
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A nanoscale chemical oscillator: reversible formation of palladium nanoparticles in ionic liquid†
Rhys W.
Lodge
,
William J.
Cull
 ,
Andreas
Weilhard
,
Stephen P.
Argent
,
Jesum
Alves Fernandes
and
Andrei N.
Khlobystov
*
,
Andreas
Weilhard
,
Stephen P.
Argent
,
Jesum
Alves Fernandes
and
Andrei N.
Khlobystov
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Andrei.Khlobystov@nottingham.ac.uk
First published on 26th March 2025
Abstract
From the theory of chaos-to-order transitions to the origins of life on Earth, oscillating chemical reactions play a fundamental role in nature. This study demonstrates how chemical oscillators can exist at the nanoscale, bridging the gap between molecules and living cells. We identify three necessary conditions for nanoscale chemical oscillations: (i) a continuous energy flux; (ii) alternating fluxes of two chemical species with opposing effects—specifically, a reductant and an oxidant; and (iii) a redox-active metal. Irradiating palladium ions dissolved in benzyl imidazolium bromide ([BnMIm]+Br−) with a 200 keV electron beam meets these requirements, resulting in the oscillating, dendritic assembly of palladium nanoparticles. By encapsulating the solution inside carbon nanotubes, we can slow down the rate of chemical oscillations, allowing for real-time analysis at the individual nanoparticle level. Our results indicate that nanoparticles in liquids are far from the thermodynamic equilibrium during transmission electron microscopy (TEM) imaging, which is an important consideration for studying nanoparticles using in situ TEM methods or employing them for catalysis in the liquid phase.
Introduction
Nanoparticles are intrinsically metastable materials. Their formation is reversible, their structure is polydisperse, and their chemical and physical properties highly depend on their size, shape, and composition.1–3 Consequently, macroscopic, ensemble averaging analysis cannot adequately describe nanoparticle dynamics, resulting in a significant impetus to understand the mechanisms of nanoparticle nucleation, growth and dissolution at the nanoscale.4–11 Among local-probe analysis methods, transmission electron microscopy (TEM) offers the best balance of spatiotemporal resolution for investigating nanoparticles in action, achieving sub-Å resolution in space and microsecond time resolution in the same experiment. Dynamic TEM measurements performed at the single-particle level hold the key to understanding nanoparticle nucleation and growth mechanisms, as well as structure–function relationships.12–17The primary drawback of local probe methods is that the probe, such as the electron beam in the case of TEM, disturbs the system under investigation. The electron beam transfers energy to the sample during observation through various mechanisms, which can be broadly categorised into processes involving the electron beam interacting with valence electrons or atomic nuclei. If TEM is to be utilised effectively to study nanoparticle dynamics it is essential to recognise that the electron beam serves a dual role as an imaging probe and a source of energy that triggers chemical reactions in the sample, a concept termed ChemTEM.18 We have previously demonstrated the use of atomically thin nano test tubes, based on single-walled carbon nanotubes, as effective platforms for studying nucleation,19 structure,20–22 dynamics and reactivity22–25 of a range of chemical species, including metal nanoparticles. If the energy and flux of the electron beam are selected correctly, the carbon nanotube can act as a passive container, providing an ideal platform for studying the innate properties of metal atoms and nanoclusters under TEM imaging conditions. Time-resolved TEM imaging can then reveal the evolution of nanoparticles along a thermodynamically driven pathway during observation, mirroring the thermally driven chemical processes that occur on a larger scale, such as metal–metal bond formation or nucleation and growth of crystals.
Recognising that nanoparticles are often synthesised and subsequently utilised in a liquid medium, using TEM to study in situ chemistry in the liquid phase is becoming increasingly popular.26–30 The critical challenges of the liquid cell are related to the material of the windows and thickness of the solvent layer preventing atomically resolved imaging of nanoparticles in solution. The use of ionic liquids (ILs) in a free-standing form offers a solution to these challenges because the low vapour pressure of ILs is compatible with the conditions of TEM without windows, which is complemented by their ability to dissolve metal compounds and stabilise metal nanoparticles. Recent examples of dynamic TEM measurements in a free-standing thin film of IL suspended on a TEM grid are particularly impressive.31–36 This approach negates the need for cell windows that decrease the resolution and interfere with the metal nanoparticle dynamics. However, it must be noted that ILs are molecular compounds themselves, and therefore are expected to react under the electron beam following the patterns known for other molecules, such as the scission of C–H bonds, free radical formation and recombination.37 Indeed, several studies have commented on the non-innocent role of IL in the electron beam.38 For example, Au nanoparticle nucleation and growth are driven by a combined effect of e-beam and heat, which appears difficult to decouple.39 Similarly, the dissolution of pre-formed Pd nanoparticles was linked to the formation of oxidative species formed from an IL under the electron beam.40 It is becoming increasingly evident that the electron beam initiates intricate chemical reactions in liquids, making it difficult to distinguish the inherent structures of nanoparticles in liquids.
Here, we show that the behaviour of metal nanoparticles in a liquid is non-equilibrium when observed using TEM. We captured the palladium nanoparticle nucleation, growth, and dissolution in real-time and direct space as a cyclic process. These nanoparticles displayed explicit oscillatory chemical behaviour due to a flow of reducing and oxidising species generated by the electron beam from the cation and anion of the IL, respectively. By confining a small volume of the reaction mixture within a carbon nanotube, we slowed down the cyclic process and examined each stage at the level of individual nanoparticles. Using an electron beam as an imaging probe and source of energy at the same time allows us to observe chemical processes with atomic resolution, including chemical oscillations at the nanoscale, which has important implications for metal catalysis in liquid reactions.
