
Self-supporting poly(3,4-ethylenedioxythiophene) and Fe3C Co-decorated electrospun carbon nanofibers as Li2S supporters for lithium–sulfur batteries†
Na
Yang‡
,
Jiarui
Xue‡
,
Yuanxiao
Ji
,
Jiyuan
Zhang
,
Weiye
Zhang
,
Xuexia
He
,
Qi
Li
 ,
Zhibin
Lei
*,
Zonghuai
Liu
and
Jie
Sun
*
,
Zhibin
Lei
*,
Zonghuai
Liu
and
Jie
Sun
*
Key Laboratory of Applied Surface and Colloid Chemistry (MOE), Shaanxi Key Laboratory for Advanced Energy Devices, Shaanxi Engineering Laboratory for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, 620 West Chang'an Street, Xi'an, Shaanxi 710119, China. E-mail: zblei@snnu.edu.cn; jiesun@snnu.edu.cn
First published on 11th February 2025
Abstract
Lithium–sulfur (Li–S) batteries have attracted significant attention in recent years owing to their high theoretical energy density (2600 W h kg−1) and specific capacity (1675 mA h g−1), abundant reserves and environmental friendliness. However, the well-known poor electrical conductivity of sulfur/Li2S, shuttle effect of lithium polysulfides (LiPSs) and in situ formation of lithium dendrites during the cycling process extremely hinder the large-scale application of Li–S batteries. In this work, we designed and prepared poly(3,4-ethylenedioxythiophene) (PEDOT) and Fe3C nanoparticle co-decorated carbon nanofiber (CNF) membranes as self-supporting Li2S hosts to improve the electrochemical performance of Li–S batteries. The PEDOT coating layer firmly connected the Fe3C nanoparticles with the CNFs to form a conductive network, significantly enhancing the conductivity of the cathode. Simultaneously, the strong chemical adsorption of PEDOT and the catalytic effect of Fe3C effectively accelerated the conversion kinetics of LiPSs, thereby improving the battery performance. As a result, the optimized PEDOT@CNFs@Fe3C-16wt%/Li2S composite cathode achieved a high initial specific capacity of 816 mA h g−1 at a current density of 0.1 C. Furthermore, the cathode exhibited only 0.033% capacity decay per cycle at 0.5 C after 1000 cycles, showing its potential as a high-performance self-supporting flexible Li–S composite cathode.
 Jie Sun | Jie Sun received his B.S. degree and Ph.D. from Northwestern Polytechnical University in Materials Science and Engineering in 2008 and 2015, respectively. During his Ph.D. research period, he had a 2-year cooperation research experience in the Australian Centre for Microscopy and Microanalysis at the University of Sydney, Australia. In December 2015, he joined Shaanxi Normal University as an associate professor. His research interests include the synthesis, phase transformation and electrochemical applications of metal–oxide-based materials. |
1 Introduction
With the continuous development and progress of the global economy and technology, the problem of energy shortage is becoming increasingly prominent. Developing new high-performance energy storage devices is an important means to solve the energy problems. Lithium–sulfur (Li–S) batteries are a new type of electrochemical energy storage device with high theoretical specific capacity (1675 mA h g−1) and energy density (2600 W h kg−1), abundant sulfur storage and environmental friendliness.1–4 Based on the electrochemical reaction mechanism between sulfur and metallic lithium, the charging and discharging processes of Li–S batteries involve a multi-step redox reaction process, which leads to a series of problems, resulting in a decrease in specific capacity and stability.5,6 These problems include the serious volume change during the transformation between sulfur and Li2S, poor electrical conductivity of both discharge and charge products, shuttling of lithium polysulfides (LiPSs) during the cycling process, and the safety problems caused by lithium dendrites.7–12 Plenty of efforts have been made to overcome these shortcomings, including cathode design,13,14 separator functionalization,15 electrolyte additive16 and anode protection.17Compared with traditional sulfur cathode, the use of fully pre-lithiated Li2S as a cathode can effectively solve some of the above problems. As the final discharge product in Li–S batteries, Li2S possesses a low density (1.66 g cm−3) and high melting point (938 °C), which can also completely avoid volume expansion during the cycling process.18–21 Moreover, the use of Li2S as a cathode is helpful for the assembly of lithium anode-free Li–S batteries.22–24 However, the extremely low electronic conductivity of Li2S (10−14 S cm−1) makes it difficult to achieve high efficiency charge transfer during the cycling process, leading to high interface impedance.19 Moreover, the complex redox reactions with slow solid/liquid phase transition kinetics25 lead to significant obstacles in the commercialization of Li2S cathodes.
Conductive polymers have been widely used in the field of energy storage owing to their excellent reversibility during redox process, high conductivity in doped states, low cost, and superior mechanical properties.26–28 Commonly used conductive polymers include polyaniline (PANi), polypyrrole (PPy), poly(3,4-ethylenedioxythiophene) (PEDOT) and their derivatives. Among them, PEDOT has relatively high conductivity, which is related to the delocalization of π electrons in its conjugated molecules. The longer conjugated chains of PEDOT (N > 15) can form effective π–π electron delocalization, enabling efficient charge transfer along the polymer chain.29,30 The commercial PEDOT currently available for sale is mainly poly(3,4-ethylenedioxythiophene):poly (styrene sulfonate) (PEDOT:PSS), which is a micelle system composed of polycations and polyanions, whose electrical conductivity is relatively poor when compared with that of pure PEDOT. The pure PEDOT can be directly prepared by using solution polymerization, electrodeposition, and gas-phase oxidation polymerization methods.31–33 During the polymerization process, the added surfactant or oxidant determines the final form of PEDOT. By adjusting the concentration of EDOT monomers and the type of surfactant, the morphology of PEDOT can also be controlled.34 For Li–S batteries, PEDOT can be used as functionalized sulfur host and separator modification materials simultaneously.35–37 The unique dendritic network structure of PEDOT can effectively accelerate the electron transfer efficiency and improve the wettability of the electrodes. Simultaneously, sulfur functional groups have an excellent affinity to LiPSs, which is helpful in the acceleration of redox reaction kinetics.38 In addition, the excellent flexibility of PEDOT has broad application prospects and research values in the preparation and application of flexible self-supporting cathodes.39,40
Transition metal carbides have been widely utilized in the fields of energy storage and catalysis,41–44 of which Fe3C with an orthorhombic structure possesses high hardness and melting point, together with excellent electrical conductivity, that has been utilized as electrocatalysts,45 electrodes46 and photocatalysts.47 In the field of Li–S batteries, Fe3C has been found to possess strong adsorption and catalytic abilities towards LiPSs.48–50 In practical use, Fe3C is often composite with carbon materials because not only its preparation method is mainly based on carbothermal reduction reaction, but also the composite structure can significantly avoid adverse reactions between Fe3C particles and electrolytes, and prevent particle aggregation during the battery cycling process simultaneously.51
In this work, we design and prepare a PEDOT-coated Fe3C nanoparticle-decorated electrospun carbon nanofibers (CNFs) as a self-supported host to load Li2S as the composite cathode for Li–S batteries. The Fe3C nanoparticles in the prepared composites possess excellent catalytic activity towards LiPSs, while the PEDOT coating can effectively connect the dispersed Fe3C nanoparticles together to obtain a highly flexible conductive network. In addition, PEDOT can fully utilize its chemical adsorption capacity on LiPSs. After the loading of Li2S, the self-supporting PEDOT@CNFs@Fe3C/Li2S composite cathode can be obtained. The synthesis route of the composites is illustrated in Scheme 1. The PEDOT coating amount can be precisely adjusted through the regulation of the EDOT monomer addition amount. Further LiPSs catalytic and electrochemical performance of the assembled cells are also investigated in detail.
