
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing diagnostics with BODIPY-bismuthene DNA biosensors†
Laura
Gutiérrez-Gálvez
a,
Estefanía
Enebral-Romero
ab,
Miguel Ángel
Valle Amores
 c,
Clara
Pina Coronado
a,
Iñigo
Torres
d,
David
López-Diego
e,
Mónica
Luna
e,
Alberto
Fraile
cf,
Félix
Zamora
dg,
José
Alemán
cf,
Jesús
Álvarez
ghij,
María José
Capitán
ik,
Encarnación
Lorenzo
abf and
Tania
García-Mendiola
*af
c,
Clara
Pina Coronado
a,
Iñigo
Torres
d,
David
López-Diego
e,
Mónica
Luna
e,
Alberto
Fraile
cf,
Félix
Zamora
dg,
José
Alemán
cf,
Jesús
Álvarez
ghij,
María José
Capitán
ik,
Encarnación
Lorenzo
abf and
Tania
García-Mendiola
*af
aDepartamento de Química Analítica y Análisis Instrumental, Universidad Autónoma de Madrid, 28049, Madrid, Spain. E-mail: tania.garcia@uam.es
bIMDEA-Nanociencia, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
cDepartamento de Química Orgánica, Universidad Autónoma de Madrid, 28049, Madrid, Spain
dDepartamento de Química Inorgánica, Universidad Autónoma de Madrid, 28049, Madrid, Spain
eInstituto de Micro y Nanotecnología IMN-CNM, CSIC (CEI UAM+CSIC), Isaac Newton 8, Tres Cantos, 28760, Madrid, Spain
fInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049, Madrid, Spain
gCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049, Madrid, Spain
hDepartamento de Física de la Materia Condensada, Universidad Autónoma de Madrid, 29049-Madrid, Spain
iFísica de Sistemas Crecidos con Baja Dimensionalidad, Universidad Autónoma de Madrid, Unidad Asociada al CSIC por el IEM, DP, Spain
jInstituto de Ciencia de Materiales “Nicolás Cabrera”, Univ. Autónoma de Madrid, 28049-Madrid, Spain
kInstituto de Estructura de la Materia IEM-CSIC, 28006-Madrid, Spain
First published on 6th March 2025
Abstract
In this work, an electrochemical biosensor is prepared based on few-layer bismuthene hexagons (FLBHs) and a water-soluble BODIPY (BDP) derivative (BDP-NaSO3) for early infection diagnosis. In particular, the detection in advance of a virus sequence in nasopharyngeal swab samples was developed. The combination of the FLBHs and BDP-NaSO3 facilitates the direct, sensitive, and specific detection of gene viruses without the need for any prior amplification step. This work demonstrates that the FLBHs provide an improved electrochemical platform for immobilizing thiolated DNA capture probes that increase the sensitivity of the biosensor, while BDP-NaSO3 serves as a newly powerful electrochemical indicator of the hybridization event. As a proof of concept, SARS-CoV-2 was selected as the model virus. The developed biosensor demonstrated selective, rapid, and straightforward detection of the specific sequence RNA-dependent RNA-polymerase (RdRp) of SARS-CoV-2 with a detection limit of 4.97 fM and a linear range from 16.6 fM to 100 fM. Furthermore, this platform successfully detects the virus directly in nasopharyngeal swab samples with a viral load of at least 19 Cts without being subjected to any prior amplification stage. Finally, the high stability of the biosensor response, which has been working under ambient conditions for over one month, the selectivity and rapidity for specific virus detection, and the requirement of low-volume samples for the determination are remarkable characteristics that make it ideal for its potential application in clinical diagnosis in point-of-care settings.
Introduction
4-Bora-3a,4a-diaza-s-indacene-based structures, commonly known as boron dipyrromethene or BODIPY (BDP), have emerged as one of the most remarkable families of fluorescent dyes since their initial synthesis by Treibs and Kreuzer in 1968.1 Structurally, the standard BDP framework is based on the complexation of the dipyrromethene unit with a boron trifluoride salt (Fig. 1, left). To understand their subsequent reactivity, the numbering and nomenclature of these compounds should be clarified. The 8-position is referred to as meso, the 3,5-positions are denoted as α, the 2,6-positions are designated as β, and the 1,7-positions are marked as β′ (Fig. 1, left).2,3 Despite their simple structure, researchers have succeeded in exploiting these multifaceted derivatives since chemical modifications of the skeleton can change their intrinsic chemical and photophysical properties: (i) enhancement of their thermal and photochemical stability, (ii) improvement of their solubility and robustness towards light, (iii) alteration of absorption/emission wavelengths (visible region in the range of 500–600 nm) that result in good redox characteristics and (iv) modification of fluorescence efficiencies. Of these properties, the latter two appear as the most essential features of these dyes for their great applicability as labelling sensors,4,5 along with their use in molecular recognition or even photocatalysis, among many other applications (right, Fig. 1).6 BDP's fluorescence exhibition depends on many factors, including types of functional groups, their position in the framework and their electronic character, and symmetry of the molecule and/or electronic conjugation with the core system, among others. Therefore, the versatility of these boron-chelated heterocycles is based on their feasible structural functionalization, especially once the central core is shaped.7–9 Basic organic chemistry is behind the numerous transformations that lead to a significant quantity of BDP scaffolds, thanks to their two-fold electronic nature and multiple positions that present interesting reactivity (Fig. 1, right). Some of the most common reactions include C–H activation, substitution reactions, additions, condensations, and cross-couplings in and/or outside the core. Despite having these structures and a multitude of applications, as far as we know, their use in the field of DNA biosensors has not been employed until now.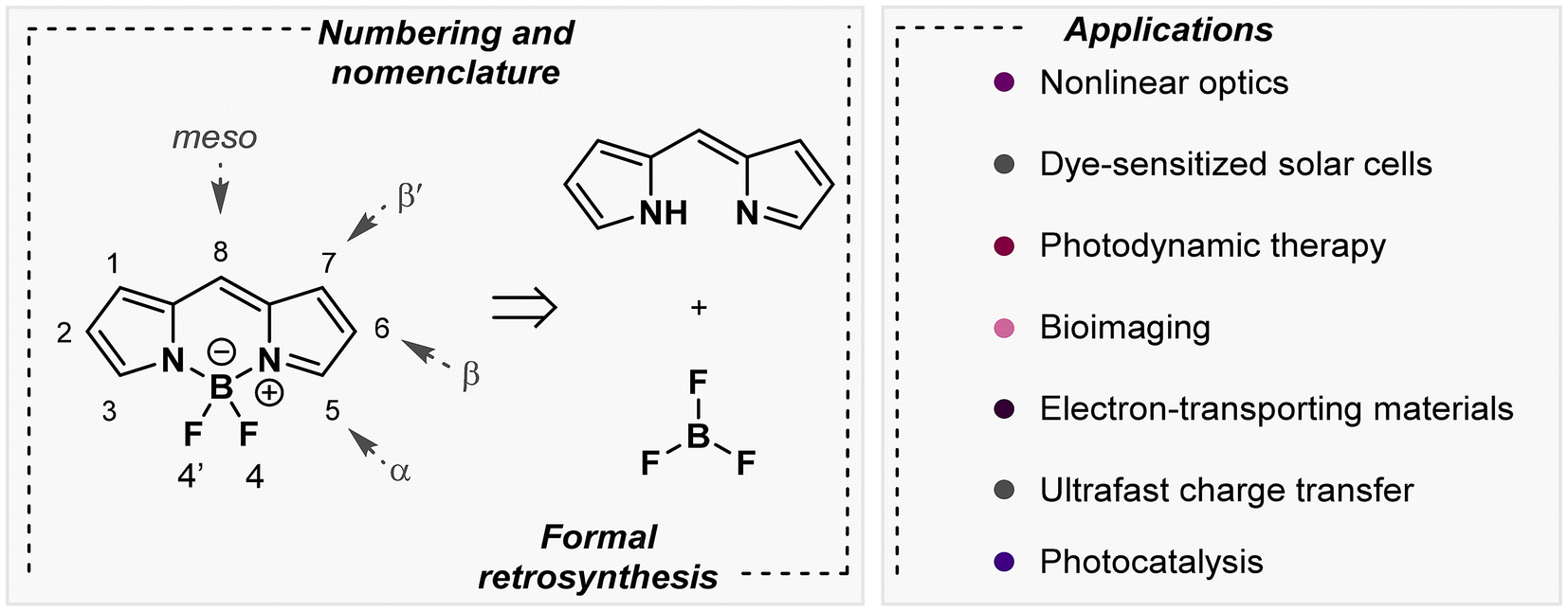 | ||
| Fig. 1 Structures of BDPs and different chemical transformations and applications. | ||
DNA biosensors are crucial tools in modern biotechnology and medical diagnostics due to their ability to detect specific DNA sequences with high sensitivity and specificity.10 These biosensors leverage the unique properties of DNA molecules, such as hybridization, to identify viruses, detect mutations linked to diseases, and monitor genetic disorders.11–13 By converting the biological interaction into a measurable signal, DNA biosensors enable rapid, accurate, and often real-time analysis. Among the different types of DNA biosensors, electrochemical ones stand out due to their interesting properties and their real-world application in point-of-care settings. They represent a significant advancement in biosensing technology, offering numerous advantages in the detection of genetic materials. The key benefits of electrochemical DNA biosensors include their rapid response time, low cost, and potential for miniaturization, which make them suitable for point-of-care diagnostics.14 Additionally, they are capable of detecting low concentrations of DNA, making them particularly valuable in early diagnosis of viral infections and avoiding their spread.15 Their robustness and ease of integration with portable devices further enhance their utility in real-world applications. One of the most attractive methodologies for hybridization detection in electrochemical DNA biosensors, due to its simplicity and low cost, is the use of electrochemical indicators.16 They are essential for translating the biological event of DNA hybridization into a measurable electrochemical signal. Still, they must meet several key conditions to serve as an effective electrochemical indicator in DNA biosensors. These include (i) specificity: the molecule must interact differently with single- or double-stranded DNA, to detect hybridization events and ensure accurate detection and minimize false signals; (ii) electrochemical activity: it should possess redox properties that allow it to undergo a well-defined and reversible electrochemical reaction, producing a clear and measurable signal; (iii) stability: the molecule must be chemically and electrochemically stable under the operating conditions of the biosensor to ensure consistent and reliable performance over time; and (iv) solubility: good solubility in the buffer or medium aqueous solutions used in the biosensor setup.17
Meeting these conditions ensures that the electrochemical indicator can effectively translate the biological event of DNA hybridization into a precise and reliable electrochemical signal, which is critical for the performance of DNA biosensors.
