DOI:
10.1039/D4NR04862H
(Paper)
Nanoscale, 2025,
17, 2860-2870
Hydride-containing Ag- and Au-rich 8-electron superatomic icosahedral cores: a DFT investigation†
Received
19th November 2024
, Accepted 14th January 2025
First published on 15th January 2025
Abstract
Following several reports on ligand-protected atom-precise nanoclusters which encapsulate hydrides as interstitial dopants within their icosahedral core, the stability, structure and bonding of MHx@Ag12 and MHx@Au12 (M = Mo–Ag; W–Au) 8-electron cores is investigated through DFT calculations. The encapsulation of up to x = 3 hydrides appears to be possible but at the cost of substantial structural distortions. In most of the computed models, the hydrides are found nearly free to move inside their icosahedral cages. Systems with one (nido-type) or two (arachno-type) missing vertices on the icosahedron are also predicted to be viable. In general, the MHx@Au12 species appear to be of lower stability than their MHx@Ag12 homologs. We believe that this the work will provide some new directions for the synthesis of hydride-encapsulating superatoms.
1. Introduction
The chemistry of atom-precise noble metal nanoclusters has become increasingly topical since the turn of the century. The major reason lies in their numerous potential applications in various fields such as photonics, health sciences, or catalysis.1–4 Moreover, their atom-precise nature makes them accurate molecular models for less well-defined nanoparticles of larger size. In recent years, researches have turned to the fine-tuning of the noble metal nanocluster properties by their controlled doping with another metal. Designing nanoclusters for their optimized properties requires in the first-place stability. This stability is ordinarily ensured through the presence of ligands such as chalcogenolates, alkynyl, phosphines, or halogenides. These ligands not only “protect” the metallic core, but also in many cases counterbalance its formal positive charge. Indeed, in most cases the structure and electron count of a nanocluster metal core can be described within the superatom model,5–9 in which they are described as having spherical electronic structures related to those of atoms, with 1S, 1P, 1D, 2S, 1F… shell ordering, and with stability ensured by a closed-shell electron configuration corresponding to one of the “magic” electron numbers of 2, 8, 18, 20, 34… In the case of noble metal nanoclusters, the electrons to be considered are those provided by their (n + 1)s valence AOs and the requirement for a “magic” number generally leads to a formally cationic [Mn]p+ core.
Hydride is one of the many ligands commonly encountered in this chemistry.10–16 This monoatomic monoanionic ligand is to some extent related to halides, with which they can sometimes be exchanged. Hydrides have however unique intrinsic characteristics, of which one is their particularly small size. Whereas ordinary ligands protect the [Mn]p+ inner core from the outside by forming a peripheral outer shell around it, it has been shown very recently that the smaller hydrides can, in some cases, get encapsulated inside the metal core.17–29 The most striking structurally characterized examples concern 8-electron superatoms, the metal core of which describes a centered icosahedron, which in addition contains one19,21,22,28 or even two23,24 hydrogen atom(s). These nanoclusters are gathered in Table 1. Note that all of them are doped silver species. Indeed, no (MHx)@Au12 relative has been isolated so far, although such species have been predicted to exist on the basis of DFT calculations.11 The encapsulated hydrides are mainly bonded to the central metal atom so that the whole cluster core is better viewed as describing a distorted (MHx)@M12 (x = 1, 2) icosahedron.
Table 1 Structurally characterized nanoclusters containing an MHx (x = 1,2) unit encapsulated within an Ag12 icosahedral cage and their 8-electron superatomic cores
| Compound |
Superatomic core |
Ref. |
| [(RhH)Ag24(SPhMe2)18]2− |
[(RhH)@Ag12]4+ |
19
|
| [(RhH)Ag20{S2P(OnPr)2}12] |
[(RhH)@Ag12]4+ |
24
|
| [(RhH)2Ag33[S2P(OnPr)2]17] |
[(RhH)@Ag12]4+ |
29
|
| [(IrH)Ag24(SPhMe2)18]2− |
[(IrH)@Ag12]4+ |
23
|
| [(PdH)Ag19{E2P(OiPr)2}12] (E = S, Se) |
[(PdH)@Ag12]5+ |
22 and 27 |
| [(PdH)Ag20{E2P(OiPr)2}12]+ (E = S, Se) |
[(PdH)@Ag12]5+ |
22 and 27 |
| [(PtH)Ag19{S2P(OnPr)2}12] |
[(PtH)@Ag12]5+ |
21
|
| [(PtH)Ag19{Se2P(OiPr)2}12] |
[(PtH)@Ag12]5+ |
21
|
| [PtHPtAg32[S2P(OnPr)2]17] |
[(PtH)@Ag12]5+ |
29
|
| [(RuH2)Ag24(SPhMe2)18]2− |
[(RuH2)@Ag12]4+ |
23
|
| [(OsH2)Ag24(SPhMe2)18]2− |
[(OsH2)@Ag12]4+ |
23
|
| [(RhH2)Ag19{S2P(OnPr)2}12] |
[(RhH2)@Ag12]5+ |
24
|
It has been shown that the 1s electron of an encapsulated hydride is to be considered as one of the eight superatomic electrons, contrary to the case where hydrides behave as ordinary outer ligands.11–29 In other words, if one assumes that a [Mn]p+ metal core possesses a given “magic” number of (n–p) electrons providing it with closed-shell stability, inserting x hydrogen atoms into it would increase its electron count by x. To maintain stability, the “magic” (n–p) electron count should be preserved, so the inserted hydrogen(s) should be, from the electron counting point of view, considered as formal proton(s), thus increasing the formal core charge to [MnHx](p+x)+. In the case the hydrides are regular external H− ligands, then the metal core should retain its p + charge, resulting in the {[Mn]Hx}(p−x)+ assembly. Of course, in both cases, the p ± x cationic charge will be, in real life, compensated by the presence of various additional peripheral anionic ligands to reach (or approach) neutrality for the whole nanocluster.
Thus, encapsulated hydrides should be considered as inclusion dopants rather than ligands, i.e., somewhat behaving as metal dopants. Because of this metal parentage, they are sometimes named “metallic hydrogens”,11,12,15,16 a designation that should not be confused with the meaning of an electron-conducting material. As any metal dopant in nanoclusters, hydrides play a crucial role in their properties. In particular, it has been shown that Au and Ag nanoclusters containing encapsulated hydrides are efficient electrocatalysts in the hydrogen evolution reaction (HER).11,12,16,23,24
Herein we report a comprehensive computational investigation on the encapsulation of one and several hydrides within the M@Ag12 and M@Au12 (M = Mo–Ag and W–Au) centered icosahedral cores, assuming the “magic” 8-electron count to be maintained. We have restricted this study to the superatomic icosahedral cores. Indeed, it has been shown previously that computing the only superatomic core of a nanocluster in its actual oxidation state, i.e. without considering its passivating shell, allows rationalizing its stability, bonding and superatomic electronic structure.30–32 On the other hand, as far as one is interested in bonding analysis only, this simplification also allows to highlight variations across the periodic table, without the blurring caused by the variation of the ligand and outer metal nature.
