
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic synergism in heterostructural Ta-doped Mo–Ni–S nanospheres: an efficient bifunctional catalyst for water splitting†
Kaichun
Gao
ab,
Yuhang
Yuan
b,
Hongbang
Zheng
b,
Yiyou
Wu
c,
Mingxin
Ye
 a and
Jianfeng
Shen
*a
a and
Jianfeng
Shen
*a
aInstitute of Special Materials and Technology, Fudan University, Shanghai 200433, China. E-mail: jfshen@fudan.edu.cn
bDepartment of Materials Science, Fudan University, Shanghai 200433, China
cHigh School Affiliated to Fudan University, Shanghai 200433, China
First published on 4th March 2025
Abstract
Designing suitable and efficient electrocatalysts for both the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is crucial for sustainable hydrogen production. To date, extensive studies have focused on transition metal chalcogenides (TMSs) due to their unique electronic structure and strong ability to be modified. Herein, we reported a Ta-doped MoS2/NiS catalyst supported on NF with a stable hierarchical nanosphere structure (Ta-MNS). The synergy between the MoS2 and NiS phases in the same plane enhanced the mechanical strength and exposed more active sites, while Ta5+ was applied to adjust the electronic structure, resulting in a well-dispersed and flexible morphology. Moreover, the formation of the –S–Ta5+–S– bridge was proposed as a significant factor in enhancing the link between components at the active phase and further promoting electron transfer. As a result, for HER, the synthesized Ta-MNS required overpotentials of only 35 and 127 mV at current densities of 10 and 100 mA cm−2, respectively. Furthermore, it demonstrated outstanding performance for OER and excellent stability, maintaining stable operation for more than 80 h. This paves a new avenue for the design of novel catalysts for water splitting.
1. Introduction
With the ever-increasing demand for traditional fossil fuels, which are causing critical environmental pollution worldwide, hydrogen is increasingly recognized as an economical and eco-friendly energy source due to its high energy density and green conversion products.1–3 Among the various methods of hydrogen production, such as water photocatalysis and carbinol pyrolysis, water electrolysis enables the production of cleaner and more efficient hydrogen.4–6 Nevertheless, the electrolysis of water suffers from sluggish kinetics for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), requiring electrocatalysts with high activity and stability to overcome these kinetic barriers.7,8 Pt (and other noble metal)-based electrocatalysts are considered the most efficient catalysts for HER, and they could have been widely adopted if not for their non-negligible manufacturing costs and poor reaction stability under alkaline conditions.9–11 Therefore, the disadvantages of Pt-based electrocatalysts have gradually stimulated considerable interest in transition metal-based catalysts,12,13 including transition metal oxides,14,15 transition metal carbides,16,17 transition metal nitrides,18,19 transition metal phosphides20,21 and transition metal chalcogenides (TMSs).22,23In particular, TMSs are certified to have strong electron-accepting abilities, making them promising electrocatalysts for water splitting. To enhance the energy efficiency and robustness of TMS catalysts for practical applications, researchers often focus on stable 3D morphologies,24,25 adjustable electronic structures,26,27 and simple fabrication strategies.28,29
Regarding stable 3D morphology, Zhang et al. successfully anchored NiS nanocrystals on porous Mo2N to prepare a Janus-type structure with a well-defined interface by forming Mo–S bonds.30 The catalyst exhibited favorable stability, continuously operating for more than 100 h without degradation at a current density of 100 mA cm−2. Moreover, Wang et al. designed a heterostructure composed of spherical hollow WS2 and petal-like MoS2,31 which enhanced mechanical strength, exposed more active sites and promoted electron transfer. This resulted in superior electrocatalytic HER activity with a low Tafel slope of 53 mV dec−1 and good durability. Even with the well-designed stable 3D morphology, researchers can still make progress on the electrocatalytic performance by adjusting the electronic structure. Consequently, various methods have been applied to obtain a more active electronic structure, including vacancy fabrication,32,33 single atom anchor,34,35 and heterostructure construction.36,37 For example, Cao et al. have reported on a 2D TaSx NSs nanocrystal catalyst with engineered sulfur vacancies on the nanosheet,38 which could serve as efficient electrocatalysts for both HER and OER. Mosallanezhad et al. successfully anchored Pt single atoms on the cobalt sulfide support (Pt@CoS),39 exhibiting a 45-fold increase in the mass current density compared to the benchmark Pt/C at 100 mV. Heterostructure construction also plays a significant role in adjusting the electronic structure since the spontaneous electron transfer at the interface between the two phases of a heterogeneous structure can modulate the chemical state of the two contacting components, resulting in high electrocatalytic performance.
Nevertheless, on account of the complex synthesis technology and insufficient chemical stability of the above methods,40–42 chemical doping is introduced to adjust the electronic structure for its strong effect on realizing multi-scale modulation and promoting charge transfer among basal plane active sites.43–45 For anion doping, Ta is outstanding among transition metal elements for its great application in capacitors and semiconductor devices.46,47 As for TMSs, the electrocatalytic activity usually concentrates on the surface, while there are abundant intrinsic S sites in the basal plane that remain inert and underutilized. When introducing Ta5+, the S sites in the basal plane become activated and the localized electrons redistribute, which can promote the charge transfer from the electrode to the reactant.48,49 Moreover, it is demonstrated that Ta5+ can serve as the electron donor to promote electron transfer, and plays an important role in the formation of the –S–Ta5+–S– bridge.50–53 These acting forces have a favorable effect on enhancing the link between components at the active phase and further promoting electron transfer. Practically, it is worth noting that the proper doping of Ta can adjust the structure of nanospheres,54,55 obtaining a more well-dispersed morphology with flexibility.
Herein, we have synthesized a Ta-doped MoS2/NiS catalyst located on NF (Ta-MNS) with the structure of hierarchical nanospheres via a simple one-step hydrothermal interface engineering-induced method, and investigated its electrocatalytic performance for HER and OER. Compared with other fabrication strategies such as liquid phase exfoliation and chemical vapor deposition, the one-step hydrothermal synthesis presents distinguished performance in achieving products with high purity, low cost, good dispersion, and controllable particle size. Meanwhile, other TM (Nb, Zr, Hf)-doped samples were also prepared to explore the effects of different transition metal ions on the water electrolysis. When used as catalysts for HER and OER, Ta-MNS exhibits small overpotentials of 35 and 199 mV for HER and OER to reach a current density of 10 mA cm−2, respectively, in an electrolyte environment of 1 M KOH. Furthermore, an electrolytic cell was formed by the identical cathode and anode of Ta-MNS, which merely required 1.627 V to drive water splitting to produce a current density of 50 mA cm−2.