In this regard, BDPs are fascinating structures with rich electrochemical properties, including different oxidation and reduction potentials, thus offering the possibility of choosing the appropriate window for biosensing. Additionally, their chemical structure can be easily modified to suit aqueous environments by incorporating suitable groups, such as sulfonate groups.
It is well known that the incorporation of nanomaterials in biosensors can improve their analytical properties and allow them to detect very low analyte concentrations. Bismuthene is a two-dimensional (2D) material derived from bismuth and has emerged as a promising candidate in the development of advanced DNA biosensors.18 Its unique structural and electronic properties, including a high surface-to-volume ratio, excellent electrical conductivity, and significant biocompatibility, make it an ideal platform for biosensing applications. The large surface area of bismuthene allows for the efficient immobilization of thiolated single-stranded DNA probes, enhancing the sensitivity of the biosensor. Additionally, its outstanding electrical properties facilitate the rapid transduction of hybridization events into detectable electrochemical signals. The biocompatibility of bismuthene ensures minimal interference with biological processes, leading to more accurate and reliable detection. Bismuthene shows strong interactions with thiol groups, enabling its use as an immobilization platform of thiolated biorecognition systems.19–22 Moreover, the 2D nature of bismuthene allows easy integration with various sensing technologies, paving the way for the development of miniaturized and highly efficient DNA biosensors. These attributes collectively highlight the potential of bismuthene to revolutionize the field of DNA biosensing, offering new avenues for early viral infection detection.
Based on the above considerations, in this work, we propose the design, synthesis, and characterization of a water-soluble BDP to be used as an electrochemical indicator in DNA biosensor development. The combination of this BDP with FLBHs will be tested to detect SARS-CoV-2 as a proof-of-concept DNA biosensor.
Results and discussion
Synthesis and characterization of the synthesized BDPs
As mentioned above, one of the objectives of this work is the synthesis of a water-soluble BDP. For this purpose, a BDP with two sulfonic groups, named BDP-NaSO3 (1), was prepared to be used as an electrochemical indicator of the hybridization event. Therefore, the first stage of this work has focused on the synthesis and characterization of BDP-NaSO3 (1). Then, BDP-NO2 (2) was first synthesized, following the route shown in Scheme 1, which consists of a BDP core built using three reaction steps in a one-pot procedure. The intermediate BDP 2 was obtained in 40% overall yield.23–25 | ||
| Scheme 1 Synthetic procedure followed to synthesize BDP-NaSO3 (1). | ||
Afterwards, to achieve BDP-NaSO3 (1), which must be water soluble, we treated BDP 2 with chlorosulfonic acid to introduce sulfonic groups, which were subsequently subjected to deprotonation in the presence of NaHCO3, thus obtaining the corresponding di-salt in quantitative yield.26 The spectroscopic data of both products agree with the reported data.23–26
To validate the results obtained from 1H NMR and ESI-HRMS (see the Experimental section), BDP-NaSO3 was further characterized using UV-visible absorption spectroscopy, fluorescence emission spectroscopy, and cyclic voltammetry. Fig. 2A presents the UV-visible absorption spectrum of BDP-NaSO3, showing an absorption maximum at 501 nm that is characteristic of these BDP structures, which typically absorb between 492 and 518 nm.27,28 The fluorescence emission spectrum of BDP-NaSO3, when excited at 470 nm (Fig. 2B), exhibits a peak at 509 nm, which aligns with the expected values for this class of compounds described in the literature.27,28 These results corroborate those previously obtained via1H NMR and ESI-HRMS analyses.
 | ||
| Fig. 2 UV-visible spectrum (A) and fluorescence emission spectrum (B) of 80.0 μM BDP-NaSO3 solution obtained after being excited at 470 nm in water. (C) The CV obtained for a bare CSPE in 20.0 mM BDP-NaSO3 solution using 0.1 M PB at pH = 7.4 as an electrolyte. Scan rate: 10 mV s−1. | ||
Taking advantage of the fact that BDPs are electroactive compounds, their electrochemical characterization was carried out using cyclic voltammetry. Fig. 2C shows the cyclic voltammogram (CV) of a carbon screen-printed electrode (CSPE) in 20.0 mM BDP-NaSO3 solution using 0.1 M phosphate buffer (PB) at pH 7.4 as a supporting electrolyte. The characteristic redox processes of these types of compounds can be observed, which usually have separations between the first and second cyclic voltammetric waves of approximately 1 V.28 The second oxidation and reduction at a potential of 0.1 V is due to the BDP core, while the first is due to the nitro group attached to benzene that is attached to the BDP core.29
Based on the results obtained with these different characterization techniques, it can be concluded that BDP-NaSO3 has been successfully synthesized and this compound shows interesting electrochemical properties in aqueous solution.
Study of the interaction between BDP-NaSO3 and DNA
One critical criterion of an electrochemical indicator is its ability to interact distinctly with DNA, particularly between double-stranded and single-stranded DNA. Therefore, the subsequent phase of this research aimed to investigate the interaction of BDP-NaSO3 with both massive calf thymus single-stranded DNA (ss-ctDNA) and double-stranded DNA (ds-ctDNA). This investigation seeks to determine its potential utility as an indicator for the electrochemical characterization of DNA hybridization events. | ||
| Fig. 3 UV-visible spectra of BDP-NaSO3 solution in the absence (BDP-NaSO3) and in the presence of ds-ctDNA (A) and ss-ctDNA (B) at different concentrations (100.0 μM, 120.0 μM, 140.0 μM, 160.0 μM, 180.0 μM, 200.0 μM, and 250.0 μM) using water as solvent. | ||
As can be seen in Fig. 3, after the addition of increasing concentrations of single- and double-stranded DNA, changes can be observed in the absorption spectrum of BDP-NaSO3. In the case of ds-ctDNA (Fig. 3A), a hypochromic effect is observed (decrease in absorbance with increasing DNA concentration), and with ss-ctDNA (Fig. 3B), a hyperchromic effect is observed (increase in absorbance with increasing DNA concentration). Both effects demonstrate that there is a clear interaction between BDP-NaSO3 and DNA due to an interaction between the aromatic rings of the synthesized compound (π–π interaction) and the base pairs of the DNA. Furthermore, the observed phenomena indicate that the interaction between BDP-NaSO3 and ss-ctDNA and ds-ctDNA is different.
From the absorbance data shown in Fig. 3, it is also possible to determine the strength of interaction between these organic compounds and the ds-ctDNA and ss-ctDNA using eqn (1) (see subsection ‘Interaction between BDP and DNA’ in the Experimental section) of Becker and Meehan.13 This equation allows the intrinsic binding constant (Kb) to be calculated by plotting [DNA]/(εa − εb) versus 1/(εa − εb). Specifically, Kb is the inverse of the slope of the plot. The Kb values obtained for ds-ctDNA and ss-ctDNA were found to be 1.00 × 104 and 3.33 × 103, respectively. These results suggest a different strength in the interaction between the ds-ctDNA or the ss-ctDNA and the BDP-NaSO3, with the interaction being stronger between BDP-NaSO3 and ds-ctDNA.