Results
Palladium acetate Pd(OAc)2 dissolved in benzyl methyl imidazolium bromide [BnMIm]+Br− (Fig. 1a) forms a viscous solution that can be directly deposited onto a TEM grid coated with a holey film of amorphous carbon. The surface tension of the IL is sufficient for it to cover the holes in the carbon film, creating pools of solution about 0.5–1 μm wide with bright-field TEM contrast different from that of the surrounding carbon film (Fig. 1b and c). These pools appear to possess a concave meniscus (Fig. S1†) and their thickness is limited by the ca. 30 nm thickness of the carbon film. When imaged using a 200 keV electron beam, the IL solution of palladium acetate appears homogeneous and featureless. However, after a few seconds under electron beam flux of ∼103 e− nm−2 s−1, a faint dendritic pattern of higher contrast material emerges. This pattern rapidly becomes more pronounced, spanning the entire width of the pool (Fig. 1c and ESI Video 1†). Palladium, the element with the highest z-number in the solution, is the only element that can form such features due to the agglomeration of atoms into nanoparticles, which then join into a dendritic structure. A close examination of the behaviour of these Pd nanoparticles shows their reversible formation and dissolution (Fig. 1d and Fig. S2†). Under the 200 keV electron beam, the initial generation of nanoparticles formed from the IL solution dissolves, leaving low contrast areas (voids), followed by the re-growth of nanoparticles in slightly different places (the second generation). This process repeats several times before coming to a halt (ESI Videos 2 and 3†). However, due to the agglomeration of nanoparticles, obtaining atomically resolved images of the palladium nanoparticles in this process is difficult, hindering the determination of their shapes and atomic planes. Furthermore, the formation and dissolution of the nanoparticles seem to occur sporadically and are highly dependent on the size of the IL pool and its proximity to the carbon film. As a result, determining the mechanisms of this cyclic growth at the single-particle level is challenging in the pool of IL. | ||
| Fig. 1 (a) A structural diagram of benzyl methyl imidazolium bromide, [BnMIm]+Br,− IL and palladium acetate, Pd(OAc)2. (b) A schematic diagram of a pool of Pd(OAc)2 IL solution formed in holes of the carbon film on a TEM grid; the thickness of the IL pool is dictated by the thickness of the carbon film. (c and d) Time series of TEM images showing the formation of a dendritic network of Pd nanoparticles in a pool of Pd(OAc)2 solution in IL under the flux of ∼103 e− nm−2 s−1 of the 200 keV electron beam. The total duration of the time series are 39 seconds and 7 seconds, for (c) and (d), respectively. Scale bars are 100 nm (c) and 50 nm (d). | ||
A similar process can be observed through TEM imaging of the same solution inside carbon nanotubes. When mixed with carbon nanotubes of 50–70 nm internal diameter, a solution of Pd(OAc)2 in IL can spontaneously diffuse into the nano test tube cavity at room temperature and atmospheric pressure due to capillary forces (Fig. 2). The nanotubes filled with the solution were drop-cast onto a TEM grid and examined by TEM imaging. Similar to the carbon film, the contrast of the IL can be clearly distinguished inside the nanotube, appearing uniform and featureless. Under the 200 keV e-beam, nucleation and growth of Pd nanoparticles directly from the solution can be observed within nanotubes, albeit at a much slower rate than in IL pools. The slower rate can be explained by the electron beam having to pass through IL with a lesser volume (Fig. S3†), meaning fewer e-beam initiated chemical processes occur in the same timeframe. Additionally, the high thermal conductivity of the nanotube allows for better heat transfer than in free-standing pools of IL, slowing the reaction rate. Similarly, carbon nanotubes allow for effective charge dissipation, preventing the charging of the IL during image acquisition. Collectively, this allows us to follow the entire process inside nano test tubes with a higher spatiotemporal resolution and to study both dynamics and mechanisms at the single-particle level.
 | ||
| Fig. 2 A schematic diagram illustrating the use of a carbon nano test tube as a vessel for the TEM imaging of oscillating reactions in an IL solution. A solution of Pd(OAc)2 in IL is spontaneously encapsulated into a carbon nanotube due to capillary force. Nanotubes filled with the solution are drop-cast onto a TEM grid and imaged by the 200 keV electron beam. The electron beam plays a dual role of an imaging probe and energy source, triggering chemical reactions in the solution, and leading to palladium nanoparticles (Pd NP) formation. After further irradiation, Pd NP begin to dissolve, with palladium returning into the IL solution in the form of Pd(II) cations. | ||
Detailed image analysis revealed diffuse areas of high contrast ranging between 3–16 nm in size forming in the first 100 s of irradiation with the 200 keV e-beam (electron flux: 1.75 × 105 e− nm−2 s−1), which began to coalesce into nanoparticles with higher contrast and sizes starting from 1 nm onwards (Fig. 3a). Between 100 and 500 s, 15 less-mobile particles remained, then decreased to 8 over the last 500 s. Analysis of lattice fringes in the disk-shaped nanoparticles formed in [BnMIm]+Br− IL in the nano test tube was consistent with the (200) and (220) planes of fcc palladium metal (Fig. 3b and c). After 1000 s some nanoparticles undergo dissolution, (Fig. 3a and ESI Video 4†). Analysis of the radius of the 15 particles formed by 500 s, as a function of time, allowed for the growth and dissolution rates of individual nanoparticles to be assessed. This analysis reveals three types of behaviour of the first-generation nanoparticles: (i) complete or (ii) partial dissolution, or (iii) growth, which establishes the driving force and the mechanism behind this process (see Discussion for details).