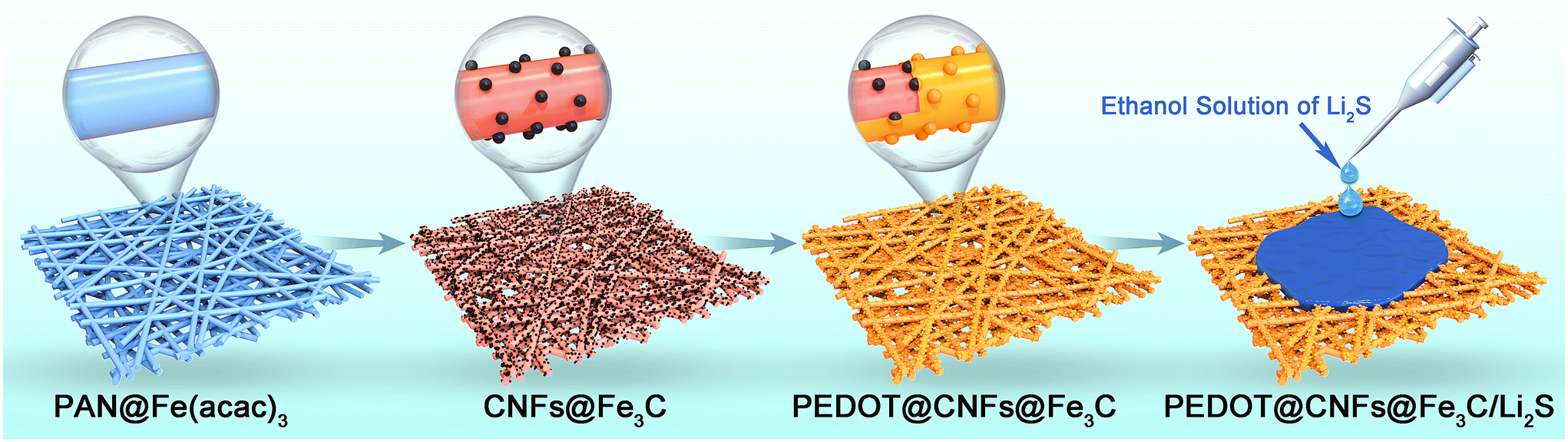 | ||
| Scheme 1 Preparation route of the PEDOT@CNFs@Fe3C/Li2S composite cathode for Li–S batteries. | ||
2 Experimental
2.1 Chemicals and materials
The 3,4-ethoxylene dioxy thiophene (97%), Ferric acetylacetonate (98%) and lithium sulfide were purchased from Shanghai Aladdin Co., Ltd. Anhydrous ethanol (AR) and hydrochloric acid (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Polyacrylonitrile (97%) was purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Acetylene black (AC) was purchased from Jiang Su XFNANO Co., Ltd. N-Methyl-2-pyrrolidone (AR) and N,N-dimethylformamide (AR) were purchased from Tianjin Kemio Chemical Reagent Co., Ltd. The polyvinylidene difluoride, Celgard 2400 separator, electrolyte for Li–S batteries and lithium metal were all purchased from Dodo Chem Co., Ltd. All the chemicals were directly used without further purification.2.2 Fabrication of CNFs@Fe3C composite fibrous membrane
The PAN/Fe(acac)3 composite nanofibers were first prepared by applying the electrospinning method. Specifically, 0.5 g polyacrylonitrile (PAN) and 0.5 g Ferric acetylacetonate (Fe(acac)3) were dissolved in 5 mL of N,N-dimethylformamide (DMF) and stirred at room temperature for 10 hours to obtain an orange red viscous spinning solution. The above spinning solution was transferred into a 10 mL syringe, followed by the performing of electrospin process. The spin rate, receiving electrode distance and receiving rate were set as 100 rpm, 16 cm and 0.12 mm min−1 respectively. Moreover, the positive and negative voltages were set to be +20 kV and −1.5 kV, respectively. After the electrospin process, the PAN@Fe(acac)3 fibrous film with orange colour could be obtained. The fibrous film was then calcinated at 200 °C for 2 hours to stabilize the structure. A further calcination process under 700 °C was carried out in a tube furnace for 1 hour under Ar atmosphere. The final CNFs@Fe3C composite fibrous membrane could then be obtained.2.3 Fabrication of PEDOT@CNFs@Fe3C composite fibrous membrane
A closed glass jar with a total volume of 500 mL was employed as the reaction vessel to prepare PEDOT@CNFs@Fe3C composite fibrous membrane. Specifically, 15 μL, 20 μL and 25 μL of EDOT monomer and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentrated hydrochloric acid were added to the beaker placed in the glass jar. The whole glass jar was then heated to 100 °C to promote the vapor polymerization of EDOT onto the surface of the CNFs@Fe3C composite membrane. After a 30 min reaction, the PEDOT@CNFs@Fe3C composite membrane can be obtained. The products were then cut into circular electrode slices with a diameter of 1 cm. The amount of PEDOT polymerization can be determined by calculating the mass difference before and after the polymerization process. For example, 15 μL EDOT monomer reacted with 50 mg of CNFs@Fe3C to obtain a final product with a total mass of 56.45 mg. The polymerized PEDOT amount was 6.45 mg with a mass ratio of 6.45 mg/56.45 mg = 11%. Such composites were named as PEDOT@CNFs@Fe3C-11wt% (P@CNFs@Fe3C-11wt%). With different EDOT monomer addition amounts, the P@CNFs@Fe3C-16wt% and P@CNFs@Fe3C-21wt% composites could also be obtained.