2. Results and discussion
2.1 The hydrogen-free 8-electron M@Ag12 and M@Au12 cores
Before investigating hydrogen encapsulation into M@Ag12 and M@Au12 cages, we first briefly review the trends within the icosahedral hydride-free 8-electron [M@Ag12]x+ and [M@Au12]x+ clusters when the second- and third-row transition-metal M varies from Group 6 to Group 11. All the optimized geometries were found to be of Ih symmetry. Corresponding selected computed data are provided in Table 2. The [Pd@Ag12]4+ example is shown in Fig. 1.
 |
| | Fig. 1 The optimized geometries of [Pd@Ag12]4+ (Ih), [PdH@Ag12]5+ (C3v), and [RuH2@Ag12]4+ (C2v). | |
Table 2 Selected computed data for the icosahedral 8-electron superatomic cores [M@Ag12]x+ and [M@Au12]x+ (M = Mo–Ag, W–Au). Interatomic distances are given in Å. WBI = Wiberg bond index; ΔEH/L = HOMO–LUMO gap; qM, qAg and qAu are atomic natural charges
| [M@Ag12]x+ |
[M@Au12]x+ |
| M |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
M |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
|
x
|
0 |
1 |
2 |
3 |
4 |
5 |
x
|
0 |
1 |
2 |
3 |
4 |
5 |
| ΔEH/L (eV) |
1.37 |
1.92 |
1.46 |
1.73 |
2.06 |
2.64 |
ΔEH/L (eV) |
1.09 |
1.46 |
1.04 |
1.73 |
2.05 |
1.95 |
| M–Ag |
2.761 |
2.753 |
2.760 |
2.783 |
2.824 |
2.899 |
M–Au |
2.757 |
2.752 |
2.757 |
2.772 |
2.797 |
2.882 |
| WBI |
0.447 |
0.397 |
0.340 |
0.262 |
0.230 |
0.230 |
WBI |
0.482 |
0.422 |
0.351 |
0.258 |
0.205 |
0.191 |
| Ag–Ag |
2.803 |
2.895 |
2.903 |
2.926 |
2.969 |
3.048 |
Au–Au |
2.899 |
2.894 |
2.899 |
2.918 |
2.942 |
3.030 |
| WBI |
0.084 |
0.085 |
0.086 |
0.090 |
0.089 |
0.084 |
WBI |
0.087 |
0.092 |
0.099 |
0.110 |
0.118 |
0.121 |
| qM |
−2.50 |
−2.22 |
−2.05 |
−1.95 |
−1.56 |
−1.30 |
q
M
|
−2.23 |
−1.93 |
−1.69 |
−1.54 |
−1.09 |
−0.69 |
| qAg |
0.21 |
0.27 |
0.34 |
0.41 |
0.46 |
0.53 |
q
Au
|
0.19 |
0.24 |
0.31 |
0.38 |
0.42 |
0.47 |
| M |
W |
Re |
Os |
Ir |
Pt |
Au |
M |
W |
Re |
Os |
Ir |
Pt |
Au |
|
x
|
0 |
1 |
2 |
3 |
4 |
5 |
x
|
0 |
1 |
2 |
3 |
4 |
5 |
| ΔEH/L (eV) |
1.68 |
2.04 |
1.61 |
1.88 |
2.17 |
2.76 |
ΔEH/L (eV) |
1.26 |
1.14 |
1.16 |
1.32 |
2.07 |
2.05 |
| M–Ag |
2.761 |
2.756 |
2.765 |
2.786 |
2.826 |
2.895 |
M–Au |
2.756 |
2.753 |
2.762 |
2.778 |
2.805 |
2.849 |
| WBI |
0.417 |
0.364 |
0.314 |
0.268 |
0.231 |
0.232 |
WBI |
0.462 |
0.404 |
0.338 |
0.281 |
0.226 |
0.205 |
| Ag–Ag |
2.904 |
2.897 |
2.908 |
2.929 |
2.971 |
3.044 |
Au–Au |
2.897 |
2.895 |
2.905 |
2.921 |
2.949 |
2.996 |
| WBI |
0.084 |
0.085 |
0086 |
0.086 |
0.086 |
0.082 |
WBI |
0.089 |
0.092 |
0.097 |
0.104 |
0.112 |
0.119 |
|
q
M
|
−3.20 |
−2.84 |
−2.56 |
−2.16 |
−1.76 |
−1.22 |
q
M
|
−2.86 |
−2.54 |
−2.24 |
−1.84 |
−1.41 |
−0.81 |
|
q
Ag
|
0.27 |
0.32 |
0.38 |
0.43 |
0.48 |
0.52 |
q
Au
|
0.24 |
0.30 |
0.35 |
0.40 |
0.45 |
0.48 |
It is known from both experimental and computational investigations that the doping of 8-electron centered Ag and Au icosahedral nanoclusters by a transition metal atom from the second and third row and situated on the left side of Ag and Au results in the occupation of the central position by this atom.33–35 The main reason for this situation originates from a stronger participation of the central atom valence nd orbitals to the nanocluster bonding. This is why in the case of the most left-sided M atoms, their significantly bonding nd shell is sometimes considered as constituting the occupied superatomic 1D level, leading such species to be described as 18-rather than 8-electron superatoms.35–37 Likewise, when both metals are from the same group 11 column, the [Au@Ag12]5+ core is observed whereas [Ag@Au12]5+ is unrealistic.34,35 Note that only the (n + 1)s electrons are considered in the 8-electron count, the M contribution being negative (M = Groups 6–9) or nul (M = Pd, Pt). All the computed models have an 1S2 1P6 closed-shell configuration secured by a significant HOMO–LUMO gap. Although this gap decreases when going from Group 11 to Group 6, it should be noted that the bare [Mo@Au12] and [W@Au12] clusters have been experimentally observed,36 right after the theoretical prediction of [W@Au12].37 This suggests that any of the cations listed in Table 2 (except [Ag@Au12]5+, see above)33,34 is likely to be stable, at least as the ligand-protected superatomic core of a larger species.
Both M–M′ (M′ = Ag, Au) and M′–M′ distances increase when M goes from Group 6 to Group to 11. Consistently, the M–M′ Wiberg bond indices (WBIs) decreases, but their variation is more marked than that of the corresponding distances. The stronger bonding for the left-sided M metals has been shown to originate from the larger participation of their nd valence orbitals to superatomic bonding, in such a way that one can include the electrons occupying this somewhat bonding nd(M) shell into the superatomic electron count, allowing [Mo@Au12] or [W@Au12], for example, to be considered as 18-electrons superatoms with 1S2 1P6 1D10 configuration.35,37 Contrarily to the M–M′ WBIs, the M′–M′ ones vary little, especially in the M′ = Ag case and they are always smaller than their M–M′ homologues.
2.2 Monohydride systems
Inserting a hydrogen atom as a dopant inside an 8-electron M@Ag12 or M@Au12 system, requires to formally add it as a proton if one wants to maintain the 8-electron count, i.e. the closed-shell stability of the whole architecture. It turns out that we were unable to converge the [(MH)@Ag12]6+ and [(MH)@Au12]6+ (M = Ag, Au) models. This may be the consequence of their large cationic charge. We note however that no example of such (AgH)- or (AuH)-centered icosahedral architecture has been reported so far. Relevant computed data of the converged monohydride models are given in Table 3. Only three of them, namely [RhHAu12]4+ and [MHAu12]5+ (M = Pd, Pt), have lost their icosahedral parentage, with the MH unit sitting inside a smaller cage (see Fig. S1†). These three models will not be discussed further in this paper.