2. Results and discussion
Scheme 1 shows the formation mechanism of Ta-MNS on NF after a simple one-step hydrothermal method. It was initially started by setting metal ions and thioacetamide dissolved in deionized water to gain a homogeneous solution with the Ni, Mo, and S precursors. Then, MNS was achieved via hydrothermal condition to form NiS–MoS2 nanosphere-based layers uniformly coated over the NF. When adding the Ta source, the agglomerated NiS–MoS2 nanosphere would gradually remodel its structure to form a smaller and more well-dispersed spherical morphology with a clearer shape, greatly ensuring the rapid reaction kinetics by increasing the exposure of the catalytic active sites. | ||
| Scheme 1 Synthetic scheme for Ta-MNS and control samples with different morphologies. | ||
SEM images from Fig. 1(a–c) and Fig. S1(a, b)† indicated that the MNS and Ta-MNS materials had similar micro-morphologies, in which MoS2–NiS and Ta-MoS2–NiS nanospheres coated the whole smooth surface of NF. Moreover, compared with MNS (Fig. S1b†), it is evident that the structure of well-dispersed nanospheres with a clear shape required an optimal Ta mass content to effectively modulate the electronic structure and enhance the linkage between the MoS2–NiS heterostructure. On the nanoscale, Fig. 1 illustrates that the surface of the nanospheres, with an average diameter of about 600 nm, is uniformly covered with abundant small 2D nanosheets, which can enlarge the electronic contact surface and increase the exposure of the catalytic active sites. The energy dispersive X-ray (EDX) element mapping results confirmed the presence of Ni, Mo, S, and Ta elements and the uniform distribution (Fig. 1(f and g)). Furthermore, the EDX spectra showed that the contents of Ni, Mo, S, and Ta in Ta-MNS were 9.7%, 14.3%, 35.5%, and 0.4%, respectively (Fig. S2†). Compared with the EDX spectra of MNS (Fig. S3†), the results showed that Ta was successfully introduced and uniformly distributed.
 | ||
| Fig. 1 (a–c) SEM images of Ta-MNS. (d and e) TEM images of Ta-MNS. (f and g) SEM image of Ta-MNS and EDS mapping for O, S, Ni, Mo, and Ta. | ||
However, as shown in Fig. S4b,† the morphology of the MoS2–NiS nanospheres partly changed into NiS nanorods with an excessive amount of Ni. Furthermore, the NF surface would crack (Fig. S4c and S5†) if the reaction time was too long. Such a phenomenon would result in a decline in mechanical properties, and further lead to a significant decrease of the catalytic active sites and block the charge transfer. Moreover, as shown in Fig. S6(a–c),† transition metal ions all had the function of adjusting the morphology to a certain extent. However, from the view of the dispersion degree of nanospheres, tantalum played a greater role than niobium than hafnium than zirconium. This might be explained in terms of the electronegativity of transition metal elements. Since the transition metal chlorides were introduced according to the molar amount of the transition metal elements and the less electronegative element would have a more polar chloride, TaCl5 had better electrolysis and dispersion in solution than NbCl5 than HfCl4 than ZrCl4. Fig. S6d† indicates that the morphology of Zr-MNS was close to that for Ta (excess)-MNS, because the excess precipitates that are difficult to disperse would instead affect the normal attachment and growth of sulfide.
High-resolution TEM (HRTEM) was employed to characterize the microscopic morphology of the synthesized catalyst. As shown in Fig. 1, the obvious hierarchical spherical structure was in accordance with the SEM results. Fig. 1e revealed a distinct dividing line, on both sides of which there were different well-resolved lattice fringes. One of them corresponded to the (102) lattice plane of NiS with a d-spacing of 0.26 nm. Meanwhile, the other corresponded to the (002) lattice plane of MoS2 with a d-spacing of 0.62 nm.56 The above results suggested the successful fabrication of the NiS–MoS2 heterostructure by the simple hydrothermal interface engineering-induced strategy. As we all know, bimetallic sulfides have been confirmed to exhibit better electrocatalytic performance than monometallic compounds due to the spontaneous electron transfer at the interface between two phases of a heterogeneous structure.57 The synergy between MoS2 and NiS phases located at the same plane could promote the rearrangement of the charges at the interface, and provide a structural basis for full exposure of the catalytic active sites without the influence of steric effects.58 Therefore, the adsorption and desorption of H* and O intermediates would be quickly expedited in water splitting progress, leading to higher electrocatalytic activity.
XRD patterns were used to further determine the phase structure in Ta-MNS samples. Fig. S7† presents clear diffraction peaks at 45.7° and 30.0°, corresponding to the (102) and (100) planes of NiS (ICDD No. 75-0613), respectively. Other diffraction peaks at 14.4°, 39.5° and 49.8° corresponded to the (002), (103) and (105) planes of MoS2 (ICDD No. 73-1508),59 respectively, and there were no diffraction peaks ascribed to the planes of tantalum sulfide. The results showed that Ta only served as an effective doping material to modulate the electronic structure without the formation of a new crystal phase, which was in accordance with our design for doping. These XRD peaks that appeared simultaneously suggested the coexistence of NiS–MoS2, indicating the successful synthesis of the Ta-MNS catalysts. Certainly, since the sample was attached to the nickel foam, Raman spectra were further measured to observe the characterization of MoS2.
Fig. 2a shows the proposed mechanisms of the dissociation of H2O and OH intermediates on the MoS2/NiS heterostructures. The constructed interfaces between NiS and MoS2 were essential to the H-chemisorption of MoS2 and the HO-chemisorption of NiS. Therefore, a special 1T MoS2/NiS heterostructure was constructed. The Raman spectra also demonstrated the coexistence of NiS–MoS2. As shown in Fig. 2b, two distinct signals at E12g (375 cm−1) and A1g (401 cm−1) were detected, corresponding to the interlayer vibrations by Mo and S atoms, as well as the intralayer vibrations by S atoms along the C-axis. Moreover, there were four vibrational peaks that appeared at the 148, 199, 283, and 334 cm−1 positions, representing the characteristic vibration modes of J1, J2, E1g, and J3 in the 1T MoS2/NiS heterostructure, respectively. Therefore, it could be demonstrated that our simple hydrothermal interface engineering-induced strategy was favorable for the generation of the 1T MoS2/NiS heterostructure, which was proved to have a more active electronic structure than the 2H MoS2/NiS heterostructure. Fig. 2b also shows the Raman spectra of MNS. The positions of those characteristic peaks were the same as that for the standard 1T MoS2/NiS,60 which was in accordance with our design for doping.
 | ||
| Fig. 2 (a) The proposed mechanisms for the dissociation of H2O and OH intermediates on the MoS2/NiS heterostructures. Green = S, blue = Mo, purple = Ni. (b) Raman spectra of MNS and Ta-MNS. | ||
Furthermore, the X-ray photoelectron spectroscopy (XPS) survey spectrum of Ta-MNS (Fig. 3) demonstrated that the chemical composition of Ni, Mo, S, and Ta was consistent with the EDX results. As shown in Fig. 3a, the Ni2+ 2p3/2 and Ni2+ 2p1/2 binding energies of the MoS2/NiS heterostructure were located at 856.23 eV and 874.02 eV, respectively, while those at 861.72 and 880.22 eV were attributed to the Ni satellite. The peaks that emerged at 853.71 eV were assigned to Ni (0), which might be from the NF base.61,62 It should be noted that the Mo 3d profiles can be divided into four peaks (Fig. 3b). The Mo4+ couplets of 3d5/2 and 3d3/2 were exhibited at 228.81 eV and 232.37 eV, respectively, with a typical peak of Mo–S at 225.92 eV for MoS2. The small peak at 235.64 eV could be ascribed to Mo6+ due to the partial oxidation.63,64 The S 2p spectra (Fig. 3c) were divided into three peaks, corresponding to the S 2p1/2, S 2p3/2, and prominent S–O bond caused by the inevitable oxidation of S2− when exposed to air.65 In the Ta 4f region (Fig. 3d), the two peaks located at 27.97 eV and 25.7 eV corresponded to Ta 4f5/2 and Ta 4f7/2,66 respectively, indicating the presence of mainly Ta5+ that could act as the electron donor to improve the electronic conductivity. In comparison, MNS (Fig. S8†) only showed one peak of O 2s, indicating that there was no other Ta source during the experiment.
 | ||
| Fig. 3 (a–d) High-resolution plots of Ni 2p, Mo 3d, S 2p, and Ta 4f, respectively, for Ta-MNS. | ||
As shown in Table S1,† the binding energies of Ni, Mo and S in different samples were similar, but the energies of Mo4+ in the Mo 3d orbital and Ni2+ in the Ni 2p orbital of Ta-MNS were shifted to a high binding energy position compared with MNS. Meanwhile, S 2p1/2 and S 2p3/2 were shifted to low binding energy positions. The same situation occurred in the comparison of MNS and NS. The Ni 2p orbital of MNS was shifted to a high binding energy position compared with NS, while the S 2p orbital was shifted to low binding energy positions. The peaks of Ni 2p and Mo 3d represented positively charged Mo and Ni,67 while the peaks of S 2p1/2 and S 2p3/2 represented negatively charged S,68 demonstrating the existence of electron transfer from metal elements to P. From NS to Ta-MNS, the binding energies of Ni 2p and Mo 3d shifted positively and the binding energies of S 2p shifted negatively. This indicated that there were more positively charged metal elements and negatively charged S on Ta-MNS, which could modulate the electronic structure of the MoS2/NiS heterogeneous structure and accelerate the water-splitting process.