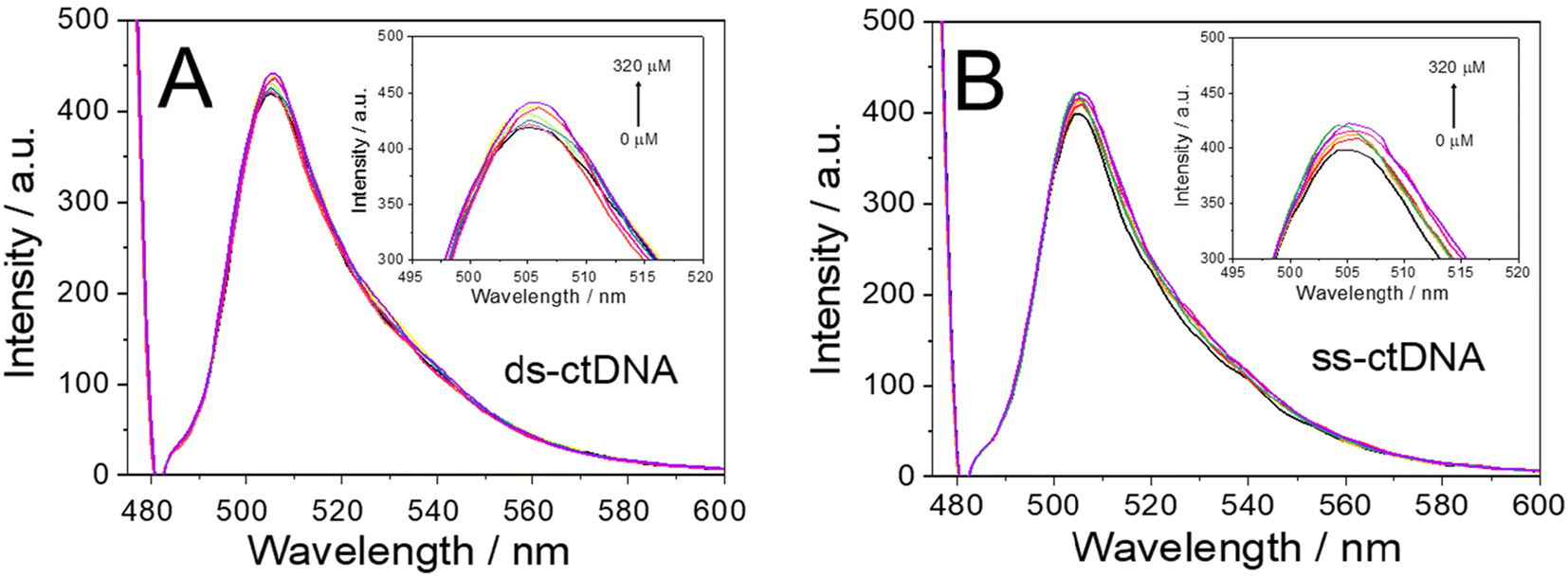 | ||
| Fig. 4 Fluorescence spectra (470 nm excitation wavelength) of BDP-NaSO3 solution in the absence (BDP-NaSO3) and in the presence of ds-ctDNA (A) and ss-ctDNA (B) at different concentrations (40.0 μM, 60.0 μM, 80.0 μM, 100.0 μM, 140.0 μM, 240.0 μM, 280.0 μM, and 320.0 μM) using water as solvent. | ||
After confirming the interaction between the BDP-NaSO3 and the DNA, the strength of this interaction was studied for both the ds-ctDNA and the ss-ctDNA by calculating the activation constant (Ksv) using the Stern–Volmer equation (eqn (2), see subsection ‘Interaction between BDP and DNA’ in the Experimental section).32 By representing F0/F against the concentration of DNA (ds-ctDNA or ss-ctDNA), the Ksν values corresponding with the slope of the plot were calculated. The Ksv values obtained for ds-ctDNA and ss-ctDNA were 2.00 × 102 and 1.0 × 102, respectively.
Using spectrophotometric titrations, the differences in the activation constant values confirm that there is a different interaction between the ds-ctDNA or the ss-ctDNA and BDP-NaSO3, and it is more significant in the case of the double-stranded DNA. These results suggest that BDP-NaSO3 could be an excellent electrochemical indicator of the hybridization event.
For this purpose, the CSPEs were modified with both ds-ctDNA and ss-ctDNA, as explained in detail in the Experimental section. Subsequently, the accumulation of 20.0 mM BDP-NaSO3 was performed on the modified electrode surface (ds-ctDNA/CSPE or ss-ctDNA/CSPE) through direct adsorption for 1 hour at room temperature. Finally, the electrodes were washed with sterilized water and differential pulse voltammograms for a bare carbon electrode (CSPE, black line), a CSPE modified with ss-ctDNA (ss-ctDNA/CSPE, blue line), and a CSPE modified with ds-ctDNA (ds-ctDNA/CSPE, red line) were recorded using 0.1 M PB at pH 7.4 as an electrolyte, as shown in Fig. 5.
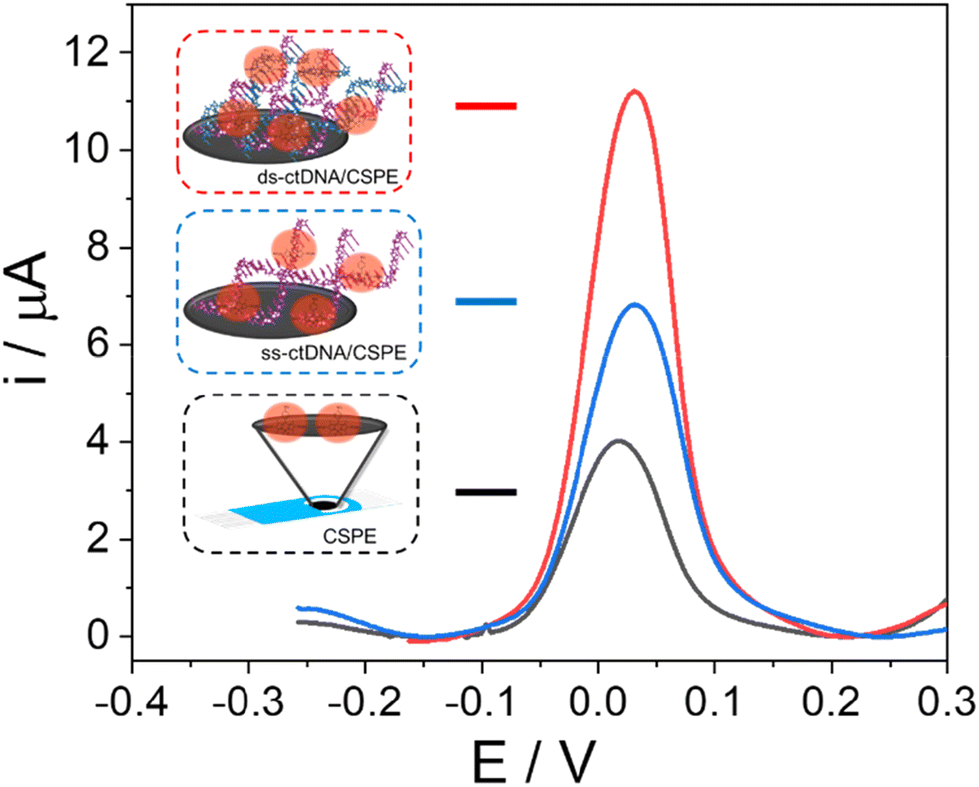 | ||
| Fig. 5 Differential pulse voltammograms obtained in 0.1 M PB solution of pH 7.4 after the accumulation of 20.0 mM BDP-NaSO3 for a bare CSPE (CSPE, black line) and a CSPE modified with ss-ctDNA (ss-ctDNA/CSPE, blue line) or with ds-ctDNA (ds-ctDNA/CSPE, red line). | ||
As can be observed, there are differences in the current intensity recorded for the oxidation of BDP-NaSO3 at a potential of around 0.3 V (a near-zero potential, ideal for a good electrochemical indicator) before and after modifying the electrode with massive DNA, indicating that there is an interaction between the organic compound and the DNA. Furthermore, differences in current intensity are observed for the ds-ctDNA/CSPE and the ss-ctDNA/CSPE, with the current intensity of the former being higher. These results confirm, on the one hand, that BDP-NaSO3 is retained on the electrode surface due to its interaction with DNA and, additionally, it exhibits a different interaction with double-stranded DNA compared to single-stranded DNA, the interaction being greater with ds-ctDNA.
The results obtained using spectrophotometric, spectrofluorimetric, and electrochemical techniques confirm that there is interaction between BDP-NaSO3 and DNA, which is greater with ds-ctDNA than with ss-ctDNA. These results demonstrate that BDP-NaSO3 could be an excellent candidate for use as an electrochemical indicator of the hybridization event in DNA biosensor development.