Table 3 Selected (averaged) computed data for the 8-electron cores [(MH)@Ag12]x+ and [(MH)@Au12]x+ (M = Mo–Pd, W–Pt). Interatomic distances are given in Å. WBI = Wiberg bond index; qM, qAg and qAu are atomic natural charges. See also Table S1† for HOMO–LUMO gaps and metal–metal distances
| [(MH)@Ag12]x+ |
[(MH)@Au12]x+ |
| M |
Mo |
Tc |
Ru |
Rh |
Pd |
M |
Mo |
Tc |
Ru |
Rha |
Pda |
|
x
|
1 |
2 |
3 |
4 |
5 |
x
|
1 |
2 |
3 |
4 |
5 |
| CSM |
0.62 |
0.57 |
0.52 |
0.44 |
0.31 |
CSM |
1.03 |
1.09 |
1.09 |
n/a |
n/a |
| M–H |
1.837 |
1.742 |
1.679 |
1.658 |
1.703 |
M–H |
1.821 |
1.719 |
1.657 |
1.637 |
1.692 |
| WBI |
0.471 |
0.428 |
0.371 |
0.297 |
0.206 |
WBI |
0.487 |
0.460 |
0.417 |
0.424 |
0.344 |
| Ag–H |
2.085 |
2.084 |
2.090 |
2.099 |
2.089 |
Au–H |
2.168 |
2.191 |
2.211 |
2.137 |
1.717 |
| WBI |
0.102 |
0.105 |
0.109 |
0.116 |
0.123 |
WBI |
0.099 |
0.099 |
0.107 |
0.171 |
0.418 |
|
q
M
|
−2.42 |
−2.13 |
−1.94 |
−1.78 |
−1.38 |
q
M
|
−1.05 |
−1.54 |
−1.80 |
−2.11 |
−0.36 |
|
q
Ag
|
0.30 |
0.35 |
0.43 |
0.50 |
0.56 |
q
Au
|
0.27 |
0.32 |
0.38 |
0.42 |
0.45 |
|
q
H
|
−0.20 |
−0.20 |
−0.21 |
−0.27 |
−0.38 |
q
H
|
−0.07 |
−0.05 |
−0.05 |
0.01 |
0.02 |
| M |
W |
Re |
Os |
Ir |
Pt |
M |
W |
Re |
Os |
Ir |
Pta |
|
x
|
1 |
2 |
3 |
4 |
5 |
x
|
1 |
2 |
3 |
4 |
5 |
|
Optimized structure having lost its icosahedral parentage.
|
| CSM |
0.69 |
0.67 |
0.64 |
0.63 |
0.66 |
CSM |
1.10 |
1.28 |
1.39 |
1.79 |
n/a |
| M–H |
1.843 |
1.754 |
1.694 |
1.655 |
1.651 |
M–H |
1.833 |
1.729 |
1.665 |
1.615 |
1.561 |
| WBI |
0.490 |
0.457 |
0.403 |
0.356 |
0.306 |
WBI |
0.504 |
0.489 |
0.451 |
0.438 |
0.584 |
| Ag–H |
2.106 |
2.114 |
2.133 |
2.168 |
2.234 |
Au–H |
2.184 |
2.233 |
2.278 |
2.376 |
3.085 |
| WBI |
0.092 |
0.092 |
0.088 |
0.094 |
0.090 |
WBI |
0.091 |
0.085 |
0.087 |
0.083 |
0.030 |
|
q
M
|
−3.00 |
−2.64 |
−2.36 |
−1.94 |
−1.49 |
q
M
|
−2.59 |
−2.26 |
−1.96 |
−1.51 |
−0.66 |
|
q
Ag
|
0.35 |
0.40 |
0.46 |
0.51 |
0.57 |
q
Au
|
0.31 |
0.36 |
0.42 |
0.46 |
0.46 |
|
q
H
|
−0.22 |
−0.20 |
−0.21 |
−0.24 |
−0.31 |
q
H
|
−0.11 |
−0.07 |
−0.04 |
−0.02 |
0.18 |
All the other structures adopt a distorted icosahedral arrangement of C3v symmetry (see the [(PdH)Ag12]5+ example in Fig. 1), with M lying approximately at its center and the hydride inside a tetrahedron made of M and an Ag3 or Au3 triangular face of the icosahedron. As a result, this face is expanded, leading to significant distortion of the whole icosahedral architecture. The amount of distortion away from ideal Ih symmetry can be evaluated with the continuous symmetry measure38 (CSM, see Tables 3 and S1†). Within the silver series, it tends to decrease when M varies from left to right, whereas a more or less opposite trend is found for the gold series. The CSM values found for the gold series are much larger than their silver counterparts, which might indicate lower stability of the (MH)-centered icosahedral Au12 architecture. Overall our results are in good qualitative agreement with the experimentally known nanoclusters that contain [(MH)@Ag12]4+ (M = Rh, Ir)19,23,24,28,29 and [(MH)@Ag12]5+ (M = Pd, Pt).21,22,28 The fact that the computed CSM values are larger than the experimental ones19,23,24 can be explained by the absence in our models of any templating outer shell that would oppose distortion.
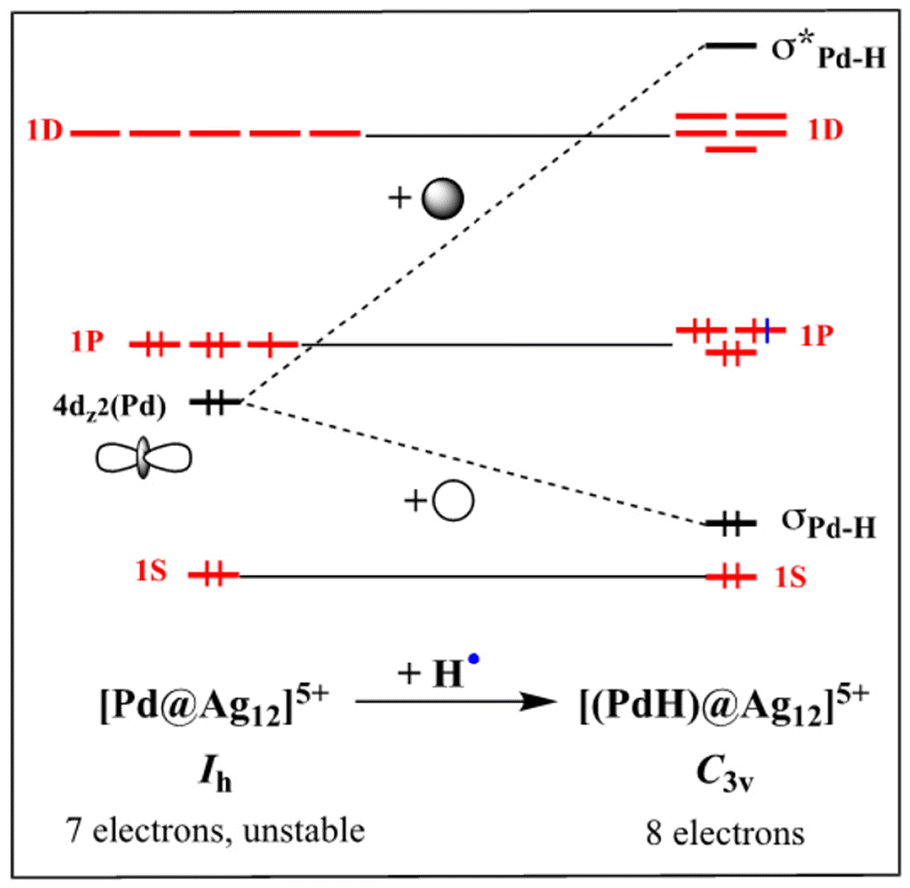
In previous investigations on nanoclusters containing an [(MH)@Ag12]5+ (M = Pd, Pt) core,21,22,25,26,28 it was found that the 1s(H) AO interacts principally with the dz2(M) orbital, creating an M–H 2-electron bond and leaving the (n + 1)s AO of M almost unperturbed and containing one electron (see Fig. 2). The superatomic orbitals being principally combinations of metallic valence s-type AOs, it follows that the 1s(H) AO is not significantly involved in their composition. From the superatomic point of view, the interstitial hydrogen provides one electron to the 5s(Pd/Pt) AO, which in turn allows the electron count to achieve the closed-shell count of eight. When M varies from Group 11 to Group 6, the participation of its nd orbitals (thus the ndz2 one) to the superatomic orbitals building increases.35,37 Moreover, the 1s(H) AO tends to interact in a somewhat larger extend with the (n + 1)s AO of M. It results that the contribution of the 1s(H) orbital to the composition of the superatomic orbitals increases. This is exemplified by the substantial increase of the M–H WBIs when going from Group 11 to Group 6 (Table 3), whereas the variation of the Ag–H WBIs is much more modest and mainly reflects the variation of the M radius. Consistently, the hydridic character of H decreases from Group 11 to Group 6.