:1 concentrated hydrochloric acid were added to the beaker placed in the glass jar. The whole glass jar was then heated to 100 °C to promote the vapor polymerization of EDOT onto the surface of the CNFs@Fe3C composite membrane. After a 30 min reaction, the PEDOT@CNFs@Fe3C composite membrane can be obtained. The products were then cut into circular electrode slices with a diameter of 1 cm. The amount of PEDOT polymerization can be determined by calculating the mass difference before and after the polymerization process. For example, 15 μL EDOT monomer reacted with 50 mg of CNFs@Fe3C to obtain a final product with a total mass of 56.45 mg. The polymerized PEDOT amount was 6.45 mg with a mass ratio of 6.45 mg/56.45 mg = 11%. Such composites were named as PEDOT@CNFs@Fe3C-11wt% (P@CNFs@Fe3C-11wt%). With different EDOT monomer addition amounts, the P@CNFs@Fe3C-16wt% and P@CNFs@Fe3C-21wt% composites could also be obtained.
2.4 Preparation of PEDOT@CNFs@Fe3C/Li2S composite cathode
The ethanol solution of Li2S was first prepared in an Ar-filled glove box (water pressure <0.01 ppm, oxygen partial pressure <1 ppm) by dissolving 120 mg Li2S (99.5% purity) into 5 ml ethanol, followed by stirring sufficiently to form a saturated solution with a Li2S concentration of 24 mg mL−1. During the loading process of Li2S, 40 μL of the prepared Li2S ethanol solution was dropped onto the surface of the P@CNFs@Fe3C supporter, followed by pumping in the small transition bin of the glove box to volatilize all ethanol. The P@CNFs@Fe3C/Li2S composite cathode could then be obtained. Under this experimental condition, the areal loading amount of Li2S was calculated as 1.2 mg cm−2.2.5 Material characterization
A Rigaku Miniflex 600 powder X-ray Diffractometer was employed to determine the phase compositions of different P@CNFs@Fe3C composites. The radiation source was selected as Cu Kα with a characteristic wavelength of λ = 1.54056 Å. The scan range was set from 5° to 80° at a scan speed of 10° min−1. The surface morphology, microstructure and corresponding elemental distribution of different composites were characterized by using a SU8020 Field Emission Scanning Electron Microscope (FE-SEM) and a JEM-2800 Field Emission Transmission Electron Microscope (TEM) operated at 200 kV. Raman spectroscopy was collected by using a Renishaw inVia Reflex Raman spectrometer to evaluate the graphitization degree of carbon materials. The wavelength of the laser light source and scan range were set to 532 nm and 800–2000 cm−1, respectively. The AXIS ULTRA X-ray Photoelectron Spectroscopy (XPS) was employed to investigate the surface valence of elements before and after Li2S6 adsorption. The X-ray source was an Al Kα source with an energy of 1486.71 eV. Owing to the extreme sensitivity of Li2S6 to the air, all the tested samples were sealed in a glove box before testing. All the peaks were first calibrated using the standard peak of C 1s at 284.8 eV before the testing process. The obtained results were all fitted using the Casa XPS software.2.6 Li2S6 visible adsorption experiment
The elemental sulfur and lithium sulfide (Li2S) were first mixed with a molar ratio of 5:1, followed by adding the bis (trifluoromethane sulfonyl) imide (LiTFSI) in a solvent mixture of 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) (1:1 in volume) solution with 2.0% LiNO3 as an additive solution as the electrolyte. The suspension was stirred in a glove box for 24 hours to finally obtain a 7.5 mM Li2S6 solution. The CNFs@Fe3C and P@CNFs@Fe3C composites with different PEDOT coating amounts were placed in different glass bottles. Then, 2 mL of the prepared Li2S6 solution was added to each bottle separately. The color change in the solution after 30 min was recorded. Moreover, the UV-visible spectra of the LiPSs solution after adsorption were collected using a PerkinElmer Lambda 950 UV-VIS-NIR Spectrophotometer.
2.7 Li2S6 symmetrical cell assembly and electrochemical performance evaluation
The CNFs@Fe3C and P@CNFs@Fe3C composites with different PEDOT coating amounts were employed as both cathode and anode to assemble the Li2S6 symmetrical cells. Specifically, the cathode, Celgard 2400 membrane separator, 40 μL prepared Li2S6 solution and anode were placed successively, followed by a sealing process to obtain the symmetrical coin cell. The Cyclic Voltammetry (CV) test was performed on a Multi Autolab M204 electrochemical workstation at a scanning speed of 1 mV s−1 and a potential ranging from −1.0 V to 1.0 V.2.8 Li2S precipitation and dissolution experiment
The elemental sulfur and lithium sulfide (Li2S) with a molar ratio of 7:1 were weighed and added to the electrolyte. After stirring in a glove box for 24 hours, a 0.2 M Li2S8 solution could be obtained. The CNFs@Fe3C and P@CNFs@Fe3C-16%wt composites were employed as the cathode, with lithium metal foil as the anode and Celgard 2400 as the separator to assemble the coin cells. The prepared Li2S8 solution (20 μL) was dropped between the cathode and the separator, followed by adding another 20 μL electrolyte to the anode side. For the Li2S precipitation experiment, the assembled cells were first discharged galvanostatically at 0.112 mA until the potential dropped to 2.11 V. Then, the mode is switched to potentiostatically discharge at 2.10 V until the current density drops below 10−5 A. The current–time curves during the whole process were collected and fitted using exponential functions to obtain the deposition capacity and nucleation time of Li2S on different electrode surfaces. For the Li2S dissolution experiment, the cells are first discharged galvanostatically to 1.7 V at 0.112 mA and then charged at a voltage of 2.4 V until the current drops below 10−5 A. The current–time curves during the process were also collected accordingly.
2.9 Li–S battery assembly and electrochemical performance evaluation
For both regular and high loading amounts of Li2S, the assembly method of the Li–S coin cells was completely the same. By placing the cathode shell, self-assembly cathode, separator, lithium anode and anode shell in an Ar-filled glove box successively, the 15 μL electrolyte (the same as that used in the Li2S6 visible adsorption experiment) was added to the cathode side and anode side. The lithium anode foil used in this work possesses a thickness of 0.6 mm and a diameter of 16 mm. After sealing carefully, the assembled cells were left in the glove box for 10 hours before electrochemical performance testing.The Cyclic Voltammetry (CV) and Electrochemical Impedance Spectroscopy (EIS) curves of the assembled cells were collected on a MUL AUTOLAB M204 electrochemical workstation. During the CV testing, the voltage window was selected as 1.6 V–2.8 V at a scan speed of 0.1 mV s−1. In addition, the EIS was collected under the frequency range of 10 mHz–100 kHz and an amplitude of 5 mV. The Galvanostatic Charge–Discharge (GCD) curves for all the cathodes were collected on a LAND battery performance testing system under current densities ranging from 0.1 C to 2 C.