Development of the biosensor for the detection of SARS-CoV-2
Once the possibility of using BDP-NaSO3 as a redox indicator of the hybridization event was established, the final stage of this work was its application for the development of an FLBH-nanostructured DNA biosensor for the electrochemical detection of the SARS-CoV-2 virus from its genetic code. The steps followed for the development of the biosensor are shown in Scheme 2. | ||
| Scheme 2 Scheme followed for the development of the biosensor. | ||
As shown in Scheme 2, the first step was the nanostructuration of the CSPE with the FLBHs. This modification was performed by spraying the nanomaterial on the electrode surface with an airbrush, as described in detail in the Experimental section. Then, probe immobilization was carried out through interactions between this 2D nanomaterial, FLBHs, and the thiol group of the probe sequence.33 To confirm that the biosensing platform was correctly developed, it was characterized using microscopic, spectroscopic and electrochemical techniques such as scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX), atomic force microscopy (AFM), fluorescence microscopy, X-ray photoelectron spectroscopy (XPS), Raman microscopy and electrochemical impedance spectroscopy (EIS).
AFM and SEM-EDX were employed to confirm the nanostructuration of the electrode and the subsequent immobilization of the capture probe. The secondary electron microscopy image of the bare carbon electrode (Fig. 6A) reveals a homogeneous and flatter surface, in contrast to the secondary electron microscopy image of the probe-SH/FLBH/CSPE sample (Fig. 6E), where bismuthene nanosheets are visible. The electron backscattered images (Fig. 6B and F) provide insights into the atomic weight distribution of the surface materials. For the bare CSPE (Fig. 6B), no contrast is observed, as only carbon is present. However, in the probe-SH/FLBH/CSPE image (Fig. 6F), a more varied contrast is detected. The darker background corresponds to the CSPE (carbon) and the lighter regions are attributed to the bismuthene nanoflakes, reflecting their respective low (carbon) and high (bismuth) atomic weights. Within the bismuthene, the contrast is not uniform, unlike the images of the FLBH/CSPE (see Fig. S4B in the ESI†), suggesting the presence of materials with lower atomic weights on the surface of the bismuthene nanoflakes, which is consistent with robe-SH functionalization. EDX analysis further highlights distinct differences. The CSPE spectrum (Fig. 6D) predominantly shows carbon, while in the spectrum of the probe-SH/FLBH/CSPE sample (Fig. 6H), new peaks corresponding to Bi and P, attributed to the FLBHs and DNA, are clearly visible. These findings suggest that the modification of the carbon electrode with the nanomaterial and the subsequent immobilization of the probe sequence has been successfully accomplished.
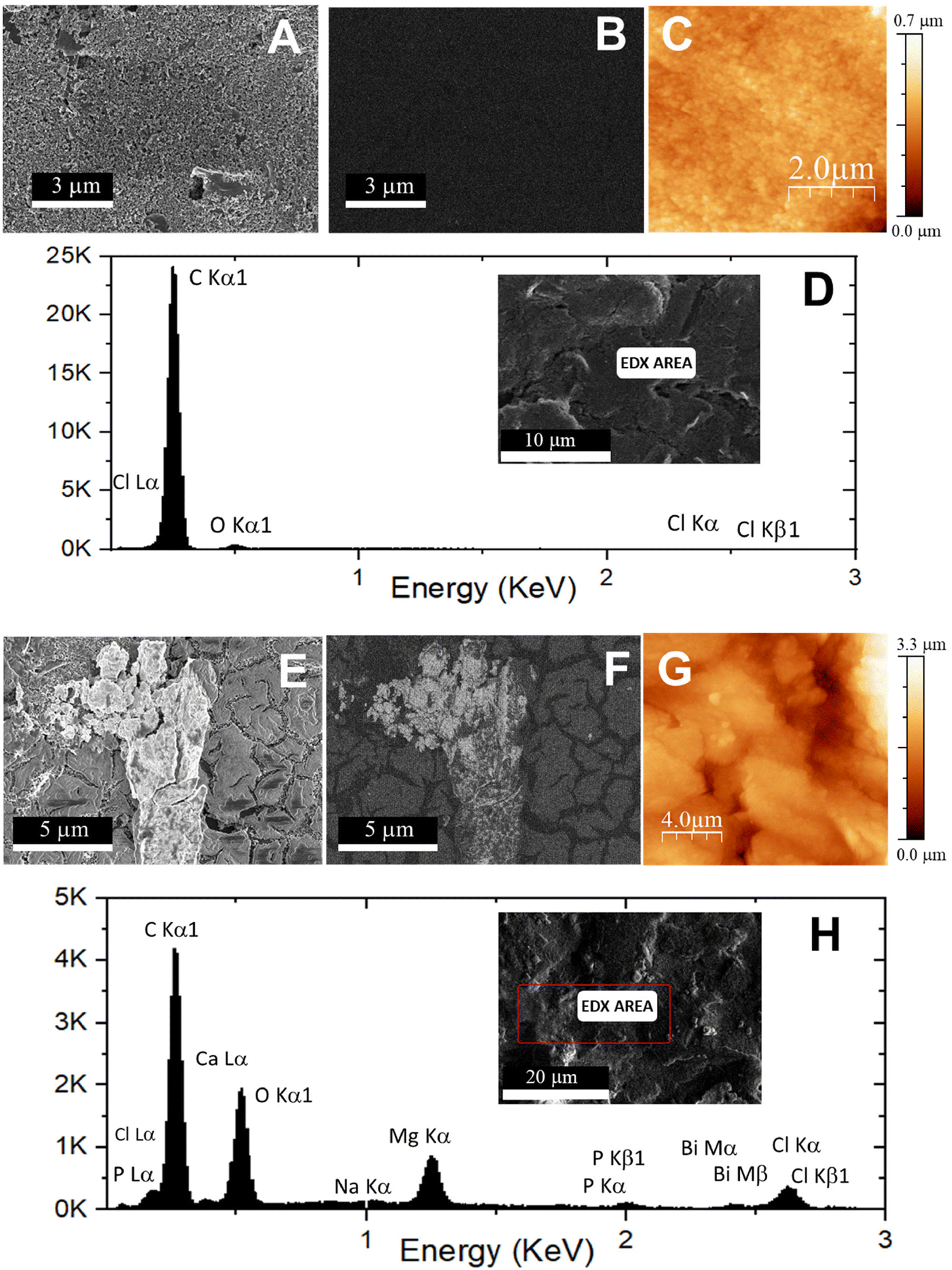 | ||
| Fig. 6 Secondary electron images (SEM, A and E), backscattered electron images (BSE, B and F), EDX images (D and H), and AFM topographic images (C and G) of the CSPE electrode (A–D) and the probe-SH/FLBH/CSPE platform (E–H). | ||
Regarding AFM characterization, the results demonstrate, as expected, a more homogeneous and flatter surface for the bare CSPE (Fig. 6C) compared to the probe-SH/FLBH/CSPE (Fig. 6G), where several bismuthene hexagons are observable. The same characterization techniques applied here were also performed for the FLBH/CSPE, with the results provided in the ESI.†
Raman characterization of the CSPE (black line), the FLBH/CSPE platform (red line), and the probe-SH/FLBH/CSPE platform (blue line) is shown in Fig. S6 of the ESI.† Due to the electrode surface, the characteristic bands of carbon at 1343 cm−1 and 1576 cm−1 can be clearly observed in all the platforms.34 Furthermore, in all spectra, a not very intense signal is observed at approximately 2680 cm−1, which corresponds to an overtone of the D band (G′ band), which indicates that the solid does not present a high structural order. In the cases of the FLBH/CSPE and the probe-SH/FLBH/CSPE, bands at 90 cm−1 and 110 cm−1, characteristic of bismuthene, are observed. Specifically, these peaks correspond to the two first-order Raman-active modes for bismuthene, Eg and A1g, respectively. Finally, for the probe-SH/FLBH/CSPE, characteristic vibration signals of the DNA phosphate backbone appear at 392 cm−1, 801 cm−1, and 1052 cm−1, although the adenine, guanine, cytosine, and thymine active Raman modes are observed at 516 cm−1, 599 cm−1, 761 cm−1, and 1151 cm−1, respectively.35
X-ray photoelectron spectroscopy (XPS) analysis was carried out (Fig. 7) to shed more light on whether the modifications performed to prepare the biosensing platform were correctly developed. This technique brought to light survey spectra of the unmodified and modified electrodes both presenting the characteristic XPS C 1s, Bi 4f, N 1s, B 1s, F 1s, or S 2p core level regions that agree with the functionalization being realized. First, the fit of the XPS C 1s surface spectrum for the commercial electrode (CSPE, Fig. 7A) afforded two main components at binding energy (BE) values of 284.4 eV and 286.8 eV, which may be assigned to a mixture of contributions from C sp2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and C sp3 (C–C, C–H) at lower BE and a CO contribution at higher BE, respectively. The incorporation of bismuthene (FLBHs) on the surface of the CSPE (FLBH/CSPE, Fig. 7B) could be easily confirmed by the appearance of two new peaks due to the splitting of Bi 4f in the fit of the XPS Bi 4f core level region (at BE of 159.8 eV with its split peak shifted by 5.3 eV). The survey analysis of the probe-SH/FLBH/CSPE (Fig. 7C) showed a peak in the fit of the XPS N 1s (at BE 400.2 eV) that could be attributed to the amide group from the incorporated DNA and two new peaks in the fit of the XPS C 1s (Fig. S7 in the ESI,† at BE values of 285.3 eV and 290.3 eV) attributed to the amide (–N–CO–) and imine (CN) groups and the π–π stacking of nucleic bases, respectively, from the anchored DNA. Finally, the presence of BDP-NaSO3 in the BDP/probe-SH/FLBH/CSPE (Fig. 7D–G), incorporated during the last step, could be determined by the detection of peaks corresponding to the nitro (–NO2, BE of 407.6 eV) and sulfonate (–SO3, BE of 169.1 eV with a split peak shifted by 1.18 eV that could be observed) groups, and the XPS peak fits of fluorine (BE of 687.0 eV) and boron (BE of 191.6 eV) atoms.