 |
| | Fig. 2 Simplified sketch illustrating the major orbital interaction (in black) resulting from the formal inclusion of a hydrogen atom within an electron-deficient [Pd@Ag12]5+ superatomic entity, to generate the stable [(PdH)@Ag12]5+ closed-shell superatom. Only one 4d(Pd) orbital is shown for clarity, the other four remaining non-bonding (and occupied) in the process. | |
2.3 Dihydride systems
As for the monohydride series discussed just above, inserting two hydrogen atoms as dopants inside an 8-electron M@Ag12 or M@Au12 system, requires to formally add them as protons in order to maintain the closed-shell 8-electron count. In the same way as illustrated in Fig. 2 for a monohydride species, the two encapsulated hydrogen atoms interact principally with two valence d-type orbitals, leaving the superatomic orbitals little perturbed. Likely because of their large cationic charges, the models [(MH2)@Ag12]7+ and [(MH2)@Au12]7+ (M = Ag, Au) as well as [(MH2)@Ag12]6+ and [(MH2)@Au12]6+ (M = Pd, Pt) could not be converged. Relevant computed data of the converged dihydride models are given in Tables 4 and S2.†
Table 4 Selected (averaged) computed data for the icosahedral 8-electron superatomic cores [(MH2)@Ag12]x+ (M = Mo–Rh, W–Ir) and [(MH2)@Au12]x+ (M = Mo–Ru, W–Os). Interatomic distances are given in Å. WBI = Wiberg bond index; qM, qAg and qAu are atomic natural charges. See also Table S2† for HOMO–LUMO gaps and metal–metal distances
| [(MH2)@Ag12]x+ |
[(MH2)@Au12]x+ |
| M |
Mo |
Tc |
Ru |
Rh |
M |
Mo |
Tc |
Ru |
|
x
|
2 |
3 |
4 |
5 |
x
|
2 |
3 |
4 |
| CSM |
1.58 |
1.55 |
1.43 |
1.37 |
CSM |
1.67 |
2.11 |
2.68 |
| M–H |
1.809 |
1.718 |
1.663 |
1.658 |
M–H |
1.990 |
1.750 |
1.686 |
| WBI |
0.483 |
0.440 |
0.377 |
0.296 |
WBI |
0.384 |
0.432 |
0.396 |
| Ag–H |
2.151 |
2.164 |
2.669 |
2.050 |
Au–H |
1.871 |
2.036 |
1.890 |
| WBI |
0.092 |
0.093 |
0.097 |
0.123 |
WBI |
0.198 |
0.145 |
0.221 |
| H–M–H (°) |
70 |
71 |
75 |
84 |
H–M–H (°) |
69 |
68 |
70 |
|
q
M
|
−2.36 |
−2.05 |
−1.81 |
−1.53 |
q
M
|
−2.12 |
−1.71 |
−1.36 |
|
q
Ag
|
0.39 |
0.45 |
0.51 |
0.59 |
q
Au
|
0.36 |
0.40 |
0.45 |
|
q
H
|
−0.16 |
−0.15 |
−0.18 |
−0.26 |
q
H
|
−0.08 |
−0.03 |
−0.02 |
| M |
W |
Re |
Os |
Ir |
M |
W |
Re |
Os |
|
x
|
2 |
3 |
4 |
5 |
x
|
2 |
3 |
4 |
| CSM |
1.71 |
1.74 |
1.69 |
1.72 |
CSM |
3.41 |
2.37 |
2.83 |
| M–H |
1.818 |
1.733 |
1.677 |
1.651 |
M–H |
1.837 |
1.722 |
1.677 |
| WBI |
0.503 |
0.469 |
0.413 |
0.358 |
WBI |
0.463 |
0.483 |
0.449 |
| Ag–H |
2.175 |
2.198 |
2.225 |
2.138 |
Au–H |
1.998 |
1.957 |
2.084 |
| WBI |
0.082 |
0.081 |
0.082 |
0.095 |
WBI |
0.148 |
0.156 |
0.137 |
| H–M–H (°) |
69 |
70 |
73 |
80 |
H–M–H (°) |
51 |
57 |
69 |
|
q
M
|
−2.81 |
−2.45 |
−2.15 |
−1.67 |
q
M
|
−2.42 |
−2.02 |
−1.70 |
|
q
Ag
|
0.43 |
0.48 |
0.54 |
0.59 |
q
Au
|
0.37 |
0.42 |
0.48 |
|
q
H
|
−0.18 |
−0.16 |
−0.17 |
−0.22 |
q
H
|
−0.03 |
0.00 |
0.00 |
It is of note that only one energy minimum was found for every computed model and all the energy minima adopt the same C2v configuration, shown in Fig. 1 for the [(RuH2)@Ag12]5+ model. In this structure, the two hydrides are approximately lying below two distant edges belonging to two edge-sharing triangular faces, as sketched in Scheme 1.
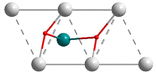 |
| | Scheme 1 A simplified 2-dimensional representation of the hydride positions in the (MH2)@M′12 (M′ = Ag, Au) computed models. Dotted lines indicate elongated M′–M′ edges. | |
Being in addition bonded to the central M atom, the hydrides are only tricoordinated, as opposed to the tetracoordination found in the monohydride models (see above). As a result of this double hydride insertion, several edges are elongated (dotted lines in Scheme 1), resulting in a substantial distortion of the icosahedral architecture, as exemplified by the large CSM values (Table 4). The H–M–H angles in Table 4 lie in the range ∼70–80°. The reported experimental values are a somewhat larger (∼90–100°).19,23,24,28,29 The discrepancy can be attributed to the effect of the peripheral shell on the superatomic core it passivates in the real compounds. Indeed, the potential energy surface associated with a variation of the H–M–H angle in our models is rather flat around its minimum. In any case, the linear H–M–H arrangement inside the Ag12 or Au12 icosahedron, as reported for other types of nanocluster19,23,24,28,29 is by far not favored.
2.4 More than two hydrides
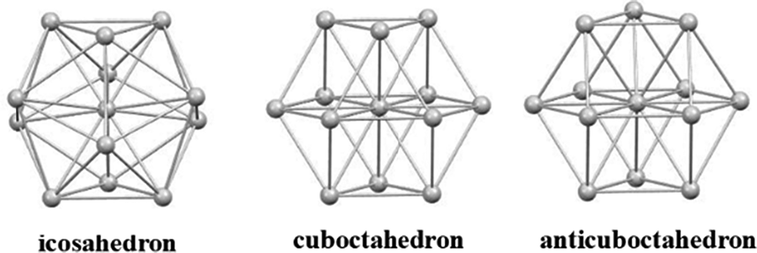
The encapsulating ability of a metal-centered icosahedral cage is obviously limited and inserting more than two hydrides in such M@Ag12 or M@Au12 templates resulted most often in extreme distortion, dismantling, or in non-converged models. This is why we also considered the possibility for hydride encapsulation in centered 12-vertex cuboctahedral and anticuboctahedral cages which have a somewhat larger volume than their icosahedral relative, while maintaining sufficient connectivity within its surface atoms (see Fig. 3). Whereas no reasonable structure could be found with four or more hydrides encapsulated within a centered 12-vertex polyhedron, with only three hydrides several (MH3)@Ag12 and (MH3)@Au12 structures could be identified.
 |
| | Fig. 3 Ideal centered 12-vertex cages. | |
Looking firstly at the silver-rich series, five M@Ag12 structures, labelled A, B, C, D and E, were characterized as true minima in the cases of M = Mo–Ru and W–Os. They are shown in Fig. 4 for the M = Mo example. In the cases of M = Tc and Re, additional secondary minima were obtained in which the hydrides are embedded within an open Ag11 cage capped by one Ag atom. These fairly distorted structures are not commented further on. In the cases of M = Ru and Os, a unique minimum of type B was found. Structures A, B, C, D and E have only approximate Cs, Cs, C3, C2 and Cs symmetry, respectively. As indicated by their large CSM values calculated with respect to the closest ideal polyhedron (Tables 5 and S3†), their Ag12 polyhedral envelope adopts configurations resulting from severe distortions away from ideal icosahedron, cuboctahedron and/or anticuboctahedron.