3 Results and discussion
3.1 Structural and morphology characterization of PAN/Fe(acac)3 and CNFs@Fe3C composite nanofiber membranes
Fig. S1† shows the structural and morphological characterization results of PAN/Fe(acac)3 and CNFs@Fe3C composite nanofiber membranes. It is noticed that the prepared PAN/Fe(acac)3 membranes exhibit a three-dimensional network structure with smooth surfaces and a diameter of approximately 200 nm, as shown in Fig. S1a and b.† After calcinating under the Ar atmosphere, the Fe(acac)3 can be successfully reduced to Fe3C with PAN transforming to CNFs simultaneously.52 A large number of nanoparticles also appear at the surface of CNFs@Fe3C after calcination, as shown in Fig. S1d and e.† The three-dimensional cross-linked network structure is still well maintained with relatively the same fiber diameter, providing a guarantee for a highly conductive self-supporting cathode in the future. X-ray Diffraction (XRD) and Raman spectroscopy characterization are further carried out to investigate the phase structure of PAN/Fe(acac)3 and CNFs@Fe3C composite nanofiber membranes. As observed from the XRD patterns in Fig. S1c,† there is a wide bulge in the patterns of PAN/Fe(acac)3 composites at around 2θ = 26°, corresponding to the PAN. Meanwhile, several diffraction peaks representing Fe(acac)3 can also be observed at around 2θ = 10°–20°. After the calcination process, the characteristic peaks representing the Fe3C (JCPDS # 06-0670) phase can be observed clearly from the XRD patterns of CNFs@Fe3C composites, suggesting the successful formation of Fe3C. Moreover, the broadening peak at around 2θ = 26° is still visible, which can now be attributed to amorphous CNFs. The Raman spectroscopy of the PAN/Fe(acac)3 and CNFs@Fe3C composite nanofiber membranes are also collected accordingly, as shown in Fig. S1f.† It is found that the PAN/Fe(acac)3 composites do not exhibit any vibration bands in the range of 800–2000 cm−1. In comparison, the CNFs@Fe3C composites show obvious D and G bands of carbon at around 1340 cm−1 and 1580 cm−1, respectively.Transmission Electron Microscopy (TEM) is further employed to characterize the microstructure of CNFs@Fe3C composites, as shown in Fig. S2.† As observed from the Bright Field (BF) image in Fig. S2a,† the interconnected network nature of the membrane with Fe3C nanoparticles decorated intensively on the surface can also be confirmed. From the High-Resolution Transmission Electron Microscopy (HRTEM) image illustrated in Fig. S2b,† it is found that the Fe3C nanoparticles with a size of approximately 15 nm are fully wrapped up by the graphite carbon. The corresponding Fast Fourier Transformation (FFT) figure taken from the red square region illustrates both diffraction spots of Fe3C and diffraction rings of graphite, as shown in Fig. S2c.† Here, subscripts “G” and “F” represent graphite and Fe3C, respectively. Using all the diffraction spots to construct the Inverse Fast Fourier Transformation (IFFT) figure, the 0.24 nm and 0.34 nm lattice spacing can be observed clearly, corresponding to the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20) plane of Fe3C and (002) plane of graphite, respectively, as shown in Fig. S2d.† The Scanning Transmission Electron Microscopy Dark-Field (STEM-DF) image and corresponding Energy Dispersive X-ray Spectroscopy (EDX) mapping scan results also clearly illustrate the successful uniform decoration of Fe3C nanoparticles on the CNFs, as shown in Fig. S2(e–h).†
20) plane of Fe3C and (002) plane of graphite, respectively, as shown in Fig. S2d.† The Scanning Transmission Electron Microscopy Dark-Field (STEM-DF) image and corresponding Energy Dispersive X-ray Spectroscopy (EDX) mapping scan results also clearly illustrate the successful uniform decoration of Fe3C nanoparticles on the CNFs, as shown in Fig. S2(e–h).†
3.2 Structural and morphology characterization of PEDOT@CNFs@Fe3C composite fibrous membranes
By employing CNFs@Fe3C composites as the substrate, PEDOT can be coated successfully using a gas-phase polymerization method to prepare P@CNFs@Fe3C composites. Fig. 1 shows the SEM images of the composites with different PEDOT polymerization amounts. As mentioned in the Experimental section, the use of 15 μL, 20 μL and 25 μL EDOT monomer correspond to the PEDOT coating amounts of 11 wt%, 16 wt% and 21 wt%, respectively. As seen from the figure, it is found that the P@CNFs@Fe3C-11wt% composite membranes exhibit quite similar morphology when compared with that of CNFs@Fe3C, corresponding to the small amount of PEDOT coating, as shown in Fig. 1a and d. With the increase in the PEDOT coating amount, Fe3C nanoparticles are gradually covered (Fig. 1b and e). When the PEDOT coating amount reaches 21 wt%, the surface exposed Fe3C nanoparticles are significantly reduced, as shown in Fig. 1c and f. Under this condition, the coating of PEDOT can connect the isolated Fe3C nanoparticles to form a continuous conductive network, thereby improving the electrical conductivity of the composite material. However, the excessive thickness of the PEDOT polymer layer may also affect Li+ transport efficiency during the charge–discharge process of the cells, which has been proved by our previous research works.53 | ||
| Fig. 1 SEM images of P@CNFs@Fe3C composite fibrous membranes with 11 wt% (a), 16 wt% (b) and 21 wt% (c) PEDOT polymerization amounts suggest the linear relationship between coating amount of PEDOT and coverage of Fe3C nanoparticles. The corresponding enlarged SEM images (d–f) clearly illustrate the different surface exposure of Fe3C nanoparticles on the surface of CNFs. | ||
Further structure analysis has also been carried out on the P@CNFs@Fe3C-16wt% composites, as shown in Fig. 2. From the BF image in Fig. 2a, there is no significant difference in the microstructure before and after PEDOT polymerization, which is consistent with the relatively lower polymerization amount. The corresponding Selected Area Electron Diffraction (SAED) patterns show two obvious polycrystalline diffraction rings, corresponding to the (002) crystal plane of graphite and the (111) crystal plane of Fe3C, respectively. From the HRTEM image shown in Fig. 2b, an obvious amorphous layer can be observed at the edge of the CNFs, which refers to the PEDOT coating layer. In addition, Fe3C nanoparticles are found to be fully wrapped by graphite carbon and the PEDOT coating. The FFT figure constructed from the enlarged HRTEM image (Fig. 2c) also illustrates the diffraction ring of graphite and several diffraction spots from Fe3C, as shown in Fig. 2d. The corresponding IFFT figure further marks the 0.24 nm lattice spacing of (110) in Fe3C and the 0.34 nm lattice spacing of graphite carbon, which exhibit similar features to those of CNFs@Fe3C composites. The high-resolution Secondary Electron Image (SEI) collected in TEM (Fig. 2f) shows a more detailed surface morphology of P@CNFs@Fe3C-16wt% composites. Specifically, the Fe3C nanoparticles are proved to be fully covered by the PEDOT conductive layer, which enables all the isolated Fe3C nanoparticles to be linked into a continuous conductive network. Finally, the corresponding EDX mapping scan results (Fig. 2g–j) indicate the distribution of Fe on the particles, while S mainly covers the surface of the fibers. Such results further demonstrate the successful fabrication of P@CNFs@Fe3C composite fibrous membranes.
