
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineered anti-HER2 drug delivery nanosystems for the treatment of breast cancer†
Silvia
Vanni‡
 a,
Tania Mariastella
Caputo‡
b,
Angela Maria
Cusano
c,
Alessandro
De Vita
*a,
Andrea
Cusano
*bc,
Claudia
Cocchi
a,
Chiara
Mulè
b,
Sofia
Principe
b,
Chiara
Liverani
a,
Giorgia
Celetti
b,
Alberto
Micco
c,
Chiara
Spadazzi
a,
Giacomo
Miserocchi
a,
Toni
Ibrahim
d,
Laura
Mercatali§
a and
Anna
Aliberti§
b
a,
Tania Mariastella
Caputo‡
b,
Angela Maria
Cusano
c,
Alessandro
De Vita
*a,
Andrea
Cusano
*bc,
Claudia
Cocchi
a,
Chiara
Mulè
b,
Sofia
Principe
b,
Chiara
Liverani
a,
Giorgia
Celetti
b,
Alberto
Micco
c,
Chiara
Spadazzi
a,
Giacomo
Miserocchi
a,
Toni
Ibrahim
d,
Laura
Mercatali§
a and
Anna
Aliberti§
b
aPreclinic and Osteoncology Unit, Biosciences Laboratory, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy. E-mail: alessandro.devita@irst.emr.it; Fax: +39-0543-739221; Tel: +39-0543-739239
bOptoelectronics Group, Department of Engineering, University of Sannio, Benevento, Italy. E-mail: a.cusano@unisannio.it; Fax: +39-0824-305840; Tel: +39-0824-305871
cCeRICTscrl Regional Center Information Communication Technology, Benevento, Italy
dOsteoncology, Bone and Soft Tissue Sarcomas and Innovative Therapies Unit, IRCCS Istituto Ortopedico Rizzoli, Bologna, Italy
First published on 21st March 2025
Abstract
Breast cancer stands as the primary cancer affecting women and the second most prevalent cause of cancer-related fatalities in developed nations. Consequently, there is a pressing demand for the advancement of therapeutic strategies that can be seamlessly integrated into clinical applications. We investigated the effectiveness of an encapsulation and decoration strategy employing biodegradable and biocompatible carriers together with 3D collagen-based culture models. Envisioning the use of nano delivery systems for localized regional release, we explored the feasibility of a light-controlled drug release, assisted by optical fibers. PLGA nanoparticles loaded or decorated with trastuzumab (TZ) were synthesized via a double emulsion protocol and characterized by dynamic light scattering, surface plasmon resonance, transmission electron microscopy, atomic force microscopy, and Fourier transform infrared spectroscopy. In vitro biological evaluation was then performed on HER2-positive breast cancer cell line BT-474, examining the effect of nanoformulations on cell viability in 2D and 3D collagen scaffold culture models. Cell cycle, apoptosis, cell morphology and distribution and protein expression were analyzed. Finally, a core-offset optical fiber was fabricated and particles release was studied in vitro using light in batch and microfluidic tests. The nanoparticles displayed uniform and spherical shape, maintaining stability in DMEM for up to seven days. The successful immobilization of TZ was verified. In vitro trials with BT-474 cells in 2D and 3D models revealed that poly(lactic-co-glycolic acid) (PLGA) nanoparticles encapsulated with TZ demonstrated similar or superior biological activity compared to free TZ. Notably, PLGA functionalized with TZ both internally and on the surface exhibited the highest effectiveness in terms of cell viability, increase of apoptosis markers, and inducing cell quiescence. This affirms the pivotal role of PLGA nanoparticles in preserving the integrity of TZ and enhancing its targeted delivery. Furthermore, we propose a breakthrough fiber-optic technology for the less invasive local delivery of PLGA-based nanocarriers that can be effectively used in clinical practice. In conclusion our studies lay the foundation for future advancements in alternative therapeutic tools for localized breast cancer treatment. The integration of advanced carriers, optical fibers, and microfluidics opens up new possibilities for innovative and targeted therapeutic approaches.
1. Introduction
Breast cancer is one of the most common tumor types worldwide and the most common cancer in women. Despite great successes in recent decades in treatment and early detection, which have reduced the mortality rate to about 8%, breast cancer is still the leading cause of cancer death in less developed countries and the second leading cause in more developed countries.1 Therefore, there is a pressing need to empower actual treatments and develop innovative therapeutic approaches which can be translated into clinical practice.Breast cancer is categorized into four main molecular subtypes: luminal A, luminal B, HER2-enriched, and basal-like, with HER2-positive being the most aggressive and unresponsive to hormone therapy.2,3 Treatment for HER2-positive cancer typically involves chemotherapy combined with immune checkpoint inhibitors and trastuzumab (TZ), especially for larger lesions. Surgery may be necessary for complete tumor removal.4 Metastatic breast cancer treatment relies on systemic drugs, often supplemented by locoregional therapy, but the five-year survival rate remains low. Bone metastases are common, negatively affecting patient outcomes and quality of life.5–7 HER2-targeted therapy, particularly TZ, has significantly improved treatment options for HER2-overexpressing breast cancer.8–10
TZ exerts its effects through multiple mechanisms, including the blockade of MAPK/PI3K signaling, leading to cell cycle arrest and reduced proliferation.11 It selectively inhibits HER2–HER3 dimerization and promotes the degradation of HER2.12 TZ also triggers immune-mediated responses, such as antibody-dependent cellular cytotoxicity (ADCC), and promotes HER2 degradation.13,14 However, many HER2-positive breast cancer patients experience resistance to TZ, often due to mechanisms like the expression of truncated HER2 forms that lack TZ-binding sites, overexpression of other growth factor receptors, or mutations that activate alternative signaling pathways.15–17 Additionally, TZ can lead to side effects, including infections, autoimmune diseases, and cardiotoxicity, along with limitations related to tumor penetration and pharmacokinetics.
Several studies focused on the development of targeted nanocarriers to improve the site specificity of TZ. In those cases, the antibody was immobilized – by physical or chemical methods – on the outer surface of the particle.18,19 The TZ–HER2 interaction induced the endocytosis of the carriers and the drug was released inside the cell. However, this approach does not guarantee the protection of the TZ from the extracellular environment.
A viable strategy to boost the efficacy and specificity of the TZ consists in the development of targeted nanocarriers in which the TZ is also encapsulated inside the nanocarrier core that, hides and protects the antibody from the external environment, improving therapeutic performance and reducing the side effects mentioned above.20–26
In this work, we investigated for the first time the effect of a monoclonal antibody targeted delivery strategy-based on PLGA nanoparticles. These nanocarriers were both loaded and externally decorated with TZ (PLGA + TZ) and tested in the human breast cancer cell line BT-474 across 2D and 3D culture models. The 3D collagen-based model, designed to mimic the extracellular matrix (ECM) microenvironment,27 effectively reproduces tumor hypoxia and allows for predictive assessments of chemotherapy response, including therapeutic efficacy, molecular adaptations, and resistance mechanisms.28
Within this experimental framework, we have studied the morphological, structural, and physicochemical characteristics of various functionalized nanoparticles, with a focus on their biological impacts on BT-474 cells in both culture settings.
In particular, we validated the efficacy of the newly developed nanoformulations analyzing the inhibition of cancer cell proliferation in relation to the increase of apoptosis markers and of cell cycle slow down. Moreover, we investigated by immunofluorescence and cytometry the effect of drugs in HER2 expression on cancer cell surface as its decrease can affect drug efficacy.
Finally, a feasibility study was performed by introducing a light-triggered fiber-optic tool for local drug delivery, with the outlook of realizing a mini-invasive device for the release of PLGA + TZ particles. Our proof-of-concept represents a new paradigm for a light-triggered drug delivery system based on a novel core-offset optical fiber architecture in combination with PLGA particles and a UV photocleavable linker. The light-triggered release of our PLGA particles was also tested in a microfluidic system as feasibility study, to effectively demonstrate the possibility to integrate the optical probe in minimally invasive clinical tools (needles, catheters and nano-endoscopes) for loco-regional drug delivery. This tool will pave the way for developing new strategies for local or metastatic relapse, in particular in hard and not accessible tissues, such as bone or brain.
2. Experimental
2.1 Reagents
PLGA (Resomer® 504 H, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 lactide:glycolide, acid terminated, MW 53200 Da), poloxamer (Pluronic F-127), Coumarin6 (C6), ethyl acetate (EtOAc), trehalose, polyvinyl alcohol (PVA), phosphate buffer saline (PBS) tablets and Paraformaldehyde (PFA) were purchased from Sigma Aldrich Co. (Merck KGaA, St Louis, MO, USA).
:50 lactide:glycolide, acid terminated, MW 53200 Da), poloxamer (Pluronic F-127), Coumarin6 (C6), ethyl acetate (EtOAc), trehalose, polyvinyl alcohol (PVA), phosphate buffer saline (PBS) tablets and Paraformaldehyde (PFA) were purchased from Sigma Aldrich Co. (Merck KGaA, St Louis, MO, USA).
Water, acetonitrile and trifluoroacetic acid (TFA) LC-MS grade were from (ROMIL Ltd, Cambridge, UK). Deionized water (18.2 MΩ cm) was purchased from a MilliQ Plus system (Merck Millipore, St Louis MO, USA) with a total organic content (TOC) < 3 ppb. Trastuzumab (TZ – Herceptin®, Roche, Basel, Switzerland) was obtained as the European Medicines Agency approved versions and formulations, available commercially to the general public and was provided by Prof. Colantuoni. All other solvents and reagents were acquired from Sigma-Aldrich Co (Merck KGaA, St Louis, MO, USA).
2.2 PLGA Particles synthesis
The PLGA particles were obtained by double emulsion technique, as also described in our previous work.29–31 Briefly, for the PLGA + TZIN particles 800 μL of an aqueous solution of TZ (13.3 nmol) and poloxamer (0.8 mol) were emulsified with 1 mL of PLGA (1.31 mol) dissolved in ethyl acetate. The first emulsion was prepared by sonicating twice for 60 seconds at an intensity of 38% with a microtip (Sonifier™ SFX150, Branson Ultrasonics, Emerson Electric Co, St Louis, MO, USA). The first emulsion was then added to 4 mL of PVA 2% w/v and sonicated as described above. Finally, 8 mL of PVA 2% w/v were added and the solution was stirred overnight (16 hours) at room temperature (RT) to assist the evaporation of the organic solvent.After stirring overnight, unencapsulated TZ was removed from the nanoparticles by centrifugation for 30 min at 4 °C and 15000 rpm (Heraeus Fresco 21; Thermo Fisher Scientific, Waltham, MA, USA). The pellet was washed three times with milliQ water. The same protocol was applied to prepare PLGA particles without TZ (N-PLGA) or loaded with coumarin 6 (PLGA + C6). Coumarin 6 was first dissolved in DMSO (2 mg mL−1), then added to the aqueous phase (final concentration 100 μg mL−1) and treated as described above. Finally, all the particles were freeze-dried with the addition of 1.25% w/v trehalose as a cryoprotectant by using Alpha 1–2 LD lyophilizator (Martin Christ, Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany). After freeze-drying, the process yield was measured as the ratio of the collected nanoparticles to the starting raw materials (TZ and polymers, cryoprotectant).