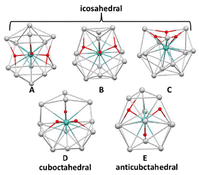 |
| | Fig. 4 The five (MH3)@Ag12 isomers exemplified in the case of [(MoH3)@Ag12]3+. | |
Table 5 Selected (averaged) computed data for the 8-electron superatomic cores [(MH3)@Ag12]x+ (M = Mo–Ru, W–Os). Interatomic distances are given in Å. WBI = Wiberg bond index; ΔE = isomer relative energy; qM, qAg and qAu are atomic natural charges. See also Table S3† for HOMO–LUMO gaps and metal–metal distances
| M |
Mo |
Tc |
Ru |
|
x
|
3 |
4 |
5 |
| Structure type |
A
|
B
|
C
|
D
|
E
|
A
|
B
|
C
|
D
|
E
|
B
|
| ΔE (kcal mol−1) |
0.0 |
2.9 |
3.1 |
7.4 |
11.5 |
0.0 |
2.1 |
2.1 |
8.0 |
10.5 |
|
| CSM |
2.32 |
2.41 |
1.88 |
6.40 |
4.91 |
2.55 |
2.37 |
1.93 |
6.49 |
6.95 |
2.25 |
| M–H |
1.791 |
1.798 |
1.797 |
1.794 |
1.808 |
1.703 |
1.707 |
1.717 |
1.733 |
1.711 |
1.659 |
| WBI |
0.492 |
0.481 |
0.481 |
0.494 |
0.489 |
0.442 |
0.436 |
0.446 |
0.435 |
0.434 |
0.615 |
| Ag–H |
2.074 |
2.085 |
2.153 |
2.055 |
2.168 |
2.065 |
2.214 |
2.167 |
2.148 |
2.067 |
2.138 |
| WBI |
0.097 |
0.098 |
0.091 |
0.113 |
0.091 |
0.098 |
0.082 |
0.096 |
0.100 |
0.093 |
0.301 |
|
q
M
|
−2.27 |
−2.30 |
−2.18 |
−2.30 |
−2.27 |
−1.97 |
−1.99 |
−1.93 |
−1.92 |
−1.97 |
−1.70 |
|
q
Ag
|
0.47 |
0.48 |
0.48 |
0.46 |
0.48 |
0.52 |
0.53 |
0.52 |
0.54 |
0.53 |
0.60 |
|
q
H
|
−0.12 |
−0.15 |
−0.15 |
−0.13 |
−0.16 |
−0.10 |
−0.13 |
−0.13 |
−0.17 |
−0.13 |
−0.15 |
| M |
W |
Re |
Os |
|
x
|
3 |
4 |
5 |
| Structure type |
A |
B
|
C
|
D
|
E
|
A
|
B
|
C
|
D
|
E
|
B
|
| ΔE (kcal mol−1) |
0.0 |
2.8 |
3.3 |
7.2 |
11.3 |
0.0 |
1.9 |
2.1 |
7.7 |
10.4 |
|
| CSM |
2.43 |
2.66 |
2.17 |
6.49 |
5.28 |
2.54 |
2.75 |
2.44 |
6.64 |
5.27 |
2.98 |
| M–H |
1.804 |
1.801 |
1.807 |
1.809 |
1.809 |
1.724 |
1.718 |
1.734 |
1.739 |
1.726 |
1.671 |
| WBI |
0.516 |
0.508 |
0.519 |
0.508 |
0.508 |
0.473 |
0.477 |
0.478 |
0.481 |
0.471 |
0.420 |
| Ag–H |
2.063 |
2.103 |
2.146 |
2.060 |
2.103 |
2.121 |
2.203 |
2.196 |
2.194 |
2.225 |
2.220 |
| WBI |
0.091 |
0.085 |
0.086 |
0.089 |
0.086 |
0.085 |
0.085 |
0.097 |
0.085 |
0.071 |
0.074 |
|
q
M
|
−2.61 |
−2.59 |
−2.51 |
−2.59 |
−2.59 |
−2.27 |
−2.27 |
−2.18 |
−2.23 |
−2.26 |
−1.94 |
|
q
Ag
|
0.50 |
0.51 |
0.50 |
0.51 |
0.51 |
0.55 |
0.55 |
0.55 |
0.56 |
0.56 |
0.61 |
|
q
H
|
−0.14 |
−0.17 |
−0.16 |
−0.17 |
−0.17 |
−0.12 |
−0.13 |
−0.14 |
−0.18 |
−0.14 |
−0.12 |
With respect to the gold-rich series, only M = Mo, W, Tc and Re provided converged systems with M@Au12 architectures and again structure-types A, B, C, D, and E were found (Table 6), but the number of isomers per M element does not exceed two.
Table 6 Selected (averaged) computed data for the 8-electron superatomic cores [(MH3)@Au12]x+ (M = Mo, W, Tc, Re). Interatomic distances are given in Å. WBI = Wiberg bond index; ΔEH/L = HOMO–LUMO gap; ΔE = isomer relative energy; qM, qAg and qAu are atomic natural charges
| M |
Mo |
W |
Tc |
Re |
|
x
|
3 |
3 |
4 |
4 |
| Structure type |
B
|
C
|
A
|
C
|
C
|
E
|
C
|
| CSM |
3.60 |
3.34 |
2.25 |
3.33 |
3.81 |
3.22 |
4.19 |
| ΔEH/L (eV) |
1.85 |
1.71 |
1.98 |
1.78 |
1.54 |
1.24 |
1.64 |
| ΔE (kcal mol−1) |
2.9 |
0.0 |
1.6 |
0.0 |
0.0 |
17.4 |
|
| M–Au |
2.977 |
3.004 |
2.924 |
2.999 |
3.039 |
2.934 |
3.031 |
| WBI |
0.347 |
0.343 |
0.354 |
0.345 |
0.296 |
0.307 |
0.301 |
| Au–Au |
2.823 |
2.816 |
2.852 |
2.816 |
2.827 |
2.873 |
2.829 |
| WBI |
0.135 |
0.137 |
0.113 |
0.133 |
0.143 |
0.123 |
0.138 |
| M–H |
1.824 |
1.791 |
1.927 |
1.806 |
1.719 |
1.846 |
1.728 |
| WBI |
0.475 |
0.494 |
0.439 |
0.526 |
0.454 |
0.357 |
0.491 |
| Au–H |
1.847 |
1.831 |
1.897 |
1.858 |
1.830 |
1.852 |
1.907 |
| WBI |
0.198 |
0.211 |
0.167 |
0.179 |
0.236 |
0.212 |
0.180 |
|
q
M
|
−1.87 |
−1.81 |
−2.43 |
−2.10 |
−1.45 |
−1.69 |
−1.75 |
|
q
Au
|
0.41 |
0.40 |
0.47 |
0.43 |
0.46 |
0.49 |
0.48 |
|
q
H
|
−0.02 |
0.01 |
−0.06 |
−0.03 |
0.03 |
−0.08 |
0.01 |
The bonding within these 8-electron superatomic models is related to that of their mono- and di-hydride relatives. Although more hypothetical (no such system experimentally reported to date), the lowest energy structures A, B and C, all of icosahedral parentage, are likely to constitute superatomic cores of viable ligand-protected nanoclusters. It is of note that, as this manuscript was under revision, an 8-electron copper-rich cluster with a (PtH3)@Cu12 core having a structure more or less related to the E anticuboctahedral arrangement was just published.39
2.5 The dynamics of the hydrides inside their icosahedral cages
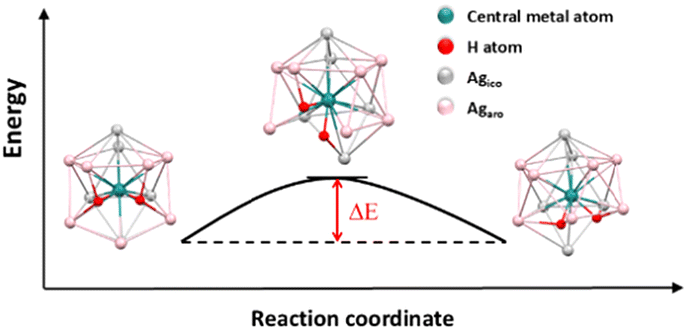
There are experimental NMR data on both mono-19,21,22,28,29 and di-hydride23,24 species indicating that the embedded hydrides move nearly freely inside their icosahedral envelopes. We have calculated the activation barrier for the hydride in (MH)@Ag12 species moving from one tetrahedral site to the next tetrahedron. This pathway is illustrated in Fig. 5 and selected computed data associated with the transition states are given in Table 7. Unsurprisingly, these transition states are of C2 symmetry, with the hydride connected to the two Ag atoms common to its starting and final tetrahedral sites. They were fully characterized as having one unique imaginary “vibrational frequency” of b1 symmetry. Owing to the flatness of the explored potential energy surfaces, some of these transition states were not easy to identify. Consistently, the energy barriers are very small (varying between 2 and 6 kcal mol−1). We have tested our results on a realistic ligand-protected nanocluster with complete outer sphere, namely the reported [(PtH)Ag19{S2P(OnPr)2}12],21 simplified in [(PtH)Ag19(S2PH2)12] for the sake of computational cost. Owing to the symmetry of this cluster (C3), there are three slightly different pathways for the hydride moving from one tetrahedral site to the next one. The one we tested has a barrier of 0.2 kcal mol−1, a value even lower than that found for the bare [PtH@Ag12]5+ core, indicating that modeling only the bare core provides results that can be safely projected on real ligand-protected species, at least at a semi-quantitative level.