C) and C sp3 (C–C, C–H) at lower BE and a CO contribution at higher BE, respectively. The incorporation of bismuthene (FLBHs) on the surface of the CSPE (FLBH/CSPE, Fig. 7B) could be easily confirmed by the appearance of two new peaks due to the splitting of Bi 4f in the fit of the XPS Bi 4f core level region (at BE of 159.8 eV with its split peak shifted by 5.3 eV). The survey analysis of the probe-SH/FLBH/CSPE (Fig. 7C) showed a peak in the fit of the XPS N 1s (at BE 400.2 eV) that could be attributed to the amide group from the incorporated DNA and two new peaks in the fit of the XPS C 1s (Fig. S7 in the ESI,† at BE values of 285.3 eV and 290.3 eV) attributed to the amide (–N–CO–) and imine (CN) groups and the π–π stacking of nucleic bases, respectively, from the anchored DNA. Finally, the presence of BDP-NaSO3 in the BDP/probe-SH/FLBH/CSPE (Fig. 7D–G), incorporated during the last step, could be determined by the detection of peaks corresponding to the nitro (–NO2, BE of 407.6 eV) and sulfonate (–SO3, BE of 169.1 eV with a split peak shifted by 1.18 eV that could be observed) groups, and the XPS peak fits of fluorine (BE of 687.0 eV) and boron (BE of 191.6 eV) atoms.
 | ||
| Fig. 7 X-ray photoelectron spectroscopy (XPS) results of a bare CSPE (A), the FLBH/CSPE platform (B), the probe-SH/FLBH/CSPE platform (C) and the BDP/probe-SH/FLBH/CSPE (D, E, F and G). | ||
EIS studies of each step in the development of the biosensing platform were performed in a solution containing 10 mM K3Fe(CN)6 and 10 mM K4Fe(CN)6 in 0.1 M PB at pH 7.0. The results obtained are represented in the Nyquist plot shown in Fig. S8,† where the diameter of the curve corresponds to the electron transfer resistance (Rct) on the electrode surface. As can be observed, the bare CSPE (black line) shows an Rct value of 109.66 Ω. After modification with FLBHs (FLBH/CSPE, red line), the Rct value increases to 235.69 Ω, due to the semiconducting character of bismuthene as a semi-metallic material, presenting a lower conductivity than carbon at the electrode surface. Another factor affecting the charge transfer between the electrode surface and the electrolyte is the increase in interfacial resistivity generated by introducing a new interface with the modification of the CSPE with FLBHs. After immobilization of the probe (probe-SH/FLBH/CSPE, blue line), a new semicircle is observed in the Nyquist plot. This occurs when there are two different layers on the electrode surface, because there has not been a complete and homogeneous coating after the immobilization of the probe. The first semicircle in the diagram corresponds to the innermost layer, which is that of the FLBHs. The second semicircle corresponds to the outermost layer and the last layer on the electrode surface, which in combination is the immobilized probe–SH probe sequence. In addition, a straight line is observed after the second semicircle, which is given by the impedance to mass transfer and thus to the migration of the redox couple to the last layer on the electrode surface.36,37 An increase in impedance of up to 459.02 Ω is observed, due to the electronegative nature of the DNA, so that an electrostatic repulsion occurs with the redox pair [Fe(CN)6]3−/4−, hindering the electronic transfer and demonstrating the correct immobilization of the probe–SH sequence. Each of the stages has been matched to the corresponding electrical circuit.
These results demonstrate that the electrode nano-constructtion with bismuthene hexagons (FLBHs) has been highly efficient, allowing immobilization of the DNA probe (probe-SH) on the electrode.
SARS-CoV-2 virus detection
After verifying the development of the correct biosensing platform, the ability of the biosensor to detect the SARS-CoV-2 virus by its genetic code was evaluated. For that, the probe-SH/FLBH/CSPE platform was incubated with the specific SARS-CoV-2 virus sequence (CoV-2C) under optimal experimental conditions. The hybridization event was electrochemically detected using differential pulse voltammetry (DPV), following the electrochemical oxidation response of BDP-NaSO3 accumulated on the platform by direct adsorption. Fig. 8 shows the bar diagram of the current intensity recorded before (probe-SH/FLBH/CSPE, blue bar) and after hybridization with the complementary sequence (CoV-2C/probe-SH/FLBH/CSPE, yellow bar) and with a non-complementary sequence (CoV-2NC/probe-SH/FLBH/CSPE, purple bar). As a control, the electrochemical response of BDP-NaSO3 on the FLBH/CSPE (grey bar) was also recorded. It can be observed that BDP-NaSO3 is hardly adsorbed on the FLBH/CSPE surface. After the immobilization of the probe on the CSPE surface (probe-SH/FLBH/CSPE), the current intensity recorded increases due to the higher accumulation of BDP-NaSO3 on the DNA probe. However, this signal is lower than the one observed after hybridization with the complementary sequence (CoV-2C/probe-SH/FLBH/CSPE), probably due to its high affinity for the double-stranded DNA. Furthermore, when hybridized with a non-complementary DNA sequence (CoV-2NC/probe-SH/FLBH/CSPE), there is no increase in the electrochemical signal compared with the one obtained before the hybridization event, confirming that no hybridization event has occurred, which verifies the selectivity of the biosensor.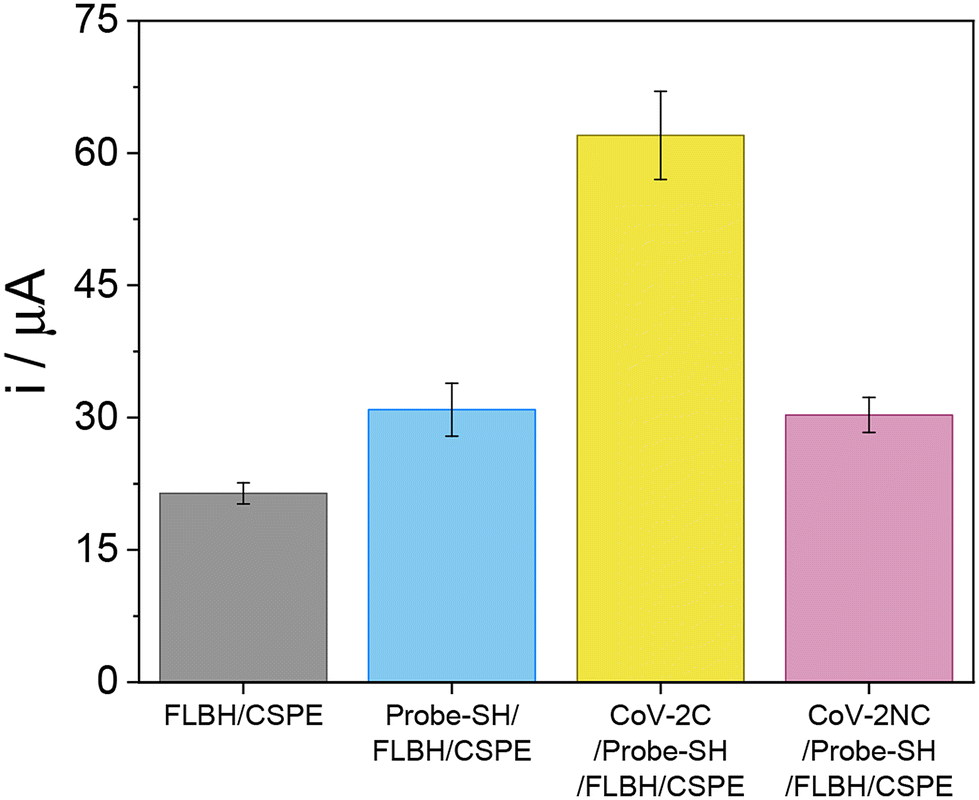 | ||
| Fig. 8 Bar diagram of the electrochemical response obtained after the accumulation of BDP-NaSO3 on a CSPE modified with FLBHs (FLBH/CSPE, grey bar) and for the biosensing platform developed before (probe-SH/FLBH/CSPE, blue bar) and after the hybridization with the SARS-CoV-2 complementary sequence (CoV-2C/probe-SH/FLBH/CSPE, yellow bar) or with a non-complementary sequence (CoV-2NC/probe-SH/FLBH/CSPE, purple bar). | ||
These results indicate that the synthesized BDP-NaSO3 clearly allows the detection of the hybridization event and, therefore, the developed biosensor can be used for the detection of a specific sequence of SARS-CoV-2.