2.3 Particles functionalization with TZ
Surface functionalization was achieved by covalent linkage of TZ on both empty and loaded particles in order to obtain PLGA + TZOUT and PLGA + TZALL particles, respectively. Carboxylic groups of PLGA were preactivated using the classical carbodiimide chemistry by incubating the aqueous solution of particles (14 mg mL−1) with 100-fold molar excess of EDC (carbodiimide crosslinker) and NHS (N-hydroxysuccinimide) for 1 hour. Then the solution was centrifuged for 15 minutes at 10000 rpm and 4 °C, to remove the unreacted EDC/NHS, and the particles were incubated with TZ (1 mg mL−1) for 1 hour at room temperature under gentle stirring. After the incubation, the solution was centrifuged to separate the unbound TZ from the functionalized particles and the pellet was washed three times in MilliQ water. The amount of TZ bound to the particle was quantified by Bradford assay and calculated as the difference between the unbound TZ recovered in the supernatant and the initial concentration used for the functionalization.
2.4 Particle characterization by dynamic light scattering
Dynamic Light Scattering (DLS) technique (Malvern Zetasizer Nano ZS device, 633 nm laser, 173° dispersion angle; Malvern, UK) was used to measure particle size and surface charge. Freeze-dried PLGA particles (N-PLGA, PLGA + TZIN, PLGA + TZOUT, PLGA + TZALL, PLGA + C6) were dissolved in MilliQ water (1 mg mL−1) gently stirring for 24 hours. The solution was then diluted (0.05 mg mL−1) and measurements were performed five times at 25 °C. The zeta potential of the nanoparticle was measured using the same instrument and a suitable cuvette.2.5 Evaluation of nanoparticles stability in phosphate buffer and cell culture medium
Particles stability was investigated under physiological conditions by DLS measurements in time course experiments. PLGA + TZALL particles (1 mg) were dispersed in 1 mL of PBS (10 mM) or DMEM (enriched with 10% of FBS) and incubated at 37 °C from 1 hour up to 14 days. All measurements were performed in triplicate.2.6 Antibody functionality validation by surface plasmon resonance
The capability of TZ to retain its functional integrity after the particle synthesis and functionalization was investigated by Surface Plasmon Resonance by using Biacore 8K (Cytiva, Marlborough, MA, USA). This Instrument is equipped with a microfluidic system with eight separate channels, each of which consisting of two flow cells (FC).On the FC2 of channel 1 (Active FC) was immobilized the protein receptor HER-2 specific for TZ, while FC1 (Reference FC) was left untreated and used as negative control. HER2 was immobilized on CM5 chips by amine coupling chemistry through the so-called high immobilization standard protocols (Cytiva). Briefly, the CM5 surface on the selected FCs was activated injecting a 1:1 (v/v) mixture of 0.4 M EDC and 0.1 M NHS solutions in water for 420 seconds. After equilibration in the buffer, the activated surfaces were exposed to solutions of HER2 protein at 30 μg mL−1 in 10 mM NaAc pH 5.0 for 7 minutes. The exceeding reactive esters were deactivated by contact with a 1 M solution of MEA at pH 8.5 for 7 minutes. The running buffer was HBS-EP + buffer. The TZ binding was evaluated by Single Cycle Kinetic assay by sequential injection of free TZ, TZ functionalized particles and naked particles at increasing concentrations (ranging from 0.016 to 0.2 μg mL−1) without regeneration steps between each sample.
The flow rate was kept at 30 μL min−1 in all analyses. Data were processed and analyzed with Biacore 8K Evaluation Software Version 3.0 (Cytiva, Marlborough, MA).
Responses from FC1 were subtracted point-by-point from those recorded on FC2.
2.7 Particles characterization: transmission electron microscopy and Atomic force microscopy analyses
Transmission electron microscopy (TEM) observations were performed on nanoparticle samples prepared by depositing 5 μL of PLGA solution on specific TEM copper grids with a carbon coated Formvar film. After evaporation of the solution, the grid was washed with deionized water and analysis was performed using a TEM Tecnai G2 Spirit TWIN (FEI, Hillsboro, OR, USA). Morphological characterization by Atomic force microscopy (AFM, Agilent Technologies 5420, Agilent Technologies, Santa Clara, CA, USA) was achieved by depositing NPs directly on a slide previously coated with a uniform Au layer. Nanoparticles film was scanned in tapping mode to avoid damage to the particles. All AFM images were acquired in the dry state and processed using Pico Image software (Keysight Technologies, Santa Rosa, CA, USA).2.8 Analysis by Fourier transform infrared spectroscopy (FTIR)
The PLGA particles (N-PLGA, PLGA + TZIN, PLGA + TZOUT, PLGA + TZALL) and the free TZ were analysed by Fourier transform infrared spectroscopy (FTIR). Spectral analysis was performed using a Spectrum 3 spectrometer (PerkinElmer Inc., Waltham, MA, USA) equipped with a total attenuated reflectance accessory (UATR). Spectra were acquired by ten scans with a resolution of 1 cm−1 and 16 scans in the spectral range of 650–4000 cm−1. A series of three replicates of each formulation was performed.2.9 TZ quantification by RP-HPLC
The quantification of TZ was performed by RP-HPLC analysis (UltiMate 3000 DIonex, Thermo-Fisher Scientific, Inc., Waltham, MA, USA) using a C4 column (BioZen Phenomenex Widepore C4, 2.1 × 100 mm, nanoparticles dimension 2.6 μm, Phenomenex Inc. Torrance, CA, USA) and monitoring the TZ absorption at 280 nm. Elution was performed at 0.3 mL min−1 setting the gradient of the mobile phases A (trifluoroacetic acid 0.08% v/v in water) and B (trifluoroacetic acid 0.08% in acetonitrile) as follows: 15% B phase for 1 minute; 15–90% B phase from 1 to 7 minutes; 90% B phase from 7 to 12 minutes; 90–15% B phase from 12 to 14 minutes; the column was finally conditioned with 15% B phase for 6 minutes. The TZ retention time was approximately 6.5 minutes. For all release analyses, a volume of 80 μL was injected in duplicate. TZ concentrations were calculated from the calibration curve generated by analysing solutions between 50–1000 ng of antibody.2.10 Encapsulation efficiency, drug loading and in vitro drug release
To calculate the encapsulation efficiency (EE), a known amount of dry particles was completely dissolved in DMSO. After 1 hour under stirring the TZ released from particles was determined by RP-HPLC after centrifugation at 16000 RCF for 20 minutes. The EE was calculated as:where Ce is the concentration of drug recovered in the supernatant after centrifugation, and Ct is the concentration of TZ added during preparation.

Likewise, the drug loading (DL) was quantified as:
 000 rpm for 20 minutes, and the supernatant was completely collected and replaced with 1 mL of fresh medium. After particle centrifugation, released TZ in each sample was analysed by RP-HPLC as described above. All experiments were performed in triplicate and the average values were taken.
000 rpm for 20 minutes, and the supernatant was completely collected and replaced with 1 mL of fresh medium. After particle centrifugation, released TZ in each sample was analysed by RP-HPLC as described above. All experiments were performed in triplicate and the average values were taken.
Release values are expressed as the cumulative percentage of drug analysed at each time point compared with the total amount encapsulated.
2.11 Fabrication and characterization of core-offset optical fibers (coOF)
The core-offset optical fiber (coOF) probe consists of a multimode fiber (Thorlabs FG105UCA) with a core diameter of 105 μm and a cladding diameter of 125 μm used as the launch fiber, and a core-offset single-mode fiber (Thorlabs SMF28) with a core diameter of 8.2 μm and a cladding diameter of 125 μm used as the scattering element.First, the OFs were stripped from the coatings and then rinsed with isopropyl alcohol. In a second step, the multimode fiber and the single-mode fiber were spliced using an arc fusion splicer (Fujikura FSM-70), which can handle different types of OFs with cladding diameters up to 125 μm and is capable of creating an offset between the core axes of the OFs, ensuring the full control of light in the cladding of the single-mode optical fiber. In the final step, the single-mode fiber was cleaved with an optical fiber cleaver (Fujikura CT-104) at a distance of 1 cm from the splice point. To obtain an optimized coOFs ensuring the correct optical power density value at the cladding/air interface, we carefully analyzed the power loss at the splicing location and the scattering efficiency. The core offset of the splicing step was optimized, resulting in a value of 10 μm.
To correctly evaluate the optical power density at the cladding interface (Scattering Power), we first evaluated through different trials the loss factor at core offset junction.
Soon after, to evaluate the total optical power emitted by the lateral surface of the single mode fiber we evaluated the scattering Power (the fabricated probe) was optically characterized using an integrating sphere (Thorlabs S140C) in conjunction with an optical power meter (Thorlabs PM100). Scattering power was first evaluated as:
| Pscatt = Pinc − Ploss − Ptip |

Experimentally, the power scattered from the lateral surface of the output single mode optical fiber was calculated as the difference between the total power measured with the integrated sphere, diminished by the power losses evaluated at the core offset junction, and the power emitted by the output tip measured by a suitable photodiode located at the fiber termination exiting the tip. The Pinc term was measured before splicing the single mode fiber to the output of the multimode fiber.
2.12 coOF functionalization with PLGA particles
The coOF was opportunely treated to covalently immobilize PLGA + C6 particles through a photocleavable linker (PC DBCO-NHS Ester I, Click chemistry tools, Cat# 1160) as already described.26 Briefly, optical fiber was hydroxylated by incubation in the piranha solution (H2O2:H2SO4 30:70) at 25 °C for 30 minutes and then immersed in 12 M HCl for 1 hour at 25 °C and 0.1 M NaOH for 1 hour at 50 °C. The hydroxylated fiber was then washed with pure ethanol and incubated in 3-azidopropyltriethoxysilane (AZTES) 67 mM overnight at 4 °C. The immobilized azide molecules present on the fiber reacted, by strain-promoted azide–alkyne cycloaddition (SPAAC), with the dibenzocyclooctyne (DBCO) of the photocleavable DBCO-PC-NHS ester linker at a concentration of 0.5 mM in DMSO at 25 °C for 16 hours. To introduce secondary amine groups into the PLGA particles, the biocompatible polyesters were activated by aminolysis with 1 mL ethylenediamine (EDA) 10 mM for 5 minutes. The particles were then centrifuged at 10000 rpm for 15 minutes at 4 °C to remove the unreacted EDA and diluted at 2.5% w/v in borate buffer 50 mM pH 8.5. The optical fiber was then immersed in the aminated PLGA particles for 30 minutes using a dip coater (KSV NIMA KN4001, Biolin Scientific Oy, Espoo, Finland – speed 1 mm min−1, speed 0.5 mm min−1). The functionalized optical fiber was carefully rinsed three times with MilliQ water and analyzed using CLSM and an optical microscope (Olympus BXFM upright).