 |
| | Fig. 5 The pathway of the hydride movement in [(RuH)Ag12]3+.(Agaro: Ag atoms around the H atom in the starting structure. Agico: Ag atoms not around the H atom in the starting structure.). | |
Table 7 Selected computed data for the (MH)@Ag12 (M = Mo–Pd, W–Pt) transition states. Interatomic averaged distances are given in Å. ΔE is its relative energy with respect to the energy minimum
| M |
ΔE (kcal mol−1) |
M–H |
Ag–H |
Ag–H–Ag (°) |
H–M–Ag (°) |
| Mo |
5.6 |
1.823 |
1.830 |
161 |
99 |
| Tc |
4.6 |
1.740 |
1.844 |
155 |
103 |
| Ru |
4.3 |
1.669 |
1.871 |
149 |
106 |
| Rh |
3.4 |
1.648 |
1.889 |
145 |
108 |
| Pd |
2.0 |
1.693 |
1.883 |
143 |
109 |
| W |
1.8 |
1.840 |
1.791 |
162 |
99 |
| Re |
5.2 |
1.743 |
1.868 |
156 |
102 |
| Os |
4.2 |
1.686 |
1.898 |
151 |
104 |
| Ir |
3.3 |
1.655 |
1.938 |
148 |
106 |
| Pt |
1.7 |
1.644 |
2.005 |
145 |
108 |
We also tested the hydride dynamics on the dihydride series (MH2)@Ag12. With two hydrides moving either sequentially or in a concerted fashion, several pathways are a priori possible. The concerted least-motion pathway illustrated in Fig. 6 was investigated. Selected computed data associated with the transition state are given in Table 8. The activation barriers remain lower than 3 kcal mol−1, except for M = Ir and Rh, with barriers of 5.1 and 7.2 kcal mol−1, respectively.
 |
| | Fig. 6 The pathway of the hydride movement in [(RuH2)Ag12]3+. | |
Table 8 Selected computed data for the (MH2)@Ag12 (M = Mo–Pd, W–Pt) transition states. Interatomic averaged distances are given in Å. ΔE is its relative energy with respect to the energy minimum
| M |
ΔE (kcal mol−1) |
M–H |
Ag–H |
Ag–H–Ag (°) |
| Mo |
2.8 |
1.794 |
1.932 |
60 |
| Tc |
2.6 |
1.706 |
1.946 |
62 |
| Ru |
2.4 |
1.650 |
1.943 |
65 |
| Rh |
7.2 |
1.659 |
2.067 |
80 |
| W |
2.8 |
1.808 |
1.948 |
60 |
| Re |
2.5 |
1.724 |
1.971 |
62 |
| Os |
2.2 |
1.668 |
1.984 |
65 |
| Ir |
5.1 |
1.645 |
2.192 |
73 |
2.6 Incomplete icosahedral cages: the nido case and beyond
Whereas the hydride-containing 8-electron superatom [HAu25(SMe)18] has been theoretically predicted to exist,11 as far as we know no gold nanocluster possessing a [(AuH)@Au12]6+ core has been structurally characterized so far. However, gold and gold-rich superatoms containing hydrides embedded in polyhedral cages having some open faces are known,12–18,40–42 although the hydride location in some of these structures remains to be clarified. This is the case of one of these species, which exhibits a Pt@(Au8Ag2) metal kernel, of which the Au8Ag2 cage can be described as an icosahedron with two unoccupied neighboring vertices.41 With respect to copper-rich superatom chemistry, hydride-containing 2-electron superatoms possessing a [(MH)@Cu11]10+ (M = Pd, Pt) core are known.25,26 In these species, the MH unit is embedded within a copper cuboctahedral cage of which one vertex is unoccupied. From the examples cited above, it appears that the same number of “magic” electrons could characterize species with complete polyhedral cages as well as incomplete cages having at least one or two unoccupied vertices. This is reminiscent to the closo, nido, arachno… parentage existing in the chemistry of the so-called Wade-Mingos-type clusters.43
Below we first investigate the 8-electron monohydride series [(MH)@Ag11]x+ and [(MH)@Au11]x+ (M = Mo–Pd, W–Pt), in which the Ag11 or Au11 cages can be viewed as incomplete icosahedra having a missing vertex. The search for energy minima was conducted starting from a M@(Ag11□) (□ = vacancy) skeleton, where the (Ag11□) shape is that of a regular icosahedron (nido-type structure). The hydride was then placed in any of the tetrahedral sites of the centered icosahedron, including those involving the □ vacancy and all these generated starting geometries where fully optimized. It turns out that the same global energy minimum of C5v symmetry was found for all the computed models, but [(PdH)@Ag11]4+. In this C5v structure, exemplified by those of [(RhH)@Ag11]3+ and [(PtH)@Ag11]4+ shown in Fig. 7, the hydride atom sits on the same side as (and close to) the missing vertex of the icosahedron. In the global minimum of [(PdH)@Ag11]4+ (Fig. 7), the hydride sits also on the same open side of the defective icosahedron, but it is now shifted towards an edge of the pentagonal face, leading to Cs symmetry. A secondary minimum, of Cs symmetry, was found for [(PdH)@Ag11]4+ (Fig. 7), where the hydride sits on the other side of the vacancy and is bonded to an icosahedron edge, forcing this part of the structure to distort.
 |
| | Fig. 7 The computed isomers of [(RhH)Ag11]3+, [(PdH)Ag11]4+, and [(PtH)Ag11]4+ with corresponding relative energies in kcal mol−1. | |
In fact, the C5v structure was found to be the unique minimum for all M dopants, except for M = Rh and Pt, for which one secondary minimum was also found (Fig. 7). The secondary minimum of [(RhH)@Ag11]3+ resembles somewhat the C2v isomer of [(PdH)@Ag11]4+, whereas that of [(PtH)@Ag11]4+ has lost its icosahedral parentage with one Ag atom capping an Ag10 cage. Selected computed data are provided in Table 9.