Meanwhile, as shown in Fig. S8 and S9,† the contrastive Nb, Zr, and Hf were also introduced successfully, demonstrating the feasibility of the doping strategy. These results strongly suggested the existence of strong electronic interactions between MoS2 and NiS, which implied the establishment of coupling interfaces. As shown in Fig. 2a, the proposed mechanisms of the dissociation of H2O and OH intermediates on the MoS2/NiS heterostructure proved that the composition of the interface between MoS2/NiS played an important role in enhancing the water splitting activities. When doping with Ta5+, there would be a number of –STa5+–S– bridges formed to strengthen the interface interaction between MoS2 and NiS. Therefore, Ta5+ could provide a strong link between the MoS2/NiS heterostructure and further make the catalyst exhibit higher electrochemical activities.
To explore the change on the chemical bonds or groups after introducing Ta5+, Ta-MNS and MNS were characterized by Fourier transform infrared spectroscopy (FTIR). As shown in Fig. S10(a),† the broad absorption band at 3437.2 cm−1 could be attributed to the stretching vibrational mode of the O–H groups, which came from adsorbed water molecules on the surface of Ta-MNS.69 The small peaks at 2856 and 2920 cm−1 were related to the stretching vibrations of the C–H bonds,70 while those at 1631.6, 1394 and 1085 cm−1 correspond to the stretching vibrations of N–O (came from NO3− produced by NH4+ ions), –CH3 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond in thioacetamide.71–73 The peaks at 1033.4 and 692 cm−1 could be assigned to the presence of Ni–S.74 Moreover, the S–Mo–S stretch was also in agreement with the IR peaks at 797.6 and 538 cm−1.75 Compared with Ta-MNS, the stretching vibrations of S–Mo–S in MNS (Fig. S10(b)†) correspond to the peaks at 871 and 615 cm−1, while the stretching vibrations of Ni–S correspond to the peaks at 1036 and 790 cm−1. It was demonstrated that when introducing Ta5+ to the system, the interaction force between the charges would make Ta5+ attract the polar S, thus elongating the bond length between Mo–S and Ni–S, leading to smaller bond energy and lower wavenumbers for Ta-MNS.76 It was also indicated that the higher bond energy in MNS would decline after doping Ta5+, meaning the energy of the system was reduced and the heterostructure was more stable in Ta-MNS.77
S bond in thioacetamide.71–73 The peaks at 1033.4 and 692 cm−1 could be assigned to the presence of Ni–S.74 Moreover, the S–Mo–S stretch was also in agreement with the IR peaks at 797.6 and 538 cm−1.75 Compared with Ta-MNS, the stretching vibrations of S–Mo–S in MNS (Fig. S10(b)†) correspond to the peaks at 871 and 615 cm−1, while the stretching vibrations of Ni–S correspond to the peaks at 1036 and 790 cm−1. It was demonstrated that when introducing Ta5+ to the system, the interaction force between the charges would make Ta5+ attract the polar S, thus elongating the bond length between Mo–S and Ni–S, leading to smaller bond energy and lower wavenumbers for Ta-MNS.76 It was also indicated that the higher bond energy in MNS would decline after doping Ta5+, meaning the energy of the system was reduced and the heterostructure was more stable in Ta-MNS.77
Moreover, as shown in Fig. S11 and Table S2,† BET/BJH analysis was employed to investigate the BET surface area, micropore volume, and average pore size. According to the results, after doping Ta, the BET surface area and the micropore volume increased, thus exposing more active sites and enhancing the catalytic capacity in HER and OER.78
To investigate the effect of the designed construction of the interface on the HER activity and durability, we conducted comprehensive electrochemical measurements in 1 M KOH with a typical three-electrode setup. From the linear scan voltammogram (LSV) polarization curves, as illustrated in Fig. 4a and Fig. S12,† Ta-MNS presented remarkable electrocatalytic activity for HER with the lowest overpotential of 35 mV to deliver 10 mA cm−2 relative to MNS (42 mV to deliver 10 mA cm−2) and NS (97 mV to deliver 10 mA cm−2). This result was ascribed to the synergetic effect between MoS2 and NiS, which played a considerable role in boosting the adsorption of the H* intermediate and lowering the overpotential for HER. Moreover, TM (Nb, Zr, Hf)-MNS (47, 61, and 56 mV to deliver 10 mA cm−2) also exhibited poor electrocatalytic activities compared with Ta-MNS, which could be explained from several aspects. Firstly, TM5+ could provide more charge carriers than TM4+, and had a smaller size to avoid blocking charge transfer. Meanwhile, TaCl5 had better electrolysis and dispersion in solution than NbCl5; thus, playing a more important role in promoting charge transfer. Moreover, the excess precipitates in Zr-MNS and Hf-MNS would instead affect the normal attachment and growth of sulfide, offsetting the positive effect of Zr4+ and Hf4+ on the electrochemical properties. Therefore, Ta-MNS had better performance than Nb-MNS than Hf-MNS than Zr-MNS. It was worth noting that when the current density exceeds 100 mA cm−2, Ta-MNS exhibited better catalytic performance than Pt–C/NF and other TM-MNS, which meant it had higher commercial potential. The response kinetics for the HER of Ta-MNS were further investigated by the Tafel plot. As shown in Fig. 4b, Ta-MNS had the lowest Tafel slope (88.75 mV dec−1), while MNS and NS exhibited Tafel slopes of 104.66 mV dec−1 and 145.09 mV dec−1, respectively, which further confirmed the synergetic effect between MoS2 and NiS in the MoS2/NiS heterostructures. Because the electrochemical desorption of the product hydrogen (Heyrovsky step) was the rate-limiting step, favorable reaction kinetics were related to the high formation rate of the hydrogen. The MoS2/NiS heterostructures could reduce the adsorption Gibbs free energy of the H* intermediate, which played an important role in improving the hydrogen evolution activity of the electrocatalyst. Compared with TM (Nb, Zr, Hf)-MNS (95.06, 112.31, 107.84 mV dec−1), Ta-MNS also had superior rapid HER rates via the Volmer–Tafel mechanism. It was revealed that doping with tantalum was beneficial to optimizing the electronic structure. Therefore, it had highly active sites for the catalytic HER.