2.13 In vitro particle release assisted by light: batch and microfluidic-assisted tests
Both batch and microfluidic methods were used to evaluate particle release. For batch tests, the sample was operated with a high-power fiber-coupled light source (Mightex FCS-0365-201) specifically designed for biological experiments. The emitting wavelength is 365 nm, and with a fiber of 1 mm core diameter and 0.39 NA, the maximum radiation flux is 360 mW. The coOF was immersed in a quartz cuvette (10 mm low-volume quartz – 400 μL) filled with PBS buffer while the laser source was on (at maximum power), so that the linker was cleaved by the light and the particles were released into the medium. The collected solution was analyzed using a fluorometer (Fluoromax-Plus from Horiba Scientific) with the following parameters: excitation wavelength 464 nm, emission wavelength from 475 to 600 nm, integration time of 0.1 s, step size of 1 nm, slit width of 5 nm bandpass for excitation and emission. After analysis, the solution was collected and replaced with fresh medium. To evaluate particle release in the microfluidic chip (Trustech, Italy), the optical fiber was inserted into the main channel of the device, which consists of three PMMA layers. The microfluidic channels are embedded in the bottom layer, while the top layer has the role of sealing the fluid system through appropriately sized screws placed on the device to prevent leaks. The microfluidic device is T-shaped and consists of two inlets and one outlet. Two nanoports were chosen for the two inlets, one of which is used to easily insert the functionalized optical fiber into the main channel, while the second serves as an interface between the side channel of the fluidic device and the buffer solution. The main channel is 4.5 cm long and 1 mm in diameter, while the side channel, which serves to keep the microgel hydrated with a buffer solution, is 1 cm long and 0.5 mm in diameter. The buffer solution flowing through the side channel is also used to eject the carriers through the outlet once the functionalized optical fiber is activated. The solutions are analyzed as described for the batch process. The microfluidic device has a custom valve that can “close” the system to prevent loss of carriers during the activation time of the functionalized optical fiber. The outlet is equipped with an adapter for a needle (or catheter) to deliver the vectors to the desired location. The entire device is 5.5 cm long and 4.5 cm wide.2.14 Collagen scaffold synthesis
All chemicals were purchased from Sigma–Aldrich (St Louis, MO, USA). The collagen scaffolds were synthesized and characterized as previously described.32–34 Type I collagen was suspended in acetic acid, precipitated to pH 5.5, and cross-linked with 1,4-butanediol diglycidyl ether. An established freezing and heating ramp (from 25 °C to −25 °C and from −25 °C to 25 °C in 50 min under vacuum conditions, p = 0.20 mmbar) produced the scaffold's porosity ensuring proper pore size, interconnectivity, and orientation. Scaffolds were sterilized in 70% ethanol for 1 hour and then washed three times in sterile Dulbecco's Phosphate-Buffered Saline (Life Technologies, Carlsbad, CA, UA). Porosity and pore size of the scaffold were determined as previously described.332.15 Cell seeding and culture
The human breast cancer cell line BT-474 was purchased from the American Type Culture Collection (Rockville, MD, USA). Cells were maintained in DMEM medium with 10% fetal bovine serum, 1% penicillin–streptomycin, and 1% glutamine (PAA, Piscataway, NJ, USA) at 37 °C in a 5% CO2 atmosphere. For monolayer cultures, 2.5 × 105 cells were seeded in 6-well plates. For 3D cultures, 2 × 105 cells suspended in 15 μL of culture medium were dropped onto the upper surface of each scaffold (1 × 5 mm) in 24 multiwell plates. Before cell seeding, scaffolds were dried out through the elimination of PBS using sterile tips. Seeding was reached by soaking the cell suspension in dried scaffolds. After cells were allowed to adhere for 1 hour at 37 °C, the culture medium was gently added in each well. The medium was replaced 24 hours after treatment (washout) and after 72 hours.2.16 Drug treatment
TZ treatment was performed in monolayer cultures or in the 3D scaffolds at the human plasma peak concentration 176 μg mL−1 and 23 μg mL−1. TZ solution was diluted in culture media. PLGA nanoparticles, both naked (vehicle, N-PLGA) and loaded with TZ (PLGA + TZIN, PLGA + TZOUT, PLGA + TZALL), were resuspended in DMEM and sonicated for 5 minutes. In brief, cells were seeded in both 6-well plates and 3D collagen-based scaffolds and were exposed to the drug or vehicle the day after. After 24 hours of treatment, culture medium was refreshed to wash out the unbound drug. The treatment was selected starting from the peak plasma concentration of TZ extrapolated from pharmacokinetic clinical data35 and adjusted taking into consideration the translation application of the study which points toward loco-regional treatment.2.17 MTT assay for cell viability
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was used to assess the efficacy of tested drug formulations. Briefly, after 72 hours or 7 days of drug exposure, controls and drug-treated samples were incubated with 0.5 mg mL−1 of MTT solution (Sigma–Aldrich) in DMEM for 2 hours at 37 °C and subsequently formazan crystals were solubilized in acidic isopropanol. Cell viability was determined by reading the absorbance at 550 nm. Survival percentages were calculated as the average absorbance of treated cells over the absorbance of untreated cells for each time point. The experiments were performed twice in three replicates.2.18 Immunofluorescence and confocal microscopy
The immunofluorescence microscopy experiments were performed as follows. Briefly, BT-474 cells were seeded on 8-chambered coverslips for 72 hours or 7 days. The cells were then fixed with 4% paraformaldehyde, for 15 minutes at RT and then permeabilized with 0.1% of TritonX 100 for 4 minutes at RT. After blocking with 1% bovine serum albumin (BSA) for 30 minutes at RT, the cells were stained with Alexa Fluor™ 488 Phalloidin and DAPI (Thermo Fisher Scientific, Waltham, MA USA) for 1 hour at RT. For collagen scaffold imaging, 3D cultured cells were incubated with the primary antibody (monoclonal antibody rabbit anti-HER2 #2165S, Cell Signaling) diluted in 1% of the blocking buffer. After appropriate washing with PBS, fluorescently labeled secondary antibody (anti-rabbit AlexaFluor 546, Thermo Fisher Scientific, Waltham, MA USA) and DAPI (Thermo Fisher Scientific, Waltham, MA USA) incubation followed for 1 hour at RT. All images were acquired with an N-SIM E laser confocal microscope (Nikon Corporation, Tokyo, Japan) and performed at 20× magnification. The TRITC filter was used for detection of HER2 and coumarin specific staining and FITC filter for phalloidin staining. DAPI specific staining was acquired with a 500 nm filter.2.19 Particle tracking/uptake
Cells cultured in monolayer were treated with coumarin only (C6), PLGA with coumarin (PLGA + C6) or PLGA containing both coumarin and TZ (PLGA + C6 + TZ). After 72 hours or 7 days of treatment, cells were fixed in 4% paraformaldehyde for 10 minutes at RT and permeabilized with 0.1% Triton for 5 minutes at RT. After washing twice with PBS, cells were blocked in 0,1% BSA for 30 minutes at RT stained with Alexa Fluor™ 488 Phalloidin and DAPI (Thermo Fisher Scientific, Waltham, MA USA) for 1 hour at 4 °C. Images were acquired with an N-SIM E laser confocal microscope (Nikon Corporation, Tokyo, Japan) and performed at 20× magnification.2.20 Western blot
Proteins were isolated with lysis Buffer (50 mm Tris/HCl pH 8, 150 mm NaCl, 1% Triton X-100, and 0.1% SDS), supplemented with the Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA USA). BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA USA) was used to determine protein content. An equal amount of protein from each sample (30 μg) was loaded on Bolt™ 10% Bis-Tris Plus Gels (Life Technologies) and transferred to polyvinylidene fluoride membranes through the trans-Blot® Turbo™ blotting system (Bio-Rad). The membranes were blocked in 5% nonfat dry milk or bovine serum albumin in PBS with 0.1% Tween 20 (Sigma-Aldrich) for 1 hour at room temperature. Then, the membranes were incubated overnight with primary antibodies at 4 °C. The following antibodies were used: Caspase-3, Caspase-9, Bax, Bcl-2 (1:1000, Cell Signaling Technology) and β-actin (1:10000; Thermo Fisher Scientific, Waltham, MA USA). After two washes, the membranes were incubated for 1 hour at room temperature with horseradish peroxidase-conjugated secondary antibody. Densitometric analysis of protein bands was performed with Quantity One software version 4.6.9 (BioRad).
2.21 Flow cytometry
For the cellular cycle analysis, cells were collected by trypsinization and pellets were fixed in cold ethanol 70% under agitation and stored at −20 °C, at least for overnight.The cells suspension was stained with a solution containing propidium iodide (PI; 200 μg ml−1), RNAse (20 mg ml−1) and NP40 (0.15%) overnight at 4 °C. The cell suspension was assayed the day after using Attune NxT Flow Cytometer (Invitrogen) and data were analyzed using FlowJo software. Samples were run in duplicate and 10000 events were recorded for each replicate. For HER2 (ErbB-2 PE-Vio 770, Miltenyi Biotec) evaluation, cells were collected at different timepoints and after treatments and stained prior to fixation according to the manufacturing protocol and incubated for 10 minutes in dark condition at 4 °C. The analyses were performed using Attune NxT Flow Cytometer (Invitrogen).
2.22 Statistics
For each experiment, at least three biologically independent replicates were performed. Data were presented as mean ± standard deviation (S.D.), or mean ± standard error of the mean (S.E.M.), as specified. The differences between groups were assessed by two-tailed Student's t-test and accepted as significant when p < 0.05.3. Results
3.1 Synthesis and characterization of PLGA nanoparticles
The synthesis of carriers for the controlled release of TZ was optimized using poly(lactic-co-glycolic acid) (PLGA) through a double water–oil–water (W/O/W) emulsion,29–31 as shown in Fig. 1. PLGA is one of the most successfully used and FDA approved biocompatible polymer for drug delivery application. It is not only compatible with tissues and cells but it is also easily converted to lactic and glycolic acid after hydrolysis and rapidly excreted. It can be easily formulated with a variety of hydrophilic or hydrophobic molecules and imparts some additional properties to drug molecules, such as protecting the drug from degradation effects, controlling the release, and modifying the surface to interact with other biological materials and achieve targeted delivery of nanoparticles. | ||
| Fig. 1 Schematic representation of the double-emulsion method for preparing PLGA nanoparticles loaded with TZ. | ||
In particular, in this study six distinct PLGA nanoparticle formulations are presented: (i) empty or “naked” nanoparticles (N-PLGA); (ii) nanoparticles with TZ encapsulated within the hydrophilic polymer core (PLGA + TZ); (iii) nanoparticles with TZ conjugated to the outer surface, leaving the core empty (PLGA + TZOUT); (iv) nanoparticles with TZ both encapsulated in the core and conjugated on the surface (PLGA + TZALL); (v) nanoparticles loaded with fluorescent coumarin 6 (PLGA + C6); and (vi) nanoparticles co-encapsulating both C6 and TZ within the core (PLGA + C6 + TZ).