 | ||
| Fig. 3 (a) A time series of TEM images showing the palladium nanoparticle growth and dissolution in the IL solution within a carbon nanotube (electron flux: 1.75 × 105 e− nm−2 s−1; total series duration: 1000 s). (b) A schematic diagram of indicating crystal facets of a Pd nanoparticle. (c) HR-TEM image of Pd NP showing the d-spacing corresponding to the Pd (200) and (220) planes with Pd (001) plane orthogonal to the electron beam. Capping of the (001) plane by Br− anions of the IL leads to preferential growth in two dimensions to form disk-shaped NPs. Scale bars in (a) and (c) are 50 and 5 nm, respectively. | ||
Discussion
In our experiments, the growth and dissolution of palladium nanoparticles is clearly driven by the electron beam. Therefore, it is essential to comprehend how the nanoparticles form and break down, through understanding how the electron beam interacts with solvent molecules. Our solvent, [BnMIm]+Br−, is a molecular compound. In general, when molecules are examined under a TEM they absorb energy through electron beam interactions with either valence electrons or atomic nuclei (Fig. 4a). The amount of energy transferred, ET, through electron beam interactions with the atomic nucleus, is described as | (1) |
| r1 = σDKOj[CH]. | (2) |
 | ||
Fig. 4 (a) List of possible interactions between the electron beam of TEM with the atomic nuclei and valence electrons irradiated materials, with atomic displacement, also known as a direct knock-on (DKO), and ionisation, also known as displacement of a valence electron (DVE), highlighted in green and red, respectively. (b) and (c) schematics describing the processes of DKO and DVE, respectively. (d) The role of DKO and DVE processes in the oscillatory reaction shown above in Fig. 3. DKO and DVE processes create fluxes of H and Br atoms from the cation and anion of the IL with the rate constants determined by the cross sections multiplied by the electron beam flux, σDKOj and σDVEj, respectively. The former acts as a reducing agent triggering the Pd(II) to Pd(0) transformation with the rate constant  and nanoparticle growth controlled by the diffusion of Pd(0) atoms in IL with a coefficient, DPd(0). Bromine atoms Br˙ act as an oxidising agent, transforming Pd(0) back to Pd(II) with the constant and nanoparticle growth controlled by the diffusion of Pd(0) atoms in IL with a coefficient, DPd(0). Bromine atoms Br˙ act as an oxidising agent, transforming Pd(0) back to Pd(II) with the constant  , which is ligated with bromide anions and diffuses back to the IL solution as [PdBr4]2− (Fig. 6), leading to complete or partial dissolution of nanoparticles. The coupling of the cyclic redox chemistry of palladium with the fluxes of oxidising and reducing species generated by the electron beam gives rise to the oscillation of nanoparticle growth and dissolution observed by TEM. , which is ligated with bromide anions and diffuses back to the IL solution as [PdBr4]2− (Fig. 6), leading to complete or partial dissolution of nanoparticles. The coupling of the cyclic redox chemistry of palladium with the fluxes of oxidising and reducing species generated by the electron beam gives rise to the oscillation of nanoparticle growth and dissolution observed by TEM. | ||
Dissociation of other bonds (C–C or C–N) is less likely due to a much lower cross-section for their DKO.
A parallel process is the electron beam – valence electron interactions where energy is transferred to a valence electron of the atom (eqn (3)):
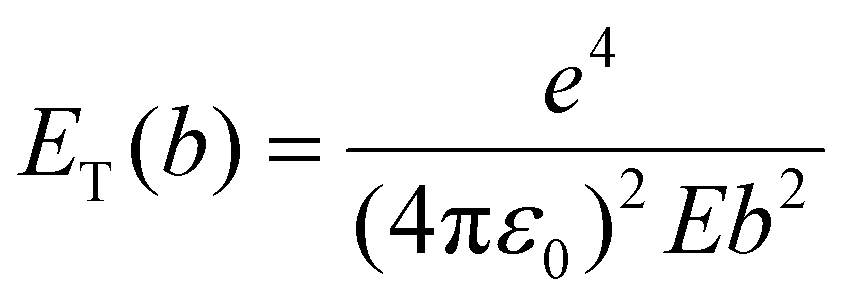 | (3) |
| r2 = σDVEj[Br−]. | (4) |
By understanding how the electron beam interacts with the IL, we can predict the most probable changes in our material when it is irradiated during TEM imaging. The material mainly consists of the organic BnMIm+ cation and bromide anion, with molar concentrations of approximately 5.5 M or 3.5 molecule per nm3. In comparison, palladium acetate is a minority component at 1.8 × 10−3 M or 0.001 molecule per nm3 (ESI, section S6†). During TEM imaging hydrogen atoms from the C–H bonds of imidazole and benzene rings, and the CH3 and CH2 groups of the BnMIm+ cation (Fig. 4b) receive the most significant amount of kinetic energy from the electron beam. As a result, a constant supply of H˙ is created with the rate of r1 (eqn (2)), where the concentration [CH] is 13 times of the concentration of IL, i.e. 71.5 M or 45.5 C–H bonds nm−3. Pd(II) cations are able to bond to radical species formed from BnMIm+, such as an N-heterocyclic carbene (NHC) complex, [Pd(BnMIm-y)2Br2] (BnMIm-y = 1-benzyl-3-methylimidazol-2-ylidene) (Fig. 5a).
 | ||
| Fig. 5 (a) Reaction scheme showing the formation of an intermediate carbene complex, [Pd(BnMIm-y)2Br2] (left) and it's structure (right), exhibiting a trans-anti square planar geometry, as determined by single crystal X-ray diffraction (Pd – cyan, Br – brown, N – blue, C – grey, H – white/pink). (b) Diffusion of Pd(0) atoms from the bulk solution to the growing nanoparticle depletes with palladium the area near the nanoparticle. (c) Diffusion of Pd(II) from the dissolving nanoparticle back to the solution restores the concentration of palladium in the solution. | ||
This complex may form transiently during the above TEM imaging experiments, manifested as the diffuse areas of high contrast observed in the first 100 seconds of the image series in Fig. 3a, as this compound contains three high z-number atoms (Pd and two Br) within the same molecule. A control experiment carried out ex situ with Pd(OAc)2 and [BnMIm]+Br− confirmed that this reaction occurs readily, upon gentle thermal activation, allowing for isolation of [Pd(BnMIm-y)2Br2]. Single crystal X-ray diffraction (XRD) analysis of this material revealed a square planar geometry of Pd centre with a trans-anti conformation of the ligands, with Pd–Br and Pd–C bond lengths commensurate with that of analogous Pd(NHC)2Br2 structures reported previously (Fig. 5a, see ESI† for further details).44 The formation of this molecule requires C–H bond dissociation, which can be achieved both by heating or directly by the electron beam (DKO). While the experimental conditions in TEM and the ex situ experiment are different, they are likely to lead to the same outcome, i.e., the formation of a Pd-carbene complex. The molecule will not remain stable in the 200 keV electron beam for a long time due to the large concentration of C–H bonds, many of which are near the metal centre. The cleavage of C–H bonds will gradually increase the amount of H-radicals around Pd(II), leading to a reduction to Pd(0) at rate r3 (eqn (5)):