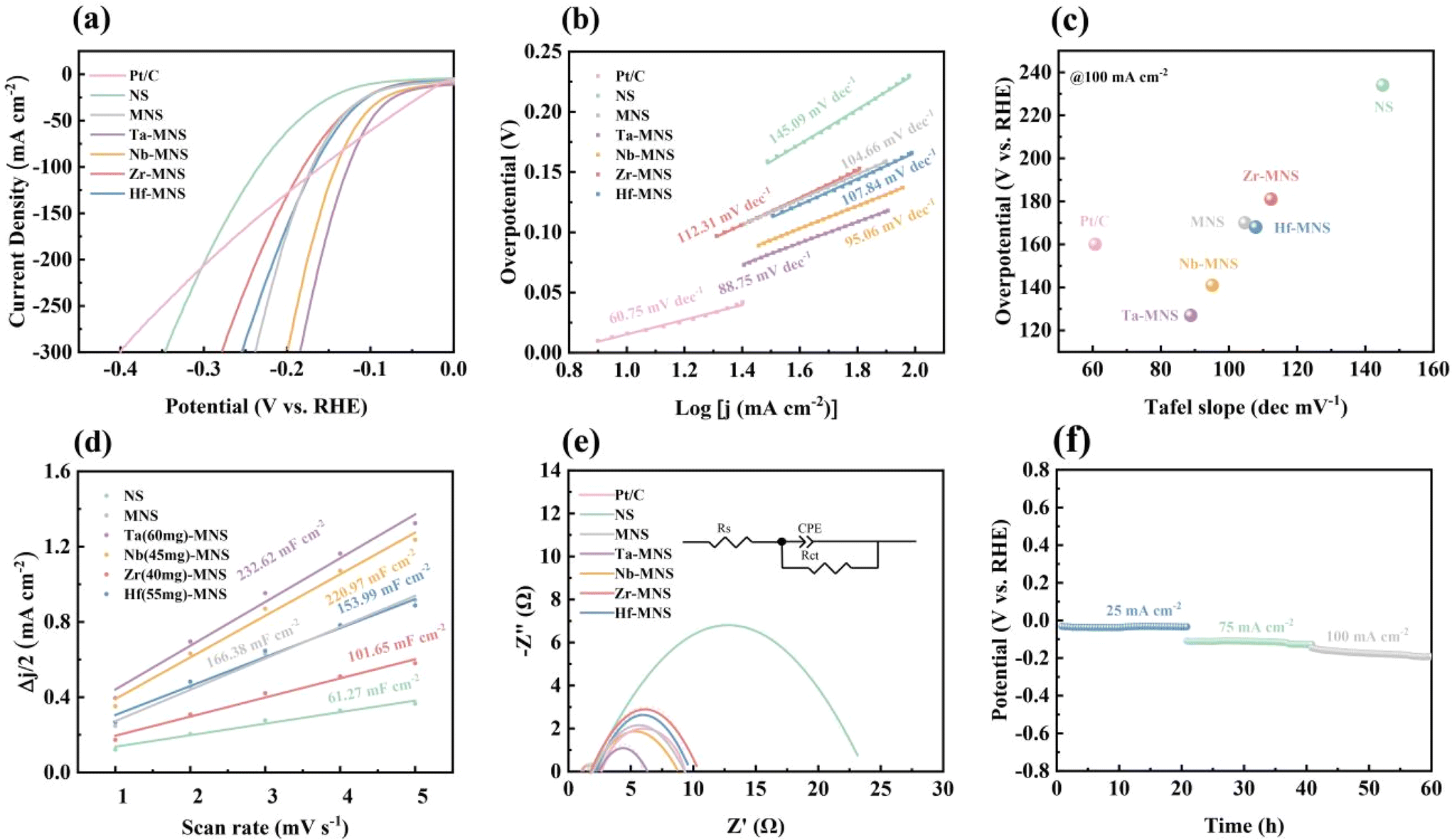 | ||
| Fig. 4 HER performance of Pt–C, NS, MNS, Ta-MNS, Nb-MNS, Zr-MNS, and Hf-MNS: (a) polarization curves. (b) Tafel plot. (c) Performance comparison. (d) Double-layer capacitances. (e) Electrochemical impedance spectra. (f) Time-dependent potential profile of Ta-MNS under a current density of 25, 75, and 100 mA cm−2 for 20 h. | ||
Moreover, the double-layer capacitance (Cdl) of catalysts was measured to estimate the electrochemical active area of a catalyst solid–liquid interface through cyclic voltammetry (CV) under different scan rates (1–5 mV s−1). As shown in Fig. 4d and Fig. S13,† the Cdl of Ta-MNS was the highest (232.62 mF cm−2) of all samples. The AECSA of Ta-MNS was also the highest (5.82 cm2), indicating that doping with tantalum exposed a large number of electrochemical active surface areas (ECSA), and therefore contributed to more active sites for the catalytic HER.
In a series of investigations, electrochemical impedance spectroscopy (EIS) was another crucial method to evaluate the charge transfer capabilities of these electrocatalysts. As shown in Fig. 4e, the faster charge transfer process reflected the smaller semicircle in the Nyquist plots. Ta-MNS possessed the smallest charge transfer resistances (Rct) in comparison with MNS and NS, demonstrating that the MoS2/NiS heterojunction was valuable in accelerating charge transfer for its built-in electric field at the interface. The doping of Ta was also considered a significant reason for improving the electrical conductivity by adjusting the electronic structure and enlarging the specific surface area.
Finally, a long-term chronopotentiometry test was further employed to estimate the stability of Ta-MNS with the current densities changed from 25 to 100 mA cm−2 for 20 h (Fig. 4f). It was inferred from the results that the favorable performance of the Ta-MNS catalyst could be maintained within a wide range of current density values.
The OER performance of the as-prepared electrodes was further examined in 1 M KOH with a typical three-electrode setup. As shown in Fig. 5a and Fig. S14,† Ta-MNS had the optimal catalytic activity for OER, requiring a small overpotential of 307 mV to reach the current densities of 50 mA cm−2, which is slightly inferior to RuO2/NF (283 mV). Moreover, the current density of Ta-MNS could reach 10 mA cm−2 with only an overpotential of 199 mV, which was 37, 66, and 208 mV lower than those of RuO2, MNS and NS, respectively, indicating that the designed heterostructures of MoS2/NiS could prominently expedite the OER process with the assistance of Ta to adjust the electronic structure.