Our optimized synthesis allows obtaining 48 ± 2% nanoparticles yield, a homogeneous diameter and distribution (PDI ∼0.1). In particular, the nanoparticles loaded with TZ (PLGA + TZIN) have a size of 159 ± 1.7 nm and a Z potential of −12 ± 0.1 mV, while the empty nanoparticles (N-PLGA) reach a diameter of 164 ± 1.3 nm and a Z potential of −29.4 ± 0.5 mV (Fig. 2A and B).
 | ||
| Fig. 2 Particles characterization by DLS: (A) Z average and (B) Z potential measurements before (full bar) and after the particle surface functionalization (pattern bar); (C) particle size stability in PBS or DMEM buffer within 14 days of incubation at 37 °C. All the measurements were performed five times with 50 μg ml−1 particles (forward scattering 173°). | ||
The smaller particle size of PLGA + TZIN compared to N-PLGA is due to the well-described packing effects caused by electrostatic interactions between the polyanionic charge of PLGA and the positive charge of the TZ. Similarly, the efficacy of TZ loading was confirmed by the significant increase in Z potential (Δ = 17.4 mV) due to the strong electrostatic interaction between the cationic amino acids of TZ and the carboxyl groups of the polymer. Subsequently, the surface of the nanoparticles (for both N-PLGA and PLGA + TZIN) was derivatized with the TZ to actively target the carriers. DLS measurements confirmed the efficiency of TZ immobilization, as shown by the increase in total diameter and Z potential, featuring 176 ± 1 nm and −12 ± 0.1 mV for PLGA-TZOUT and 180 ± 5.1 nm and −4.9 ± 0.2 mV for PLGA-TZALL, respectively. Finally, to assess the uptake of nanoparticles by cells, the labelled PLGA + C6 and PLGA + TZ + C6 nanoparticles have diameters of 188 ± 3.14 nm and 167 ± 1.68 nm, respectively.
The physical stability of nanoparticles was evaluated by measuring particle size, PDI, in PBS and in the media used for the cell viability, cell cytotoxicity, and cell uptake studies at 37 °C in a time point experiment over 14 days. As shown in Fig. 2C, no significant changes in the diameter of the nanoparticles were observed until day 4 in both PBS and DMEM at 37 °C. From day 7 to day 14, a slight fluctuation in the particle size was observed in DMEM. However, the particles do not undergo phenomenon of aggregation which can affect a drug delivery nanosystem performance. Atomic force microscopy (AFM) (Fig. 3A) and transmission electron microscopy (TEM) (Fig. 3B) were performed to morphologically characterize the particles, because these features can have a great influence on the final quality of the carriers. The images acquired by AFM and TEM reveal that the particles have a spherical shape, homogeneous distribution and are free from defects such as cracks and aggregates. The PLGA particles were further analyzed by FTIR (Fig. 3C). The infrared spectrum of all PLGA particles exhibits an intense peak at 1756 cm−1 associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the polymeric ester groups, as well as bands at 3348 cm−1 due to the OH stretching (Fig. 3D above) and at 3000–2944 cm−1 caused by the C–H stretching. In the PLGA + TZIN, PLGA + TZOUT and PLGA + TZALL spectra, the amide I bond region (1700–1600 cm−1, Fig. 3D below) is clearly visible, corresponding to the CO stretching vibrations of the amide group, the N–H bond and the C–N stretching also visible in the free TZ spectrum (Fig. 3C yellow line).
O stretching vibrations of the polymeric ester groups, as well as bands at 3348 cm−1 due to the OH stretching (Fig. 3D above) and at 3000–2944 cm−1 caused by the C–H stretching. In the PLGA + TZIN, PLGA + TZOUT and PLGA + TZALL spectra, the amide I bond region (1700–1600 cm−1, Fig. 3D below) is clearly visible, corresponding to the CO stretching vibrations of the amide group, the N–H bond and the C–N stretching also visible in the free TZ spectrum (Fig. 3C yellow line).
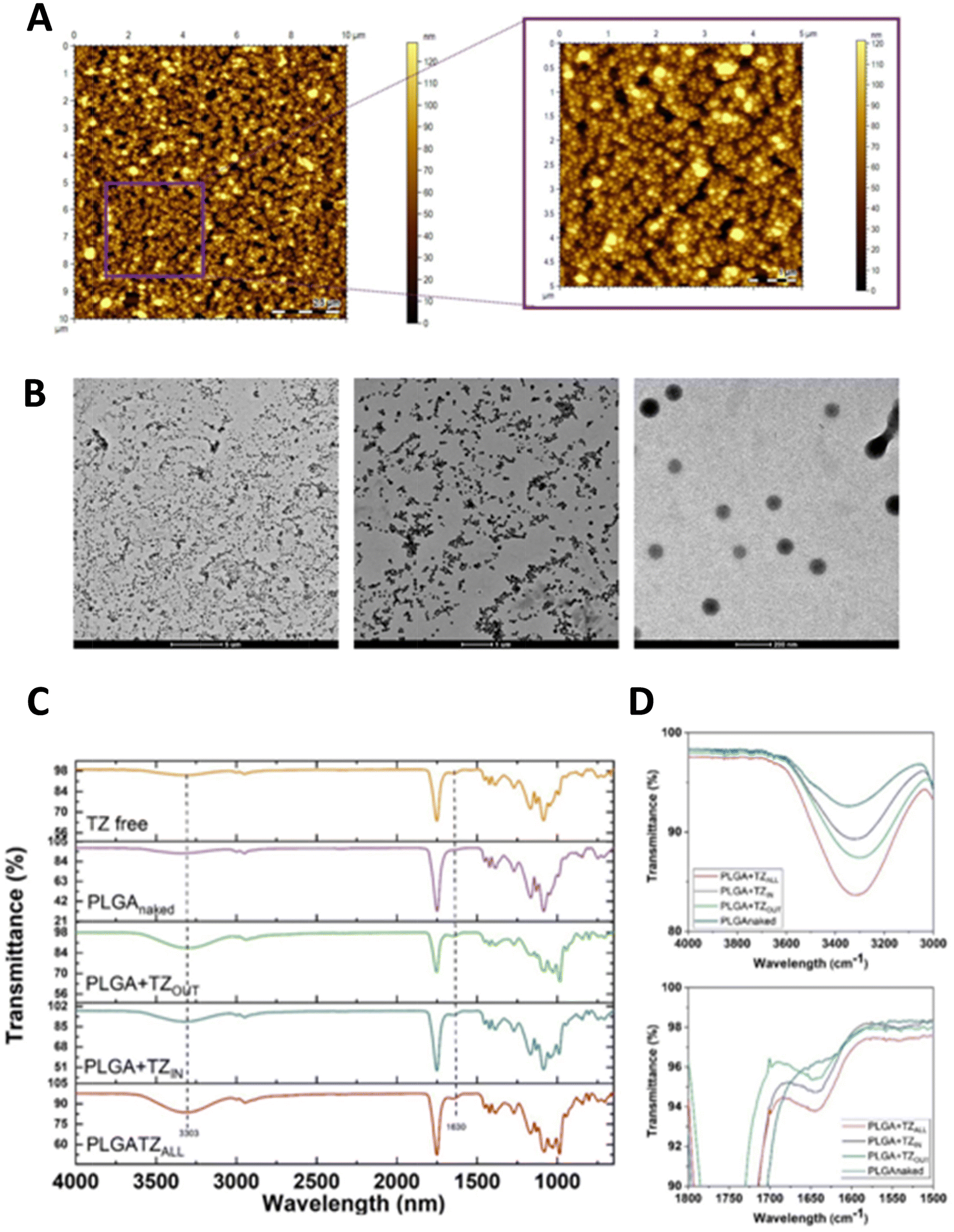 | ||
| Fig. 3 Morphological characterization of PLGA nanoparticles. (A) AFM analyses of PLGA particles deposited on a gold surface (10 μm × 10 μm and zoomed region); (B) transmission electron microscope images of PLGA particles (scale bar 5 μm, 1 μm and 200 nm); (C) FTIR analysis of PLGA particles before (violet and blue lines) and after the particle surface functionalization (green and red lines) compared with free TZ (yellow line); (D) spectrum zoom between 40000–3000 cm−1 (above) and 1800–1500 cm−1 (below). | ||
The TZ encapsulated in the hydrophilic core of the nanoparticles was quantified chromatographically as described in the material section. After optimization of the synthesis procedure, the encapsulation efficiency (EE) and the percentage of loaded drug (%DL) reached 58.7 ± 3.5% and 1.4 ± 0.1%, respectively. Moreover, both the naked and loaded nanoparticles were additionally decorated with the TZ. The amount of TZ covalently bound to the particle surface, quantified by the Bradford assay, was 24 ± 2.7 μg mL−1 for one milligram of nanoparticles.
In this study, a random covalent strategy was used for the immobilization of TZ on the nanoparticle surface. Since the functionality of an antibody is closely related to the integrity of its functional domain, it was essential to investigate whether the coupling procedure did not affect the functional integrity of the bound TZ. To this end, we used surface plasmon resonance (SPR), a powerful label-free technique that allows real-time analysis of molecular interactions. The HER2 protein was immobilized on the chip (as described in material and method section) using the classical amine coupling protocol, at a concentration of 30 μg mL−1 and achieving an immobilization level of 7543 RU (Fig. S1†). Single cycle kinetic (SCK) curves were then generated by injecting increasing concentrations of PLGA + TZOUT ranging from 0.026 to 2.16 μg mL−1 (Fig. S1B†). Data for each nanoparticle concentration were compared with the free TZ and the naked preparation under identical conditions. The PLGA + TZOUT successfully interacted with HER-2 and resulted in an increase in ΔRU at all injected concentrations, allowing to generate dose–response curves. This result demonstrate that the functionalization procedure adopted for the TZ surface immobilization does not affect its recognition properties and thus is suitable for the particle surface decoration. However, at all concentrations tested, PLGA + TZOUT resulted in lower ΔRU signals compared with free TZ (Fig. S1A†). This predictable result was caused by the nanoparticles’ steric hindrance, which made the HER2 sites spatially less inaccessible to the antibody compared with its free form. The naked PLGA nanoparticles did not elicit significant sensing responses and produced extremely low ΔRU signals, likely due to nonspecific adsorption (Fig. S1B†).