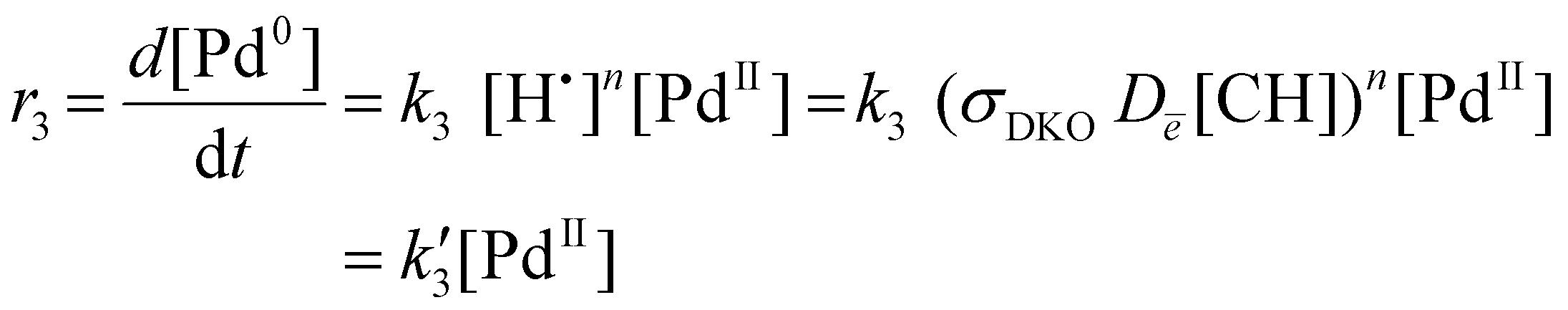 | (5) |
Where Dē is the electron beam dose (defined as a product of electron beam flux j and time the sample has been irradiated), n is the reaction order with respect to hydrogen atoms. Under our TEM imaging conditions, with the electron beam spread across a large area, each area of the IL pool receives the same flux of electrons, j, such that Pd0 atoms are produced at the same rate, r3, throughout the IL. The concentration of palladium atoms [Pd0] remains uniform until a nanoparticle nucleates and starts growth, leading to a gradient of concentration between the bulk solution and surface of the nanoparticle, [Pd0]–[Pd0]s. Because the reduction reaction takes place in the bulk solution, the rate of nanoparticle growth under these conditions is expected to be diffusion-controlled:
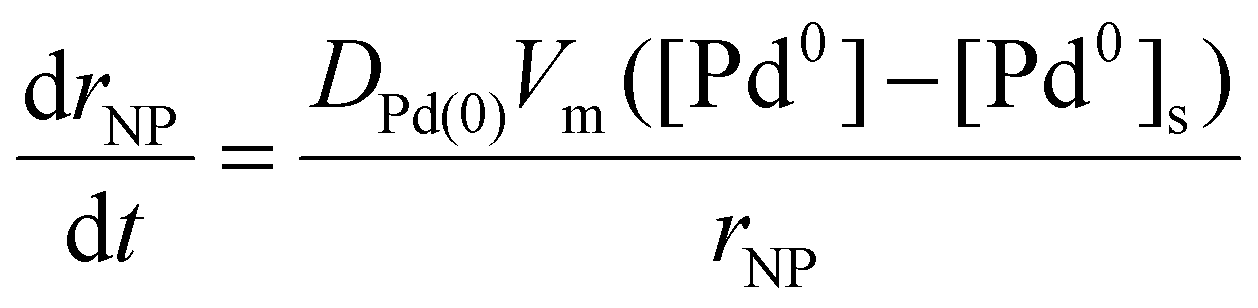 | (6) |
Simultaneously, the bromide anion, present at the same high molar concentration as the cation of the IL, supplies highly reactive oxidative Br˙ radicals (atomic bromine). These radicals can react with Pd(0) to oxidise them back to Pd(II) (Fig. 4d). Initially, the reaction rate is slow due to the low amount of Pd(0) atoms in the system. However, as the concentration of Pd(0) increases, the oxidation rate of palladium metal with bromine, r4, accelerates (eqn (7)), leading to the onset of the dissolution of the nanoparticles.
 | (7) |
As the nanoparticle re-dissolves, Pd(II) cations diffuse back into the solution (Fig. 5c), restoring the concentration of [PdII], thus accelerating the reduction process r3, leading to the formation of the second generation of nanoparticles (Fig. 4d). This becomes particularly apparent after approximately 700 seconds of e-beam irradiation in a carbon nanotube. This observation is consistent with a previous report that Br˙ radicals can oxidise pre-formed palladium nanoparticles to Pd(II) under scanning TEM (STEM) imaging conditions utilising the 200 keV electron beam.40 The result of the latter is the conversion of palladium metal to soluble forms of PdBr2 or [PdBr4]2−, which are rapidly dissolved into the IL, leading to the observed etching and dissolution of nanoparticles after 700 seconds of irradiation in our conditions inside the nano test tube (Fig. 6a). In a more open system, such as a solution of Pd(OAc)2 in [BnMIm]+Br− pool deposited directly onto the TEM grid, the nanoparticle dissolution process is faster than in carbon nanotube and proceeds to completion (Fig. 1d and S1†). The termination of the oscillatory process at the nanoparticle formation stage suggests that the flow of oxidising bromine atoms ceases before the flow of reducing hydrogen atoms. This can be attributed to the higher number density of hydrogen compared to bromine in the IL, with the H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br ratio of 13:1. Due to the dendritic nature of palladium growth, determination of the exact shape and structure of individual nanoparticles in the pool of IL is challenging due to complex aggregation and over-growth in subsequent generations of nanoparticles. In parallel, radicals formed from IL cations are likely to cross-link to form a polymer, which explains the solidification of the IL seen as voids left by dissolving Pd particles (Fig. 1D). Continued irradiation beyond the timescale of our measurements is likely to convert IL into nitrogen-doped carbon/graphene.45
:Br ratio of 13:1. Due to the dendritic nature of palladium growth, determination of the exact shape and structure of individual nanoparticles in the pool of IL is challenging due to complex aggregation and over-growth in subsequent generations of nanoparticles. In parallel, radicals formed from IL cations are likely to cross-link to form a polymer, which explains the solidification of the IL seen as voids left by dissolving Pd particles (Fig. 1D). Continued irradiation beyond the timescale of our measurements is likely to convert IL into nitrogen-doped carbon/graphene.45
 | ||
| Fig. 6 (a) Oxidation of Pd(0) to Pd(II) by bromine atoms leads to full or partial dissolution of nanoparticles. [PdBr4]2− is a soluble form of Pd(II), which can be recycled to grow the next generation of nanoparticles. Reduction of Pd(II) to Pd(0) by hydrogen radicals leads to nanoparticle growth with bromide anions adsorbed on the crystal facets, thus controlling the nanoparticle shape. (b) TEM image highlighting the 15 nanoparticles in IL solution in nanotube present after 500 s of 200 keV electron beam irradiation, the numbers n–m indicate ‘number of nanoparticle’ – ‘time at which the particle was last observed’ (a value of m = 1000 indicates that the particle did not undergo dissolution within the 1000 s timeframe). (c) A graph of particle radius plotted against time shows that the Pd NP generally grew at a similar rate for the first 500 s (driven by the reduction of Pd(II) to Pd(0) through DKO) after which some particles dissolved very suddenly (dashed lines; driven by Pd(0) to Pd(II) oxidation by Br˙ radicals through DVE), and others experienced decreased growth. (d) The rate of growth against the normalised particle size, rmean, allows for the trends to be observed, indicating that the change in particle size was more likely to be due to monomeric addition or migration and coalescence (a minority of cases) over Ostwald ripening. Scale bar is 50 nm. | ||
