The samples (SP-1, SP-2, BC-1 and BC-2) were very briefly surface treated (<10 s) with oleic acid and then subjected to high humidity levels (∼76% RH). The passivation occurs by binding the carboxyl group (–COO−) of oleic acid to surface Pb2+ and/or CH3NH3+ sites, depending on the surface termination of the perovskite film. We opted for such a brief chemical treatment to investigate the efficacy of the OA molecule's adherence to the film's surface only, thus mainly shielding the top layer.
Fig. 2 illustrates the changes in UV-Vis absorbance and PL spectra of the samples over time. Results show an enhancement in the stability of the treated samples, except BC-1, which fully degraded through exposure to humidity within one week and no longer retained the characteristic absorption onset around 770 nm, representative of the MAPbI3 bandgap. BC-2 samples were fully degraded after four months; meanwhile, the spin-coated samples were fully degraded after six months of continuous exposure. All untreated control samples degraded after one week (Fig. S1†). We theorize that the difference in crystal size might have a critical role in the viability of surface passivation treatment, especially a brief treatment, allowing for trapped active spots that can interact with humidity and initiate degradation at a much faster pace. Not only that, when closely examining the SEM images of the BC-2 versus the spin-coated samples, the grains while similar in size are protruding through the surface and exhibit more fissures than the smooth spin-coated films, resulting in a faster degradation within 4 months in place of 6 months as indicated by Fig. 2d.
 | ||
| Fig. 2 UV-Vis absorbance and PL spectra of MAPbI3 films treated with oleic acid and aged at ≈76% RH (a) SC-1, (b) SC-2, (c) BC-1, (d) BC-2. (e) A schematic illustration of the blue shift in PL after passivation. | ||
We note that all samples after passivation registered higher PL intensity with a nominal repeatable blue-shifted peak. After OA treatment, the PL peaks shifted from 780, 771, 786, and 778 nm to 776, 768, 779.5 and 775 nm for SC-1, SC-2, BC-1, and BC-2 samples, respectively. A consistent observed blue shift indicates the passivation of radiative trap states at the surface level of the samples, as illustrated in Fig. 2e.35,41 Such a phenomenon was described in previous works, as the spontaneous radiative recombination from defects resulting in a red shift in the emission peak, and the passivation of these defects leading to a blue-shifted emission peak denoting a radiative band edge transition.35 Peaks from all untreated films vanished after 1 week of continuous exposure to humidity, consistent with the absorbance results, including that of the treated one-step blade coating film. The aged, treated films of SC-1, SC-2 and BC-2 showed PL spectra with peaks at 768, 768 and 773 nm, respectively. A slight blue shift in the aged peaks of SC-1 and SC-2 samples could be either due to degradation and the formation of PbI2 or due to an additional passivation effect from water molecules that are not yet enough to induce degradation but rather in small quantity reported to be beneficial.17,42 Upon further exposure, the films showed complete degradation after six months for the SC-1 and SC-2 films and after 4 months for the BC-2 films. This is in line with our earlier observations; while crystal and grain size matter, morphological roughness also plays a role.