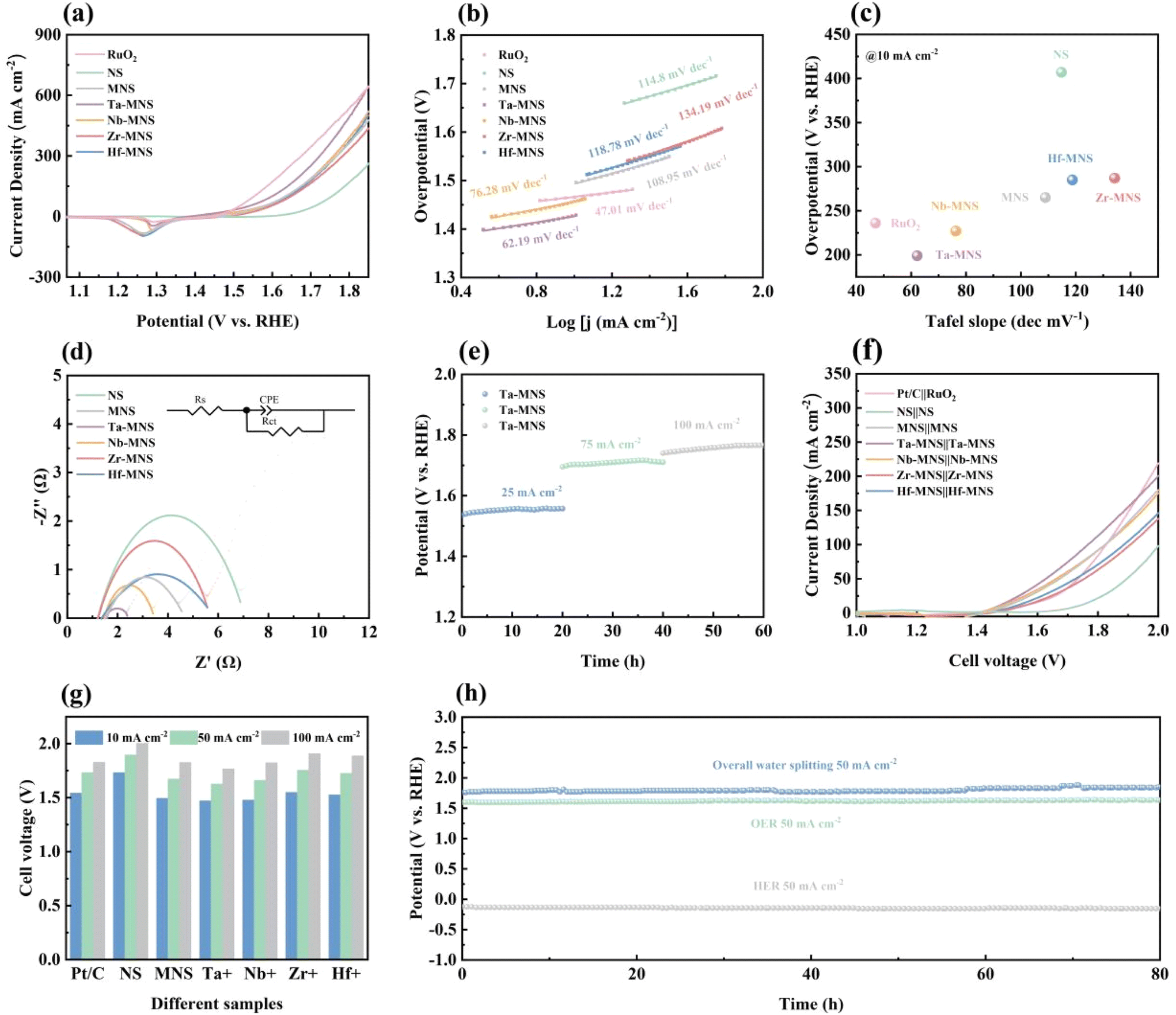 | ||
| Fig. 5 OER performance of RuO2, NS, MNS, Ta-MNS, Nb-MNS, Zr-MNS, and Hf-MNS: (a) polarization curves. (b) Tafel plot. (c) Performance comparison. (d) Electrochemical impedance spectra. (e) Time-dependent potential profile of Ta-MNS under a current density of 25, 75, and 100 mA cm−2 for 20 h. (f) LSV curves of RuO2||Pt/C, NS||NS, MNS||MNS, Ta-MNS||Ta-MNS, Nb-MNS||Nb-MNS, Zr-MNS||Zr-MNS and Hf-MNS||Hf-MNS as bifunctional catalysts in 1.0 M KOH solution for overall water splitting. (g) Cell voltage at 10, 50, and 100 mA cm−2. (h) Time-dependent potential profile of Ta-MNS under a current density of 50 mA cm−2 for 80 h in HER and OER, and cell voltage of Ta-MNS||Ta-MNS in overall water splitting at 50 mA cm−2 for 80 h. | ||
The Tafel slope of Ta-MNS (Fig. 5(b and c)) was identified as 62.19 mV dec−1, which was noticeably lower than those of NS (114.8 mV dec−1), MNS (108.95 mV dec−1) and TM (Nb, Zr, Hf)-MNS (76.28, 134.19, 118.78 mV dec−1), evidencing the significantly boosted OER kinetics. Particularly, the drop in the Tafel slop of the MNS after doping with tantalum also indicated an advantageous change in the chemisorption of OH−, namely the rate-limiting step for OER, owing to the strong interfacial electronic interaction by the function of Ta5+. The Nyquist plots (Fig. 5d) further showed that the formation of the MoS2/NiS heterostructures and the doping of tantalum significantly reduced the charge transfer resistance. Hence, it promoted the charge transfer efficiency during the OER process.
Moreover, the outstanding long-term OER stability of Ta-MNS was cooperatively confirmed by the chronopotentiometry test with a current density change from 25 to 100 mA cm−2 for 20 h (Fig. 5e), demonstrating the durable electrocatalytic activity for the electrochemical catalyst.
Given the excellent HER and OER electrocatalytic performance, Ta-MNS was used as the cathode and anode, respectively, for overall water splitting. The LSV curves (Fig. 5(f and g)) show the notable catalytic activity of Ta-MNS, requiring a small cell voltage of 1.627 and 1.767 V to attain a current density of 50 and 100 mA cm−2, respectively, which was superior to that of Pt/C||RuO2 (1.733 and 1.827 V).
Additionally, the chronopotentiometry test for HER, OER, and OWS with a current density of 50 mA cm−2 for 80 h (Fig. 5h) was also carried out to demonstrate the outstanding durability and excellent mass transport properties of Ta-MNS. It was inferred from the results that the favorable performance of the Ta-MNS catalyst could be maintained for at least 80 h.
Moreover, to investigate the mass content of Ni and Ta sources for enhanced HER and OER performance, several samples with different amounts of TaCl5 (0, 30, 60, 90, 120 mg) and different sizes of nickel foam (1 × 1, 1 × 2, 2 × 2 cm2) were prepared. As shown in Fig. S15 and S16,† with the adjustment of the amount of Ni and Ta, the activity and durability of the catalyst greatly changed, indicating the favorable charge transfer capabilities owing to a balanced mass content of Ni and Mo, under which the special structure of MoS2/NiS could come into play. The results suggested that the embedding of Ta5+ induced the expansion of the MoS2/NiS lattice and the electron transfer at the interface, thus regulating the electronic structure of the basal plane. The result also suggested that Ta5+ could serve as electron donors at the proper concentration. By contrast, if there were superfluous TaCl5 in the solution, the contact area between Ni and other contents would decrease, leading to more difficulty for the MoS2/NiS structure to grow on the surface of NF. Additionally, it could be observed from the samples of different reaction times (5, 6, 7 h) in Fig. S17† that the degree of vulcanization was a key factor in the electrocatalytic activity of Ta-MNS for HER and OER.
The XPS survey spectrum of Ta-MNS after the HER chronopotentiometry test for 20 h showed peaks for the Ni, Mo, S, and Ta elements, indicating good stability of the electrocatalyst (Fig. 6(a–d)). The Mo 3d region showed well-defined peaks for the Mo4+ ions, while the peak intensity of Mo6+ ions increased, indicating the stable morphology of the catalyst and a few surface oxidations of MoS2.79 Meanwhile, conversely, the Ta 4f region exhibited no obvious oxidation or change after the chronopotentiometry test. The XPS survey spectrum of Ta-MNS after the OER chronopotentiometry test for 20 h showed peaks for Ni, Mo, S, and Ta elements. The peaks of Mo 3d (Fig. 6b) and S 2p (Fig. 6c) became relatively weak and inconspicuous after the OER stability test, which might be attributed to the dissolution of Mo and S atoms during the OER process.80 Furthermore, the slight Ta5+ under the surface of the catalyst almost disappeared (Fig. 6d), indicating the surface reconstruction of sulfides due to the extremely oxidizing OER environment, which was consistent with the previously reported literature.81,82 Nevertheless, the synergetic effect between these sulfides and oxidized species formed during the OER progress could expedite the kinetics for OER by offering more active sites, which was attributed to the increasing movement of the oxyhydroxides, oxides, and electrons in the surface layer. Consequently, Ta-MNS could maintain remarkable electrocatalytic activity over at least 80 h for OER. Moreover, as shown in Table S3,† the Mo4+ in the Mo 3d orbital and Ni2+ in the Ni 2p orbital of Ta-MNS were shifted to a low binding energy position on average after the reaction. Meanwhile, S 2p1/2, S 2p3/2, and S–O were shifted to high binding energy positions, which meant electrons were transferred from S to the metal atoms during the HER and OER process, confirming the role of positively charged metal elements and negatively charged S in getting electrons and providing electrons to protons.