The formation and dissolution of nanoparticles inside the nanotube is a cleaner process than in the IL pool. We can follow transformations at the single-particle level with much higher spatiotemporal resolution in the former. Firstly, this enables the examination of the shape and atomic planes of the nanoparticles, revealing a disk-shaped morphology faceted with (200) and (220) planes. This result correlates well with the previously observed phenomenon of bromide ligand capping the (001) Pd plane, leading to preferential crystal growth in the (200) and (220) planes and, hence, anisotropic, disk-shaped nanoparticles.46–48 The total number of Pd(0) atoms in NPs at specific points in time was estimated and plotted in Fig. S4.†
Notably, the growth and dissolution processes are significantly slowed down inside carbon nanotubes compared to pools of free IL, enabling detailed analysis of the evolution of the radii of the 15 particles over 500 s as a function of time. This allows for individual nanoparticles’ growth and dissolution rates to be assessed (Fig. 6b). When the growth rate is plotted against a normalised particle radius, rmean, determined by the mean particle radius at t = 100 s for the 100–500 s data set and t = 500 s for the 500–1000 s data set, trends in the growth can be visualised (Fig. 6c).49 The four quadrants show whether a particle was larger or smaller than the mean, and whether or not it was increasing or decreasing in size over the periods of 100–500 s and then 500–1000 s. Quadrant 1 – smaller than average particles that were increasing in size; Quadrant 2 – larger than average particles that were increasing in size; Quadrant 3 – smaller than average that were decreasing in size; and Quadrant 4 – larger than average particles that were decreasing in size (Fig. 6d). For the 100–500 s data set, all measured nanoparticles had a positive derivative, indicating growth (average 0.0046 nm s−1); however, for the 500–1000 s data set, the seven particles that dissolved had negative derivatives and the remaining eight still had positive derivatives (average 0.0039 nm s−1). As the growth or dissolution of nanoparticles was independent of their size, with several larger-than-average nanoparticles dissolving precipitously (dotted lines Fig. 6c) and with all four quadrants being populated (Fig. 6d), the dynamics of the Pd nanoparticles under the 200 keV e-beam is not driven by Ostwald ripening (the expected trend would result in the predominant population of quadrants 2 and 3 with smaller particles getting smaller and larger particles getting larger)50,51 but is an interplay between oscillatory dissolution and growth, with occasional coalescence events (e.g. nanoparticle number 12 between 300–350 s). This picture aligns with the non-equilibrium reaction conditions explained above, involving fluxes of both reducing and oxidising species cycling the oxidation state of palladium. We expect that future developments in this area will focus on quantitatively determining the rates of processes involved in the oscillatory growth of nanoparticles. This can be accomplished through experimental or modelling methods, which will allow for the prediction and design of new systems similar to those discussed here.
Our time-resolved imaging using the electron beam of TEM both as a stimulus of chemical reactions and an imaging probe (ChemTEM approach)18 has been effectively applied previously to follow reactions of molecules in real-time and direct space. Reactions of polycondensation, cyclisation, cycloaddition, as well as metal crystal nucleation in ChemTEM always led to a thermodynamic products, as the reacting molecules were in an inert and stable environment (e.g. carbon nanotube or graphene support). In the current case, however, IL solvent is not a passive spectator but an active participant in the chemistry we observe. Our results show that when exposed to 200 keV electron beam irradiation, the IL solvent produces continuous fluxes of reducing H˙ and oxidising Br˙ species simultaneously, resulting in the cyclic growth and dissolution of Pd nanoparticles. The reaction pathways switch between the reduction and oxidation of palladium as a function of oscillating local concentrations of Pd(II) and Pd(0) species, controlling the rates of these reactions, respectively (Fig. 6a). These conditions can be compared to the Belousov–Zhabotinsky reaction, occurring under non-equilibrium thermodynamics, where the oxidation state of cerium ions oscillates between +3 and +4, forming evolving patterns of colour in the solution. The fractal-like patterns we observed in the IL pool during the oscillatory nanoparticle growth may also result from the non-equilibrium condition created by the electron beam in our nanoscale system (Fig. 1c).
In general, during a TEM investigation of metal nanoparticles in a liquid (e.g. in water, any organic solvents, including ILs), it is important to consider the impact of the electron beam on the molecules of the solvent. For example, a prior study reported oscillations of the Ostwald ripening in pairs of bismuth nanoparticles in dichlorobenzene with overlapping diffusion layers at 180 °C, which was attributed to the precursor-dependant chemical potential as a driving force.6 However, our earlier works showed that both C–H and C–Cl bonds on aromatic rings can be readily broken by the electron beam, so dichlorobenzene may play an active chemical role in TEM experiments.41,52 In our current study, it is clear that reactive species created by the electron beam from the halogen- and hydrogen-containing solvent, such as [BnMIm]+Br−, can reduce metal cations to nanoparticles and oxidise nanoparticles to metal cations, resulting in the growth and dissolution of the nanoparticles during the same TEM experiment. We expect that this phenomenon may also be relevant to other solvents, such as halogenated organics or even water, which can generate both strongly reducing and oxidising species when subjected to electron beam irradiation. However, this would be less applicable to hydrocarbon solvents that do not form oxidising species under the electron beam. Therefore, it is essential to consider these factors when interpreting the dynamics of the nanoparticles observed in TEM images.