 | ||
| Fig. 2 TEM BF image of P@CNFs@Fe3C-16wt% nanocomposites and the corresponding SAED patterns (a) suggest the cross-linked fibrous morphology of the CNFs with Fe3C nanoparticles decorated. The corresponding HRTEM image (b) confirms the amorphous nature of the PEDOT coating layer, which is fully covered on the surface of CNFs. The enlarged HRTEM image (c) and the corresponding FFT (d) and IFFT (e) figures further confirm the existence and distribution of graphite carbon and Fe3C nanoparticles. The high resolution SEI (f) confirms the uniform coverage of the PEDOT layer at the surface of CNFs@Fe3C composites. The corresponding EDX mappings(g–j) clearly illustrate the uniform distribution of Fe, S and C elements. | ||
3.3 Lithium polysulfide adsorption and catalytic behavior of PEDOT@CNFs@Fe3C composite fibrous membranes
A visualized adsorption experiment was carried out on the P@CNFs@Fe3C composites to investigate the adsorption performance of Lithium Polysulfides (LiPSs), as shown in Fig. 3. As observed from the optical photograph in Fig. 3a, it is noticed that different P@CNFs@Fe3C composites exhibit varying degrees of color fading after standing for 30 min. Among the 4 different composites, the P@CNFs@Fe3C-16wt% shows the most obvious adsorption effect, suggesting its best adsorption performance on LiPSs. Moreover, the UV-visible spectra of the LiPS solutions after adsorption are also collected, as shown in Fig. 3b. It is easy to find that the Li2S6 solution containing P@CNFs@Fe3C-16wt% composites exhibit the lowest adsorption peak among all the electrodes, demonstrating their best adsorption capacity. To further investigate the chemical interaction among PEDOT, Fe3C and LiPSs, the XPS is employed to investigate the surface chemical states of the samples before and after Li2S6 adsorption. Fig. 3c shows the S 2p core level spectra of P@CNFs@Fe3C-16wt% before adsorption. It is found that the experimental data can be fitted to 4 independent peaks, in which the binding energies at 164.3 eV and 165.6 eV belong to S 2p1/2 spin splitting peaks, while the binding energies at 164.8 eV and 163.3 eV belong to those of S 2p3/2. After adsorption, the peaks representing sulfate and thiosulfate appear at 170.5 eV and 169.3 eV, respectively, which may be due to the partial oxidation of LiPSs. In addition, the relative peak intensities of S 2p1/2 and S 2p3/2 decrease significantly with the binding energy shifted toward a higher energy direction at the same time, as shown in Fig. 3e. Owing to the presence of sulfur in PEDOT, this binding energy shift indicates a strong chemical interaction between PEDOT and polysulfide ions, demonstrating its excellent anchoring effect on LiPSs. Fig. 3d shows the Fe 2p core energy level spectra before the adsorption process, in which 6 characteristic peaks can be observed, including two satellite peaks (730.5 eV and 717.3 eV), two characteristic peaks representing Fe3+ (726.4 eV and 712.8 eV) and two characteristic peaks representing Fe2+ (724.3 eV and 710.8 eV). After adsorption, the peak intensities of the 6 characteristic peaks also weakened, together with a binding energy shift toward a higher energy direction, as shown in Fig. 3f. This further proves the enhancement of Fe–S bonding, which originated from the chemical interaction between Fe3C and LiPSs. | ||
| Fig. 3 Visible adsorption experimental results (a) and corresponding UV-visible spectra (b) of different electrodes suggest the best LiPSs adsorption performance of P@CNFs@Fe3C-16wt% composites. The XPS S 2p core level spectra before (c) and after adsorption (e) show an obvious peak shift toward a high binding energy direction and the appearance of the characterization peaks representing sulfate and thiosulfate, suggesting the strong interaction between PEDOT and LiPSs. The XPS Fe 2p core level spectra before (d) and after adsorption (f) also reveal the peak shift toward a high energy direction together with a decrease in peak intensity, demonstrating the chemical interaction between Fe3C and LiPSs. | ||
By assembling Li2S6 symmetrical cells using CNFs@Fe3C and different P@CNFs@Fe3C composites for both cathode and anode, the LiPSs catalytic performance can be evaluated through CV test, as shown in Fig. 4a–c. As depicted in Fig. 4a, it is noticed that all the 3 different P@CNFs@Fe3C composites exhibit significant oxidation–reduction peaks within the selected voltage range except CNFs@Fe3C composites, further suggesting the obvious LiPS catalytic performance of the PEDOT coating layer. Among them, the P@CNFs@Fe3C-16wt% electrode exhibits the maximum response current and peak area, indicating the best catalytic activity. From the CV curves of the first 5 cycles of P@CNFs@Fe3C-16wt% electrode under a scan rate of 1 mV s−1, it is found that there is no significant change in the oxidation–reduction peak during cycling, demonstrating its optimized electrochemical stability, as shown in Fig. 4b. By changing the scan rates from 1 mV s−1 to 3 mV s−1, the oxidation–reduction peaks can always be observed, further proving its excellent catalytic stability on LiPSs, as shown in Fig. 4c.
 | ||
| Fig. 4 CV curves of the Li2S6 symmetric cells of different electrodes (a) exhibit the best electrochemical activity of the P@CNFs@Fe3C-16wt% electrode. The first 5 cycles of the CV curves of Li2S6 symmetric cells under a scan rate of 1 mV s−1 (b) suggest the excellent cyclic stability of the P@CNFs@Fe3C-16wt% electrode. CV curves collected under different scan rates (c) also reveal the superior catalytic stability of the P@CNFs@Fe3C-16wt% electrode. The potentiostatic i–t curves (d–g) of the two electrodes further demonstrate the largest Li2S nucleation and dissolution capacity of the P@CNFs@Fe3C-16wt% electrode. | ||
The Li2S precipitation and dissolution experiments are carried out to further evaluate the catalytic effect of different electrodes on the solid–liquid and liquid–solid conversion processes of LiPSs, as shown in Fig. 4d–g. For the precipitation experiment, it is found that the P@CNFs@Fe3C-16wt% composites exhibit a higher precipitation peak current and larger Li2S nucleation capacity (402.4 mA h g−1) when compared with those of CNFs@Fe3C composites (312.8 mA h g−1). Moreover, the Li2S nucleation time is shorter for P@CNFs@Fe3C-16wt% (850 s) than that of CNFs@Fe3C electrode (1300 s), further demonstrating the catalytic activity of the PEDOT coating on the conversion from LiPSs to Li2S. Furthermore, the Li2S dissolution experiment results confirm the short dissolution time (470 s) and larger dissolution amount (1488.2 mA h g−1) of P@CNFs@Fe3C-16wt% electrode, as shown in Fig. 4f and g.