X-ray diffraction analysis was performed to investigate the humidity-induced structural changes of the perovskite films; Fig. 3 depicts the difference in their crystal structure, with surface passivation of oleic acid, during the aging process in high humidity conditions. All freshly prepared samples showed the characteristic perovskite peaks at 2θ around 14.14°, 28.50°, and 43.34° assigned to the (110), (220), and (330) lattice planes, indicating a tetragonal crystal structure.30 The results indicated the slight formation of hexagonal PbI2 with a signature peak at 2θ around 12.65° by the end of the first month in SC-1 and BC-2 samples. The SC-2 sample did not show any rise of the PbI2 peak for the first month of humidity exposure. Nevertheless, with continuous exposure, the PbI2 characteristic diffraction peak gradually increased in these samples, while MAPbI3 peaks decreased. Additionally, BC-1 was converted to a mixture of hydrated intermediate phase, as evidenced by the peaks at 8.22° and 10.79°, and MAPbI3 after three days of aging, and by the end of the first week, it degraded completely to 100% intermediate phase.12,43 The peak at 8.22° has been reported for forming 2D phases in perovskite, which is consistent with the formation of low-dimension hydrated phases.17,44 The diffraction patterns of the untreated samples are shown in Fig. S2:† they all completely degraded to PbI2 except for the one-step blade coating, which degraded to a mixture of intermediate phase and PbI2. This implies that deprotonation of the organic cation is the dominant degradation mechanism for the samples, except for BC-1, which decomposes via bulk hydration. Based on the results above, oleic acid improved the stability of the prepared films, except for those coated using the one-step blade method. To further investigate the structural changes in the films during aging, we analyzed the GIWAXS patterns of the aged films treated, as shown in Fig. 4. The samples exhibit lattice planes of (110) and (220) peaks at a momentum transfer q ≈ 1.0 and 2.0 Å−1.45 The spin-coated films display homogeneous arcs, indicating isotropic distribution. In contrast, the blade-coated films exhibit a higher degree of preferential crystallographic orientation, characterized by partial arc segments and more pronounced Bragg spots.30 After aging in humid conditions, the one-step blade-coated film completely degraded in one week, with the only patterns being strong arcs at q ≈ 0.5 and 0.75 Å−1 assigned to the hydrated intermediate phase.46–48 The two-step blade-coated, and the spin-coated samples were the most stable after 1 month of exposure. The one step spin coated sample showed a slight increase in PbI2 (100) peak intensity at q ≈ 0.9 Å−1 and the two-step blade-coated sample showed a more distinct peak. To test the reversibility of this intermediate phase of BC-1 sample, we heated the samples that degraded to 100% intermediate phase on a hotplate at 100 °C for 30 minutes in air. As seen in Fig. 5a, the degraded treated film did not show a significant change in the UV-vis absorbance spectrum after heating, just a minute recovery of the perovskite absorption onset as seen in the inset, implying very limited reversibility. The XRD results in Fig. 5b show that upon heating, the untreated film converts to mostly PbI2 with residues of perovskite, while the treated film converts to only residues of perovskite with no PbI2 formation. This indicates that oleic acid fully inhibits the deprotonation process: most of the perovskite leached out of the film during the hydration phase, and only the remnants were recovered upon heating.