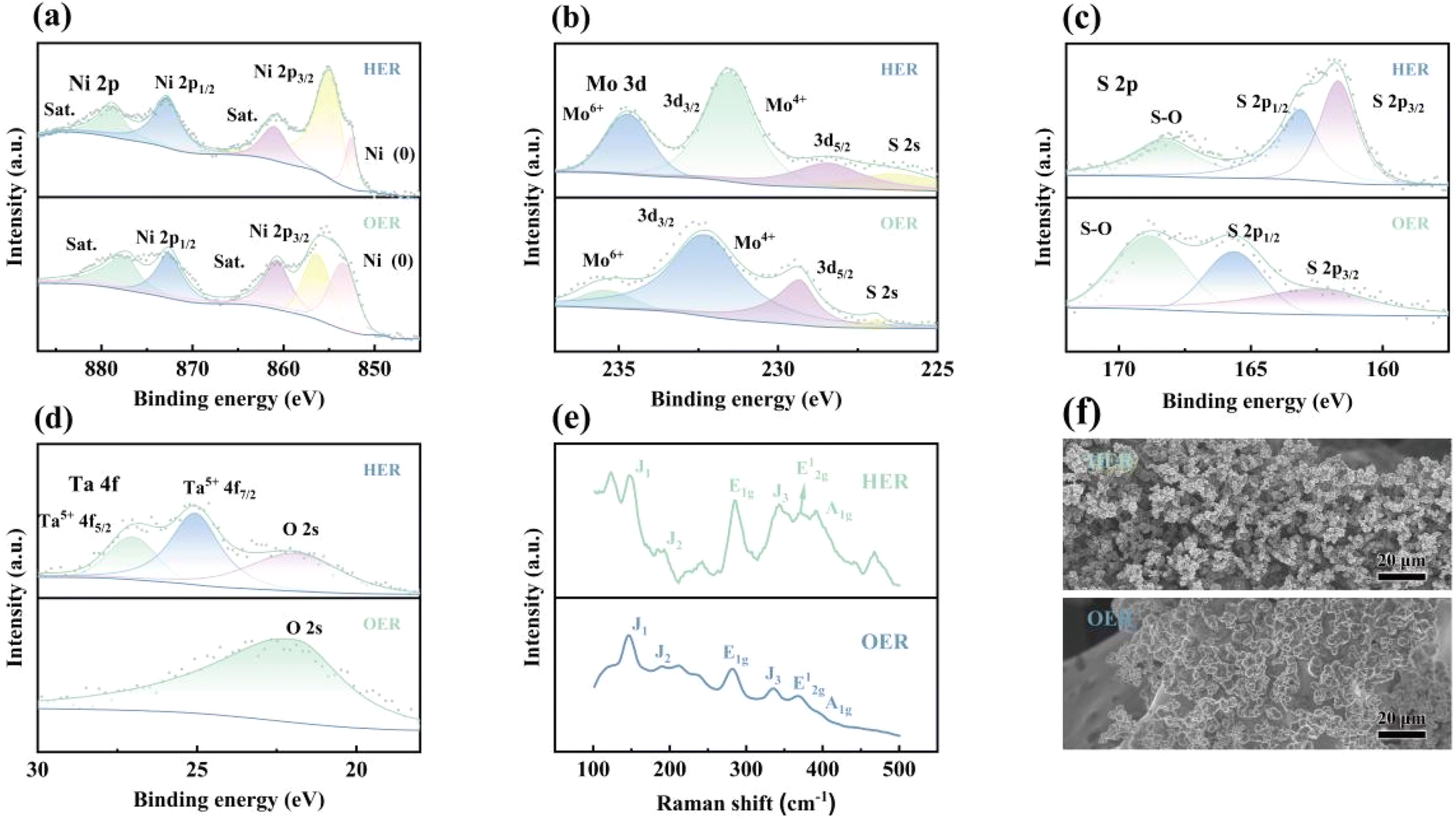 | ||
| Fig. 6 (a–d) XPS spectra of Ta-MNS (Ni 2p, Mo 3d, S 2p, Ta 4f) after HER or OER tests for 20 h. (e) Raman spectra of Ta-MNS after HER or OER tests for 20 h. (f) SEM images of Ta-MNS after HER or OER tests for 20 h. | ||
The Raman spectra and SEM images (Fig. 6(e and f)) showed the morphology of Ta-MNS after a HER and OER chronopotentiometry test for 20 h, respectively, which hardly changed compared to the untested materials, indicating the outstanding structural stability of the catalyst.
Finally, as shown in Table S4,† we compared the overall water-splitting driving voltage and the HER, as well as OER overpotentials, of Ta-MNS in 1 M KOH solution with the performance of some recently reported transition metal-based electrocatalysts to show the remarkable performance of the sample.
3. Conclusion
In summary, a Ta-doped MoS2/NiS catalyst located on NF (Ta-MNS) with the structure of hierarchical nanospheres was synthesized via a simple one-step hydrothermal interface engineering-induced method. It exhibited multifunctional outstanding electrocatalytic performance due to the spontaneous electron transfer at the interface between two phases of a MoS2/NiS heterogeneous structure. A stable 3D morphology was another preponderance of Ta-MNS, which could not only effectively prevent the stacking and aggregation of 2D nanosheets, ensuring rapid reaction kinetics by expediting electron transfer, but also enhance the mechanical strength, improving the stability. Moreover, compared with other transition metal elements (Nb, Zr, Hf), the introduction of Ta was favorable for adjusting the morphology of nanospheres, modulating the electronic structure, and enhancing the link between components by the –S–Ta5+–S– bridge. This work provided a new idea of doping, combined with heterogeneous construction, for the reasonable design and application of multifunctional catalysts for water splitting and energy conversion.Data availability
The data supporting this article are included within the article and in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52222315).References
- Q. Hassan, S. Algburi, M. Jaszczur, A. K. Al-Jiboory, T. J. Al Musawi, B. M. Ali, P. Viktor, M. Fodor, M. Ahsan, H. M. Salman and A. Z. Sameen, Process Saf. Environ., 2024, 184, 1069–1093 CrossRef CAS.
- F. Li, D. Liu, K. Sun, S. H. Yang, F. Z. Peng, K. X. Zhang, G. D. Guo and Y. Si, Sustainability, 2024, 16, 1890 CrossRef CAS.
- A. Maka and M. Mehmood, Clean Energy, 2024, 8, 1–7 CrossRef.
- K. Ham, S. Bae and J. Lee, J. Energy Chem., 2024, 95, 554–576 CrossRef CAS.
- R. R. Deng, B. Zhang and Q. B. Zhang, ChemCatChem, 2024, 16, e202301165 Search PubMed.
- H. M. Yang, C. M. Ni, X. N. Gao, S. H. Lin, X. Y. He, L. Tian and Z. Li, ChemSusChem, 2024, 17, e202400977 CrossRef CAS PubMed.
- Z. Y. Yang, J. Zhu, X. L. Xu, L. Wang, G. B. Zhou, Z. Yang and Y. F. Zhang, RSC Adv., 2023, 13, 4056–4064 RSC.
- S. O. Hanslin, H. Jónsson and J. Akola, Phys. Chem. Chem. Phys., 2023, 25, 32541–32548 RSC.
- P. Y. Kuang, Z. R. Ni, B. C. Zhu, Y. Lin and J. G. Yu, Adv. Mater., 2023, 35, 2303030 CrossRef CAS PubMed.
- Z. X. Wu, P. F. Yang, Q. C. Li, W. P. Xiao, Z. J. Li, G. R. Xu, F. S. Liu, B. H. Jia, T. Y. Ma, S. H. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 Search PubMed.