3.4 Electrochemical performance evaluation on P@CNFs@Fe3C/Li2S composite cathode
After the loading of Li2S onto the surface of different P@CNFs@Fe3C composites, the coin cells are further assembled to evaluate the electrochemical performance of different composite cathodes, as shown in Fig. 5. Fig. 5a shows the CV curves of the battery assembled with 4 different composite cathodes at a scan rate of 0.1 mV s−1. It should be mentioned that Li2S can only achieve complete conversion to LiPSs when initially charged above 3.26 V.54 In this case, the potential range during the CV test is selected to be 1.5–3.5 V. As observed from the figure, it is found that P@CNFs@Fe3C-16wt%/Li2S electrode exhibits 3 reduction peaks located at 2.49 V, 2.28 V, and 1.97 V, corresponding to the reduction process from S8 to long chain LiPSs and the following reduction to Li2S2/Li2S.55 In addition, one obvious oxidation peak can be found at around 2.51 V, which corresponds to the reverse conversion from Li2S2/Li2S to LiPSs and S8. Moreover, the distance between the oxidation and reduction peaks is the smallest for P@CNFs@Fe3C-16wt%/Li2S composite cathode, suggesting its excellent redox activity and minimal electrochemical polarization. From the enlarged CV curves illustrating the oxidation peaks in Fig. 5b, the above mentioned largest peak intensity and smallest oxidation potential can also be proved. The first 3 cycles of the CV curves for P@CNFs@Fe3C-16wt%/Li2S composite cathode (Fig. 5c) suggest a gradually increasing peak intensity, which corresponds to the increased charge and discharge capacity. Electrochemical Impedance Spectroscopy (EIS) is further employed to investigate the reaction kinetics and interface characteristics of the cells, as shown in Fig. 5d. It is noticed that the charge transfer resistance in the high-frequency region of different cathodes is significantly reduced as the PEDOT coating amount increases, which proves the gradually enhanced electrical conductivity. In contrast, the slope of the straight line in the low-frequency region gradually decreases with the increase in PEDOT polymerization amount, which demonstrates that a thicker PEDOT layer may increase the resistance of Li+ ion transport. | ||
| Fig. 5 CV curves of CNFs@Fe3C/Li2S and P@CNFs@Fe3C/Li2S composite cathodes with different PEDOT coating amounts (a) suggest the highest peak current and smallest electrochemical polarization of the P@CNFs@Fe3C-16wt%/Li2S cathode. The enlarged CV curves (b) and the first 3 cycles of the CV curves (c) further illustrate excellent redox activity and cyclic stability of the P@CNFs@Fe3C-16wt%/Li2S cathode. The EIS curves (d) suggest a positive correlation relationship between PEDOT coating thickness and electronic conductivity and ion diffusion resistance. Cyclic performance curves (e) together with corresponding galvanostatic charge–discharge curves (f) under 0.1 C demonstrate the best cyclic performance of the P@CNFs@Fe3C-16wt%/Li2S cathode. The rate performance (g) and corresponding galvanostatic charge–discharge curves under different current densities (h) also prove the best rate performance of the P@CNFs@Fe3C-16wt%/Li2S cathode. The long cyclic performance under 0.5 C for 1000 cycles (i) exhibits a 0.033% capacity decay per cycle for the P@CNFs@Fe3C-16wt%/Li2S cathode. The discharge plateaus (j) can always be observed clearly during the whole cycling process. | ||
The cyclic performance of the cells using CNFs@Fe3C/Li2S and P@CNFs@Fe3C/Li2S composite cathodes with different PEDOT coating amounts is displayed in Fig. 5e. The initial discharge capacities of the CNFs@Fe3C/Li2S, P@CNFs@Fe3C-11wt%/Li2S, P@CNFs@Fe3C-16wt%/Li2S and P@CNFs@Fe3C-21wt%/Li2S composite cathodes are confirmed to be 489 mA h g−1, 639 mA h g−1, 816 mA h g−1 and 705 mA h g−1, respectively. From the corresponding galvanostatic charge–discharge curves shown in Fig. 5f, it is found that the P@CNFs@Fe3C-16wt%/Li2S composite cathode exhibits the smallest electrochemical polarization, which is consistent with the CV results. After 100 cycles, the capacity retention of the above 4 composite cathodes can be calculated to be 38%, 48%, 71% and 62%, respectively, further demonstrating the best cyclic stability of P@CNFs@Fe3C-16wt%/Li2S cathode. The rate performance of the 4 composite cathodes is also evaluated in Fig. 5g. The optimized P@CNFs@Fe3C-16wt%/Li2S composite cathode can achieve the initial discharge capacity of 860 mA h g−1, 685 mA h g−1, 543 mA h g−1, 426 mA h g−1 and 338 mA h g−1 under the current densities of 0.1 C, 0.2 C, 0.5 C, 1 C and 2 C, respectively, which are highest among all the 4 composite cathodes. The corresponding galvanostatic charge–discharge curves in Fig. 5h exhibit the clearly visible discharge plateaus with the increase of discharge rates. When the current density returns to 0.1 C, the discharge capacity can also recover to 857 mA h g−1, demonstrating the excellent rate performance of P@CNFs@Fe3C-16wt%/Li2S composite cathode. Fig. 5i illustrates the long cyclic performance of CNFs@Fe3C/Li2S and P@CNFs@Fe3C-16wt%/Li2S composite cathode. Under the current density of 0.5 C, the initial capacity of the cells with P@CNFs@Fe3C-16wt%/Li2S cathode can reach up to 546 mA h g−1. After 1000 cycles, it can still remain at 366 mA h g−1 reversible capacity, corresponding to a capacity decay of 0.033% per cycle. The corresponding GCD curves at different cycles show clear discharge platforms, further proving the excellent cyclic stability of P@CNFs@Fe3C-16wt%/Li2S cathode, as shown in Fig. 5j. The surface morphology of the P@CNFs@Fe3C-16%/Li2S cathode after 1000 cycles has further been characterized by SEM, as shown in Fig. S3.† No obvious cracks or detachment of materials can be observed, illustrating the excellent structural stability of the cathode. Furthermore, the P@CNFs@Fe3C-16%/Li2S cathode is confirmed to display an areal capacity of 1.62 mA h cm−2 with a Li2S loading amount of 3.6 mg cm−2 and an E/Li2S ratio of 8.3 μL mg−1 under a current density of 0.2 C, demonstrating the application potential under high Li2S loading amount conditions, as shown in Fig. S4.† All the above results indicate that P@CNFs@Fe3C/Li2S cathode possesses both high electrical conductivity and LiPSs catalytic activity, which can effectively improve the specific capacity and cycling stability of Li–S batteries. Table S1† compares the electrochemical performance of the recently reported Li2S-based cathodes in Li–S batteries.