Afterwards, the ability of the developed biosensor to detect the SARS-CoV-2 virus and the selectivity of the device were tested. For that, as described in detail in the Experimental section, the probe-SH/FLBH/CSPE platform was incubated with different concentrations of up to 100 fM of the specific SARS-CoV-2 sequence (CoV-2C). As can be observed in Fig. 9A, the biosensor response increases as the concentration of the SARS-CoV-2 sequence increases. This increase is linear to the SARS-CoV-2 concentration and fits the linear equation i = 1.53 ± 0.05 × [CoV-2C] + 44.7 ± 2.6 (R = 0.992). The analytical properties of the biosensor were obtained from the linear plot of the calibration curve. The limit of detection (LOD) and quantification (LOQ) were estimated using the 3 Sb m−1 and 10 Sb m−1 criteria, respectively, where Sb is the standard deviation of the background signal (probe-SH/FLBH/CSPE). The LOD and LOQ values were calculated to be 5.10 and 17.0 fM, respectively. A sensitivity of 1.57 μA fM−1 and a linear range of 17.0–100 fM were obtained. A relative standard deviation (RSD) of 2% was estimated for a 10.0 fM concentration of CoV-2C under reproducibility conditions. We also evaluated the repeatability of the biosensor by measuring its response 5 times and it was found to be 0.30%.
 | ||
| Fig. 9 Calibration plot (A) of the current intensity recorded versus the SARS-CoV-2 RdRp gene (CoV-2C) concentration (up to 100 fM). Bar diagram (B) of the biosensor's response for 10.0 μL of non-amplified nasopharyngeal swab samples from a SARS-CoV-2 infected patient (Cts 19, dark blue bar) and a non-infected patient (negative control, light blue bar). Data are presented as the mean ± standard deviation (n = 3). Statistical analysis was performed using Student's t-test with a p value of < 0.05 (sig.). | ||
It is worth mentioning the high stability of the biosensor response, which remains nearly stable for one month under ambient conditions. The selectivity of the biosensor was also assessed by recording the response to samples containing the SARS-CoV-2 sequence (10.0 fM) in the absence and presence of other viral sequences, such as SARS-CoV-1 and influenza A (H7N9) (each at a final concentration of 10.0 fM). As shown in Fig. S9 in the ESI,† the presence of these potentially interfering viral sequences does not affect the biosensor's response to the SARS-CoV-2 virus.
SARS-CoV-2 direct detection in COVID-19 patient samples
Building on the successful detection of synthetic sequences, we advanced our methodology to detect SARS-CoV-2 directly in nasopharyngeal swab samples from COVID-19 patients without any amplification process. These samples were previously treated and analyzed using RT-qPCR at Hospital Ramón y Cajal, providing a benchmark for validating the biosensor. Specifically, two different nasopharyngeal samples from an infected patient (Cts 19) and a non-infected patient (negative control) were used. Fig. 9B shows the signals obtained for both samples. The biosensor response was significantly higher for the infected patient sample (12.6 ± 0.6 μA) compared to the non-infected patient sample (1.0 ± 0.1 μA). Statistical analysis confirmed that the infected patient samples were significantly different from the control with a Student's t-test p-value of 0.0006234, indicating a confidence interval of 95%. Hence, we believe that the proposed biosensor has adequate sensitivity to be employed as a screening method to rapidly discriminate between infected (viral load at least 19 Cts) and non-infected patient samples without the need for any amplification process.Table S1 in the ESI† shows biosensors described in the literature for the specific detection of the SARS-CoV-2 virus. The analytical parameters of the electrochemical biosensor developed in this work compare well with those reported in the literature for similar platforms, achieving better limit of detection values and having the applicability of detecting the SARS-CoV-2 virus in human samples without the need for amplification, proving that it could be a rapid, simple, sensitive and great practical alternative for virus detection.
Experimental
Reagents and apparatus
Synthetic DNA SARS-CoV-2 sequences used in this work are listed in Table 1 and comprise the capture probe, a complementary sequence (CoV-2C), a non-complementary (CoV-2NC) sequence, and potential interferent sequences from other viruses such as influenza A (H7N9) and SARS-CoV-1. The capture probe is a single-stranded DNA sequence complementary to the analyte, a specific DNA sequence from the RNA-dependent RNA polymerase (RdRp) gene. Prior to use, stock solutions of thiol-modified probes (probe-SH) were treated with DTT and then purified by elution through a NAP-10 column of Sephadex G-25. Afterwards, the stock solutions of the thiol probes were prepared at a final concentration of 10.0 μM in 10.0 mM phosphate buffer (PB) at pH 7.0. The stock solutions of the analyte sequences were prepared in 10.0 mM phosphate buffer (pH 7.0) with 0.4 M NaCl. Aliquots of a few microliters of all stock solutions were stored at −20 °C.
| DNA sequences | Named | |
|---|---|---|
| Thiolated probe | 5′-SH-GCATCTCCTGATGAGGTTCCACCTG | Probe-SH |
| Analyte | 5′-CAGGTGGAACCTCATCAGGAGATGC | CoV-2C |
| Non-complementary | 5′-ACACTAGCCATCCTTACTGCGCTTCG | CoV-2NC |
| Interferent 1 | 5′-CCAGGT GGAAC ATCAT CCGGT GATGC | SARS-CoV-1 |
| Interferent 2 | 5′-TTAGTCATCTGCGGGAATGCAGCATTATCT | Influenza A |
The samples were obtained with the consent of all participants and approved by “Comité de Ética de la Investigación con Medicamentos del Hospital Universitario Ramón y Cajal”. Reference: 127-21.
Fluorescence spectra were recorded using a Cary Eclipse Varian spectrofluorimeter.
Electrochemical experiments were performed using an Autolab (PGSTAT 30) potentiostat attached to a PC with appropriate software (GPES and FRA) for the total control of the experiments and data acquisition. A screen-printed electrode connector (DropSens) was used as an interface. Screen-printed carbon electrodes (CSPEs) used as transducers were supplied by Metrohm. CSPEs contain a carbon working electrode, a silver pseudo-reference electrode, and a carbon counter electrode.
Spectra from high-resolution mass spectrometry (HRMS) were obtained using an Agilent Technologies 6120 Quadrupole LC/MS coupled with an SFC Agilent Technologies 1260 Infinity Series instrument for ESI (electrospray ionization)-MS. MassWorks software version 4.0.0.0 (Cerno Bioscience) was used for formula identification. MassWorks is an MS calibration software that calibrates isotope profiles to achieve high mass accuracy and enables elemental composition determination using conventional mass spectrometers of unit mass resolution, allowing highly accurate comparisons between calibrated and theoretical spectra.23–25
Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker AV-300 spectrometer, running at 300 MHz for 1H. Chemical shifts (δ) are reported in ppm relative to residual solvent signals (CDCl3: 7.26 ppm for 1H NMR). Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet), coupling constant (Hz) and integration. Solid-state 13C (100.61 MHz) CPMAS NMR spectra were obtained using a Bruker AV-400 WB spectrometer at 300 K with a 4 mm triple-channel probe head (BL4 X/Y/1H). Samples were carefully packed in a 4 mm diameter cylindrical zirconia rotor with Kel-F end-caps. Operating conditions involved 2.75 μs 90° 1H pulses and decoupling the field strength of 90.9 kHz achieved by using a TPPM sequence. The rotor spin rate was set at 10 kHz. A relaxation delay of 4 s and a contact time of 3 ms were used. 13C spectra were originally referenced to an adamantane sample and then the chemical shifts were recalculated to Me4Si [for the CH2 group, δ(adamantane) = 29.5 ppm].
X-Ray powder diffraction (XRPD) was used for the characterization of the FLBHs. PXRD patterns were recorded using a Bruker D8 Advance instrument with Cu Kα radiation and a rapid detector (lynxeye).
Raman spectroscopic characterization of the FLBHs was carried out using a confocal Raman microscope with a spectral resolution of 0.02 cm−1 coupled with an AFM instrument (Witec ALPHA 300RA) with laser excitation at 532 nm and a 100× objective lens (NA = 0.95). The incident laser power was 0.5 mW. The optical diffraction resolution was about 200 nm laterally and 500 nm vertically. The samples were mounted on a piezo-driven scan platform with 4 nm lateral and 0.5 nm vertical positioning accuracy, and also equipped with an active vibration isolation system (0.7–1000 Hz). The spectra and images were processed and analyzed with WiTec Project Plus 2.08 software.
Transmission electron microscopy (TEM) images of the FLBHs were obtained using a JEOL JEM 2100 FX TEM system with an accelerating voltage of 200 kV. The microscope has an ORIUS SC1000 multi-scan charge-coupled device (CCD) camera and an OXFORD INCA X-ray energy dispersive spectroscopy (XEDS) microanalysis system. For the preparation of TEM samples, the product obtained by centrifugation was dispersed in CHCl3 and deposited on lacey formvar/carbon copper grids (300 mesh).