Finally, the release kinetics of TZ was evaluated by the prepared formulations. PLGA is slowly hydrolyzed in lactic acid and glycolic acid in aqueous buffers, resulting in the release of the loaded drug in a time window of several weeks to months. In our study, the release of TZ in both PLGA + TZIN and PLGA + TZOUT was observed for up to 14 days in PBS buffer at 37 °C. As shown in Fig. S2A,† approximately 30% of the encapsulated TZ is released from PLGA + TZIN in the first 30 minutes. Thereafter, a slow and sustained release is measured, reaching 52 ± 4.6% after two weeks of incubation. Similarly, the TZ bound to the surface of the particle (PLGA + TZOUT) was released from the medium as a result of polymer hydrolysis (Fig. S2B†). Specifically, 25.5% of TZ was recovered into the medium after 1 hour of incubation and reached 69.8 ± 0.1% after 14 days of incubation. Drug release experiments were conducted only at pH 7.4, as previous studies indicated that while alkaline and strongly acidic conditions accelerate PLGA degradation, the difference between slightly acidic and neutral pH is minimal due to autocatalysis by the polymer's carboxylic end groups.36–40 Since the pH of the microenvironment of breast cancer tumors is typically between 6.97 and 8, we assume that this slight deviation will not significantly affect the release of the drug.
3.2 Biological evaluation of PLGA nanoparticles efficacy
Further, we focused on studying the biological activity of PLGA nanoparticles prepared with TZ. For this purpose, we used BT-474 cell line, a breast cancer cell line derived from solid invasive ductal carcinoma expressing HER2. We investigated the in vitro efficacy of both free TZ and in various PLGA preparations, after 72 hours or 7 days in BT-474 cells cultured either in a standard monolayer culture or in a 3D culture with collagen-based scaffolds.3.3 Experimental setting based on the cytotoxic effect of free TZ in the 2D and 3D culture model
To determine the optimal experimental design, we first tested different concentrations of the drug (free TZ) in both 2D standard monolayer and 3D cultures. We found that plasma peak concentration of free TZ (176 μg μL−1) resulted in an inhibition of cell proliferation of about 40% in 2D and 30% in 3D cultures after 72 hours, while the inhibition of cell proliferation reached about 75% in 2D and 55% in 3D cultures after 7 days of treatment (Fig. S3†). This trend is consistent with our previously published data showing increased drug resistance of cells cultured in 3D compared to 2D cultures, suggesting that our 3D culture model is a more informative tool for drug screening compared to standard monolayer cultures. Moreover, these results are also consistent with the duplication time of the BT-474 cell line, which is approximately 90 hours in standard monolayer cultures. However, considering the translational application of our study, which consists in locoregional therapy, and in order to determine a concentration compatible with the efficiency of encapsulation by PLGA nanoparticles, we also tested lower concentrations of the drug and selected 23 μg μL−1 and 11.5 μg μL−1 in agreement with previous studies. As shown in Fig. S3,† there was no striking difference between the two drug concentrations tested in terms of cell survival, except for the 2D culture at 7 days. Indeed, cancer cell proliferation at 72 hours for both concentrations was inhibited at approximately 35% in 2D and 30% in 3D cultures. After 7 days, the inhibition was about 40% for the lowest concentration tested and about 60% for the highest in 2D culture, while in 3D culture it was about 60% for both concentrations tested. Considering that the 3D culture model is closest to the in vivo conditions, these data suggest that the lower drug dose is sufficient to effectively inhibit cell proliferation.After these initial optimization steps, we selected 11.5 μg mL−1 as the optimal effective concentration for treatment-which corresponds to 23 μg mL−1 of total drug, assuming that the nanoparticles release 50% of the encapsulated drug (see Fig. S2†). Furthermore, according to these preliminary data, we identified 7 days as the most representative time point for subsequent analyses, also considering the non-linear half-life of TZ, which can range from a few days to several weeks.35
3.4 PLGA nanoparticle internalization
In order to assess the internalization of PLGA nanoparticles in the selected cells we performed particle tracking experiments with confocal microscopy. We took advantage of coumarin (C6), which is a natural fluorescent dye which emits in the red spectrum, loaded both on empty PLGA particles (PLGA + C6) and on PLGA + TZ particles (PLGA + C6 + TZ). As shown in Fig. 4 and 5, PLGA particles are effectively internalized by BT-474 cells as suggested by co-localization of C6 (TRITC, red) and phalloidin (green), particularly at 7 days (Fig. 5), while C6 alone (C6 only) showed a more diffuse and non-specific pattern. Moreover, we can also observe an altered morphology of cells treated with PLGA + C6 + TZ in comparison to others, indicating the retained efficacy of TZ and, likely, indirectly suggesting its internalization along C6 dye. These results are in line with previously published works in which the TZ decoration of TPGS-g-chitosan-based41 nanoparticles and magnetic core–shell-based nanoparticles42 increased their tumor targeting ability. Moreover, PLGA-nanoparticles delivering TZ with an increased antitumor efficacy compared to the anti-HER2 drug have been already described.23,29 In this regard, to the best of our knowledge, this is the first time in which it is described a PLGA-nanoparticle loaded with an anti – HER2 monoclonal antibody and decorated with the same molecule. The improved antitumor activity observed with our nanosystem compared to the one with only the decoration underlines its potential role as a promising cutting edge technology.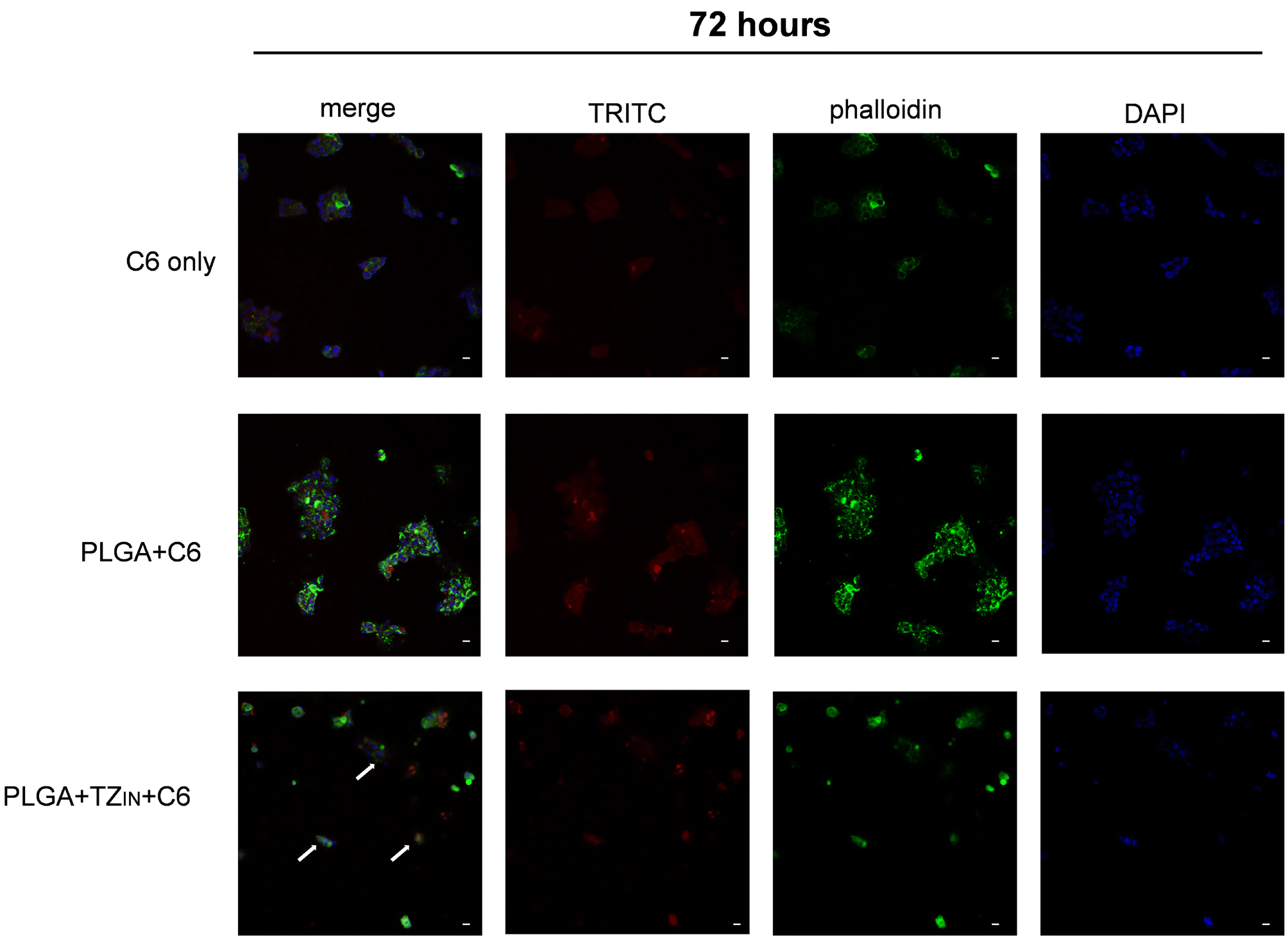 | ||
| Fig. 4 Internalization of PLGA particles in BT-474 cell line after 72 hours of treatment in 2D culture. Red signal coumarin/PLGA particles, green signal phalloidin/cytoskeleton, blue signal DAPI/nuclei. 20× magnification, scale bar 20 μm. | ||
 | ||
| Fig. 5 Internalization of PLGA particles in BT-474 cell line after 7 days of treatment in 2D culture. Red signal coumarin/PLGA particles, green signal phalloidin/cytoskeleton, blue signal DAPI/nuclei. 20× magnification, scale bar 20 μm. | ||
3.5 Cytotoxic effect of PLGA + TZ nanoparticles in 2D and 3D culture model
We investigated the cytotoxic effect of PLGA nanoparticles measuring the inhibition of cancer cell proliferation in all the developed formulations. First, N-PLGA showed no significant toxicity in either 2D or 3D cultures at either time point tested (Fig. S4†), confirming the safety of this formulation for drug treatment.We then explored the efficacy of different forms of TZ delivery with PLGA particles. In addition to PLGA particles loaded with TZ (PLGA + TZIN), we also tested PLGA particles decorated with TZ on the external surface (PLGA + TZOUT) and PLGA particles having TZ both inside and outside (PLGA + TZALL). As shown in Fig. 6, all PLGA + TZ nanoformulations showed higher efficacy compared with free TZ at both time points tested, suggesting that delivery of TZ through PLGA based nanoparticles is more effective. This result is likely due to less degradation of the drug and a consequent increase in the amount of TZ that can reach HER2 target receptors. In particular, free TZ inhibited cancer cell proliferation of about 60%, while, PLGA + TZOUT of about 70%. Of note, PLGA + TZALL showed the highest efficacy in inhibiting cell proliferation after both 72 hours and 7 days of treatment in 2D cultures, achieving about 80% inhibition, suggesting a potential synergistic effect of dual functionalization, with TZ acting as both a targeting factor and a therapeutic molecule. In contrast, in the 3D culture model, all PLGA + TZ nanoformulations showed similar efficacy in inhibiting cell proliferation, with an average inhibition rate of 40–50% at 72 hours and 70% after 7 days post-treatment, similar to free TZ.