- Y. P. Zhu, K. Fan, C. S. Hsu, G. Chen, C. S. Chen, T. C. Liu, Z. Z. Lin, S. X. She, L. Q. Li, H. M. Zhou, Y. Zhu, H. M. Chen and H. T. Huang, Adv. Mater., 2023, 35, 2301133 CrossRef CAS PubMed.
- Y. Wang, X. P. Li, Z. Huang, H. Z. Wang, Z. L. Chen, J. F. Zhang, X. R. Zheng, Y. D. Deng and W. B. Hu, Angew. Chem., Int. Ed., 2023, 62, e202215256 Search PubMed.
- H. Su, X. D. Pan, S. Q. Li, H. Zhang and R. Q. Zou, Carbon Energy, 2023, 5, e296 CrossRef CAS.
- L. Li, J. M. Chen, Z. J. Xiao, X. Y. Zhang, S. K. Kwak, D. L. Tian and J. M. Lee, Adv. Sustainable Syst., 2024, 2400587, DOI:10.1002/adsu.202400587.
- K. Kim, C. Kim, S. M. Bak, C. Y. Nam and J. H. Moon, Chem. Eng. J., 2024, 488, 150976 CrossRef CAS.
- P. F. Wan and Q. Tang, Mater. Chem. Front., 2024, 8, 507–527 RSC.
- S. Cho, C. H. Lee and S. U. Lee, ACS Mater. Lett., 2024, 6, 2304–2310 CrossRef CAS.
- P. F. Tan, Y. Wang, L. Yang, X. Q. Zhang and J. Pan, Diamond Relat. Mater., 2023, 136, 109974 CrossRef CAS.
- Y. Lu, W. L. Weng, Y. H. Lu, X. X. Pang, N. X. Hu and B. H. Peng, J. Solid State Chem., 2024, 335, 124701 Search PubMed.
- S. Shah, N. A. Khan, M. Imran, M. Rashid, M. K. Tufail, A. U. Rehman, G. Balkourani, M. Sohail, T. Najam and P. Tsiakaras, Membranes, 2023, 13, 113 Search PubMed.
- K. Jiang, J. Q. Li, Z. L. Zheng, T. T. Zhang, G. Q. Wang, C. Shi and X. Y. Hou, ACS Appl. Energy Mater., 2024, 7, 7895–7905 Search PubMed.
- Z. Z. Wang, X. Z. Zhou, J. E. Wu, Y. M. Wei, Y. B. Cui, Y. L. Xia, W. M. Xu, S. C. Mu and J. X. Cui, Macromol. Rapid Commun., 2025, 46, 2400740 Search PubMed.
- A. Farhan, M. Murad, W. Qayyum, A. Nawaz, M. Sajid, S. Shahid and M. A. Qamar, J. Solid State Chem., 2024, 329, 124445 CrossRef CAS.
- Y. H. Guan, L. Dong, L. C. Qiu, P. Yan and X. Y. Yu, ChemCatChem, 2023, 15, e202300845 Search PubMed.
- S. F. Zai, Y. H. Wu, Y. T. Zhou, Z. T. H Li and C. Bin Guo, J. Colloid Interface Sci., 2023, 643, 350–359 CrossRef PubMed.
- R. R. Fu, X. G. Jiao, J. A. Yu, Q. Z. Jiao, C. H. Feng and Y. Zhao, J. Electroanal. Chem., 2023, 930, 117137 CrossRef CAS.
- Y. P. Li, W. T. Wang, M. Y. Cheng, Y. F. Feng, X. Han, Q. Z. Qian, Y. Zhu and G. Q. Zhang, Adv. Mater., 2023, 35, 2206351 CAS.
- C. H. Jin, R. J. Fu, L. Q. Ran, W. H. Wang, F. X. Wang, D. Z. Zheng, Q. Feng and G. X. Wang, RSC Adv., 2023, 13, 13985–13990 RSC.
- X. J. Niu, Y. J. Wang, G. H. Gao, T. D. Yang, J. W. Mei, Y. C. Qi, R. Z. Tian and J. S. Li, J. Colloid Interface Sci., 2023, 652, 989–996 Search PubMed.
- Y. Zang, S. C. Huang, B. P. Yang, G. Chen, X. H. Liu and N. Zhang, Appl. Surf. Sci., 2023, 611, 155656 CrossRef CAS.
- D. Z. Wang, L. Yang, R. Q. Liu, T. Guo, H. Fei and Z. Z. Wu, Trans. Nonferrous Met. Soc., 2023, 33, 1540–1549 Search PubMed.
- C. Q. Gu, T. Sun, Z. Y. Wang, S. S. Jiang and Z. H. Wang, Small Methods, 2023, 7, 2201529 Search PubMed.
- D. Chodvadiya, M. H. Dalsaniya, N. N. Som, B. Chakraborty, D. Kurzydlowski, K. J. Kurzydlowski and P. K. Jha, Int. J. Hydrogen Energy, 2023, 48, 5138–5151 CrossRef CAS.
- X. Zhao, W. P. Li, Y. H. Cao, A. Portniagin, B. Tang, S. X. Wang, Q. Liu, D. Yu, X. Y. Zhong, X. R. Zheng and A. L. Rogach, ACS Nano, 2024, 18, 4256–4268 CrossRef CAS PubMed.
- I. Ahmed, R. Biswas, M. Iqbal, A. Roy and K. K. Haldar, ChemNanoMat, 2023, 9, e202200550 CrossRef CAS.
- W. An, W. Y. Lu, L. Ma, W. Li, C. C. Yan, U. H. Prova, C. X. Wang and G. Y. Huang, Int. J. Hydrogen Energy, 2023, 48, 35953–35961 Search PubMed.
- L. L. Zhang, X. X. Shi, A. J. Xu, W. W. Zhong, J. T. Zhang and S. J. Shen, Nano Res., 2024, 17, 3693–3699 CrossRef CAS.
- Y. W. Cao, L. H. Li, X. X. Yu, M. Tahir, Z. Y. Xiang, W. Kong, Z. H. Lu, X. R. Xing and Y. L. Song, ACS Appl. Mater. Interfaces, 2022, 14, 56725–56734 Search PubMed.
- A. Mosallanezhad, C. Wei, P. A. Koudakan, Y. Y. Fang, S. W. Niu, Z. A. Bian, B. Liu, T. Huang, H. G. Pan and G. M. Wang, Appl. Catal., B, 2022, 315, 121534 CrossRef CAS.
- D. Yue, T. L. Feng, Z. C. Zhu, S. Y. Lu and B. Yang, ACS Catal., 2024, 14, 3006–3017 Search PubMed.
- H. Zhang, F. Wan, X. G. Li, X. H. Chen, S. L. Xiong and B. J. Xi, Adv. Funct. Mater., 2023, 33, 2306340 CrossRef CAS.
- X. Wang, J. Wu, Y. W. Zhang, Y. Sun, K. K. Ma, Y. Xie, W. H. Zheng, Z. Tian, Z. Kang and Y. Zhang, Adv. Mater., 2023, 35, 2206576 CrossRef CAS PubMed.
- Y. X. Ren, X. Y. Miao, J. X. Zhang, Q. D. Lu, Y. Chen, H. B. Fan, F. Teng, H. F. Zhai, X. X. He, Y. Long, C. M. Zhang and P. Hu, J. Mater. Chem. A, 2023, 11, 2690–2697 RSC.
- A. Das, D. Roy, B. K. Das, M. I. Ansari, K. K. Chattopadhyay and S. Sarkar, Catal. Today, 2023, 423, 113921 CAS.
- P. T. Liu, J. Y. Zhu, J. Y. Zhang, P. X. Xi, K. Tao, D. Q. Gao and D. S. Xue, ACS Energy Lett., 2017, 2, 745–752 Search PubMed.