Scanning electron microscopy (SEM) images of the FLBHs were obtained using an FE-SEM Hitachi S-4700 instrument operating at an accelerating voltage of 20 kV.
X-ray photoelectron spectroscopy (XPS) experiments reported in this paper were carried out under ultra-high vacuum (UHV) conditions. The experimental chamber has a base pressure of 2 × 10−10 mbar and is equipped with an Mg anode X-ray source whose Kα emission line produces photons of energy hν = 1253.6 eV, which are used for X-ray photoemission spectroscopy (XPS) measurements. For the analysis of the XPS peaks, the contribution of the Mg Kα intrinsic line has been eliminated by deconvoluting the peaks with an iterative Richardson–Lucy algorithm that was applied until reaching a maximum in the Shannon entropy.38 The Mg Kα satellites have also been removed with the help of an automated algorithm employing constrained penalized spline fitting.39 The deconvoluted XPS spectra obtained in this way are equivalent to the spectra acquired with a monochromatic source. A hemispherical energy analyzer (LEYBOLD LHS10) was used. The pass energy of the analyzer was set to 50 eV for the XPS measurements to resolve 0.7 eV.
Procedures
Synthesis of 5,5-difluoro-1,3,7,9-tetramethyl-10-(4-nitrophenyl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinine (2)26.
4-Nitrobenzaldehyde (0.6 g, 4.0 mmol) and 2,4-dimethylpyrrole (0.76 g, 8.0 mmol) were dissolved in 350 mL of CH2Cl2 under an Ar atmosphere. One drop of TFA was added and the solution was stirred at room temperature overnight. When TLC monitoring (alumina; CH2Cl2) showed complete consumption of the aldehyde, a solution of DDQ (0.91 g, 4.0 mmol) in CH2Cl2 was added and further stirring was continued for 20 min. The reaction mixture was washed with water and the organic layer was separated, dried over MgSO4, filtered, and evaporated under reduced pressure. The compound was purified by flash column chromatography over alumina (CH2Cl2). A brown powder was thus obtained and 5 mL of N,N-diisopropylethylamine was dissolved in 200 mL of toluene under an Ar atmosphere. Then, 5 mL of BF3·Et2O was added and the solution was stirred at room temperature for 30 min. The reaction mixture was washed with water and the organic layer was separated, dried over MgSO4, filtered, and evaporated. The compound was purified by silica gel column chromatography (Cy

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 1:1) to give an orange solid (590 mg, 40% yield). This non-water-soluble BDP is called BDP-NO2 in this work.
:CH2Cl2 = 1:1) to give an orange solid (590 mg, 40% yield). This non-water-soluble BDP is called BDP-NO2 in this work.
Spectroscopic data are in concordance with those reported in the literature.26
1 H NMR (300 MHz, CDCl 3 ) δ: 8.39 and 7.54 (AA′BB′ system, 2H), 6.02 (s, 2H), 2.57 (s, 6H), 1.36 (s, 6H). ESI-HRMS calculated for C19H19BF2N3O2+ (M + H)+: 370.1533; found: 370.1462.
Synthesis of sodium 5,5-difluoro-1,3,7,9-tetramethyl-10-(4-nitrophenyl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinine-2,8-disulfonate (1)27.
A solution of chlorosulfonic acid (36 μ L, 0.54 mmol) in dry CH2Cl2 was added dropwise to a solution of BDP (2) (100 mg, 0.27 mmol) in dry CH2Cl2 (2 mL) for 10 min under N2 at −40 °C. An orange precipitate was formed as the solution mixture warmed slowly to room temperature. The disulfonic acid was isolated by vacuum filtration and treated with water. The aqueous solution was neutralized with NaHCO3 (49 mg, 0.54 mmol) (pH = 6), concentrated to 2 mL, and treated with brine. The desired product was reprecipitated afterwards to provide an orange powder (155 mg, quant. yield). This water-soluble BDP is called BDP-NaSO3 in this manuscript.

Spectroscopic data are in concordance with those reported in the literature.27
1 H NMR (300 MHz, D 2 O) δ: 8.50 and 7.72 (AA′BB′ system, 2H), 2.77 (s, 6H), 1.64 (s, 6H). ESI-HRMS calculated for C19H17BF2N3Na2O8S2+ (M + H)+: 574.0308; found: 574.0282.
The intrinsic binding constants, Kb, were determined using eqn (1):40–42
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εa − εf) | (1) |
The magnitude of hypochromicity in the presence of DNA has been correlated with the overall binding strength and was calculated as:
| Hypochromicity % = (εb − εf/εf) × 100 |
On the other hand, the activation constant, Ksv, values were calculated using the Stern–Volmer equation (eqn (2)):32
| F0/F = 1 + Ksv·[DNA] | (2) |
Electrochemical studies were carried out by modifying CSPEs via drop-casting onto them 10.0 μL of 1.00 mM ds-ctDNA or ss-ctDNA solution, followed by air-drying. Afterwards, the resultant modified electrodes were soaked in sterilized water for 30 min and rinsed with water to remove any un-adsorbed DNA. These modified electrodes are denoted in the text as ds-ctDNA/CSPE or ss-ctDNA/CSPE, respectively.
Electrode modification. Carbon screen-printed electrodes (CSPEs) were nanostructured with the FLBHs by spraying them onto the electrode surface with an airbrush for 60 s. During the process, the electrode was heated at 90 °C on a hot plate, which allows for the fast evaporation of the solvent. Afterwards, 10.0 μL of the corresponding capture probe (probe-SH) of 10.0 μM concentration was deposited on the FLBH/CSPE by drop-casting and kept at room temperature for 24 h. Finally, it was soaked in Milli-Q water for 30 min. The resulting modified electrodes are named probe-SH/FLBH/CSPE.
Hybridization and electrochemical detection. The modified electrode was hybridized with 10.0 μL of the analyte solution at different concentrations, using 10.0 mM phosphate buffer (PB) at pH 7.0 supplemented with 0.4 M NaCl as buffer solution for 1 h at 40 °C in a humidity chamber under stirring. The analyte solution may contain a complementary sequence corresponding to SARS-CoV-2 (CoV-2C) or a non-complementary sequence used as a control (CoV-2NC). Electrochemical detection was carried out using the synthesized BDP-NaSO3 as the electrochemical indicator. 10.0 μL of 20.0 mM BDP-NaSO3 was dropped on the surface of the modified and hybridized electrode for 60 min. Finally, the electrodes were rinsed with sterile water, placed in 0.1 M PB at pH 7.0, and differential pulse voltammograms (DPVs) were immediately recorded.
For the detection process, 5.00 μL of the RNA sample was deposited on the electrode modified with the capture probe (probe-SH/FLBH/CSPE) and allowed to hybridize for 1 hour at 40 °C. Following hybridization, BDP-NaSO3 was accumulated on the hybridized DNA layer as previously described and differential pulse voltammetry (DPV) measurements were performed.