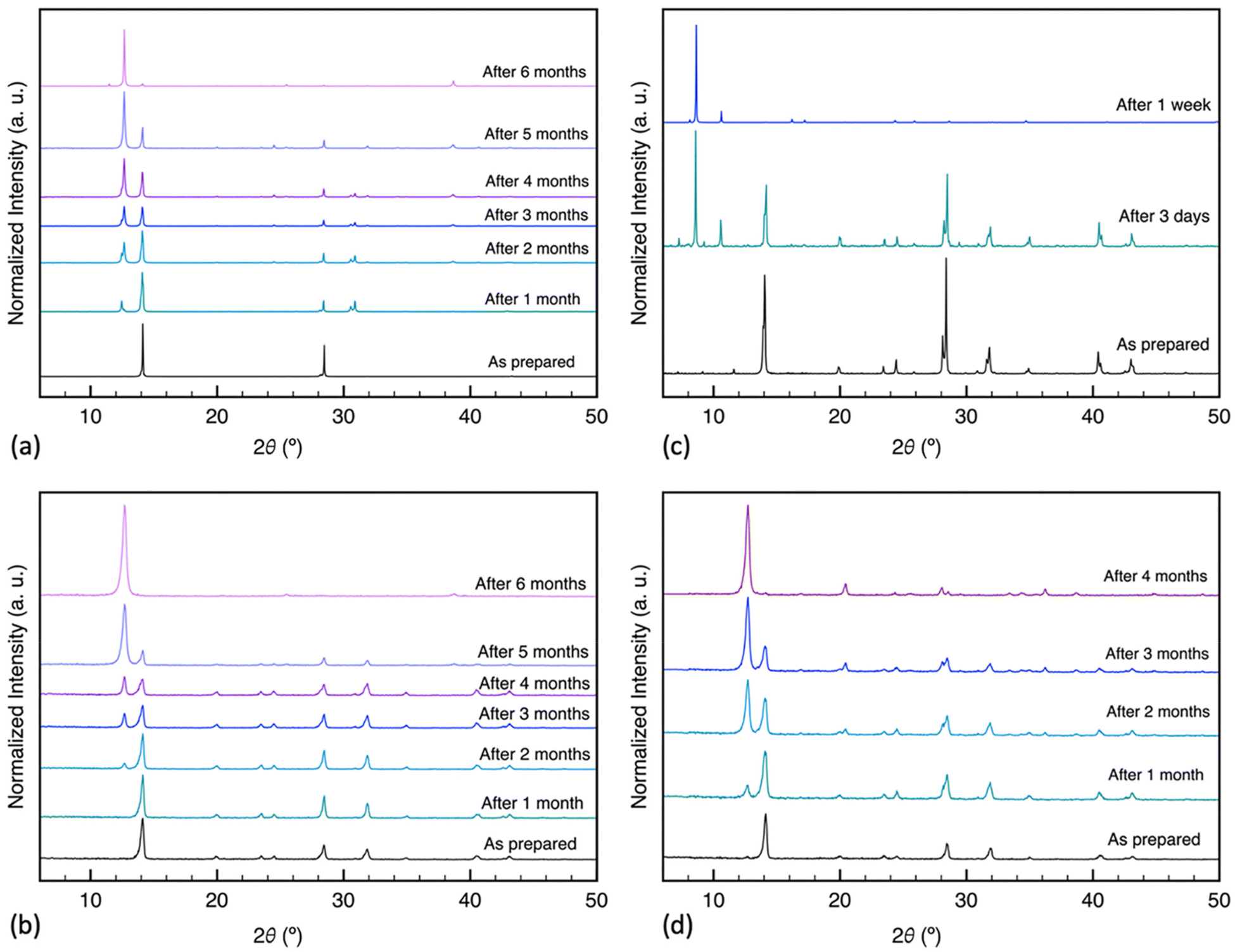 | ||
| Fig. 3 X-ray diffraction patterns (λ = 1.5406 Å) of MAPbI3 films prepared via different deposition methods, treated with oleic acid and aged at ≈76% RH: (a) SC-1, (b) SC-2, (c) BC-1, (d) BC-2. | ||
 | ||
| Fig. 4 GIWAXS patterns of MAPbI3 films treated with oleic acid ((i) as prepared, and (ii) aged) prepared via different deposition methods and aged at ≈76% RH: (a) SC-1, (b) SC-2, (c) BC-1, (d) BC-2. The incidence angle θ was 0.15° for all measurements. | ||
 | ||
| Fig. 5 (a) UV-Vis absorbance of a BC-1 sample treated with oleic acid before and after the degradation process and then the subsequent heating process. (b) X-ray diffraction patterns (λ = 1.5406 Å) of the heated BC-1 samples after the degradation process and then the subsequent heating process. | ||
To probe the chemical composition of the hydrated phase, the molecular structure of the samples under degradation was investigated by examining their FTIR spectra. The pristine samples showed strong vibrational bands at 717 and 900 cm−1 for CH2 stretching and CH3 rocking respectively.49,50 Other weaker bands were observed at 1468, 1564, 2110 and 3100 cm−1 assigned to C–H scissoring, N–H bending, C–N stretching and N–H stretching respectively.51–54 Samples prepared via blade coating showed bands in the range between 1300 to 1700 cm−1 more prominently than the spin-coated films, which could be due to the difference in the preparation processes and surface morphology. The samples treated with oleic acid showed vibrational bands at 1280, 1720, 2850 and 2920 cm−1 assigned to C–O stretching, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, symmetrical CH2 stretching and asymmetrical CH2 stretching respectively.55 The FTIR spectrum of oleic acid is provided in the ESI (Fig. S3).† The results in Fig. 6 indicate that all untreated samples showed significantly reduced intensity in vibrational bands characteristic of the organic cation, which is consistent with the degradation mechanism of the MA+ from the perovskite structure.56 All untreated samples, except the one-step blade coating film, showed no new vibrational bands with humidity exposure. The aged pristine one-step blade coating film showed both decreased intensity of the organic cation bands and the creation of new bands at 1425, 1925, 2464, 2550, 2906, 3485 and 3612 cm−1 due to the formation of hydrated intermediate compounds such as –COO, and O–H stretching modes.57 The treated films, except one-step blade coating, did not show significant change with humidity exposure. The treated one-step blade coating (BC-1) film showed decreased band intensities of the vibrational modes of the CH2 and CH3 functional groups; however, it showed stronger intensities of O–H in plane stretching at 1452 cm−1, the carbonyl functional group at 1705 cm−1, –COO− stretching at 1582 cm−1 and N–H stretching at 2857 and 2928 cm−1.58 In