- J. M. de Oliveira, I. A. Anes, J. L. Coleti, D. Espinosa, M. S. de Carvalho and J. Tenório, Can. J. Chem. Eng., 2023, 101, 1743–1761 Search PubMed.
- H. L. Li, Z. G. Yao, J. Zhang, X. J. Cai, L. Li, G. Liu, J. J. Liu, L. Cui and J. H. Huang, SN Appl. Sci., 2020, 2, 671 Search PubMed.
- X. H. Xia, G. Y. Zhao, Q. Yan, B. Wang, Q. F. Wang and H. J. Xie, ACS Sustainable Chem. Eng., 2022, 10, 182–193 Search PubMed.
- Z. L. Zheng, L. Yu, M. Gao, X. Y. Chen, W. Zhou, C. Ma, L. H. Wu, J. F. Zhu, X. Y. Meng, J. T. Hu, Y. C. Tu, S. S. Wu, J. Mao, Z. Q. Tian and D. H. Deng, Nat. Commun., 2020, 11, 3315 Search PubMed.
- K. C. Ok, Y. Park, K. B. Chung and J. S. Park, Appl. Phys. Lett., 2013, 103, 213501 Search PubMed.
- R. Niishiro, H. Kato and A. Kudo, Phys. Chem. Chem. Phys., 2005, 7, 2241–2245 Search PubMed.
- C. J. Lv, J. Yang, Y. Peng, X. C. Duan, J. M. Ma, Q. H. Li and T. H. Wang, Electrochim. Acta, 2019, 297, 258–266 Search PubMed.
- K. Ahanjan, M. Shamsipur, A. Taherpour and A. Pashabadi, Electrochim. Acta, 2022, 433, 141249 Search PubMed.
- X. L. Jiang, X. Q. Yue, Y. X. Li, X. J. Wei, Q. J. Zheng, F. Y. Xie, D. N. Lin and G. X. Qu, Chem. Eng. J., 2021, 426, 130718 Search PubMed.
- W. L. Yu, Z. Chen, X. L. Jiang, W. P. Xiao, B. Dong and Z. X. Wu, Int. J. Hydrogen Energy, 2022, 47, 14414–14421 Search PubMed.
- M. X. Chen, J. J. Tao, L. H. Zhang, C. H. Yang, H. Luo, X. H. Kong and D. W. Zhang, ACS Appl. Energy Mater., 2024, 7, 4454–4461 CrossRef CAS.
- X. Li, M. Zhang, Y. Liu, X. Sun, D. Li, B. Liu, M. Yang, H. Chen, S. Ding and Z. Lin, Appl. Catal., B, 2024, 359, 124530 Search PubMed.
- B. Chen, P. Hu, F. Yang, X. J. Hua, F. F. Yang, F. Zhu, R. Y. Sun, K. Hao, K. S. Wang and Z. Y. Yin, Small, 2023, 19, 2207177 Search PubMed.
- Y. A. Liu, Q. Li, Y. Zhu, X. Y. Chen, F. Xue, M. Lyu, Q. H. Li, X. Chen, J. X. Deng, J. Miao, Y. L. Cao, K. Lin and X. R. Xing, Dalton Trans., 2023, 52, 8530–8535 CAS.
- H. L. Wei, A. D. Tan, W. B. Liu, J. H. Piao, K. Wan, Z. X. Liang, Z. P. Xiang and Z. Y. Fu, Catalysts, 2022, 12, 947 Search PubMed.
- X. W. Zhao, J. H. Bao, Y. M. Zhou, Y. W. Zhang, X. L. Sheng, B. Wu, Y. Y. Wang, C. J. Zuo and X. H. Bu, New J. Chem., 2022, 46, 5505–5514 CAS.
- L. K. Gao, X. Cui, Z. W. Wang, C. D. Sewell, Z. L. Li, S. Liang, M. Y. Zhang, J. Li, Y. J. Hu and Z. Q. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023421118 Search PubMed.
- S. Ni, H. N. Qu, H. F. Xing, Z. H. Xu, X. Y. Zhu, M. L. Yuan, M. Rong, L. Wang, J. M. Yu, Y. Q. Li, L. R. Yang and H. Z. Liu, Chin. J. Chem. Eng., 2022, 41, 320–328 CrossRef CAS.
- X. Liu, Z. H. Yin, M. Cui, L. G. Gao, A. M. Liu, W. N. Su, S. R. Chen, T. L. Mac and Y. Q. Li, J. Colloid Interface Sci., 2022, 610, 653–662 CrossRef CAS PubMed.
- L. Yang, X. Q. Yuan, R. X. Song and W. Liang, J. Phys. Chem. Solids, 2023, 181, 111540 CrossRef CAS.
- M. Tayebi, Z. Masoumi, B. Seo, C. S. Lim, C. H. Hong, H. J. Kim, D. Kyung and H. G. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 26107–26120 CrossRef CAS PubMed.
- H. Liu, X. Zhou, C. Ye, M. Ye and J. Shen, Appl. Catal., B, 2024, 343, 123560 Search PubMed.
- Y. Men, P. Li, F. Yang, G. Cheng, S. Chen and W. Luo, Appl. Catal., B, 2019, 253, 21–27 CrossRef CAS.
- T. Dippong, E. A. Levei, O. Cadar, I. G. Deac, M. Lazar, G. Borodi and I. Petean, J. Alloys Compd., 2020, 849, 156695 CrossRef CAS.
- M. M. S. Sanad, M. M. Farahat and M. A. A. Khalek, Adv. Powder Technol., 2021, 32, 1573–1583 Search PubMed.
- I. Barnes, K. H. Becker and J. Starcke, J. Phys. Chem., 1991, 95, 9736–9740 CrossRef CAS.
- T. Dippong, E. A. Levei, O. Cadar, A. Mesaros and G. Borodi, J. Anal. Appl. Pyrolysis, 2017, 125, 169–177 CrossRef CAS.
- W. J. Dong, X. B. Wang, B. J. Li, L. N. Wang, B. Y. Chen, C. R. Li, X. A. Li, T. R. Zhang and Z. Shi, Dalton Trans., 2011, 40, 243–248 RSC.
- T. V. Sathisha, B. E. K. Swamy, S. Reddy, B. N. Chandrashekar and B. Eswarappa, J. Mol. Liq., 2012, 172, 53–58 CAS.
- A. V. Murugan, M. Quintin, M. H. Delville, G. Campet, A. K. Viswanath, C. S. Gopinath and K. Vijayamohanan, J. Mater. Res., 2006, 21, 112–118 CrossRef CAS.
- S. W. Arafat, Z. K. Heiba, M. Sanad, H. Elshimy, H. S. Alsalem, A. M. Alenad and T. A. Taha, Opt. Mater., 2022, 129, 112561 CrossRef CAS.
- P. Wang and J. H. Wang, Curr. Appl. Phys., 2022, 36, 51–62 CrossRef.
- B. Farahani, M. Giahi, M. H. Ghorbani, R. Fazaeli and O. Moradi, J. Nanostruct. Chem., 2023, 13, 303–320 CrossRef CAS.
- D. Balaji, J. Madhavan, V. Vinesh, B. Neppolian, M. S. Alsalhi and S. Prasad, Mater. Chem. Phys., 2022, 280, 125839 CrossRef CAS.
- C. J. Gu, G. Y. Zhou, J. Yang, H. Pang, M. Y. Zhang, Q. Zhao, X. F. Gu, S. Tian, J. B. Zhang, L. Xu and Y. W. Tang, Chem. Eng. J., 2022, 443, 136321 CrossRef CAS.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. H. Dong, S. H. Liu, X. D. Zhuang and X. L. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- L. K. Gao, X. Cui, C. D. Sewell, J. Li and Z. Q. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05475j |
| This journal is © The Royal Society of Chemistry 2025 |