Table 9 Selected (averaged) computed data for the nido-type 8-electron superatomic cores [(MH)@Ag11]x+ (M = Mo–Pd, W–Pt), of C5v structure except for [(PdH)@Ag11]4+ (Cs symmetry). Interatomic distances are given in Å. WBI = Wiberg bond index; ΔEH/L = HOMO–LUMO gap; qM, qAg and qAu are atomic natural charges
| M |
Mo |
Tc |
Ru |
Rh |
Pd |
W |
Re |
Os |
Ir |
Pt |
|
x
|
0 |
1 |
2 |
3 |
4 |
0 |
1 |
2 |
3 |
4 |
| ΔEH/L (eV) |
1.01 |
1.06 |
1.24 |
1.62 |
1.93 |
1.65 |
1.16 |
1.37 |
1.73 |
2.04 |
| M–Ag |
2.765 |
2.758 |
2.765 |
2.791 |
2.840 |
2.770 |
2.765 |
2.775 |
2.801 |
2.850 |
| WBI |
0.435 |
0.376 |
0.308 |
0.231 |
0.198 |
0.417 |
0.359 |
0.298 |
0.250 |
0.214 |
| Ag–Ag |
2.892 |
2.886 |
2.894 |
2.920 |
2.977 |
2.894 |
2.890 |
2.900 |
2.924 |
2.971 |
| WBI |
0.092 |
0.093 |
0.093 |
0.094 |
0.092 |
0.090 |
0.091 |
0.091 |
0.090 |
0.087 |
| M–H |
1.740 |
1.669 |
1.617 |
1.592 |
1.669 |
1.756 |
1.693 |
1.643 |
1.611 |
1.605 |
| WBI |
0.575 |
0.529 |
0.471 |
0.400 |
0.255 |
0.592 |
0.548 |
0.482 |
0.430 |
0.379 |
| Ag–H |
2.569 |
2.541 |
2.536 |
2.555 |
2.031 |
2.585 |
2.562 |
2.562 |
2.585 |
2.644 |
| WBI |
0.051 |
0.059 |
0.067 |
0.075 |
0.170 |
0.047 |
0.054 |
0.061 |
0.064 |
0.063 |
|
q
M
|
−2.28 |
−2.03 |
−1.82 |
−1.63 |
−1.12 |
−2.78 |
−2.49 |
−2.22 |
−1.83 |
−1.41 |
|
q
Ag
|
0.21 |
0.278 |
0.35 |
0.43 |
0.49 |
0.26 |
0.33 |
0.39 |
0.45 |
0.51 |
|
q
H
|
−0.08 |
−0.05 |
−0.06 |
−0.09 |
−0.27 |
−0.13 |
−0.09 |
−0.07 |
−0.09 |
−0.15 |
In all the structures of Fig. 7, the hydride is strongly bonded to M and weakly to Ag. In the C5v structure, the M–Ag distances are particularly long and the associated WBIs particularly weak (Table 9). Nevertheless, this is the dominant low-energy structure in the [(MH)@Ag11]x+ series. It is somewhat reminiscent to that of the 8-electron [HAu9(PMe3)]2+ cluster, where the hydride sits over an open face of the Au@Au8 metal kernel.12 Conversely, it is at variance with the 2-electron nido-type nanoclusters [MHCu11{S2P(OiPr)2}6(CCPh)4] (M = Pd, Pt) of cuboctahedral parentage, where the hydride sits at a tetrahedral site opposite to the vacancy.23,24 It is also noteworthy that in the C5v structure the hydride contribution to the superatomic orbitals becomes nonnegligible. In the case of [(PtH)@Ag11]4+ it contributes 35% and 5% to the 1S and 1Pz orbitals respectively. By comparison, in the Cs minimum of [(PdH)@Ag11]4+ (Fig. 7), its contributions are only 2% and 1%, respectively. Thus, in the C5v structure, the hydride tends to some extend to replace the missing Ag atom on the icosahedron and play the role of a superatom constituent, whereas in the other structures it behaves similarly as in the closo-type relatives, i.e. as an inserted dopant.23
Switching from the Ag-rich species to their gold relatives yielded fairly different results. The global minima found for the 8-electron models [(MH)@Au11]x+ (M = Mo–Pd, W–Pt) are shown in Fig. 8. Only five of them have retained their icosahedral parentage, of which only two (corresponding to M = Tc and Re) are of C5v symmetry. Those corresponding to M = Mo, W and Ru derive from the C5v structure by the opening of one edge of the open pentagonal face. The potential energy surfaces of these species were found more complex that in the case of the Ag-rich series, leading to a larger number of isomers. In addition, their lower HOMO–LUMO gaps suggest lower stability, at least in their considered form of unligated superatomic cores. This is why this series will not be discussed further on. In any case, as already found in the other investigated series, gold-rich clusters are more flexible than their silver homologues, leading to a more complex and less predictable structural chemistry.
 |
| | Fig. 8 The lowest energy isomers of the 8-electron models [(MH)@Au11]x+ (M = Mo–Pd, W–Pt) with their HOMO–LUMO gap (eV) below. | |
Unfortunately, exploring the possibility for 8-electron arachno-type systems with (MH)@Ag10 or (MH)@Au10 architectures was not possible, owing to the huge number of potential isomeric structures to be considered. However, there is one example of such a species, which has been structurally characterized (except with respect to the hydride location) namely [PtH(AgNO3)2(AuPPh3)8]+.41 Its Ag2Au8 polyhedron can be viewed as an icosahedron having two neighboring vertices unoccupied. We thus simplified the superatomic core of this nanocluster into the corresponding bimetallic arachno model [PtHAu10]3+. Only one energy minimum (of C2v symmetry) was found, with the hydride located at the open side of the uncomplete icosahedron, (Fig. 9). Again, the Pt–H bond is strong (1.569 Å, WBI = 0.509), whereas the two shortest Ag–H contacts are fairly long (2.632 Å, WBI = 0.083). The participation of the hydride in the 1S and 1Pz orbitals is non-negligible (10% and 6%, respectively). Very similar results were also found for [PdHAg10]3+. These results suggest that hydride-encapsulating arachno species are also potentially viable cores for ligand-protected superatoms susceptible to be isolated.
 |
| | Fig. 9 The optimized geometry the 8-electron arachno model [PtHAu10]3+. | |
3. Conclusion
Our calculations indicate that (MHx)Ag12 (M = Group 6 to (at least) Group 10; x = 1–3) architectures are viable as constituting the superatomic core of ligand-protected 8-electron nanoclusters. Such entities are expected to more or less retain the icosahedral arrangement of their non-hydridic parents, with some cuboctahedral and/or anticuboctahedral features in the case of x = 3. In all these species, the hydrides are much strongly bonded to the central M atom than to their nearest Ag neighbors. This is why the encapsulated hydrides are relatively free to move inside the Ag12 cage, at least in most of the mono- and di-hydride cases for which particularly small activations barriers are computed. Thus, it is not excluded that the hydride hopping from one tetrahedral site to a next one is accompanied by tunnelling effect through the small barrier. Nido-type monohydride species, i.e. MH units encapsulated within an uncomplete Ag11 icosahedron, are also expected to be stable for the 8-electron count. Interestingly, in most of the computed models, the hydride prefers to be located on the vacancy side, tending to replace the missing Ag atom(s) on the icosahedron, including in the participation of its 1s AO to the superatomic orbitals. In general, the (MHx)Au12 species exhibit similar structural features as their isoelectronic (MHx)Ag12 homologues, but with smaller HOMO–LUMO gaps and more complicated potential energy hypersurfaces, suggesting lower stability and/or more complex structural chemistry. Moreover, for the most right-sided M elements, the Au12 cage tends to get unstable with respect to smaller Aun (n < 12) cages capped by 12 − n Au atoms.