addition, new bands formed including C–O stretching at 1258 cm−1, antisymmetric stretching –COO− at 1617 cm−1, N–H stretching modes at 2464, 2551, 2766 cm−1, and O–H stretching modes at 3157, 3455 cm−1.57–60 Such FTIR band changes of a hydrated perovskite are in agreement with a previous study performed by Gan et al.57 These results demonstrate that the one-step blade coating samples degrade by forming hydrated intermediate compounds with and without the surface passivation treatment. In addition, BC-1 samples show a weak peak around 1020–1030 cm−1 assigned to SO stretching band that is characteristic to DMSO, denoting the slight formation of MAI-PbI2-DMSO intermediate phase with the preparation step, concurring with the XRD results.61,62 These results are consistent with our previous conclusion that SC-1, SC-2, and BC-2 films indeed degrade directly to PbI2 through the evaporation of the organic cation, while BC-1 degrades through the formation of hydrated and metastable intermediate compounds.
O stretching, symmetrical CH2 stretching and asymmetrical CH2 stretching respectively.55 The FTIR spectrum of oleic acid is provided in the ESI (Fig. S3).† The results in Fig. 6 indicate that all untreated samples showed significantly reduced intensity in vibrational bands characteristic of the organic cation, which is consistent with the degradation mechanism of the MA+ from the perovskite structure.56 All untreated samples, except the one-step blade coating film, showed no new vibrational bands with humidity exposure. The aged pristine one-step blade coating film showed both decreased intensity of the organic cation bands and the creation of new bands at 1425, 1925, 2464, 2550, 2906, 3485 and 3612 cm−1 due to the formation of hydrated intermediate compounds such as –COO, and O–H stretching modes.57 The treated films, except one-step blade coating, did not show significant change with humidity exposure. The treated one-step blade coating (BC-1) film showed decreased band intensities of the vibrational modes of the CH2 and CH3 functional groups; however, it showed stronger intensities of O–H in plane stretching at 1452 cm−1, the carbonyl functional group at 1705 cm−1, –COO− stretching at 1582 cm−1 and N–H stretching at 2857 and 2928 cm−1.58 In addition, new bands formed including C–O stretching at 1258 cm−1, antisymmetric stretching –COO− at 1617 cm−1, N–H stretching modes at 2464, 2551, 2766 cm−1, and O–H stretching modes at 3157, 3455 cm−1.57–60 Such FTIR band changes of a hydrated perovskite are in agreement with a previous study performed by Gan et al.57 These results demonstrate that the one-step blade coating samples degrade by forming hydrated intermediate compounds with and without the surface passivation treatment. In addition, BC-1 samples show a weak peak around 1020–1030 cm−1 assigned to SO stretching band that is characteristic to DMSO, denoting the slight formation of MAI-PbI2-DMSO intermediate phase with the preparation step, concurring with the XRD results.61,62 These results are consistent with our previous conclusion that SC-1, SC-2, and BC-2 films indeed degrade directly to PbI2 through the evaporation of the organic cation, while BC-1 degrades through the formation of hydrated and metastable intermediate compounds.