4. Conclusions
PEDOT is successfully polymerized onto the surface of CNFs@Fe3C composite nanofibers prepared by electrospinning combined with carbothermal reduction reaction. The polymerization amount can be precisely adjusted by changing the added amount of EDOT monomer. Through careful structural characterization, the optimized PEDOT polymerization parameters are confirmed to be 20 μL EDOT monomer and polymerized at 100 °C for 30 min. The obtained P@CNFs@Fe3C-16wt% composites exhibit uniform morphology and superior adsorption and catalytic performance on LiPSs. Using different P@CNFs@Fe3C composites to load Li2S as the cathodes to assemble Li–S batteries, it is found that the P@ CNFs@Fe3C-16wt%/Li2S cathode possesses the highest initial specific capacity of 816 mA h g−1 at the current density of 0.1 C, and a capacity decay rate per cycle of 0.033% after 1000 cycles at 0.5 C. With Li2S loading amount of 3.6 mg cm−2 and E/Li2S ratio of 8.3 μL mg−1 under the current density of 0.2 C, an areal capacity of 1.62 mA h cm−2 can also be achieved for the P@ CNFs@Fe3C-16wt%/Li2S cathode.Author contributions
Na Yang: writing-review and editing, writing-original draft, investigation, formal analysis. Jiarui Xue: supervision, formal analysis, writing-review and editing, investigation. Yuanxiao Ji: investigation, formal analysis. Jiyuan Zhang: validation, visualization. Weiye Zhang: data curation, validation. Xuexia He: resources, software. Qi Li: supervision, software. Zhibin Lei: methodology, resources, funding acquisition. Zonghuai Liu: resources, supervision, methodology. Jie Sun: conceptualization, formal analysis, funding acquisition, methodology, resources, supervision, writing-review and editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI file.† Source data are provided in this paper. A request can be made to the corresponding author for accessing the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Natural Science Basic Research Plan of Shaanxi Province (2021JM-191, 2019JLP-12), the funds of Shaanxi Sanqin Scholars Innovation Team and the Central University Foundation of Shaanxi Normal University (GK202302005).References
- J. Wang, H. Wang, S. Jia, Q. Zhao, Q. Zheng, Y. Ma, T. Ma and X. Li, J. Energy Storage, 2023, 72, 108372 CrossRef.
- J. Li, L. Gao, F. Pan, C. Gong, L. Sun, H. Gao, J. Zhang, Y. Zhao, G. Wang and H. Liu, Nano-Micro Lett., 2023, 16, 12 CrossRef.
- Y. Guo, Q. Niu, F. Pei, Q. Wang, Y. Zhang, L. Du, Y. Zhang, Y. Zhang, Y. Zhang, L. Fan, Q. Zhang, L. Yuan and Y. Huang, Energy Environ. Sci., 2024, 17, 1330–1367 RSC.
- P. Yu, S. Sun, C. Sun, C. Zeng, Z. Hua, N. Ahmad, R. Shao and W. Yang, Adv. Funct. Mater., 2024, 34, 2306939 CrossRef CAS.
- L. Zhou, D. L. Danilov, F. Qiao, J. Wang, H. Li, R.-A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2022, 12, 2202094 CrossRef CAS.
- M. Zhao, H.-J. Peng, B.-Q. Li and J.-Q. Huang, Acc. Chem. Res., 2024, 57, 545–557 CAS.
- L. Huang, J. Li, B. Liu, Y. Li, S. Shen, S. Deng, C. Lu, W. Zhang, Y. Xia, G. Pan, X. Wang, Q. Xiong, X. Xia and J. Tu, Adv. Funct. Mater., 2020, 30, 1910375 CrossRef CAS.
- D. M. Brieske, A. Warnecke and D. U. Sauer, Energy Storage Mater., 2023, 55, 289–300 CrossRef.
- W. Yao, J. Xu, L. Ma, X. Lu, D. Luo, J. Qian, L. Zhan, I. Manke, C. Yang, P. Adelhelm and R. Chen, Adv. Mater., 2023, 35, 2212116 CrossRef CAS PubMed.
- X. Kang, T. He, R. Zou, S. Niu, Y. Ma, F. Zhu and F. Ran, Small, 2024, 20, 2306503 CrossRef CAS PubMed.
- R. Guo, Y. Yang, X. L. Huang, C. Zhao, B. Hu, F. Huo, H. K. Liu, B. Sun, Z. Sun and S. X. Dou, Adv. Funct. Mater., 2024, 34, 2307108 CrossRef CAS.
- S. Feng, J. Wang, J. Wen, X. Li, Z. Wang, Y. Zeng and J. Xiao, ACS Sustainable Chem. Eng., 2023, 11, 8544–8555 CrossRef CAS.
- Y. Zhang, H. W. Song, K. R. Crompton, X. Yang, K. Zhao and S. Lee, Nano Energy, 2023, 115, 108756 CrossRef CAS.
- Q. Yang, J. Cai, G. Li, R. Gao, Z. Han, J. Han, D. Liu, L. Song, Z. Shi, D. Wang, G. Wang, W. Zheng, G. Zhou and Y. Song, Nat. Commun., 2024, 15, 3231 CrossRef CAS PubMed.
- J. Feng, C. Zhang, W. Liu, S. Yu, L. Wang, T. Wang, C. Shi, X. Zhao, S. Chen, S. Chou and J. Song, Angew. Chem., Int. Ed., 2024, 63, e202407042 CrossRef CAS PubMed.
- C. Geng, W. Qu, Z. Han, L. Wang, W. Lv and Q.-H. Yang, Adv. Energy Mater., 2023, 13, 2204246 CrossRef CAS.
- T. Lai, A. Bhargav and A. Manthiram, Adv. Funct. Mater., 2023, 33, 2304568 CrossRef CAS.
- H. Liu, P. Zeng, Y. Li, H. Yu, M. Chen, Z. Luo, C. Miao, G. Chen and X. Wang, J. Alloys Compd., 2020, 835, 155421 CrossRef CAS.
- G. Gao, X. Yang, J. Bi, W. Guan, Z. Du and W. Ai, J. Mater. Chem. A, 2023, 11, 26318–26339 RSC.
- Q. Fan, Y. Si, F. Zhu, W. Guo and Y. Fu, Angew. Chem., Int. Ed., 2023, 62, e202306705 CrossRef CAS PubMed.