Statistical analysis
The results are presented as the mean ± standard deviation of three different measurements (n = 3). Statistical analysis was performed using Excel software. For comparison of the means of two groups, a Student's t-test for independent samples with a confidence interval of 95% was performed.Conclusions
In this study, we demonstrated that: (i) the FLBHs serve as an excellent platform for integrating biocompatible elements, significantly enhancing electrochemical sensitivity; (ii) thiolated single-stranded DNA attaches efficiently to FLBHs, enabling effective recognition of its complementary sequence; (iii) BDP-NaSO3 can be incorporated into DNA interacting as an intercalator via π–π stacking within the DNA structure, resulting in a notable enhancement of electrochemical performance; and (iv) the construction of an electrochemical biosensor that integrates FLBHs and a thiolated capture probe with BDP-NaSO3 produces a highly efficient biosensor. This biosensor exhibits a stable response under ambient conditions for over one month and shows excellent selectivity for the detection of a specific virus. Consequently, the fabrication of real-world sensor devices using this approach appears to have high potential.Author contributions
Laura Gutiérrez-Gálvez: methodology, formal analysis, data curation, experimental investigation, and writing (original draft preparation, review and editing). Estefanía Enebral-Romero: formal analysis, data curation, experimental investigation, and writing (original draft preparation, review, and editing). Miguel Ángel Valle Amores: methodology, formal analysis, data curation, experimental investigation, and writing (review and editing). Clara Pina Coronado: data curation, formal analysis, and experimental investigation. Íñigo Torres: methodology, data curation, formal analysis, experimental investigation, and writing (review and editing). David López-Diego: experimental investigation and writing (review and editing). Mónica Luna: resources, writing (review and editing), supervision, project administration and funding acquisition. Alberto Fraile: resources, writing (review and editing), supervision, project administration and funding acquisition. Félix Zamora: resources, writing (review and editing), supervision, project administration and funding acquisition. José Alemán: resources, writing (review and editing), supervision, project administration and funding acquisition. Jesús Álvarez: experimental investigation, resources, writing (review and editing), supervision, project administration and funding acquisition. María José Capitán: experimental investigation, resources, writing (review and editing), supervision, project administration and funding acquisition. Encarnación Lorenzo: resources, writing (review and editing), supervision, project administration and funding acquisition. Tania García-Mendiola: conceptualization, formal analysis, resources, data curation, writing (original draft preparation, review, and editing), supervision, project administration and funding acquisition.Data availability
The data supporting the findings of this study are available from the corresponding author upon request. Source data are provided in the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Spanish Ministry of Economy and Competitiveness (PID2020-116728RB-I00, RED2022-134120-T, PID2023-150844OB-I00, PID2022-138908NB-C31, and PID2021-122299NB-I00), the Community of Madrid (REACT-UE NANOCOV-CM and Y2020/NMT6469), the Spanish Ministry of Science, Innovation and Universities (TED2021-129738B-I00, TED2021-130470B-I00, and TED2021-129999B-C32) and the “María de Maeztu” Programme for Units of Excellence in R&D, CEX2023-001316-M. Laura Gutiérrez-Gálvez gratefully acknowledges the financial support of a Formación del Profesorado Universitario (FPU) grant from the Spanish Ministry of Universities (FPU19/06309). Estefanía Enebral-Romero received financial support from the “Nanotecnología para detección del SARS-CoV-2 y sus variantes, NANOCOV” project and a contract as a pre-doctoral researcher funded by a grant CEX2020-001039-S, supported by MCIN/AEI/10.13039/501100011033. We also acknowledge María U. González and Raquel Alvaro for their help with fluorescence microscopy and EDAX mapping, respectively, and the services of the MiNa Laboratory at IMN and funding from CM (project S2018/NMT-4291 TEC2SPACE), MINECO (project CSIC13-4E-1794) and the EU (FEDER, FSE). We also acknowledge the “Servicio de Microbiología, Hospital Universitario Ramón y Cajal and Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS)” for providing the clinical samples. We acknowledge the support from the “(MAD2D-CM)-UAM” project funded by Comunidad de Madrid by the Recovery, Transformation and Resilience Plan and by NextGenerationEU from the European Union.References
- A. Treibs and F. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- R. G. Clarke and M. J. Hall, in Advances in Heterocyclic Chemistry, Elsevier, 2019, vol. 128, pp. 181–261 Search PubMed.
- G. M. Ziarani, R. Moradi, N. Lashgari and H. G. Kruger, Metal-free synthetic organic dyes, Elsevier, Amsterdam, Natherlands, 2018 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 2021, 1809–1824 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 2015, ejoc.201590085 CrossRef.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- A. Kowalczyk, Curr. Opin. Electrochem., 2020, 23, 36–41 CrossRef CAS.
- C. Pina-Coronado, Á. Martínez-Sobrino, L. Gutiérrez-Gálvez, R. Del Caño, E. Martínez-Periñán, D. García-Nieto, M. Rodríguez-Peña, M. Luna, P. Milán-Rois, M. Castellanos, M. Abreu, R. Cantón, J. C. Galán, T. Pineda, F. Pariente, Á. Somoza, T. García-Mendiola, R. Miranda and E. Lorenzo, Sens. Actuators, B, 2022, 369, 132217 CrossRef CAS PubMed.
- E. Martínez-Periñán, T. García-Mendiola, E. Enebral-Romero, R. del Caño, M. Vera-Hidalgo, M. Vázquez Sulleiro, C. Navío, F. Pariente, E. M. Pérez and E. Lorenzo, Biosens. Bioelectron., 2021, 189, 113375 CrossRef PubMed.
- T. García-Mendiola, S. Requena-Sanz, E. Martínez-Periñán, I. Bravo, F. Pariente and E. Lorenzo, Electrochim. Acta, 2020, 353, 136522 CrossRef.
- P. Williamson, P. Piskunen, H. Ijäs, A. Butterworth, V. Linko and D. K. Corrigan, ACS Sens., 2023, 8, 1471–1480 CrossRef CAS PubMed.
- R. Del Caño, T. García-Mendiola, D. García-Nieto, R. Álvaro, M. Luna, H. A. Iniesta, R. Coloma, C. R. Diaz, P. Milán-Rois, M. Castellanos, M. Abreu, R. Cantón, J. C. Galán, T. Pineda, F. Pariente, R. Miranda, Á. Somoza and E. Lorenzo, Microchim. Acta, 2022, 189, 171 CrossRef PubMed.
- T. García, M. Revenga-Parra, H. D. Abruña, F. Pariente and E. Lorenzo, Anal. Chem., 2008, 80, 77–84 CrossRef PubMed.
- Y. Xie, L. Huang, Y. Qi, J. Hu, L. Song and H. Feng, Green Chem., 2022, 24, 1978–1982 RSC.
- L. Gutiérrez-Gálvez, D. García-Fernández, M. D. Barrio, M. Luna, Í. Torres, F. Zamora, C. Navío, P. Milán-Rois, M. Castellanos, M. Abreu, R. Cantón, J. C. Galán, Á. Somoza, R. Miranda, T. García-Mendiola and E. Lorenzo, Talanta, 2024, 269, 125405 CrossRef PubMed.
- M. Adamovski, A. Zając, P. Gründler and G.-U. Flechsig, Electrochem. Commun., 2006, 8, 932–936 CrossRef CAS.
- T. Romann, V. Grozovski and E. Lust, Electrochem. Commun., 2007, 9, 2507–2513 CrossRef CAS.
- N. Yu, Z. Wang, J. Zhang, Z. Liu, B. Zhu, J. Yu, M. Zhu, C. Peng and Z. Chen, Biomaterials, 2018, 161, 279–291 CrossRef CAS PubMed.
- G. Alessio Verni, B. Long, F. Gity, M. Lanius, P. Schüffelgen, G. Mussler, D. Grützmacher, J. Greer and J. D. Holmes, RSC Adv., 2018, 8, 33368–33373 RSC.
- Y. Wang and M. Gu, Anal. Chem., 2010, 82, 7055–7062 CrossRef CAS PubMed.
- N. Ochiai, K. Sasamoto and K. MacNamara, J. Chromatogr., A, 2012, 1270, 296–304 CrossRef CAS PubMed.
- H. Ho, R. Lee, C. Chen, S. Wang, Z. Li and M. Lee, Rapid Commun. Mass Spectrom., 2011, 25, 25–32 CrossRef CAS PubMed.
- T. Matsumoto, Y. Urano, T. Shoda, H. Kojima and T. Nagano, Org. Lett., 2007, 9, 3375–3377 CrossRef CAS PubMed.
- L. Li, J. Han, B. Nguyen and K. Burgess, J. Org. Chem., 2008, 73, 1963–1970 CrossRef CAS PubMed.
- A. B. Nepomnyashchii and A. J. Bard, Acc. Chem. Res., 2012, 45, 1844–1853 CrossRef CAS PubMed.
- P. Chauhan, K. Chu, N. Yan and Z. Ding, J. Electroanal. Chem., 2016, 781, 181–189 CrossRef CAS.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- J. B. Chaires, N. Dattagupta and D. M. Crothers, Biochemistry, 1982, 21, 3933–3940 CrossRef CAS PubMed.
- P. Paul and G. Suresh Kumar, J. Fluoresc., 2012, 22, 71–80 CrossRef CAS PubMed.
- I. Torres, A. M. Villa-Manso, M. Revenga-Parra, C. Gutiérrez-Sánchez, D. A. Aldave, E. Salagre, E. G. Michel, M. Varela, J. Gómez-Herrero, E. Lorenzo, F. Pariente and F. Zamora, Appl. Mater. Today, 2022, 26, 101360 CrossRef.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- B. Prescott, W. Steinmetz and G. J. Thomas, Biopolymers, 1984, 23, 235–256 CrossRef CAS PubMed.
- Z. O. Uygun and H. D. Ertuğrul Uygun, Sens. Actuators, B, 2014, 202, 448–453 CrossRef CAS.
- T. Yang, S. Wang, H. Jin, W. Bao, S. Huang and J. Wang, Sens. Actuators, B, 2013, 178, 310–315 CrossRef CAS.
- J. Verbeeck and G. Bertoni, Ultramicroscopy, 2009, 109, 1343–1352 CrossRef CAS PubMed.
- J. J. De Rooi, N. M. Van Der Pers, R. W. A. Hendrikx, R. Delhez, A. J. Böttger and P. H. C. Eilers, J. Appl. Crystallogr., 2014, 47, 852–860 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- D. E. V. Schmechel and D. M. Crothers, Biopolymers, 1971, 10, 465–480 CrossRef CAS PubMed.
- T. Meehan, H. Gamper and J. F. Becker, J. Biol. Chem., 1982, 257, 10479–10485 CrossRef CAS PubMed.
- D. T. Breslin, C. Yu, D. Ly and G. B. Schuster, Biochemistry, 1997, 36, 10463–10473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05258g |
| This journal is © The Royal Society of Chemistry 2025 |