 | ||
| Fig. 6 FTIR spectra of MAPbI3 films prepared via different deposition methods and aged at ≈76% RH: (a) SC-1, (b) SC-2, (c) BC-1, (d) BC-2. The graphs marked with (i) denote the untreated control samples, and (ii) denotes samples treated with oleic acid. | ||
To further understand the inadequacy of the surface passivation treatment in one-step blade-coated perovskite films, we proceeded to analyze the surface properties of our films. Rather than merely considering crystal and grain size as key factors, we investigated whether bond terminations affected the treatment efficacy. We quantified the atomic concentration percentages of the elements at the perovskite surface level via XPS measurements presented in Fig. 7. The XPS survey spectra (see ESI, Fig S6†) for the MAPbI3 perovskite films reveal peaks for O, N, and C at binding energies of approximately 532, 401, and 284.6 eV, respectively. Both I and Pb exhibit doublet peaks due to spin–orbit splitting, appearing at around 618, 630, 137, and 143 eV, respectively.63 The oxygen peak can be explained by the propensity of MAPbI3 films to undergo surface oxidation and the formation of PbO/Pb(OH)2 like states with terminated Pb dangling bonds, as reported by Rocks et al.63 However, no secondary phase of PbO was detected in the XRD measurements, which aligns with prior studies reporting an O peak in XPS without evidence in XRD.63,64 The results shown in Fig. 7 indicated a significant difference between the one-step blade-coated (BC-1) film and the other films, in their as-prepared condition, displaying notably a lower C concentration and higher Pb and I concentrations, indicating more PbI2 termination. This concurs with the study suggesting that annealing at high temperatures promotes MAI desorption at the surface level.19 This may explain the ineffectiveness of oleic acid in adequately passivating the iodine-rich surface. After passivation, we observed a sharp increase in the C and O atomic concentrations due to the presence of oleic acid (C18H34O2). The formation of the surface passivation layer decreased the atomic concentrations of N, Pb, and I through the few nanometers tested through XPS. We thought to tailor the treatment to increase its efficiency on BC-1 films: we know that BC-1 films are prone to form thicker films with larger and rougher crystal sizes with deeper grain boundaries, as shown in Fig. 1e–h and S5, S6† and we know that its surface is not the ideal location for OA anchoring. In fact, humidity can seep into the deeper boundaries, creating hydrated intermediates with the deeper MAI sites of grain boundaries of the films despite being surface terminated by PbI2. Hence, as detailed in the Experimental section, we experimented with a more extensive and prolonged treatment by dipping the BC-1 films in the oleic acid solution instead of spin coating. The stability of the films was significantly enhanced, as shown in Fig. 8. As oleic acid molecules bind to Pb2+ or MAI+ sites, the formation of hydrated intermediates and the deprotonation of MA are hindered, causing these degradation processes to slow down dramatically.
 | ||
| Fig. 7 Atomic concentration percentages of C, O, N, Pb, and I at the surface level of the MAPbI3 films, derived from X-ray photoelectron spectra. | ||
 | ||
| Fig. 8 (a) UV-Vis absorbance and PL spectra and (b) FTIR spectra of BC-1 film treated with oleic acid dipping method, and aged at ≈76% RH. | ||
In summary, using oleic acid for surface treatment is a cost-effective and efficient method to improve the stability of perovskite films and enhance their charge transport properties by passivating surface defects. However, this treatment must be customized to account for the specific film-forming properties of various deposition techniques. Our research showed that applying oleic acid through spin coating yields superior results on smoother perovskite films with finer grain boundaries, in contrast to textured films with larger crystals and deeper grain boundaries. Furthermore, surface termination significantly influences treatment effectiveness; while extensive research has focused on enhancing the stability and efficiency of spin-coated films, there has been limited exploration into films produced with large-scale manufacturing-compatible techniques. Given the challenges of scaling up high-efficiency devices from small to large formats, it is essential to re-evaluate strategies to facilitate a smoother transition to commercial production.
Data availability
All data relevant to the manuscript submitted is included in the main text and ESI† documents. If there is any more data needed, we will be happy to make it readily available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Natural Sciences and Engineering Research Council of Canada through the Discovery Grant Programand the Collaborative Research and Training Experience program, the support of Canada Foundation for Innovation through John R. Evans Leaders Fund, and the support through the government of Canada's New Frontier in Research Fund, the support through Canada Research Chairs Program, and the support of Research Nova Scotia. This work was also supported by the Killam Predoctoral Scholarship. We are extremely grateful for the help from Dr Ian Hill, Charlotte Clegg, and Andy George in the Department of Physics and Atmospheric Science at Dalhousie University for acess to FTIR, PL and XPS measurements. We acknowledge Pat Scallion and Eric Moreau's great contribution to collecting SEM and XRD data at the FIBSEM Facility in Mechanical Engineering. GIWAXS measurements were performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.References
- Y. Chen, Z. Yue, S.-W. Tsang and Y. Cheng, Nano Energy, 2025, 137, 110782 CrossRef CAS.