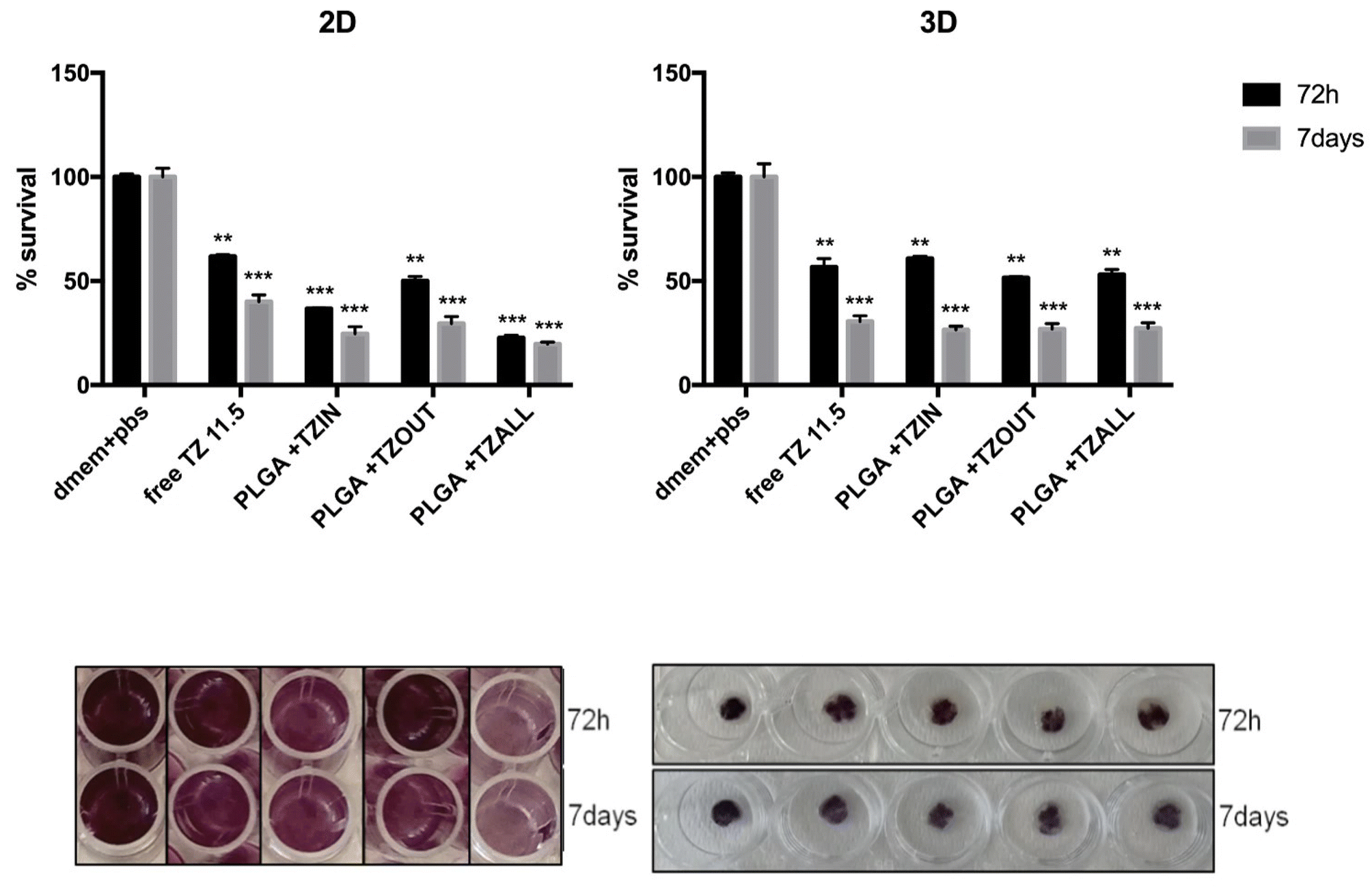 | ||
| Fig. 6 Comparison between free TZ and different PLGA + TZ nanoparticles in inhibiting cell proliferation in BT-474 cell line after 72 hours or 7 days of treatment in 2D and 3D culture models. **p < 0.01, ***p < 0.001 compared to untreated controls. | ||
These data are in line with our previous results showing more aggressive and chemoresistance-prone behavior in 3D-grown cells compared with standard monolayer cultures. This is likely due to several events, such as the formation of a hypoxic core, increased cell plasticity leading to the presence of different cell phenotypes, decreased drug penetration, and a less homogeneous distribution of cells in the matrix, all of which typically resemble tumor conditions.
3.6 Effect of PLGA + TZ nanoparticles on HER2 expression
In order to highlight the mechanism of action of free TZ or loaded PLGA nanoparticles, we focused our attention on the changes in HER2 expression and localization on breast cancer cells due to the drug. In particular, as HER2 is the target of TZ, it is relevant to investigate if the treatment determines the block of HER2 pathway also by a decrease of its expression on cancer cell surface.To corroborate our hypothesis we next analyzed the expression of HER2 receptor and LAMP1, a lysosome marker, in BT-474 cell line in 2Dtreated with free TZ or PLGA + TZ after 72 hours or 7 days by flow cytometry. With respect to LAMP1, we did not detect any signal, suggesting that the lysosomal pathway is not involved in TZ degradation. HER2 positivity was lower (green) in all treated samples compared with controls, both 72 hours and 7 days after treatment (Fig. S5,† and Table 1). Overall, we observed a 35–45% reduction in HER2 positivity at 72 hours and a 15–20% reduction at 7 days in all treated samples. These data are in part corroborated by previous observations showing a variable colocalization of TZ with LAMP1. In particular, in low-expressing HER2 breast cancer cells, TZ seems to be localized in the lumen of transferrin receptor-positive endosomes which is known to enter in the lysosomal pathway. On the other hand, in high-expressing HER2 breast cancer cells, TZ seems just sequestered in an area known as limiting membrane of transferrin receptor-positive endosomes.14
| HER2% | 72 hours | 7 days |
|---|---|---|
| CTRL | 93.02 | 87.97 |
| free TZ | 42.58 | 60.64 |
| PLGA + TZIN | 45.48 | 64.68 |
| PLGA + TZOUT | 29.49 | 69.86 |
| PLGA + TZALL | 28.44 | 64.96 |
To characterize the effect of TZ on BT-474 cell line3D culture, either free or delivered by PLGA nanoparticles, we examined HER2 expression after 72 hours or 7 days of treatment by confocal microscopy. It was found that treatment with TZ resulted in a decrease in HER2 receptor (red signal) for each formulation and at each time point tested (Fig. 7), consistent with the mechanism of action of the TZ and the previously reported data. Interestingly, the decrease is more pronounced with PLGA + TZIN and PLGA + TZALL than with free TZ, supporting our hypothesis that these nanoformulations are the most effective.
 | ||
| Fig. 7 Confocal microscopy imaging of HER2 expression in BT-474 cell line after 72 hours or 7 days of treatment with free TZ or PLGA + TZ nanoparticles in 3D collagen-based cultures. HER2 (red), DAPI (blue, nuclei). 20× magnification. | ||
3.7 Apoptosis and cell cycle progression
We analyzed the expression of key proteins involved in the apoptosis pathway either 72 hours or 7 days after treatment to evaluate the involvement of this mechanism in the cell death process after TZ treatment. As shown in Fig. 8, an increase in the expression levels of the pro-apoptotic proteins such as the initiator caspase Caspase-9 and the effector caspase Caspase-3 was observed in all treated samples compared with the control after 72 hours. Notably, the increase is more pronounced in the PLGA + TZALL sample, suggesting that this formulation is potentially most effective in targeting the HER2 receptor and preserving TZ integrity. At the same time, we observed a decrease in the anti-apoptotic protein Bcl-2, indicating the involvement of the intrinsic apoptotic pathway in the cell death triggered by TZ treatment. On the other hand, we did not detect any significant change in the expression of these proteins after 7 days of treatment, suggesting that apoptotic induction was already concluded. | ||
| Fig. 8 Expression levels of proteins involved in apoptotic pathway after treatment of BT-474 cell line with free TZ or with different PLGA + TZ nanoparticles after 72 hours or 7 days in 2D. | ||
We also investigated the role of free TZ and various PLGA + TZ particles in cell cycle progression. As shown in Fig. 9 and Table 2, we observed an increase in the number of cells in G0/G1 phase at both 72 hours and 7 days after treatment and also a decrease of cells in S phase after 7 days compared with controls, indicating an increase in resting cells and a decrease in actively replicating cells. This is in line with cell viability experiments and with the mechanism of action of TZ, which is known to block the cell cycle by inhibiting MAPK/PI3K signaling as well as by directly downregulating downstream cyclins. In addition, TZ has been previously reported in the literature to induce a reduction in proliferating cells as well as a prolongation of G1 phase and a great increase of quiescent BT-474 cells.43
 | ||
| Fig. 9 Cell cycle analysis on BT-474 cell line treated with free TZ or with PLGA + TZ nanoparticles after 72 hours or 7 days. | ||
| 72 hours | G0/G1 | S | G2/M |
|---|---|---|---|
| CTRL | 68.79% | 7.76% | 23.44% |
| Free TZ | 85.87% | 8.75% | 5.38% |
| PLGA + TZIN | 86.04% | 9.66% | 4.30% |
| PLGA + TZOUT | 87.86% | 8.35% | 3.79% |
| PLGA + TZALL | 86.23% | 9.36% | 4.40% |
| 7 days | G0/G1 | S | G2/M |
|---|---|---|---|
| CTRL | 69.66% | 23.06% | 7.28% |
| Free TZ | 87.18% | 7.97% | 4.85,% |
| PLGA + TZIN | 88.39% | 7.82% | 3.80% |
| PLGA + TZOUT | 86.65% | 10.07% | 3.29% |
| PLGA + TZALL | 85.45% | 11.07% | 3.48% |
3.8 Proof of concept: “light triggered release of nanocarriers by core-offset optical fibers (coOFs) to control the spatial and temporal delivery of drugs”
The use of light as a trigger stimulus for drug release is a promising approach with several advantages, such as precise control of parameters (i.e., wavelength and power density), which can be tailored to specific applications. However, there are still challenges and limitations associated with the use of light for drug delivery, especially in the clinical setting. In this context, optical fibers (OFs) can be a valuable tool for the development of a smart device for local drug delivery, thanks to their size, biocompatibility, and ability to be easily integrated into needles, catheters, and nanoendoscopes.44–48It's interesting to note that although OFs have been used as intrinsically light-coupled platforms for light-mediated drug delivery, they are subject to limitations, particularly in terms of payload capacity. To address this issue, methods are needed to immobilize drug carriers on the cylindrical surface of optical fibers, which provide a larger surface area for drug loading. This approach requires special light coupling mechanisms to efficiently guide light from the fiber core to the cladding and activate carrier release. To this end, various coupling methods have been developed, including fiber Bragg gratings (FBGs) with tapers, long period gratings (LPGs), tilted FBGs, etc.49–52
Our previous work has explored side-emitting fibers (seOFs) as a coupling method to efficiently scatter light to the outer surface of the optical fiber and activate PLGA particles through a suitable photocleavable linker.26 Considering the issues with the availability and high cost of seOF, in this work, we propose a new optical fiber assisted architecture featuring the integration of core-offset optical fibers (coOFs) to enhance the light scattering at the fiber–fiber interface, thereby promoting the activation of a photocleavable component to release PLGA nanoparticles (Fig. 10). The proposed device integrates two types of optical fibers: a multimode fiber used as launch fiber and a single-mode fiber connected by a core offset. This architecture allows for spread light in the fiber cladding and achieve the correct light power density to promote the nanoparticles release through the photo-cleaving activation. A core offset splicing technique was thus selected to connect the multimode fiber and the single mode fiber, in which the cores of the two fibers are intentionally misaligned of 10 μm.
 | ||
| Fig. 10 Light triggered release of nanoparticles by optical fiber: (A) schematic of the optical fiber probe; (B) image of the illuminated probe immersed in a quartz cuvette and nanoparticles cumulative release graph measured for experiment in batch solution; (C) image of the illuminated placed in the microfluidic chip and nanoparticles cumulative release graph measured for experiment in chip. | ||
Drug carriers are bonded to the fiber surface using amine-reactive crosslinker chemistry. The carriers are attached to the fiber via a spacer arm containing a photocleavable component. Upon irradiation with low intensity UV light (wavelength 365 nm, 1–5 mW cm−2), the photocleavable component is activated, resulting in efficient release of the drug carriers. This method allows precise control of drug carrier release with low-intensity UV light, offering the advantage of using readily available optical fiber types and a core offset splicing technique that can provide a more accessible, flexible and cost-effective alternative to seOFs for the same purpose.
The coOFs probes used in the experiments are fabricated according to the technique described in the Methods section with a scattered single-mode fiber element length of 1 cm and an offset between the two fiber types of 10 μm, resulting in a scattering efficiency (and power density) of 24.9 ± 3.3% (4.64 ± 0.7 mW cm−2). The method was optimized resulting in highly reproducible of all prepared samples.
The amount of nanoparticles released after UV activation was calculated based on the fluorescence emission of coumarin 6 encapsulated in the nanoparticles. As shown in Fig. 10, the amount of nanoparticles released can be precisely controlled (magenta square) by tuning the illumination time. Thus, after 15 seconds of light activation, 40.28 ± 5.27 ng mL−1 of particles were released in the medium, reaching 208.69 ± 50.03 ng mL−1 after 60 s and 658.82 ± 50.03 ng mL−1 after 330 s.
The release triggered by the coOFs was monitored for 40 min, and the maximum released particle concentration was 1055.39 ng ml−1. To confirm that the release was triggered by the UV light, optical fibers functionalized with the nanoparticles were immersed in the buffer and fluorescence was monitored at the same time points without light activation. The black circle in Fig. 10 shows that without UV light activation, 18.44 ± 0.93 ng mL−1 and 36.60 ± 6.89 ng mL−1 of particles were found in the medium after 60 s and 330 s, respectively (10% and 5.6%, respectively, compared to the positive control), while 66.66 ± 10.24 ng mL−1 were measured after 40 minutes of immersion (6.3% compared to the light-activated control). Subsequently, the functionalized optical fiber was integrated in a microfluidic device and the release of nanoparticles was monitored as in the batch experiments. For the optofluidic system, the experiment was conducted for a maximum of 30 minutes, resulting in nanoparticle release of 700 ng mL−1 (data in magenta in Fig. 10). As a control, the same steps were performed for a not -UV activated coOF. As shown in Fig. 10, the amount of nanoparticles is 100 ng mL−1 (14% compared to the positive control). The difference in the amount of nanoparticles released between batch and microfluidic methods could be due to variations in efficiency and power density between the two coOFs used in the experiments. Additionally, the adsorption of nanoparticles onto the surface of the microfluidic chip may further reduce the amount of nanoparticles released in the microfluidic setup.
4. Discussion
In the landscape of solid tumors, breast cancer represents the first major cause of death worldwide, with more than 7 million women affected by this disease.53 Although the use of monoclonal antibody TZ has revolutionized the treatment of HER2 + patients, the ultimate goal of eradicating residual disease remains a challenge. The advent of immune checkpoint inhibitors in combination with standard chemotherapy has altered the natural history of this disease, as demonstrated by the Cleopatra trial.54,55 In this context, several clinical trials are currently underway to investigate the role of immune checkpoint inhibitors and HER-targeted treatment combinations in both early or advanced and metastatic breast cancer.56 Despite these advances, disease control in breast cancer has not yet been fully achieved. Moreover, patients treated with anti-HER2 therapies often rapidly develop resistance.57 In this regard, several potential resistance mechanisms involving HER2 signaling networks have been investigated. One of them is related to the masking of TZ binding sites. Indeed, approximately 30% of HER2-positive tumors express a number of truncated carboxy-terminal forms of HER2 – resulting from proteolytic shedding or impaired translation – that lack the TZ-binding domain and cannot be inhibited by TZ anymore.16 Another mechanism relies on the overexpression of other growth factor receptors such as c-Met and IGF-1R, which may have similar effects on cell proliferation even when HER2 signaling is inhibited by TZ. Finally, intracellular changes in HER2 signaling due to newly emerged activating mutations may lead to PTEN deficiency and/or constitutive PI3K/Akt activation, rendering these cells capable of triggering other downstream signaling pathways even when HER2 is blocked by TZ or PTEN is active.17Several mechanisms have been proposed for TZ action, ranging from blockade of MAPK/PI3K signaling – leading to an increase in cell cycle arrest and suppression of cell growth and proliferation – to direct downregulation of downstream mediators of cell cycle progression.11 These effects are achieved through selective binding to the extracellular domain of HER2 receptor, leading to the inhibition of HER2–HER3 dimerization.12 In this way, TZ can also induce immune-mediated responses by activating immune effector cells, resulting in antibody-dependent cellular cytotoxicity (ADCC)13 and promote HER2 internalization and degradation. In addition, TZ is internalized via endocytosis in recycling vesicles rather than in degrading ones, therefore it is recycled to the plasma membrane in a short period of time.
A promising strategy to overcome the common drawbacks of TZ-based therapy relies on the development of targeted drug delivery systems (t-DDS). Among the plethora of t-DDS, PLGA nanoparticles have attracted considerable attention due to their attractive properties of enhancing cargo stability, promoting transport across the cell membrane and increasing safety and efficacy.19–21,58
Indeed, encapsulation in nanoparticles not only improves the solubility and stability of drugs but also allows to protect drugs from premature degradation, increasing the amount of active drug that reaches the target site, therefore allowing to reduce concentration and systemic side effects.
Several studies have limited the use of TZ for guided treatment approaches,42,59–62 in fact, there are only two examples of TZ-encapsulated nanoparticles in the literature.23,29 Colzani et al. have shown that double emulsion (w/o/w) is a good method to preserve the native structure of TZ antibody, but the obtained particles are characterized by limited encapsulation efficiency and lack of targeted delivery.
In our previous work we have already proved that encapsulation allows to remarkably improve the TZ cytotoxic effect on the SKBR3 cell line.29 This approach prevents the particles from being entrapped by the reticuloendothelial systems, but also supports their accumulation in tumor tissues through the EPR effect (enhanced permeability and retention), thereby improving delivery efficiency and therapeutic efficacy and minimizing toxic effects.63,64 Nevertheless, in that study a notable limitation in real clinical application persisted due to the absence of an active targeting strategy to specifically deliver cytotoxic drugs to HER2-expressing tumors. Thus, in the current work, we report the first example of a synergistic approach, where the TZ is both encapsulated in the core and immobilized on the surface of PLGA nanoparticles and their effect was tested on BT-474 2D and 3D culture models. The proposed strategy allows: (i) stabilizing and protecting the encapsulated TZ from the external environment, and (ii) achieving a “site-specific” nanoparticle delivery. Regarding the first aspect, our optimized PLGA formulation shows improved EE % (+21%) and DL % (+0.5%) compared to those previously reported by other groups. Usually the second feature is achieved by decorating the surface of the nanoparticles with an appropriate amount of monoclonal antibody by chemical functionalization or surface adsorption.23 A common chemical strategy to covalently immobilize the monoclonal antibody on the nanoparticle surface (i.e., functionalized liposomes) relies on amide65 or disulfide66 bonds to stabilize the targeted nanoparticles in blood until they are cleaved in tumor cells.67–69 In our study, PLGA + TZALL are obtained by non-oriented chemical conjugation of TZ on the nanoparticle surface through the formation of an amide bond. The efficiency of conjugation, i.e., the amount of bound TZ per mg of nanoparticle, is indirectly determined by measuring the amount of unbound TZ (μg mg−1 nanoparticle). The efficiency of functionalization reported in other studies on targeted delivery systems70–77 shows that the amount of TZ falls in a wide range (ng–μg of TZ), indicating that the targeting efficiency is not strictly correlated with the TZ density of functionalization. This strongly suggests that various parameters such as the type of nanoparticles, the response of the biological system, the chemistry of antibody conjugation, and the preservation of its recognition properties should be considered. Indeed, the chemical bond between the antibody and the nanoparticle is one of the crucial factors for maintaining the activity of the antibody and the integrity of the nanodelivery system in the extracellular environment. The effectiveness of the functionalization process must be evaluated in terms of maintaining the structural and functional properties of the TZ after conjugation. In the panorama of TZ-decorated nanocarriers for targeted therapy, the structural integrity is usually verified by SDS page or circular dichroism analysis, which indirectly confirms the integrity of the antibodies.78,79 In the present work, the biological function of TZ, when immobilized on the surface of nanoparticles, was directly assessed by SPR experiments through the evaluation of its recognition ability against its cognate, HER2. Surface plasmon resonance revealed a specific interaction of PLGA + TZALL with the immobilized HER2 protein compared with uncoated nanoparticles, as shown by the binding curves (see Fig. S1†). In a real clinical scenario, after entering the human body, nanoparticles encounter various environments that can lead to changes in their surface composition. This process results in the formation of a layer of adsorbed proteins, known as the protein corona. This protein corona formation is a critical consideration as it can influence the cells internalization process and the drug release mechanisms of nanoparticles.80 Thus, to conduct a more comprehensive stability characterization and investigate whether a protein corona was forming on the surfaces of our nanoparticles (PLGA + TZALL) we decided to expose them to a culture medium containing proteins. This exposure did not reveal any significant increase in the mean particle size of the NPs. The lack of significant size change suggests that (i) the surfaces of the NPs does not suffer from substantial adsorption of biomolecules contained in the biological media; and (ii) the TZ anchored on the surface, which is crucial for specific targeting, has not been displaced by other proteins like albumin. All these evidences endorse the integrity and efficacy of our nanoparticles formulation.