Conclusions
We demonstrate that a nanoparticle in liquid under TEM conditions is an open chemical system far from equilibrium. It behaves as a nanoscale chemical oscillator driven by the fluxes of oxidative and reductive species generated by the electron beam from the solvent. This factor creates a fundamental hurdle if TEM imaging aims to understand the structure and composition of static nanoparticles. We alter the structure of nanoparticles in a liquid by the act of observation in TEM due to the dual role of the electron beam – an imaging probe and a stimulus of chemical reactions. However, this observer effect gives a unique opportunity to study nanoparticles in action. Specifically, in this work, we have shown that ionic liquid (IL) solvent within carbon nano test tubes provides an excellent experimental platform for investigating nucleation, growth, and dissolution dynamics of metal nanoparticles in the liquid phase that can be tracked at the single-particle level. The electron beam activates both the cation (C–H bond cleavage) and anion (valence electron displacement from Br−) parts of the IL, which trigger Pd(II) to Pd(0) reduction and Pd(0) to Pd(II) oxidation, respectively. This gives rise to a nanoscale chemical oscillator realised in palladium nanoparticles cycling between the growth and dissolution regimes, driven by the electron beam creating parallel fluxes of reducing (H˙) and oxidising (Br˙) species. The dynamics of palladium nanoparticles in liquids, imaged at the atomic scale, are significant for the mechanistic understanding of crystal nucleation and growth, and the role of the interactions and bonding with anions/cations, being crucial for catalysis,53–58 including the emergence of unusual conditions where the boundary between heterogeneous and homogeneous catalysis becomes blurred.53 Moreover, our research has identified the atomic mechanisms and the kinetic framework that describe how the electron beam can be used as a precise tool to create nanostructures in liquids reversibly. This knowledge complements recent advances in creating nanostructures with atomic precision and in specific locations using the electron beam.59–63 Studying chemical oscillations at the nanoscale, as observed in TEM experiments, has enabled us to identify the conditions under which nanoparticles exhibit cyclic behaviour that resembles living organisms—e.g. birth, growth, and decay, followed by the next generation of nanoparticles repeating the cycle. Future research into these fascinating dynamic systems may help bridge the gap between molecules and living cells.Experimental
General
Carbon nanotubes (PR19) were purchased from Applied Science, USA and were thermally annealed at 400 °C for 1 h in air before use. No further opening procedures were required, as these nanotubes do not possess end caps, allowing for filling of their internal cavity with IL. The average length of these CNTs was previously recorded as 15.34 ± 12.10 μm.64 All other chemicals were purchased from Sigma Aldrich and used without any further purification. Holey carbon film on copper mesh TEM grids were purchased from Agar Scientific Ltd, UK. TEM analysis was performed on a JEOL 2100F FEG-TEM with an accelerating voltage 200 kV (field emission electron gun source, information limit 0.19 nm). Images were acquired using the ‘acquire series’ function in Gatan Digital Micrograph suite v2 with acquisitions at 10 s intervals during which sample drift was manually corrected and the electron beam flux was maintained at 3.68 × 105 e− nm−2 s−1. The resulting stack was aligned using the “Stack Alignment Gatan” script (version: 20150524, v2.0) in Gatan Digital Micrograph v3 (D. R. G. Mitchell 2015). EDX spectroscopy was performed using an Oxford Instruments XMax 80 T silicon drift detector with INCA Energy 250 Microanalysis system. NMR spectra were collected in DMSO-d6 using a Bruker AV400 spectrometer at room temperature. Attenuated total reflectance Infrared spectroscopy was carried out using a Bruker FT-IR spectrometer over the range of 600–4000 cm−1. ESI-MS performed on Bruker microTOFII in positive reflection mode. CHN microanalysis performed on a Exeter Analytical CE-440 Elemental Analyzer.Synthesis of benzyl methyl imidazolium bromide
The [BnMIm]+Br− was synthesised by the following procedure: to 1-methylimidazole (10.0 mL, 125 mmol, 0.93 eq.) was added benzylbromide (16.0 mL, 135 mmol, 1.00 eq.) and acetonitrile (50.0 mL) and the mixture heated to reflux with stirring for 24 h. The solvent was then removed under reduced pressure to give crude [BnMIm]+Br− as an orange liquid. To crude [BnMIm]+Br− was added water (50 mL) and the mixture was washed with ethyl acetate (4 × 50 mL). The aqueous layer was then dried using anhydrous magnesium sulphate and evaporated under reduced pressure to give [BnMIm]+Br− (23.5 g, 93 mmol, 74%) as a pale-yellow liquid. 1H NMR spectroscopy (400 MHz, DMSO-d6 ppm): 9.31 (s, 1H), 7.83 (s, 1H), 7.74 (s, 1H), 7.44 (m, 5H), 5.45 (s, 2H), 3.86 (s, 3H); 13C NMR (101 MHz, DMSO-d6 ppm): 136.6 (s, 1C), 134.9 (s, 1C) 129.0 (s, 2C), 128.7 (s, 1C), 128.2 (s, 2C), 124.0 (s, 1C), 122.3 (s, 1C), 51.8 (s, 1C), 35.9 (s, 1C); ESI-MS [BnMIm]+ calc. m/z: 173.1079 found m/z: 173.1078; ATR-IR (νmax/cm−1): 3388, 3056, 2089, 1600, 1453, 1157, 720.TEM sample preparation
To palladium acetate (0.2 mg, 8.9 × 10−4 mmol) was added heat-treated carbon nanotubes (0.2 mg) and [BnMIm]+Br− (0.5 mL) and the mixture sonicated for 10 s before ethanol (0.5 mL) was added and the mixture was sonicated for a further 10 s to create a homogeneous carbon nanotube (CNT) dispersion. Phase separation occurred within seconds and an aliquot of the ethanolic mixture (top) was then deposited dropwise onto the TEM grid. The ethanol was left to dry under ambient conditions to leave the ((Pd(OAc)2)@[BnMIm]+Br−)@CNT sample.See ESI† file for synthetic method and characterisation of [Pd(BnMIm-y)2Br2].
Author contributions
The manuscript was written through contributions of all authors.Data availability
Other data information can be obtained from the corresponding author on request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the support of the Advanced Molecular Materials Interdisciplinary Research Cluster, University of Nottingham. The authors would like to thank the Nanoscale and Microscale Research Centre (nmRC), University of Nottingham, for access to TEM facilities, and the Engineering and Physical Sciences Research Council (EPSRC; programme grant MASI EP/V000055/1), and Royal Society for funding. We would also like to thank Tong Lui for conducting the CHN microanalysis. W. J. C. acknowledges funding from the Leverhulme Trust, grant number RPG-2022-300: Taming the radicals: highly reactive species incarcerated in carbon cages. The Authors thank Stephen T. Skowron for helpful discussion on e-beam interactions with metal nanoparticles.References
- M. Iqbal, Y. V. Kaneti, J. Kim, B. Yuliarto, Y. M. Kang, Y. Bando, Y. Sugahara and Y. Yamauchi, Small, 2019, 15, 1–27 CrossRef PubMed.