- Y. Ji, J. Zhang, N. Yang, J. Xue, W. Zhang, X. He, Q. Li, Z. Lei, Z. Liu and J. Sun, Appl. Surf. Sci., 2025, 679, 161263 CrossRef CAS.
- M. A. Weret, S.-K. Jiang, K. N. Shitaw, C.-Y. Chang, T. M. Tekaligne, J.-C. Chiou, S.-C. Yang, N. T. Temesgen, Y. Nikodimos, S.-H. Wu, C.-C. Wang, W.-N. Su and B. J. Hwang, ACS Energy Lett., 2023, 8, 2817–2823 CrossRef CAS.
- J. Offermann, A. Paolella, R. Adelung and M. Abdollahifar, Chem. Eng. J., 2024, 157920 CrossRef CAS.
- Y. Zhao, L. Huang, D. Zhao and J. Yang Lee, Angew. Chem., Int. Ed., 2023, 62, e202308976 CrossRef CAS.
- T. Meng, J. Gao, J. Zhu, N. Li, M. Xu, C. M. Li and J. Jiang, J. Mater. Chem. A, 2020, 8, 11976–11985 RSC.
- A. Moyseowicz, D. Minta and G. Gryglewicz, ChemElectroChem, 2023, 10, e202201145 CrossRef CAS.
- C. Li, K. Zhang, X. Cheng, J. Li, Y. Jiang, P. Li, B. Wang and H. Peng, Prog. Polym. Sci., 2023, 143, 101714 CrossRef CAS.
- S. K. Pati, D. Patra, S. Muduli, S. Mishra and S. Park, Small, 2023, 19, 2300689 CrossRef CAS PubMed.
- I. Petsagkourakis, E. Pavlopoulou, E. Cloutet, Y. F. Chen, X. Liu, M. Fahlman, M. Berggren, X. Crispin, S. Dilhaire, G. Fleury and G. Hadziioannou, Org. Electron., 2018, 52, 335–341 CrossRef CAS.
- L. R. Dalton, J. Thomson and H. S. Nalwa, Polymer, 1987, 28, 543–552 CrossRef CAS.
- X. Han, J. Sun, Q. Li, X. He, L. Dang, Z. Liu and Z. Lei, ACS Sustainable Chem. Eng., 2023, 11, 2938–2948 CrossRef CAS.
- F. Niu, X. Han, H. Sun, Q. Li, X. He, Z. Liu, J. Sun and Z. Lei, ACS Sustainable Chem. Eng., 2021, 9, 4146–4156 CrossRef CAS.
- T. Yang, D. Xin, N. Zhang, J. Li, X. Zhang, L. Dang, Q. Li, J. Sun, X. He, R. Jiang, Z. Liu and Z. Lei, J. Mater. Chem. A, 2024, 12, 10137–10147 RSC.
- W. A. El-Said, M. Abdelshakour, J.-H. Choi and J.-W. Choi, Molecules, 2020, 25, 307 CrossRef CAS PubMed.
- X. Chen, C. Zhao, K. Yang, S. Sun, J. Bi, N. Zhu, Q. Cai, J. Wang and W. Yan, Energy Environ. Mater., 2023, 6, e12483 CrossRef CAS.
- Q. Huang, G. Zha, Z. Hu, H. Liu, S. Agarwal and H. Hou, Phys. Chem. Chem. Phys., 2023, 25, 23579–23587 RSC.
- S. A. Abbas, M. A. Ibrahem, L.-H. Hu, C.-N. Lin, J. Fang, K. M. Boopathi, P.-C. Wang, L.-J. Li and C.-W. Chu, J. Mater. Chem. A, 2016, 4, 9661–9669 RSC.
- A. Sharma, G. Andersson, J. Rivnay, J. F. Alvino, G. F. Metha, M. R. Andersson, K. Zuber and M. Fabretto, Adv. Mater. Interfaces, 2018, 5, 1800594 CrossRef.
- M. Zhang, K. Amin, M. Cheng, H. Yuan, L. Mao, W. Yan and Z. Wei, Nanoscale, 2018, 10, 21790–21797 RSC.
- M. Zhang, Q. Meng, A. Ahmad, L. Mao, W. Yan and Z. Wei, J. Mater. Chem. A, 2017, 5, 17647–17652 RSC.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef PubMed.
- K. N. Dinh, Q. Liang, C.-F. Du, J. Zhao, A. I. Y. Tok, H. Mao and Q. Yan, Nano Today, 2019, 25, 99–121 CrossRef CAS.
- K. He, E. Campbell, Z. Huang, R. Shen, Q. Li, S. Zhang, Y. L. Zhong, P. Zhang and X. Li, Small Struct., 2022, 3, 2200104 CrossRef CAS.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed.
- G. Li, J. Liu, C. Xu, H. Chen, H. Hu, R. Jin, L. Sun, H. Chen, C. Guo, H. Li and Y. Si, Energy Storage Mater., 2023, 56, 394–402 CrossRef.
- D. Yang, T. Lv, J. Song, J. Chen, L. Hao, Q. Tian and L. Cui, Chem. Eng. J., 2024, 496, 153844 CrossRef CAS.
- C. Liao, W. Jing, F. Wang and Y. Liu, Mater. Today Catalysis, 2023, 3, 100030 CrossRef.
- H. Li, S. Ma, H. Cai, H. Zhou, Z. Huang, Z. Hou, J. Wu, W. Yang, H. Yi, C. Fu and Y. Kuang, Energy Storage Mater., 2019, 18, 338–348 CrossRef.
- R. Chen, Y. Zhou and X. Li, Small, 2024, 20, 2308261 CrossRef CAS PubMed.
- K. Shi, Y. Sun, Z. Xiong, J. Li, H. Nan, Y. Lin, Z. Wei and Q. Liu, Chem. Eng. J., 2023, 460, 141794 CrossRef CAS.
- S. Wang, X. Liu, H. Duan, Y. Deng and G. Chen, Chem. Eng. J., 2021, 415, 129001 CrossRef CAS.
- R. Chen, Y. Zhou and X. Li, Nano Lett., 2022, 22, 1217–1224 CrossRef CAS PubMed.
- N. Yang, J. Zhang, Y. Ji, W. Zhang, J. Xue, X. He, Q. Li, Z. Lei, Z. Liu and J. Sun, ACS Sustainable Chem. Eng., 2025, 13, 1378–1390 CrossRef CAS.
- G. Zhou, J. Sun, Y. Jin, W. Chen, C. Zu, R. Zhang, Y. Qiu, J. Zhao, D. Zhuo, Y. Liu, X. Tao, W. Liu, K. Yan, H. R. Lee and Y. Cui, Adv. Mater., 2017, 29, 1603366 CrossRef PubMed.
- K. Cai, M.-K. Song, E. J. Cairns and Y. Zhang, Nano Lett., 2012, 12, 6474–6479 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04989f |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |