
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoding in-plane orientation in cellulose nanopapers hybridized with tailored polymeric nanoparticles†
Åsa
Jerlhagen
 ab,
Korneliya
Gordeyeva
ab,
Martina
Cattaruzza
a,
Louise
Brandt
ac,
Benedikt
Sochor
de,
Sarathlal
Koyiloth Vayalil
df,
Stephan V.
Roth
acd,
Lars
Wågberg
abc and
Eva
Malmström
*abc
ab,
Korneliya
Gordeyeva
ab,
Martina
Cattaruzza
a,
Louise
Brandt
ac,
Benedikt
Sochor
de,
Sarathlal
Koyiloth Vayalil
df,
Stephan V.
Roth
acd,
Lars
Wågberg
abc and
Eva
Malmström
*abc
aKTH Royal Institute of Technology, Department of Fiber and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Teknikringen 56, SE-100 44 Stockholm, Sweden
bFibRe − Centre for Lignocellulose-based Thermoplastics, KTH Royal Institute of Technology, Department of Fiber and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Teknikringen 56, SE-100 44 Stockholm, Sweden
cWallenberg Wood Science Center, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
dDeutsches Elektronen-Synchrotron DESY, Notkestrasse 85, 22607 Hamburg, Germany
eAdvanced Light Source, Lawrence Berkeley National, Laboratory, 6 Cyclotron Rd, Berkeley, CA 94720, USA
fApplied Science Cluster, University of Petroleum and Energy Studies UPES, 248007 Dehradun, India
First published on 5th March 2025
Abstract
Biobased cellulose nanofibrils (CNFs) constitute important building blocks for biomimetic, nanostructured materials, and considerable potential exists in their hybridization with tailorable polymeric nanoparticles. CNFs naturally assemble into oriented, fibrillar structures in their cross-section. This work shows that polymeric nanoparticle additives have the potential to increase or decrease orientation of these cellulose structures, which allows the control of bulk mechanical properties. Small amounts of these additives (<1 wt%) are shown to promote the alignment of CNFs, and the particle size is found to determine a tailorable maximum feature size which can be modified. Herein, X-ray scattering allows for the quantification of orientation at different length scales. This newly developed method of measuring cross-sectional orientation allows for understanding the influence of nanoparticle characteristics on the CNF network structure at different length scales in hybrid cellulose-nanoparticle materials, where previously quantitative description has been lacking. It thus constitutes an important foundation for further development and understanding of nanocellulose materials on the level of their nanoscale building blocks and their interactions, which in turn are decisive for their macroscopic properties.
Introduction
Since the advent of the nano-world in the 20th century, the importance of nanostructures in determining material properties has become increasingly evident. Controlled assembly of nanostructures enables materials with remarkable properties, as seen in natural systems like nacre and wood, where toughness arises from precise nanoscale architecture.1,2 In synthetic materials, fine-tuning of nanostructures holds significant potential for achieving novel properties, where advanced characterization techniques are key to elucidate structure–property relationships.3,4In recent years, nanomaterials derived from cellulose have garnered significant attention due to their inherent biobased nature and advantageous properties such as high strength, stiffness, durability and chemical functionality, which allow for versatile design possibilities and applications such as thin barriers, flexible electronics substrates and optoelectronic materials.5–10 Cellulose nanofibrils (CNFs) are fibrous nanomaterials with diameters around 3 nm and micrometer-scale lengths, dispersed in aqueous media with charged surfaces ensuring colloidal stability.11 These charges facilitate assembly strategies such as layer-by-layer adsorption,12 or by mediating interactions during drying by the addition of small-molecule or polymeric additives.13–15 Tailored additives are particularly promising for controlled structure formation and property tuning through simple methods like evaporation or vacuum filtration.16,17
Much of the research on CNF-based films focuses on tailoring mechanical properties.18,19 High strength and stiffness are achieved by dewatering CNFs under conditions that maintain colloidal stability,18 while polymeric additives often increase ductility at the expense of stiffness.20,21 Polymers with room-temperature glass transition temperatures provide the greatest improvements in toughness,22 while polymers like polyacrylamide (PAM) enhance modulus through strong intercomponent bonding.23,24
There is an expanding body of literature where polymeric nanoparticle additives show synergistic property-improvements, but where the structure–property relationships remain poorly understood.25–29 High loadings (25 wt%) of rigid cationic particles significantly improve ductility (>200%) through new energy dissipation modes,25,26 but at low loading (<1 wt%) the particles have a stiffening effect.28 These nanoparticle additives are likely to induce major structural changes in cellulosic ordering, which may be the driver of macroscopic property impact.
Cellulose nanopapers naturally form anisotropic structures, with nanoscopic fibrils aligning perpendicular to the filtration direction during assembly.30 These structures are described with various terminologies such as layered, laminar, aligned, stratified, or anisotropic, and are widely documented by electron microscopy (Fig. 1).31–35 However, these methods have limitations, including sample preparation artifacts, limited bulk visualization and lack of quantitative information. X-ray diffraction (XRD) and Raman spectroscopy are bulk techniques used to quantify crystallite alignment, but significant differences in alignment between different materials are rarely observed.32,33,36–38
 | ||
| Fig. 1 Schematic of the investigated cellulose nanopaper and FE-SEM images showing (a) the surface and (b) the cryofractured cross-section. | ||
The hybridization of CNFs with colloidally stable polymeric nanoparticles likely leads to ordering and disordering on multiple length scales. To understand this orientation disorder and its impact on properties, it is necessary to evaluate structures across multiple length scales. In this study, we employ wide- and ultra-small-angle X-ray scattering (WAXS and USAXS) to probe hybrid nanocellulose materials and quantify orientation parameters across scales, linking them to bulk mechanical properties.
Experimental
Materials
Never-dried sulfite pulp was kindly donated by Domsjö. N,N-Dimethylamino ethyl methacrylate (DMAEMA) (98%) containing 700–1000 ppm monomethyl ether hydroquinone (MEHQ), and methyl methacrylate (MMA) (99%) containing ≤30 ppm MEHQ were purchased from Sigma Aldrich, and the inhibitor was removed prior to polymerization by passing over basic alumina. Basic alumina (aluminium oxide 90 active basic (0.063–0.200 mm) activity stage I) was purchased from Merck Millipore. Dioxane anhydrous (99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA) (98%) and 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA, 97%) were purchased from Sigma Aldrich and used as received. Acetone and hydrochloric acid (HCl) (37%) were purchased from VWR and used as received.Preparation of CNFs
TEMPO-mediated oxidized CNFs were prepared according to previously described procedures.11,39 Never-dried softwood sulfite pulp (Domsjö) was treated by TEMPO-mediated oxidation at pH 10, and fibrillated in a high-pressure homogenizer (Microfluidizer M-110EH, MicrofluidicsCorp, USA) by one pass through large chambers (400 μm and 200 μm, 800 bar) and four passes through smaller chambers (200 μm and 100 μm, 1600 bar) to produce a CNF gel with dry content ∼10 g L−1. CNF dispersions (∼2 g L−1) were prepared by Ultraturrax homogenization, tip sonication and centrifugation.39 The total charge of CNFs was determined by conductometric titration as 448 ± 16 μmol g−1.Synthesis of polymeric nanolatex with RAFT-mediated polymerization-induced self-assembly (PISA)
The chain-transfer agent CTPPA was synthesized according to previously published protocol.40The water-soluble PDMAEMA block (macroCTA) was synthesized by solution RAFT polymerization. DMAEMA monomer (6.74 mL, 40 mmol, 25 eq.), ACVA (44.85 mg, 0.16 mmol, 0.1 eq.), CTPPA (444 mg, 1.6 mmol, 1 eq.) were dissolved in dioxane (13.26 mL, [M]DMAEMA = 2.0 M) and degassed by Argon bubbling for 15 minutes at 0 °C. The reaction proceeded at 70 °C for 24 hours (conversion ∼99% from 1H-NMR). The crude reaction was precipitated in acetone with concentrated HCl (1.6 ml, 20 mmol), decanted and dried under reduced pressure at 50 °C overnight.
RAFT-mediated emulsion PISA was performed with MMA in water. The large and small particles were formulated for degree of polymerization 200 and 500 of the central MMA block. The PDMAEMA macroCTA (158.7 mg, 0.038 mmol, 1 eq.), AIBA (1.24 mg, 0.0046 mmol, 0.12 eq.) and MMA (2.01 ml, 18.9 mmol, 500 eq.) were dissolved in distilled water (total batch size 20 ml). Dry content 10 wt%. The mixture was degassed by Argon bubbling for 15 minutes at 0 °C. The reaction proceeded at 70 °C for 3 hours (gravimetric conversion >99%). The crude dispersion was purified by dialysis (12–14 kDa cutoff) in distilled water.
Preparation of hybrid cellulose nanopapers
Phosphate buffer was prepared by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between K3PO4 and K2HPO4. The polymeric nanoparticles were diluted to 0.2 wt% in 10 or 100 mM phosphate buffer. The nanoparticle dispersion was added drop-wise under vigorous stirring to a CNF dispersion of 0.2 wt%. After complete addition the hybrid dispersion was stirred vigorously for 30 minutes, and then vacuum filtered over PVDF filter membranes overnight. The wet cakes were dried for 2 days in holders to prevent buckling in a room conditioned to 50% relative humidity.
:1 molar ratio between K3PO4 and K2HPO4. The polymeric nanoparticles were diluted to 0.2 wt% in 10 or 100 mM phosphate buffer. The nanoparticle dispersion was added drop-wise under vigorous stirring to a CNF dispersion of 0.2 wt%. After complete addition the hybrid dispersion was stirred vigorously for 30 minutes, and then vacuum filtered over PVDF filter membranes overnight. The wet cakes were dried for 2 days in holders to prevent buckling in a room conditioned to 50% relative humidity.
Nuclear magnetic resonance
1H-NMR was used to determine conversion of DMAEMA by comparing intensity of the residual vinyl peak at 6.0 ppm versus a polymer peak at 4.0 ppm. 1H-NMR was performed on a Bruker Avance NMR at 400 MHz using CDCl3 as solvent.Size exclusion chromatography (SEC)
SEC was performed on a TOSOH EcoSEC HLC-8320GPC equipped with EcoSEC RI detector and three columns (PSS GRAM 10 μm; Microguard, 100 Å, and 1000 Å) from PSS GmbH (resolving range 100–1000000 g mol−1), using DMF with 0.01 M LiBr as mobile phase, at 50 °C, and with a flow rate of 1 ml min−1. Calibration was performed using PMMA standards ranging from 100 to 1000000 g mol−1, and toluene was used as flow rate marker.
Dynamic light scattering (DLS)
The hydrodynamic diameter (DH), polydispersity index (PdI) and zeta potential (ζ) of the polymeric nanoparticles were determined using a Malvern Zetasizer NanoZS at 0.1 wt% in distilled water at 25 °C. The standard for size correlation was polystyrene latex.Polyelectrolyte titration (PET)
PET was performed on a Stabino unit (Particle Metrix GmbH, Germany). The cationic nanoparticles were titrated against poly(vinyl sulfate) (KPVS) to find the surface charge.Conductometric titration
Conductometric titration of TEMPO-mediated oxidized pulp was performed following the protocol described elsewhere.41Table-top small-angle X-ray scattering (SAXS)
SAXS was performed on a Mat:Nordic (SAXSLAB/Xenox) with a Rigaku 003+ high brilliance microfocus Cu–K radiation source (λ = 1.5406 Å) and Pilatus 300K detector. The nanoparticle dispersions (1 wt% in distilled water) were measured in borosilicate glass capillaries (1.5 mm diameter, wall thickness 0.01 mm) with an exposure time of 3600 s.Field-emission scanning electron microscopy (FE-SEM)
FE-SEM was performed on a Hitachi S-4800. The materials were fractured in liquid nitrogen or in the tensile testing rig, mounted and coated with a thin layer of Pt/Pd before imaging.Atomic force microscopy (AFM)
Diameter and length of CNFs where assessed with an AFM (Multimode 8, Bruker, USA) in tapping mode in air. A CNF dispersion (0.3 wt%) was diluted to 0.0005 wt% and homogenized for 10 min at 10000 rpm using Ultraturrax (IKA T25D, Sweden). Remaining aggregates were removed by centrifugation for 1 hour at 4000 rpm. The supernatant was mixed on a Vortex Genie 2 (Scientific Industries Inc., USA) for 5 min prior to adsorption. Freshly cleaved mica was treated with (3-aminopropyl)triethoxysilane for 30 seconds, before adsorption of the CNF dispersion for another 30 seconds.
X-ray scattering experiments (WAXS and USAXS)
X-ray scattering experiments were performed at beamline P0342 with a sample to detector distance of 9.41 m for USAXS (PILATUS 2M detector, pixel size 172 μm) and 36.89 cm for WAXS (Lambda 9M detector, pixel size 55 μm). Sample to detector distances were calibrated using standard calibrants. The beam size was 22 × 30 μm, as focused on the sample. A 0.3 μm Al absorber was used with 1 s exposure. The nanocomposites were sliced with a sharp scalpel to ∼1.5 mm. Because slices of material are relatively thick in the beam direction (∼1.5 mm), the surface deformation caused by slicing will give negligible scattering as compared to the bulk material scattering.Tensile testing
Tensile testing was performed using an Instron 5944 with 500 N load cell at 23 °C and 50% RH. Strips were cut with a punch cutter tool to 6 mm in width, and stored under the same conditions for at least 48 h prior to testing. The gauge length was set to 30 mm and strained at 10% min−1. Minimum 5 specimens per sample were tested.Results and discussion
Synthesis of hybrid materials
The structure-forming additives in this study are tailored block copolymers synthesized according to the principles of polymerization-induced self-assembly (PISA), employing reversible addition–fragmentation chain-transfer (RAFT) polymerization.43,44 Briefly, poly(2-(N,N-dimethylamino) ethyl methacrylate) (PDMAEMA) with degree of polymerization 25 was chain extended with methyl methacrylate (MMA) in aqueous emulsion polymerization. This yielded spherical particles with rigid PMMA core and tertiary-amine PDMAEMA functional shell (Fig. 2b). This shell stabilizes the particles in aqueous dispersion, and acts as anchoring block in the adsorption to cellulose.45 The rigid core is chosen due to a hypothesized structure-forming effect, as rigid core-additives have previously shown positive synergistic effects on mechanical properties in cellulose nanocomposites.26 | ||
| Fig. 2 To a dispersion of anionic cellulose nanofibrils (CNFs) (a) is added cationic nanolatex particles (b), either large (L) ∼90 nm or small (S) ∼30 nm. The light blue cloud around particles indicates the size of the extended shell polymer and associated water, as measured by DLS. The hybrid dispersion (c) is stirred vigorously to ensure component adsorption, and is then vacuum filtered to produce materials with oriented structures (d). The nanoparticle amount (np) and size (L or S) impact the assembly upon water removal. | ||
Two particles of different sizes were synthesized, hence termed large (L) and small (S) (Table 1). The particle cores are ∼90 and ∼30 nm in diameter. The particles are spherical, with no higher morphologies (worms, vesicles) present as shown by field emission scanning electron microscopy (FE-SEM) imaging (Fig. S3†). Dynamic light scattering (DLS) yields the hydrodynamic volume DH, which takes into account the size of the extended shell polymer (PDMAEMA) and its associated water when the nanoparticle is in the dispersed state. Both the size of the nanoparticle in dry state (core size) and in the wet state (DH) are important characteristics regarding the structure-forming capabilities. The structure and charge of the wet particle in dispersion will control the extent of interaction and potential wrapping with the oppositely charged CNF. Conversely the dry size (core size) of the particle will determine which size of naturally-occurring void space it can occupy in the assembling CNF-matrix. The two nanoparticles have relatively similar zeta potentials, and the particles have surface charge of 470 and 950 μmol g−1 respectively. The adsorption of PDMAEMA-functional particles to cellulose has been found to be strongly dependent on the size and charge, where smaller size and lower charge increases adsorption.45
| Name |
D
SEMa (nm) |
D
SAXSb (nm) |
D
Hc (nm) |
PdIc |
ζd (mV) |
Surface chargee (μmol g−1) |
|---|---|---|---|---|---|---|
| a FE-SEM imaging of nanoparticles adsorbed to plasma-treated silicon wafer. b SAXS of nanoparticles in dispersion fit to spherical model. c Hydrodynamic diameter by dynamic light scattering (DLS). d Zeta potential. e Surface charge by polyelectrolyte titration. | ||||||
| D25-M500 (L) | 79 ± 13 | 88 ± 0.003 | 119 ± 0.6 | 0.029 ± 0.025 | 66 ± 0.2 | 470 ± 30 |
| D25-M200 (S) | 26 ± 5 | 27 ± 0.025 | 40 ± 0.1 | 0.076 ± 0.004 | 54 ± 3.3 | 950 ± 30 |
Anionic CNFs of surface charge 450 μmol g−1 were used for this work (Fig. 2a). TEMPO-mediated oxidation of softwood pulp was employed to introduce carboxylate charges on the nanofibril surface, and the CNFs were separated through homogenization.46 The block copolymer particles were diluted to 2 g L−1 in 100 mM or 10 mM phosphate buffer, and adsorbed to cellulose nanofibrils (CNFs) by slow addition to a 2 g L−1 CNF dispersion during vigorous stirring (Fig. 2b) resulting in a homogeneous hybrid dispersion (Fig. 2c). The buffer is added to avoid localized differences in pH, which helps in reducing visible aggregation during mixing. The buffer should also maximize adsorption of the components, as the increase in ionic strength decreases the Debye length and thus intercomponent repulsion (Fig. 2c).47,48 The hybrid dispersion is then vacuum filtered to produce cellulose-rich nanocomposites with aligned structures on different length scales (Fig. 2d).
In this work, the nanoparticles were buffered to two different molarities, to investigate whether buffer impacts assembly and material properties. The samples are hence referred to as LBnp or SBnp where S/L denotes small or large particles, np is the nanoparticle amount in wt%, and B denotes nanoparticles in 100 (high) or 10 mM (low) phosphate buffer molarity. B refers to the molarity of buffer in the nanoparticle dispersions before they are added to CNF. The total buffer molarities in the hybrid dispersions are thus between 0.4 and 21.9 mM and vary with nanoparticle amount np. Reference materials without particles are referred to as CNFB. All hybrid materials are detailed in Table S2.† No macroscopic aggregation is seen in these hybrid dispersions, when nanoparticles are added to CNFs, likely because of the stabilizing effect high aspect ratio CNFs have on colloidal systems, as shown earlier.49 With the addition of these particles, it is visible in SEM images that the assembled layered structure of neat CNF is altered (Fig. 3).
 | ||
| Fig. 3 Cross-sections of hybrid cellulose nanopapers with increasing amounts of PISA nanoparticles. Cross-sections are prepared by cryo-fracture in liquid N2 and imaged by FE-SEM. Left: CNF reference without nanolatexes. Upper row: Hybrid materials with large nanolatexes. Lower row: Hybrid material with small nanolatexes. | ||
FE-SEM imaging of cross-sections
The classical method of assessing cross-section structuring is imaging by FE-SEM of cross-sections fractured by different methods. Fig. 3 shows the cross-sections of hybrid cellulose nanopapers with increasing amounts of PISA nanoparticles. In the CNF reference, we clearly see the appearance of layers of different sizes in the material, most visible at sizes of 100 and 200 nm. Fig. 3 shows representative images of cross-sections of hybrid materials produced with high buffer. Cross-sectional images of low-buffer samples (Slownp and Llownp) show no significant differences vs. high buffered samples Lhighnp and Shighnp (Fig. S5†). The particles retain their spherical shape after hybridization with CNFs (Fig. S6a†).It is clear that the addition of nanoparticles to CNF has an impact on cross-sectional structures formed during dewatering and assembly (Fig. 3). The overall set of images in Fig. 3 shows that increasing amounts of nanoparticles disorder the CNF matrix, and at 25 wt% particle content the layered structures are no longer visible. Surfaces fractured at 50% relative humidity (during tensile testing) also show clearly increased disorder at the highest additive amounts (Fig. 4). the impact of 25 wt% large (L) and small (S) particles on the scattering distribution, indicating differences in material alignment.
 | ||
| Fig. 4 Fracture surfaces of hybrid cellulose nanopapers with increasing amounts of PISA nanoparticles. Fractured at 50% relative humidity, and imaged by FE-SEM. Left: CNF reference without nanolatexes. Upper row: Hybrid materials with large nanolatexes. Lower row: Hybrid material with small nanolatexes. | ||
However, from these types of images it is not possible to quantitatively describe the structuring, and thus we cannot assess well the impact of nanoparticle size and concentration. Additionally, with these types of images we only visualize ordering of relatively large structures well (>100 nm). In these images we can clearly see that CNF layering is impacted by the addition of particles, but to quantify the effects we turn to WAXS and USAXS.
Scattering in the cross-section of nanopapers
The hybrid nanopapers were sliced to 1.5 mm strips which were placed in-line to the X-ray beam (Fig. 5a). To ensure that the incoming X-ray beam is perfectly in-line with the layered structures inside the material, the specimen was rotated to find the highest scattering path length, before acquiring images on the detectors. | ||
| Fig. 5 Geometry of the scattering set-up in the cross-section direction (a) of hybrid cellulose nanopapers, as studied by simultaneous WAXS (b) and USAXS (c). The detector images show raw data from the reference CNF0. Radial cuts in the WAXS detector (d) show the cross-section (Wz) and in-plane (Wy) scattering response, and the region chosen for azimuthal integration over the (200) scattering plane is indicated as W200. Radial cuts in the USAXS detector (e) show the cross-section (USz) and in-plane (USy) scattering response, with three q-ranges chosen for azimuthal integration; US100, US20 and US10, with d-spacing ∼100, 20 and 10 nm respectively. (f) Azimuthal integration of the US100q-range, showing the impact of 25 wt% large (L) particles on the scattering distribution, indicating differences in material alignment through the S parameter. | ||
The scattering patterns show anisotropic features in WAXS and USAXS, characterized by increased scattering intensity in the z-direction versus the y-direction (Fig. 5b and c). The cross-sectional scattering in WAXS (Wz) show distinct crystalline cellulose peaks at q ∼1.1 and ∼1.6 Å−1 corresponding to scattering from the (200) and the combined (110) and (1–10) crystalline planes.50 Conversely, the in-plane scattering (Wy) is low and no crystalline signals are discerned. This is indicative of a highly aligned system on the crystalline scale. The inclusion of nanoparticles in the hybrid materials does not greatly influence the features of the WAXS signals in these radial cuts, as the nanoparticles themselves are amorphous and show no crystalline peaks. USAXS cuts in the cross-section (USz) and in-plane (USy) directions are also indicative of a highly aligned system on larger length scales (Fig. 5c). The signal from pure cellulose is smeared/featureless, with intensity in the USz direction being larger than in the USy direction. This both indicates some ordering,51 and a relatively high dispersity in feature sizes. From SEM images we see clearly features of around 100–200 nm, see Fig. 3 and 4. If these structures were highly well-defined, a plateau region in the lower q region of this data would be expected. The data thus suggests that the layers have high dispersity in feature sizes.
The addition of nanoparticles yields USAXS patterns indicative of monodisperse features, with fringes in the intensity clearly visible (Fig. 5e). These patterns are in agreement with particles of sizes ∼100 and ∼20 nm. The appearance of higher order oscillations indicates that the particles are well dispersed in the cellulosic matrix. It corroborates that large-scale agglomeration of particles does not take place while mixing and drying of the cellulose-nanoparticle hybrid dispersion into sheets, but rather that these colloidally stable building blocks remain discrete through the drying process. This indeed supports the earlier statement that macroscopic aggregation can be avoided during mixing of the different components. The data in Fig. 5d and e shows clearly the need for both WAXS and USAXS to characterize assembled structures found in the cross-section of these materials.
To quantify alignment and ordering at different length scales, cuts for azimuthal integration are chosen for the crystalline regions of the (200) plane (Fig. 5d), as well as in USAXS at size ranges 100, 20 and 10 nm, corresponding to shaded regions in Fig. 5e. The q ranges are found in Table S3† and representative image with these cuts in Fig. S7.† These cuts are chosen to probe size ranges similar to the large particles, small particles and CNFs. The azimuthal integrations at the chosen q ranges yield distributions that describe the alignment in the material at these certain length scales. It is obvious from inspection of the distribution curves that the incorporation of nanoparticles impacts the alignment throughout the bulk of the material by a clear broadening of I(χ) (Fig. 5f). In the literature, several different mathematical strategies have been used to reduce these distributions to orientation values.52,53 The most commonly employed calculates the Herman's order parameter S, which is generated from the orientation distribution function (ODF) according to the second Legendre polynomial (eqn (1)), where χ is the scattering angle. S is computed from the integration over background-subtracted two-dimensional WAXS and USAXS according to eqn (2) and normalization according to eqn (3).
The orientation parameter S is a value between 0 and 1 that describes the alignment of structures in the field of view. When S = 0 the structures are anisotropic with no alignment, and when S = 1 the structures are perfectly aligned. Some relevant values of the S parameter are schematically illustrated in Fig. 6b. It is crucial to remember that in this experiment the S parameter only describes alignment in the field of view of the incoming X-ray beam (see Fig. 5a), and does not describe alignment in 3 dimensions.
 | (1) |
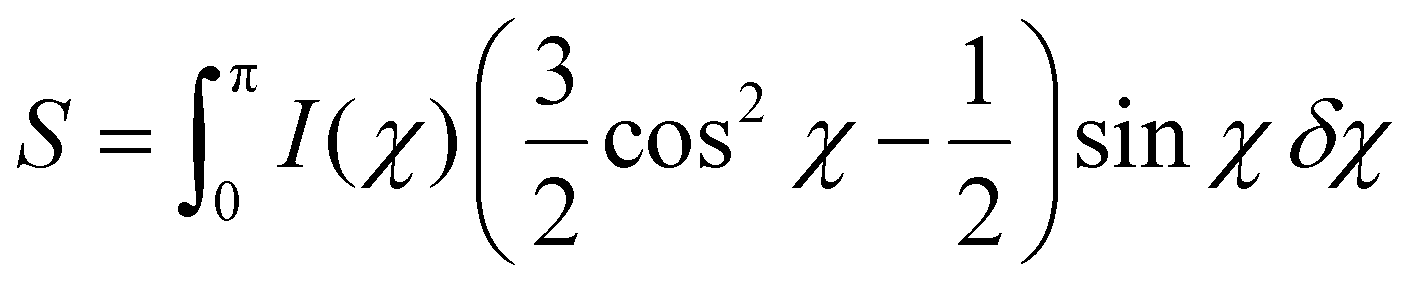 | (2) |
 | (3) |
 | ||
| Fig. 6 (a) Orientation parameters (S) for neat CNF0 describing orientation at four different size ranges, (b) schematic representation of some relevant values for S, (c) orientation parameter (S) changes with nanoparticle addition (np) when adding large nanoparticles (Lnp), (d) orientation parameter (S) changes with nanoparticle addition (np) when adding small nanoparticles (Snp). | ||
Influence of nanoparticles on orientation parameters
The extracted S-parameters for all produced materials provide valuable insight into material structuring on different length scales. The alignment at different length scales is seen for the pure CNF material in Fig. 6a. Firstly, S for WAXS equals 0.26 for the pure CNF material, which correlates well to XRD literature values (0.12–0.34).32,36 In the USAXS cuts we see that anisotropy occurs in all investigated length scales, but alignment is highest at a length scale of 100 nm. This corroborates SEM imaging, where layering at length scales 100 nm and above is observed, see Fig. 3. Thus, our results in Fig. 6a provide a quantitative and bulk description of the ∼100 nm structures reported via SEM imaging in the literature. These trends are also corroborated in a CNF-drying study where the orientation index is found to be larger in the low q-region.54 In Fig. 6b we visualized the arrangement of high aspect ratio particles (such as CNFs) organized according to relevant S-parameters.The impact of nanoparticles on the alignment of cellulosic structures is shown in Fig. 6c and d. For large particles (diameter ∼90 nm), there is an increase in alignment with low (0.5 wt%) nanoparticle content (Fig. 6c). This effect is mirrored with small nanoparticles (diameter ∼30 nm), and is seen up to 1 wt% (Fig. 6d). We have previously found that addition of such small amounts of these nanoparticles in CNF-networks leads to stiffening of the material, an effect that was attributed to nanoparticles acting as cross-linking points when they are few and far apart.28 These new findings instead indicate that the stiffening effect stems from the particles inducing an increased alignment of cellulosic structures during assembly. The dehydration effect is further corroborated by a sharp increase in density of the nanopapers containing low amounts of nanoparticles (Fig. S10†).
The increase in alignment that is attained by colloidal nanoparticle additives is an effect that is very interesting to probe further, as it provides a way to tailor material structure by modification of additives rather than fabrication method. One way to understand these results is to visualize that colloidal polymeric nanoparticles can fit into naturally occurring/forming pores inside the CNF network. The nanoparticle additives could pull CNFs closer during water removal, leading to an increased packing and alignment. Cationic nanoparticles are known to dehydrate cellulose model surfaces, as electrostatically driven adsorption between anionic CNFs and cationic nanoparticles release counterions and bound water at the adsorbing surfaces.45 Calorimetry has shown that adsorption of various species to cellulose is generally driven by release of bound water at the surface.55 This dehydration effect could allow the tighter packing of CNFs in a hybrid mixture where polymeric nanoparticles can fit inside naturally forming pores.
CNF-networks are known to be nanoporous and with a broad distribution of pore sizes around an average value of 40 nm.16,56 Our results indicate that the larger nanopores which can accommodate nanoparticles of size ∼90 nm (L) are filled between 0.5 < np < 1 wt%. Such a nanopore is visible in Fig. 3 for the pure CNF (see also Fig. S6b†). Conversely, nanopores suitable for accommodating nanoparticles of size ∼30 nm (S) are filled between 1 < np < 5 wt%. We thus show that with the addition of polymeric nanoparticles of well-defined sizes, it could be possible to block pores of certain size and size ranges. Additional to tailoring mechanical properties of the composites, this might be relevant for applications where CNF films are used as filtration membranes or barrier materials.57,58 The critical nanoparticle concentration npcrit is shown as a dashed line in Fig. 6c and d.
At nanoparticle content above npcrit, alignment is reduced as particles that cannot fit into natural pores act as steric barriers for CNF assembly. For large nanoparticle additives, S decreases on all length scales at np > npcrit. For small nanoparticles however, alignment is not impacted at 100 nm but on all smaller length scales. This indicates that the small particles can fit inside the 100 nm features formed during assembly and drying, so that these structures can still form. To conclude, large particles (90 nm) are large enough to hamper structure formation at all length scales, but small particles (30 nm) are not (schematic in Fig. S8†). Low buffered materials (Llownp and Slownp) result in nearly identical values for S, corroborating that the buffer does not impact alignment, as seen in SEM images (Fig. S9†).
Influence of buffer in neat CNF
The full range of hybrid materials discussed above were fabricated with nanoparticles buffered in 10 or 100 mM phosphate buffer, and compared to CNF0 with no added buffer. To investigate whether the buffer alone can have an impact on cellulosic alignment we prepared neat cellulose materials with buffer concentrations equal to Llow25 and Lhigh25 (1.1 and 21.9 mM). The corresponding S-parameters calculated from these materials show that the structuring in cellulose is indeed impacted by buffer concentration when no nanoparticles are added (Fig. 7a). On the crystalline scale (W200 and US10), significant increases in alignment cannot be concluded. However, at larger size scales of 20 and 100 nm (US20 and US100), we see an increase in alignment with increased buffer concentration. These results are mirrored in the structures observed in cryo-fractured cross-sections in the FE-SEM (Fig. 7b).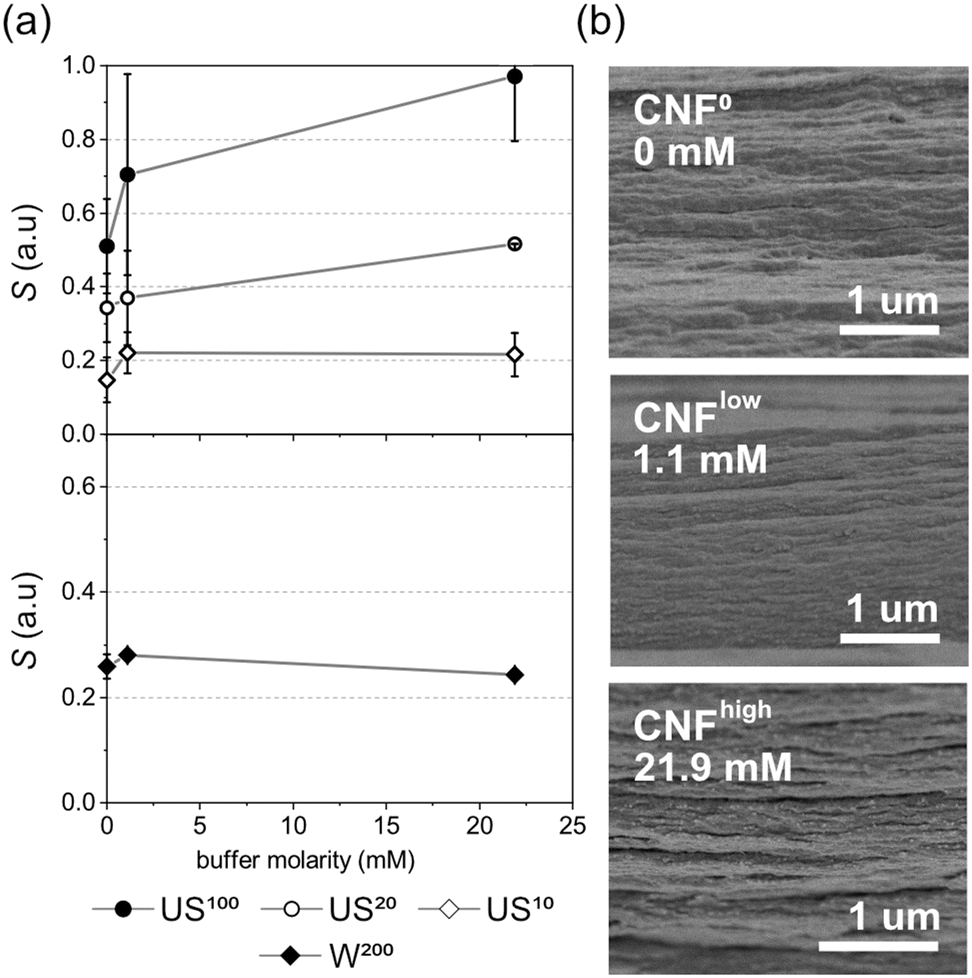 | ||
| Fig. 7 (a) Impact of added buffer, in neat CNF samples, on orientation parameters and (b) cryo-fractured cross-sections imaged by FE-SEM show the increased appearance of oriented layers when higher concentration of buffer is used. | ||
It is known that the assembly of CNFs during drying is impacted by colloidal stability. Charges on the CNFs surface repel one another, and thus as water is removed during drying, their continued repulsion prevent aggregation to produce homogeneous nanofibrillar material.59 Increase in ionic strength has been linked to a decrease in viscosity and increase in dewatering rate as a result of a decreased colloidal stability.60 If charges are neutralized/protonated or shielded by the increase of ionic strength, CNFs are expected to associate earlier in the drying process.61 When the CNF concentration is high enough the fibrils will form gels or colloidal glasses due to their large anisotropy and macroscopic aggregation are avoided. The buffer concentrations applied here are not high enough to aggregate the CNFs at the initial concentration of 2 g L−1. However, as CNF dry content increases during water removal, one can visualize that the onset of association is reached earlier in the drying process, giving rise to more well-defined layers and increase in alignment of these. Our results indicate that the layer formation and alignment characterized in this work is formed by a concentration-driven association of CNFs during water removal, which is tightly linked to the colloidal stability of CNFs.62
Within the hybrid materials with CNFs and polymeric nanoparticles however, buffer does not impact material alignment (Fig. S9†). This shows that addition of the nanoparticles dominates the colloidally-driven nanostructuring detected in neat CNFs. These ‘structure-forming’ additives thus seem to function via a steric mechanism rather than a mechanism based on charge or van der Waals driven interactions.
Impact of CNF alignment on tensile properties
The range of hybrid nanopapers Lhighnp and Shighnp were subjected to standardized tensile testing (50% relative humidity, 23 °C). Representative curves (Fig. 8a and b) show the plasticizing effect at high nanoparticle addition. Nanoparticle addition reduces the elastic (E) and plastic (P) moduli (Fig. 8c and d). Similarly, the nanoparticle addition reduces alignment (Fig. 6c and d). Hence, mechanical properties and alignment show similar trends to nanoparticle addition. Especially in the plastic modulus, we observe a slight increase in resistance to strain at low nanoparticle addition (0.5 wt%). These effects corroborate our previous findings, where small amounts of similar additives were associated with increases in both elastic and plastic moduli.28 | ||
| Fig. 8 Representative stress–strain curves show plasticizing effect of (a) large and (b) small particles. The elastic (c) and plastic (d) moduli are extracted from the entire datasets, showing qualitatively the plasticizing effect. (e) The plastic modulus (P) looks correlated to the orientation parameters (S) on different length scales. (f) The Pearson's correlation coefficient r shows high correlation between elastic modulus and orientation on all length scales. | ||
Hence, we investigate a potential correlation between stiffness and alignment. When plotting E and P versus alignment, we see clear correlations (Fig. 8e and Fig. S11†). To further assess correlation strength, the Pearson's correlation coefficient r is calculated and plotted in Fig. 8f.
The elastic modulus (E) describes small-strain deformation behaviour. For the range Lhighnp strong correlation (>0.5) is observed on all length scales. Orientation of structures on all length scales is thus responsible for small-strain deformation behaviour. Interestingly, the hybrid materials with small particles Shighnp shows very poor correlation at the 100 nm range. This indicates that the structures at this size range are not responsible for small-strain deformation behaviour in these materials. The plastic modulus (P) describes large-scale deformation behaviour. Plastic behaviour in CNF-materials is often described by interfibrillar sliding, which is dominated by the friction coefficient between fibrils.63 Correlations between S and P appear similar to S and E, but the plastic modulus shows consistently stronger correlation (Fig. 8f). This indicates that large-scale deformation behaviour has an even stronger dependency on cross-sectional orientation.
The dependency of modulus on orientation in the crystalline size range has been shown using XRD in cold-drawn nanocellulose materials.36 Cold-drawing of cellulose wet-cakes led to increase in crystallite alignment both in the cross-sectional and in-plane directions. This behaviour was associated with increases in both modulus and strength. Our work reports the correlation between modulus and alignment also on much larger size scales (<100 nm). We can thus propose a method for tuning cross-section alignment by the use of tailored additives.
Conclusions
Cellulose nanofibrils assemble into oriented structures upon dewatering, and it is shown that a polymeric nanoparticle additive can be used to control alignment of these structures to impact bulk mechanical properties. A method is developed for quantifying orientation in the cross-section of CNF materials on different length scales using WAXS and USAXS. Small amounts (<1 wt%) of cationic polymeric nanoparticles are found to increase alignment, which lead to increased resistance to plastic deformation. Larger amounts of particles misalign CNF-structures at length scales of the particle diameter and smaller. Calculated orientation parameters correlate well to bulk moduli. This work constitutes an important step forward in the understanding of structure–property relationships in CNF materials hybridized with structure-forming additives, which is useful in barrier, filtration and advanced optoelectronic applications.Author contributions
Å.J. performed sample preparation, carried out all characterization, analyzed the data, and drafted and revised the manuscript. K.G. performed sample preparation, carried out the X-ray characterization, assisted in data analysis, and revised the manuscript. M.C. and L.B. carried out the X-ray characterization. B.S. and S.K.V. carried out the X-ray characterization, and assisted in data analysis. S.V.R, L.W. and E.M. supervised the project and revised the manuscript. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Swedish Research Council (2020-05486), the Knut and Alice Wallenberg foundation (KAW) through the Wallenberg Wood Science Centre (WWSC) (2018.0452), and FibRe; a “Vinnova-funded Competence Centre for Design for Circularity: Lignocellulose-based Thermoplastics” (2019-00047). M.C. wishes to acknowledge funding from the Swedish Energy Agency (grant #48488).We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank J. Rubeck for assistance in using beamline P03. Beamtime was allocated for proposal I-11014778. The authors also wish to acknowledge N. Venkatraman at Chalmers University of Technology for the table-top SAXS measurements, and A. Riazanova at KTH Royal Institute of Technology for assistance with FE-SEM.
References
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- A. Finnemore, P. Cunha, T. Shean, S. Vignolini, S. Guldin, M. Oyen and U. Steiner, Nat. Commun., 2012, 3, 966 CrossRef PubMed.
- Z. Tang, N. A. Kotov, S. Magonov and B. Ozturk, Nat. Mater., 2003, 2, 413–418 Search PubMed.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- L. Valencia, S. Kumar, E. M. Nomena, G. Salazar-Alvarez and A. P. Mathew, ACS Appl. Nano Mater., 2020, 3, 7172–7181 CrossRef CAS.
- J. R. G. Navarro, J. Rostami, A. Ahlinder, J. B. Mietner, D. Bernin, B. Saake and U. Edlund, Biomacromolecules, 2020, 21, 1952–1961 CrossRef CAS PubMed.
- C. Harder, A. E. Alexakis, Y. Bulut, S. Xiong, B. Sochor, G. Pan, H. Zhong, K. Goordeyeva, M. A. Reus, V. Körstgens, A. Jeromin, T. F. Keller, L. D. Söderberg, E. Malmström, P. Müller-Buschbaum and S. V. Roth, Adv. Opt. Mater., 2023, 11, 2203058 CrossRef CAS.
- J. Rostami, K. Gordeyeva, T. Benselfelt, E. Lahchaichi, S. A. Hall, A. V. Riazanova, P. A. Larsson, G. C. Ciftci and L. Wågberg, Mater. Today, 2021, 48, 47–58 CrossRef CAS.
- T. Abitbol, A. Rivkin, Y. Cao, Y. Nevo, E. Abraham, T. Ben-Shalom, S. Lapidot and O. Shoseyov, Curr. Opin. Biotechnol., 2016, 39, 76–88 CrossRef CAS PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- E. Karabulut and L. Wågberg, Soft Matter, 2011, 7, 3467 RSC.
- E. S. Forti, S. M. El Awad Azrak, X. Y. Ng, W. Cho, G. T. Schueneman, R. J. Moon, D. M. Fox and J. P. Youngblood, Cellulose, 2021, 28, 6449–6465 CrossRef CAS.
- M. Shimizu, T. Saito, H. Fukuzumi and A. Isogai, Biomacromolecules, 2014, 15, 4320–4325 CrossRef CAS PubMed.
- A. J. Benítez and A. Walther, Biomacromolecules, 2017, 18, 1642–1653 CrossRef PubMed.
- H. Sehaqui, Q. Zhou, O. Ikkala and L. A. Berglund, Biomacromolecules, 2011, 12, 3638–3644 CrossRef CAS.
- Y. Qing, R. Sabo, Y. Wu, J. Y. Zhu and Z. Cai, Cellulose, 2015, 22, 1091–1102 CrossRef CAS.
- A. J. Benítez and A. Walther, J. Mater. Chem. A, 2017, 5, 16003–16024 RSC.
- L. A. Berglund and T. Peijs, MRS Bull., 2010, 35, 201–207 CrossRef CAS.
- H. Sehaqui, Q. Zhou and L. A. Berglund, Soft Matter, 2011, 7, 7342 RSC.
- A. J. Svagan, M. A. S. Azizi Samir and L. A. Berglund, Biomacromolecules, 2007, 8, 2556–2563 CrossRef CAS PubMed.
- A. J. Benítez, F. Lossada, B. Zhu, T. Rudolph and A. Walther, Biomacromolecules, 2016, 17, 2417–2426 CrossRef.
- T. Kurihara and A. Isogai, Cellulose, 2015, 22, 499–506 CrossRef CAS.
- T. Kurihara and A. Isogai, Cellulose, 2014, 21, 291–299 CrossRef CAS.
- J. Engström, F. L. Hatton, L. Wågberg, F. D'Agosto, M. Lansalot, E. Malmström and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- J. Engström, A. M. Jimenez and E. Malmström, Nanoscale, 2020, 12, 6462–6471 Search PubMed.
- M. Wang, A. Olszewska, A. Walther, J. M. Malho, F. H. Schacher, J. Ruokolainen, M. Ankerfors, J. Laine, L. A. Berglund, M. Osterberg and O. Ikkala, Biomacromolecules, 2011, 12, 2074–2081 CrossRef CAS.
- A. E. Alexakis, Å. Jerlhagen, M. R. T. Leggieri, A. Eliasson, T. Benselfelt and E. Malmström, Macromol. Chem. Phys., 2023, 224, 2200249 CrossRef CAS.
- V. Kumar, E. Lazarus, P. Salminen, D. Bousfield and M. Toivakka, Nord. Pulp Pap. Res. J., 2016, 31, 333–340 CrossRef CAS.
- W. Liu, K. Liu, H. Du, T. Zheng, N. Zhang, T. Xu, B. Pang, X. Zhang, C. Si and K. Zhang, Nano-Micro Lett., 2022, 14, 104 CrossRef CAS PubMed.
- A. J. Benítez, J. Torres-Rendon, M. Poutanen and A. Walther, Biomacromolecules, 2013, 14, 4497–4506 CrossRef.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. LindströM and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- M. Zhao, F. Ansari, M. Takeuchi, M. Shimizu, T. Saito, L. A. Berglund and A. Isogai, Nanoscale Horiz., 2018, 3, 28–34 RSC.
- M. Larsson, A. Johnsson, S. Gårdebjer, R. Bordes and A. Larsson, Mater. Des., 2017, 122, 414–421 CrossRef CAS.
- J. J. Kaschuk, Y. Al Haj, J. Valdez Garcia, A. Kamppinen, O. J. Rojas, T. Abitbol, K. Miettunen and J. Vapaavuori, Carbohydr. Polym., 2024, 332, 121877 CrossRef CAS PubMed.
- H. Sehaqui, N. Ezekiel Mushi, S. Morimune, M. Salajkova, T. Nishino and L. A. Berglund, ACS Appl. Mater. Interfaces, 2012, 4, 1043–1049 CrossRef CAS.
- X. Han, P. Chen, L. Li, Y. Nishiyama and X. Yang, Cellulose, 2023, 30, 8151–8159 Search PubMed.
- T. Hirano, K. Mitsuzawa, S. Ishioka, K. Daicho, H. Soeta, M. Zhao, M. Takeda, Y. Takai, S. Fujisawa and T. Saito, Front. Chem., 2020, 8, 68 CrossRef CAS.
- T. Kaldéus, M. Nordenström, A. Carlmark, L. Wågberg and E. Malmström, Carbohydr. Polym., 2018, 181, 871–878 CrossRef PubMed.
- Å. Jerlhagen, O. Wilson and E. Malmström, ACS Macro Lett., 2024, 565–570, DOI:10.1021/acsmacrolett.4c00112.
- S. Katz, R. P. Beatson and M. Anthony, Sven. Papperstidn., 1984, 87, 48–53 Search PubMed.
- A. Buffet, A. Rothkirch, R. Döhrmann, V. Körstgens, M. M. Abul Kashem, J. Perlich, G. Herzog, M. Schwartzkopf, R. Gehrke, P. Müller-Buschbaum and S. V. Roth, J. Synchrotron Radiat., 2012, 19, 647–653 CrossRef CAS.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef.
- A. E. Alexakis, M. R. Telaretti Leggieri, L. Wågberg, E. Malmström and T. Benselfelt, J. Colloid Interface Sci., 2023, 634, 610–620 CrossRef PubMed.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS.
- S. Utsel, E. E. Malmström, A. Carlmark and L. Wågberg, Soft Matter, 2010, 6, 342–352 RSC.
- F. Xie, T. Nylander, L. Piculell, S. Utsel, L. Wågberg, T. Åkesson and J. Forsman, Langmuir, 2013, 29, 12421–12431 CrossRef CAS PubMed.
- M. Nordenström, T. Benselfelt, R. Hollertz, S. Wennmalm, P. A. Larsson, A. Mehandzhiyski, N. Rolland, I. Zozoulenko, D. Söderberg and L. Wågberg, Carbohydr. Polym., 2022, 297, 120046 Search PubMed.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS.
- K. M. O. Håkansson, A. B. Fall, F. Lundell, S. Yu, C. Krywka, S. V. Roth, G. Santoro, M. Kvick, L. Prahl Wittberg, L. Wågberg and L. D. Söderberg, Nat. Commun., 2014, 5, 4018 CrossRef.
- K. Li, C. M. Clarkson, L. Wang, Y. Liu, M. Lamm, Z. Pang, Y. Zhou, J. Qian, M. Tajvidi, D. J. Gardner, H. Tekinalp, L. Hu, T. Li, A. J. Ragauskas, J. P. Youngblood and S. Ozcan, ACS Nano, 2021, 15, 3646–3673 CrossRef CAS PubMed.
- T. Rosén, C. Brouzet, S. V. Roth, F. Lundell and L. D. Söderberg, J. Phys. Chem. C, 2018, 122, 6889–6899 CrossRef.
- L. Li, P. Chen, L. Medina, L. Yang, Y. Nishiyama and L. A. Berglund, ACS Nano, 2023, 17, 15810–15820 CrossRef CAS PubMed.
- S. Lombardo and W. Thielemans, Cellulose, 2019, 26, 249–279 CrossRef CAS.
- C. J. Brett, N. Mittal, W. Ohm, M. Gensch, L. P. Kreuzer, V. Körstgens, M. Månsson, H. Frielinghaus, P. Müller-Buschbaum, L. D. Söderberg and S. V. Roth, Macromolecules, 2019, 52, 4721–4728 CrossRef CAS.
- A. Mautner, K.-Y. Lee, T. Tammelin, A. P. Mathew, A. J. Nedoma, K. Li and A. Bismarck, React. Funct. Polym., 2015, 86, 209–214 Search PubMed.
- J. Xia, Z. Zhang, W. Liu, V. C. F. Li, Y. Cao, W. Zhang and Y. Deng, Cellulose, 2018, 25, 4057–4066 CrossRef CAS.
- M. Nordenström, A. Fall, G. Nyström and L. Wågberg, Langmuir, 2017, 33, 9772–9780 CrossRef.
- A. Fall, M. Henriksson, A. Karppinen, A. Opstad, E. B. Heggset and K. Syverud, Cellulose, 2022, 29, 7649–7662 CrossRef CAS.
- A. B. Fall, S. B. Lindström, O. Sundman, L. Ödberg and L. Wågberg, Langmuir, 2011, 27, 11332–11338 CrossRef CAS PubMed.
- T. Benselfelt, J. Engström and L. Wågberg, Green Chem., 2018, 20, 2558–2570 RSC.
- F. Chen, W. Xiang, D. Sawada, L. Bai, M. Hummel, H. Sixta and T. Budtova, ACS Nano, 2020, 14, 11150–11159 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04381b |
| This journal is © The Royal Society of Chemistry 2025 |