- M. N. O'Brien, M. R. Jones and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11717–11725 CrossRef PubMed.
- E. D. Goodman, J. A. Schwalbe and M. Cargnello, ACS Catal., 2017, 7, 7156–7173 CrossRef CAS.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- S. A. C. Carabineiro, L. M. D. R. S. Martins, M. Avalos-Borja, J. G. Buijnsters, A. J. L. Pombeiro and J. L. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS.
- H. L. Xin and H. Zheng, Nano Lett., 2012, 12, 1470–1474 CrossRef CAS.
- B. Van Vaerenbergh, J. Lauwaert, P. Vermeir, J. W. Thybaut and J. De Clercq, React. Chem. Eng., 2020, 5, 1556–1618 RSC.
- J. H. Kim, D. Shin, J. Lee, D. S. Baek, T. J. Shin, Y. T. Kim, H. Y. Jeong, J. H. Kwak, H. Kim and S. H. Joo, ACS Nano, 2020, 14, 1990–2001 CrossRef CAS PubMed.
- X. Yang, Q. Li, E. Lu, Z. Wang, X. Gong, Z. Yu, Y. Guo, L. Wang, Y. Guo, W. Zhan, J. Zhang and S. Dai, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- W. L. Vrijburg, E. Moioli, W. Chen, M. Zhang, B. J. P. Terlingen, B. Zijlstra, I. A. W. Filot, A. Züttel, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 7823–7839 CrossRef CAS.
- R. W. Lodge, G. A. Rance, M. W. Fay and A. N. Khlobystov, Nanoscale, 2018, 10, 19046–19051 RSC.
- D. S. Gavhane, H. van Gog, B. Thombare, G. Lole, L. Christiaan Post, M. A. More and M. A. van Huis, npj 2D Mater. Appl., 2021, 5, 1–9 CrossRef.
- C. Zhu, S. Liang, E. Song, Y. Zhou, W. Wang, F. Shan, Y. Shi, C. Hao, K. Yin, T. Zhang, J. Liu, H. Zheng and L. Sun, Nat. Commun., 2018, 9, 1–7 CrossRef CAS.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS PubMed.
- Z. Ying, J. Diao, S. Wang, X. Cai, Y. Cai, H. Liu and N. Wang, Mater. Charact., 2020, 170, 110706 CrossRef CAS.
- J. Wu, H. Shan, W. Chen, X. Gu, P. Tao, C. Song, W. Shang and T. Deng, Adv. Mater., 2016, 28, 9686–9712 CrossRef CAS.
- P. T. Z. Adibi, F. Mazzotta, T. J. Antosiewicz, M. Skoglundh, H. Grönbeck and C. Langhammer, ACS Catal., 2015, 5, 426–432 CrossRef.
- S. T. Skowron, T. W. Chamberlain, J. Biskupek, U. Kaiser, E. Besley and A. N. Khlobystov, Acc. Chem. Res., 2017, 50, 1797–1807 CrossRef CAS PubMed.
- K. Cao, J. Biskupek, C. T. Stoppiello, R. L. McSweeney, T. W. Chamberlain, Z. Liu, K. Suenaga, S. T. Skowron, E. Besley, A. N. Khlobystov and U. Kaiser, Nat. Chem., 2020, 12, 921–928 CrossRef CAS PubMed.
- K. Cao, S. T. Skowron, C. T. Stoppiello, J. Biskupek, A. N. Khlobystov and U. Kaiser, Angew. Chem., Int. Ed., 2020, 59, 22922–22927 CrossRef CAS PubMed.
- K. Cao, T. W. Chamberlain, J. Biskupek, T. Zoberbier, U. Kaiser and A. N. Khlobystov, Nano Lett., 2018, 18, 6334–6339 CAS.
- W. J. Cull, S. T. Skowron, R. Hayter, C. T. Stoppiello, G. A. Rance, J. Biskupek, Z. R. Kudrynskyi, Z. D. Kovalyuk, C. S. Allen, T. J. A. Slater, U. Kaiser, A. Patane and A. N. Khlobystov, ACS Nano, 2023, 17, 6026–6072 Search PubMed.
- K. Cao, T. Zoberbier, J. Biskupek, A. Botos, R. L. McSweeney, A. Kurtoglu, C. T. Stoppiello, A. V. Markevich, E. Besley, T. W. Chamberlain, U. Kaiser and A. N. Khlobystov, Nat. Commun., 2018, 9, 3382 CrossRef PubMed.
- T. Zoberbier, T. W. Chamberlain, J. Biskupek, M. Suyetin, A. G. Majouga, E. Besley, U. Kaiser and A. N. Khlobystov, Small, 2016, 12, 1649–1657 CAS.
- I. Cardillo-Zallo, J. Biskupek, S. Bloodworth, E. S. Marsden, M. W. Fay, Q. M. Ramasse, G. A. Rance, C. T. Stoppiello, W. J. Cull, B. L. Weare, R. J. Whitby, U. Kaiser, P. D. Brown and A. N. Khlobystov, ACS Nano, 2024, 18, 2958–2971 CrossRef CAS PubMed.
- F. Tao and P. A. Crozier, Chem. Rev., 2016, 116, 3487–3539 CAS.
- X. Wang, J. Yang, C. M. Andrei, L. Soleymani and K. Grandfield, Commun. Chem., 2018, 1, 80 CrossRef.
- J. Yang, J. Koo, S. Kim, S. Jeon, B. K. Choi, S. Kwon, J. Kim, B. H. Kim, W. C. Lee, W. B. Lee, H. Lee, T. Hyeon, P. Ercius and J. Park, J. Am. Chem. Soc., 2019, 141, 763–768 CrossRef CAS PubMed.
- L. Zheng, X. Zhang, K. C. Bustillo, Y. Yao, L. Zhao, M. Zhu, W. Li and H. Zheng, Nanoscale, 2018, 10, 11281–11286 RSC.