- Best Research-Cell Efficiency Chart | Photovoltaic Research | NREL, https://www.nrel.gov/pv/cell-efficiency.html, (accessed March 9, 2025).
- Y. Cheng and L. Ding, Energy Environ. Sci., 2021, 14, 3233–3255 RSC.
- N. Li, X. Niu, Q. Chen and H. Zhou, Chem. Soc. Rev., 2020, 49, 8235–8286 RSC.
- L. Duan, D. Walter, N. Chang, J. Bullock, D. Kang, S. P. Phang, K. Weber, T. White, D. Macdonald, K. Catchpole and H. Shen, Nat. Rev. Mater., 2023, 8, 261–281 CrossRef CAS.
- P. Zhu, C. Chen, J. Dai, Y. Zhang, R. Mao, S. Chen, J. Huang and J. Zhu, Adv. Mater., 2024, 36, 2307357 CrossRef CAS PubMed.
- E. H. Balaguera and J. Bisquert, Small, 2025, 21, 2409534 CrossRef PubMed.
- S. J. Kim, I. H. Im, J. H. Baek, S. Choi, S. H. Park, D. E. Lee, J. Y. Kim, S. Y. Kim, N.-G. Park, D. Lee, J. J. Yang and H. W. Jang, Nat. Nanotechnol., 2025, 20, 83–92 CrossRef CAS PubMed.
- J. Chen, X. Wang, T. Wang, J. Li, H. Y. Chia, H. Liang, S. Xi, S. Liu, X. Guo, R. Guo, Z. Jia, X. Yin, Q. Zhou, Y. Wang, Z. Shi, H. Zhou, D. Lai, M. Zhang, Z. Xing, W. R. Leow, W. Yan and Y. Hou, Nat. Energy, 2025, 10, 181–190 CAS.
- M. H. Miah, M. B. Rahman, M. Nur-E-Alam, M. A. Islam, M. Shahinuzzaman, M. R. Rahman, M. H. Ullah and M. U. Khandaker, RSC Adv., 2025, 15, 628–654 RSC.
- D. B. Khadka, M. Yanagida and Y. Shirai, Sol. Energy Mater. Sol. Cells, 2025, 281, 113319 CrossRef CAS.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- Z. Song, A. Abate, S. C. Watthage, G. K. Liyanage, A. B. Phillips, U. Steiner, M. Graetzel and M. J. Heben, Adv. Energy Mater., 2016, 6, 1600846 CrossRef.
- S. Cheng and H. Zhong, J. Phys. Chem. Lett., 2022, 13, 2281–2290 CrossRef CAS PubMed.
- H. Baishya, R. D. Adhikari, M. J. Patel, D. Yadav, T. Sarmah, M. Alam, M. Kalita and P. K. Iyer, J. Energy Chem., 2024, 94, 217–253 CrossRef CAS.
- Z. Song, N. Shrestha, S. C. Watthage, G. K. Liyanage, Z. S. Almutawah, R. H. Ahangharnejhad, A. B. Phillips, R. J. Ellingson and M. J. Heben, J. Phys. Chem. Lett., 2018, 9, 6312–6320 CrossRef CAS PubMed.
- Y. Gao, D. Lin, P. Liu, T. Shi and W. Xie, Mater. Chem. Front., 2024, 8, 785–799 RSC.
- Y. Lu, Z. Si, H. Liu, Y. Ge, J. Hu, Z. Zhang, X. Mu, K. Selvakumar and M. Sui, Chem. – Eur. J., 2021, 27, 3729–3736 CrossRef CAS PubMed.
- B. Chen, S. Wang, Y. Song, C. Li and F. Hao, Chem. Eng. J., 2022, 430, 132701 CrossRef CAS.
- D. Zhang, D. Li, Y. Hu, A. Mei and H. Han, Commun. Mater., 2022, 3, 1–14 CrossRef.