Computational details
Calculations were performed at the Density Functional Theory (DFT) level using the Gaussian (R) 16 program.44 The BP86 functional was used45,46 together with the valence triple-zeta polarization functions (def2-TZVP) basis set.47,48 All the structures were optimized without any symmetry constraint and confirmed as true minima on their potential energy surface by analytical vibration frequency calculations. Natural atomic orbital (NAO) populations and Wiberg bond indices (the sum of quadratic non-diagonal elements of the density matrix between two atoms) were computed with the natural bond orbital NBO 6.0 program49 implemented in the Gaussian (R) 16 package. The compositions of the molecular orbitals were calculated using Multiwfn software.50,51 In this paper, the meaning of non-convergence during geometry optimization refers to a geometry evolution which leads to dissociation/fragmentation of the considered cluster.
Data availability
The data supporting the findings of this study are available within the article and its ESI.† Complementary raw data are available from the corresponding authors, upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr J. Wei (Ningxia University) is acknowledged for inspiring discussions. H. L. thanks the China Scholarship Council for a Ph.D. grant.
References
- C. M. Aikens, R. Jin, X. Roy and T. Tsukuda, J. Chem. Phys., 2022, 156, 170401 CrossRef CAS.
- J. Yang, F. Yang, C. Zhang, X. He and R. Jin, ACS Mater. Lett., 2022, 4, 1279 CrossRef CAS.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443 RSC.
- M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372 CrossRef CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157 CrossRef CAS.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847 RSC.
- H. Häkkinen, Adv. Phys.: X, 2016, 1, 467 Search PubMed.
- F. Gam, J. Wei, S. Kahlal, J.-Y. Saillard and J.-F. Halet, Struct. Bonding, 2021, 188, 69 CrossRef CAS.
- Z. Lin, Chin. J. Struct. Chem., 2024, 43, 100254 CrossRef CAS.
- R. S. Dhayal, W. E. van Zyl and C. W. Liu, Acc. Chem. Res., 2016, 49, 86 CrossRef CAS PubMed.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D.-e Jiang, Chem. Mater., 2017, 29, 4840 CrossRef CAS.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 8380 CrossRef CAS PubMed.
- S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Acc. Chem. Res., 2018, 51, 3074 CrossRef CAS PubMed.
- W. E. van Zyl and C. W. Liu, Chem. – Eur. J., 2022, 28, e202104241 CrossRef CAS.
- S. Maity, S. Takano, S. Masuda and T. Tsukuda, J. Phys. Chem. C, 2024, 128, 19 CrossRef CAS.
- T.-H. Chiu, J.-H. Liao, R. P. B. Silalahi, M. N. Pilla and C. W. Liu, Nanoscale Horiz., 2024, 9, 675 RSC.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 12314 CrossRef CAS PubMed.
- H. Hirai, S. Takano and T. Tsukuda, ACS Omega, 2019, 4, 7070 CrossRef CAS.
- H. Yi, S. M. Han, S. Song, M. Kim, E. Sim and D. Lee, Angew. Chem., Int. Ed., 2021, 60, 22293 CrossRef CAS PubMed.
- C. Zhu, T. Duan, H. Li, X. Wei, X. Kang, Y. Pei and M. Zhu, Inorg. Chem. Front., 2021, 8, 4407 RSC.
- T.-H. Chiu, J.-H. Liao, F. Gam, Y.-Y. Wu, X. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2022, 144, 10599 CrossRef CAS.
- Y.-R. Ni, M. N. Pillay, T.-H. Chiu, Y.-Y. Wu, S. Kahlal, J.-Y. Saillard and C. W. Liu, Chem. – Eur. J., 2023, 29, e202300730 CrossRef CAS.
- H. Yi, S. Song, S. M. Han, J. Lee, W. Kim, E. Sim and D. Lee, Angew. Chem., Int. Ed., 2023, 62, e202302591 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, Y.-Y. Wu, J.-Y. Chen, Y.-J. Chen, W. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2023, 145, 16739 CrossRef CAS.
- R. P. B. Silalahi, Y. Jo, J.-H. Liao, T.-H. Chiu, E. Park, W. Choi, H. Liang, S. Kahlal, J.-Y. Saillard, D. Lee and C. W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301272 CrossRef PubMed.
- R. P. B. Silalahi, H. Liang, Y. Jo, J.-H. Liao, T.-H. Chiu, Y.-Y. Wu, X. Wang, S. Kahlal, J.-Y. Saillard, Q. Wang, W. Choi, D. Lee, J.-Y. Saillard and C. W. Liu, Chem. – Eur. J., 2024, 30, e202303755 CrossRef PubMed.
- Y.-R. Ni, M. N. Pillay, T.-H. Chiu, J. Rajaram, Y.-Y. Wu, S. Kahlal, J.-Y. Saillard and C. W. Liu, Inorg. Chem., 2024, 63, 2766 CrossRef CAS PubMed.
- S. M. Han, S. Song, H. Yi, E. Sim and D. Lee, Nanoscale, 2024, 16, 4851 RSC.
- T.-H. Chiu, M. N. Pillay, Y.-Y. Wu, Y. Niihori, Y. Negishi, J.-Y. Chen, Y. J. Chen, S. Kahlal, J.-Y. Saillard and C. W. Liu, Chem. Sci., 2024, 15, 14660 RSC.
- R. S. Dhayal, J.-H. Liao, Y.-C. Liu, M.-H. Chiang, S. Kahlal, J.-Y. Saillard and C. W. Liu, Angew. Chem., Int. Ed., 2015, 54, 3702 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2019, 141, 12957 CrossRef CAS.
- F. Gam, C. W. Liu, S. Kahlal and J.-Y. Saillard, Nanoscale, 2020, 12, 20308 RSC.
- F. Alkan, P. Pandeya and C. M. Aikens, J. Phys. Chem. C, 2019, 123, 9516 CrossRef CAS.
- F. Gam, I. Chantrenne, S. Kahlal, T.-H. Chiu, J.-H. Liao, C. W. Liu and J.-Y. Saillard, Nanoscale, 2022, 14, 196 RSC.
- J. Wei, D. MacLeod Carey, J.-F. Halet, S. Kahlal, J.-Y. Saillard and A. Muñoz-Castro, Inorg. Chem., 2023, 62, 3047 CrossRef CAS.
- X. Li, B. Kiran, J. Li, H.-J. Zhai and L.-S. Wang, Angew. Chem., Int. Ed., 2002, 41, 4786 CrossRef CAS PubMed.
- P. Pyykkö and N. Runeberg, Angew. Chem., Int. Ed., 2002, 41, 2174 CrossRef.
- H. Zabrodsky, S. Peleg and D. Avnir, J. Am. Chem. Soc., 1992, 114, 7843 CrossRef CAS.
- A. He, D. Zuo, G. Jiang, X. Tang, L. Wang, L. Feng, Z. Zhao, J. Wei, N. Zheng and H. Shen, Sci. Adv., 2025, 11, eads4488 CrossRef.
- J. J. Bour, P. P. J. Schlebos, R. P. E. Kanters, M. F. J. Schoondergang, H. Addens, A. Overweg and J. J. Steggerda, Inorg. Chim. Acta, 1991, 181, 195 CrossRef CAS.
- T. G. M. M. Kappen, P. P. J. Schlebos, J. J. Bour, W. P. Bosman, G. Beurskens, J. M. M. Smits, P. T. Beurskens and J. J. Steggerda, Inorg. Chem., 1995, 34, 2121 CrossRef CAS.
- T. G. M. M. Kappen, J. G. M. Van der Linden, A. M. Roelofsen, J. J. Bour, P. P. J. Schlebos and J. J. Steggerda, Inorg. Chim. Acta, 1996, 245, 133 CrossRef CAS.
-
D. M. P. Mingos and D. J. Wales, Introduction to Cluster Chemistry, Prentice Hall, Englewood Cliffs, NJ, 1990 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- A. Schaefer, H. Horn and R. J. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS.
- A. Schaefer, C. Huber and R. J. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS.
- J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41 CrossRef.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Jean-Yves
Saillard
*b and
Jean-Yves
Saillard