- R. Hufschmid, E. Teeman, B. L. Mehdi, K. M. Krishnan and N. D. Browning, Nanoscale, 2019, 11, 13098–13107 RSC.
- D. Keller, T. R. Henninen and R. Erni, Micron, 2019, 117, 16–21 CrossRef CAS PubMed.
- B. Fritsch, A. Hutzler, M. Wu, S. Khadivianazar, L. Vogl, M. P. M. Jank, M. März and E. Spiecker, Nanoscale Adv., 2021, 3, 2466–2474 RSC.
- T. Xu and L. Sun, Superlattices Microstruct., 2016, 99, 24–34 CrossRef CAS.
- V. V. Chaban and O. V. Prezhdo, ACS Nano, 2014, 8, 8190–8197 CrossRef CAS PubMed.
- Y. Yamada, K. Taguchi, T. Ikuta, A. Horibe and K. Takahashi, J. Phys. Chem. C, 2018, 122, 21910–21918 CrossRef CAS.
- C. M. Wang, J. Mater. Res., 2015, 30, 326–339 CrossRef CAS.
- R. F. Egerton, Micron, 2019, 119, 72–87 CrossRef CAS.
- B. J. Mincher and J. F. Wishart, Solvent Extr. Ion Exch., 2014, 32, 563–583 CrossRef CAS.
- J. M. Grogan, N. M. Schneider, F. M. Ross and H. H. Bau, Nano Lett., 2014, 14, 359–364 CrossRef CAS PubMed.
- Y. Jiang, G. Zhu, F. Lin, H. Zhang, C. Jin, J. Yuan, D. Yang and Z. Zhang, Nano Lett., 2014, 14, 3761–3765 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622–629 CrossRef CAS PubMed.
- T. Miyata and T. Mizoguchi, Ultramicroscopy, 2017, 178, 81–87 CrossRef CAS PubMed.
- J. M. Seymour, E. Gousseva, A. I. Large, C. J. Clarke, P. Licence, R. M. Fogarty, D. A. Duncan, P. Ferrer, F. Venturini, R. A. Bennett, R. G. Palgrave and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2021, 23, 20957–20973 RSC.
- M. S. Szulmanowicz, A. Gniewek, W. Gil and A. M. Trzeciak, ChemCatChem, 2013, 5, 1152–1160 CrossRef CAS.
- A. Turchanin, Ann. Phys., 2017, 529, 1–13 CrossRef.
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed.
- W. Gao, A. O. Elnabawy, Z. D. Hood, Y. Shi, X. Wang, L. T. Roling, X. Pan, M. Mavrikakis, Y. Xia and M. Chi, Nat. Commun., 2021, 12, 1–10 Search PubMed.
- M. Liu, Y. Zheng, L. Zhang, L. Guo and Y. Xia, J. Am. Chem. Soc., 2013, 135, 11752–11755 CrossRef CAS PubMed.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Catal., 2011, 281, 147–155 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- R. Van Den Berg, T. E. Parmentier, C. F. Elkjær, C. J. Gommes, J. Sehested, S. Helveg, P. E. De Jongh and K. P. De Jong, ACS Catal., 2015, 5, 4439–4448 CrossRef CAS.
- K. L. Y. Fung, S. T. Skowron, R. Hayter, S. E. Mason, B. L. Weare, N. A. Besley, Q. M. Ramasse, C. S. Allen and A. N. Khlobystov, Phys. Chem. Chem. Phys., 2023, 9092–9103 Search PubMed.
- I. Cano, A. Weilhard, C. Martin, J. Pinto, R. W. Lodge, A. R. Santos, G. A. Rance, E. H. Åhlgren, E. Jónsson, J. Yuan, Z. Y. Li, P. Licence, A. N. Khlobystov and J. Alves Fernandes, Nat. Commun., 2021, 12, 2–7 CrossRef PubMed.
- V. M. Chernyshev, O. V. Khazipov, D. B. Eremin, E. A. Denisova and V. P. Ananikov, Coord. Chem. Rev., 2021, 437, 213860 CrossRef CAS.
- S. Yao, H. Sun and D. Ma, J. Phys. D: Appl. Phys., 2021, 54, 144001 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- T. Welton, Biophys. Rev., 2018, 10, 691–706 Search PubMed.
- T. Uematsu, M. Baba, Y. Oshima, T. Tsuda, T. Torimoto and S. Kuwabata, J. Am. Chem. Soc., 2014, 136, 13789–13797 CrossRef CAS PubMed.
- M. Tripathi, A. Mittelberger, N. A. Pike, C. Mangler, J. C. Meyer, M. J. Verstraete, J. Kotakoski and T. Susi, Nano Lett., 2018, 18, 5319–5323 CrossRef CAS PubMed.
- C. Su, M. Tripathi, Q. B. Yan, Z. Wang, Z. Zhang, C. Hofer, H. Wang, L. Basile, G. Su, M. Dong, J. C. Meyer, J. Kotakoski, J. Kong, J. C. Idrobo, T. Susi and J. Li, Sci. Adv., 2019, 5, 1–11 Search PubMed.
- R. Mirzayev, K. Mustonen, M. R. A. Monazam, A. Mittelberger, T. J. Pennycook, C. Mangler, T. Susi, J. Kotakoski and J. C. Meyer, Sci. Adv., 2017, 3, 6–11 Search PubMed.
- A. Chuvilin, J. C. Meyer, G. Algara-Siller and U. Kaiser, New J. Phys., 2009, 11, 083019 CrossRef.
- T. Susi, J. C. Meyer and J. Kotakoski, Ultramicroscopy, 2017, 180, 163–172 CrossRef CAS PubMed.
- M. Aygün, C. T. Stoppiello, M. A. Lebedeva, E. F. Smith, M. D. C. Gimenez-Lopez, A. N. Khlobystov and T. W. Chamberlain, J. Mater. Chem. A, 2017, 5, 21467–21477 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Time series summarizing the Pd growth/dissolution seen in ESI Video S3, visualization of the less dense areas of IL inside the CNTs, details of the synthesis and crystal structure determination of [Pd(BnMIm-y)2Br2] and calculation of molar reactants. ESI Videos 1–3 show Pd nanoparticle formation in free-standing films of IL and ESI Video 4 shows Pd nanoparticle formation inside IL inside a carbon nanotube. CCDC 2124145. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr04150j |
| This journal is © The Royal Society of Chemistry 2025 |