- A. Mahapatra, D. Prochowicz, M. M. Tavakoli, S. Trivedi, P. Kumar and P. Yadav, J. Mater. Chem. A, 2019, 8, 27–54 RSC.
- S. Abicho, B. Hailegnaw, G. A. Workneh and T. Yohannes, Mater. Renew. Sustain. Energy, 2022, 11, 47–70 CrossRef.
- S. Liu, Y. Guan, Y. Sheng, Y. Hu, Y. Rong, A. Mei and H. Han, Adv. Energy Mater., 2020, 10, 1902492 CrossRef CAS.
- G. Abdelmageed, H. R. Sully, S. B. Naghadeh, A. E. Ali, S. A. Carter and J. Z. Zhang, ACS Appl. Energy Mater., 2018, 1, 387–392 CrossRef CAS.
- M. Roy, M. Sykora and M. Aslam, Top. Curr. Chem., 2024, 382, 9 CrossRef CAS PubMed.
- J. Zhang, R. W. Crisp, J. Gao, D. M. Kroupa, M. C. Beard and J. M. Luther, J. Phys. Chem. Lett., 2015, 6, 1830–1833 CrossRef CAS PubMed.
- A. Maxwell, H. Chen, L. Grater, C. Li, S. Teale, J. Wang, L. Zeng, Z. Wang, S. M. Park, M. Vafaie, S. Sidhik, I. W. Metcalf, Y. Liu, A. D. Mohite, B. Chen and E. H. Sargent, ACS Energy Lett., 2024, 9, 520–527 CrossRef CAS.
- M. M. Hasan, C. Clegg, M. Manning, A. El Ghanam, C. Su, M. D. Harding, C. Bennett, I. G. Hill and G. I. Koleilat, ACS Photonics, 2020, 7, 57–67 CrossRef CAS.
- B. T. Smith, S. T. Thornton, G. Abdelmageed, R. F. Kahwagi, R. Elsebai, V. Chiriac, C.-Y. Kim, S. Hinds and G. I. Koleilat, Adv. Photonics Res., 2023, 4, 2200088 CrossRef CAS.
- M. Wang, Y. Feng, J. Bian, H. Liu and Y. Shi, Chem. Phys. Lett., 2018, 692, 44–49 CrossRef CAS.
- T.-S. Su, T.-E. Fan, H.-K. Si, D.-A. Le, N. Perumbalathodi and T.-C. Wei, Sol. RRL, 2021, 5, 2100109 CrossRef CAS.
- L. Greenspan, J. Res. Natl. Bur. Stand., Sect. A, 1977, 81, 89–96 CrossRef.
- A. Wexler and S. Hasegawa, J. Res. Natl. Bur. Stand., 1954, 53, 19 CrossRef CAS.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- A. F. G. Leontowich, A. Gomez, B. D. Moreno, D. Muir, D. Spasyuk, G. King, J. W. Reid, C.-Y. Kim and S. Kycia, J. Synchrotron Radiat., 2021, 28, 961–969 CrossRef CAS PubMed.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- H. Xie, X. Liu, L. Lyu, D. Niu, Q. Wang, J. Huang and Y. Gao, J. Phys. Chem. C, 2016, 120, 215–220 CrossRef CAS.
- G. Greczynski and L. Hultman, J. Appl. Phys., 2022, 132, 011101 CrossRef CAS.
- M. He, B. Li, X. Cui, B. Jiang, Y. He, Y. Chen, D. O'Neil, P. Szymanski, M. A. Ei-Sayed, J. Huang and Z. Lin, Nat. Commun., 2017, 8, 16045 CrossRef CAS PubMed.
- J. Xia, C. Liang, S. Mei, H. Gu, B. He, Z. Zhang, T. Liu, K. Wang, S. Wang, S. Chen, Y. Cai and G. Xing, J. Mater. Chem. A, 2021, 9, 2919–2927 RSC.
- H. Kim, J. Lee, B. Kim, H. R. Byun, S. H. Kim, H. M. Oh, S. Baik and M. S. Jeong, Sci. Rep., 2019, 9, 15461 CrossRef PubMed.
- J. A. Christians, P. A. M. Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- K. Yao, X. Wang, F. Li and L. Zhou, Chem. Commun., 2015, 51, 15430–15433 RSC.
- A. Z. Chen, B. J. Foley, J. H. Ma, M. R. Alpert, J. S. Niezgoda and J. J. Choi, J. Mater. Chem. A, 2017, 5, 7796–7800 RSC.
- N.-K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S.-W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2017, 7, 4645 CrossRef PubMed.
- H.-H. Huang, Z. Ma, J. Strzalka, Y. Ren, K.-F. Lin, L. Wang, H. Zhou, Z. Jiang and W. Chen, Cell Rep. Phys. Sci., 2021, 2, 100395 CrossRef CAS.
- M. Qin, H. Xue, H. Zhang, H. Hu, K. Liu, Y. Li, Z. Qin, J. Ma, H. Zhu, K. Yan, G. Fang, G. Li, U.-S. Jeng, G. Brocks, S. Tao and X. Lu, Adv. Mater., 2020, 32, 2004630 CrossRef CAS PubMed.
- G. Abdelmageed, L. Jewell, K. Hellier, L. Seymour, B. Luo, F. Bridges, J. Z. Zhang and S. Carter, Appl. Phys. Lett., 2016, 109, 233905 CrossRef.
- Y. Zheng, Y. Cui and W. Wang, Minerals, 2018, 8, 341 CrossRef.
- J. Yang, Q. Hong, Z. Yuan, R. Xu, X. Guo, S. Xiong, X. Liu, S. Braun, Y. Li, J. Tang, C. Duan, M. Fahlman and Q. Bao, Adv. Opt. Mater., 2018, 6, 1800262 CrossRef.
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky, A. Pucci and R. Lovrinčić, J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS PubMed.
- G. Abdelmageed, C. Mackeen, K. Hellier, L. Jewell, L. Seymour, M. Tingwald, F. Bridges, J. Z. Zhang and S. Carter, Sol. Energy Mater. Sol. Cells, 2018, 174, 566–571 CrossRef CAS.
- T. Vincent, C. Vincent, Y. Barré, Y. Guari, G. L. Saout and E. Guibal, J. Mater. Chem. A, 2014, 2, 10007–10021 RSC.
- W. A. P. J. Premaratne, W. M. G. I. Priyadarshana, S. H. P. Gunawardena and A. A. P. D. Alwis, J. Sci. Univ. Kelaniya, 2014, 8, 33–48 CrossRef.
- D. Yerezhep, Z. Omarova, A. Aldiyarov, A. Shinbayeva and N. Tokmoldin, Molecules, 2023, 28, 1288 CrossRef CAS PubMed.
- Z. Gan, Z. Yu, M. Meng, W. Xia and X. Zhang, APL Mater., 2019, 7, 031107 CrossRef.
- J. Ibarra, J. Melendres, M. Almada, M. G. Burboa, P. Taboada, J. Juárez and M. A. Valdez, Mater. Res. Express, 2015, 2, 095010 CrossRef.
- M. Z. Hossain, A. K. Jhawar, M. B. I. Chowdhury, W. Z. Xu, W. Wu, D. V. Hiscott and P. A. Charpentier, Energy Fuels, 2017, 31, 4013–4023 CrossRef CAS.
- J. J. Nájera and A. B. Horn, Phys. Chem. Chem. Phys., 2009, 11, 483–494 RSC.
- H. Chen, X. Ding, P. Xu, T. Hayat, A. Alsaedi, J. Yao, Y. Ding and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 1781–1791 CrossRef CAS PubMed.
- A. D. Sheikh, A. P. Patil, S. S. Mali, C. K. Hong and P. S. Patil, J. Mater. Sci., 2019, 54, 10825–10835 CrossRef CAS.
- C. Rocks, V. Svrcek, P. Maguire and D. Mariotti, J. Mater. Chem. C, 2017, 5, 902–916 RSC.
- Z. Ahmad, M. A. Najeeb, R. A. Shakoor, A. Alashraf, S. A. Al-Muhtaseb, A. Soliman and M. K. Nazeeruddin, Sci. Rep., 2017, 7, 15406 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00325c |
| This journal is © The Royal Society of Chemistry 2025 |