Finally, the PLGA + TZ preparations were evaluated for their internalization capability, targeting efficiency, cell cytotoxicity, and their effect on cell cycle progression and apoptosis.
To partially recapitulate the tumor niche microenvironment in terms of mechanical stimuli and biological properties, we combined our 3D collagen scaffold with a HER2-positive cell line. Using this integrated approach, we observed 40% inhibition of cell proliferation at 72 hours and 65% inhibition at 7 days in 3D cultures treated with PLGA nanoparticles encapsulating TZ (PLGA + TZIN), demonstrating that this nanoformulation is able to effectively maintain the integrity and efficacy of TZ. Moreover, we demonstrated that the decoration of PLGA nanoparticles with TZ on the surface (PLGA + TZALL) led to a further increase in TZ efficacy, with a reduction of cell proliferation by 80% in 2D and 75% in 3D 7 days after treatment. In addition, cell cycle analysis showed a significant increase in quiescent cells and a decrease in actively replicating cells in the treated samples, consistent with the results of the viability experiments and with the known mechanism of action of TZ, thus confirming the maintenance of the drug's activity. Overall, these data suggest that our dual functionalization allows a more efficient delivery of TZ to its target, a hypothesis that was also confirmed by confocal microscopy and western blot analyzes, which showed that treatment with PLGA + TZALL resulted in a strong decrease in HER2 expression and an increase in Caspase-3 and Caspase-9 expression. In addition, cytometric analysis confirmed the reduction in HER2 signaling after TZ treatment, a reduction that was particularly evident in PLGA + TZALL-treated cells at 72 hours. These results are corroborated by several lines of evidence in the literature that TZ is able to reduce downstream HER2 signaling by internalizing and/or degrading the receptor.81 In addition, nanoparticles have been reported to enhance endocytosis, compensate for poor water solubility, and prolong the residence time of therapeutic agents in blood, thereby improving intracellular accumulation of encapsulated TZ in tumors.82 However, we were unable to detect a signal of LAMP1 labeling in cytometry, suggesting that degradation pathways other than lysosomal-mediated may be a possibility.
Overall, these results confirm that (i) the encapsulation strategy greatly enhances the cytotoxic effect of TZ by protecting the antibody from the external environment, and (ii) the TZ surface decoration of nanocarriers further increases their efficacy. Indeed, nanoscale drug delivery systems could take advantage of enhanced permeability and retention effect and provide a passive strategy to improve the therapeutic potential of various drugs and bioactive molecules.83 Further opportunities to improve the therapeutic efficacy of monoclonal antibodies as targeting ligands relies on the use Fab fragments, or Aptamers, that due to their properties (smaller size, easier modification, immobilization properties, and better stability) can more easily target tumor cells (on-targets) while avoiding healthy cells (off-targets).84,85
Based on the great potential of PLGA + TZ carriers, for in vivo applications, studies focusing on their combination with engineered devices are needed to improve tumor targeting while minimizing chemotherapy-induced systemic toxicity. For the above we have developed a fiber-optic device integrated with PLGA nanoparticles chemically engineered to be responsive to external stimuli. Specifically, a new coOF-based system was developed with the primary aim of demonstrating the feasibility of controlling the spatial and temporal release of drug carriers using a miniaturized light-driven device. The release of nanocarriers is controlled by modifying the surface of coOFs through the conjugation of a heterobifunctional UV-cleavable linker. When the UV source is activated, the light guided by the optical fiber is scattered by the cladding of the coOFs, triggering the cleavage of the linker and triggering the release of the nanoparticles. The results show a slight decrease in the scattering efficiency of the coOF probes when compared to the seOFs, developed in our previous study.26 Despite this modest efficiency reduction, the actual optical probe involves low cost and commercially available optical fibers when compared with the seOFs, featuring higher costs and limited availability.
Furthermore, the coOF has been coupled into a microfluidic device to demonstrate the feasibility to build up optimized light triggered loco-regional drug delivery nanosystems, through the suitable integration of engineered optical fiber probes in the working channels of minimally invasive clinical tools, as needles catheters and nano-endoscopes enabling the precise control of delivered nanocarriers to the desired location in the human body. The integrated microfluidic coOF system achieves the same efficiency in drug particle release observed using the conventional batch method, demonstrating the effectiveness of the proposed approach. Moreover, the size of the optical fibers involved in this study, also, suggests the potential use of multiple optical fiber probes to increase the amount of the released drug or to delivery multi-drug assisted therapy for personalised medicine.
Nevertheless, in order to move from the conceptualization of the proposed device to its practical application in a clinical setting, further tests are needed.
One limitation of our study resides in the lack of in vivo activity evaluation of our system. In this regard, further studies will be needed in order to better validate our data. These assessments will encompass a progression from our initial 3D model to the implementation of in vivo models. This iterative process will be essential to ensure the device's effectiveness and operability, when used in real-world clinical setting.
5. Conclusions
In this study, we investigated, for the first time, a novel drug delivery system based on PLGA nanoparticles in which TZ is not only encapsulated into the core but also immobilized on the outer surface of the carrier. Moreover, we also propose a platform based on optical fiber for the locoregional precise delivery of these nanocarriers.This approach boosts the efficacy of the therapy since the drug is protected from external stimuli and actively targeted to the tumor cells. The nanocarrier performance was evaluated in a 3D collagen-based culture model, designed to mimic cellular interactions within the ECM microenvironment. Furthermore, as proof-of-concept, we have demonstrated a light-triggered fiber optic platform for localized drug delivery, using particles embedded within specially engineered optical fibers (coOFs). This platform allows for seamless integration into needles and nanoendoscopes, achieving precise, light-activated drug release at targeted sites.
Future in vivo experiments will be essential to assess important pharmacological factors such as pharmacokinetics, systemic toxicity, and biodistribution. In this regard, murine models represent the gold standard for the preclinical assessment of antitumor therapies. Indeed, this model is able to recapitulate both the tumor microenvironment and tridimensional structural, as well as the crosstalk among different cell populations. However, recently an alternative valid approach is emerging. Zebrafish model indeed allows to overcome both the complexity to culture ex vivo samples and the low engraftment rates obtained with PDX murine models.86 Another advantage of this model is represented by the low number of cells necessary for each injection (typically 100–500 cells per embryo), or the small tissue fragments necessary for each single transplantation.87
Considering the promising safety profile of FDA-approved PLGA nanoparticles as suitable nanocarriers for human applications, it could be conceivable to propose the clinical use of TZ-loaded PLGA nanoparticles for innovative locoregional therapies on local and metastatic relapse of breast cancer. Such innovations could transform clinical practice by minimizing side effects and boosting therapeutic efficacy compared to standard drug administration methods.
Abbreviations
| AFM | Atomic force microscopy |
| C6 | Coumarin |
| CLSM | Confocal laser scan microscopy |
| DLS | Dynamic light scattering |
| DMEM | Dulbecco's Modified Eagle Medium |
| ECM | Extracellular matrix |
| EE | Encapsulation efficacy |
| FTIR | Fourier transform infrared spectroscopy |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate buffer saline |
| PFA | Paraformaldehyde |
| PLGA | Poly(lactic-co-glycolic acid) |
| PVA | Polyvinyl alcohol |
| RP-HPLC | Reversed-phase high performance liquid chromatography |
| SPR | Surface plasmon resonance |
| TEM | Transmission electron microscopy |
| TZ | Trastuzumab |
Author contributions
SV: conceptualization, methodology, formal analysis, writing – original draft preparation. TMC: conceptualization, methodology, formal analysis, writing – original draft preparation. AMC: data curation, experiment investigation, writing—Original draft preparation. ADV: conceptualization, supervision, writing—review & editing. AA: conceptualization, supervision, writing—review & editing. CC: data curation, experiment investigation. CM: data curation, experiment investigation, writing – original draft preparation. SP: experiment investigation, prepared figures, writing. CL: conceptualization, supervision, writing—review & editing. GC: experiment investigation, prepared figures, writing. AM: conceptualization, writing—original draft preparation. CS: data curation, experiment investigation. GM: data curation, experiment investigation. TI: project administration, supervision, funding acquisition, writing – review & editing. LM: project administration, supervision, funding acquisition, writing – review & editing. AC: project administration, supervision, funding acquisition, writing – review & editing. All authors read and approved the final version of the manuscript.Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request. Supplementary Information available at https://www.researchsquare.com/article/rs-3891320/v1.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by Italian Ministry of University and Research (PON- NeON ARS01_00769).References
- N. Harbeck and M. Gnant, Breast cancer, Lancet, 2017, 389, 1134–1150, DOI:10.1016/S0140-6736(16)31891-8.
- C. M. Perou, T. Sørlie and M. B. Eisen, et al., Molecular portraits of human breast tumours, Nature, 2000, 406, 747–752, DOI:10.1038/35021093.
- T. Sørlie, C. M. Perou and R. Tibshirani, et al., Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10869–10874, DOI:10.1073/pnas.191367098.
- G. P. Dowling, S. Keelan and S. Toomey, et al., Review of the status of neoadjuvant therapy in HER2-positive breast cancer, Front. Oncol., 2023, 13, 1066007, DOI:10.3389/fonc.2023.1066007.
- P. S. Steeg, Tumor metastasis: mechanistic insights and clinical challenges, Nat. Med., 2006, 12, 895–904, DOI:10.1038/nm1469.
- F. Bray, J. Ferlay and I. Soerjomataram, et al., Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 2018, 68, 394–424, DOI:10.3322/caac.21492.
- R. E. Coleman, P. I. Croucher and A. R. Padhani, et al., Bone metastases, Nat. Rev. Dis. Primers, 2020, 6, 83, DOI:10.1038/s41572-020-00216-3.
- S. Ali, J. Hendry and D. Le, et al., Efficacy of adjuvant trastuzumab in women with HER2-positive T1a or bN0M0 breast cancer: a population-based cohort study, Sci. Rep., 2022, 12, 1068, DOI:10.1038/s41598-022-05209-8.
- M. M. Moasser, The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis, Oncogene, 2007, 26, 6469–6487, DOI:10.1038/sj.onc.1210477.
- M. A. Cobleigh, C. L. Vogel and D. Tripathy, et al., Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease, J. Clin. Oncol., 1999, 17, 2639–2648, DOI:10.1200/JCO.1999.17.9.2639.
- J. Cheng, M. Liang and M. F. Carvalho, et al., Molecular Mechanism of HER2 Rapid Internalization and Redirected Trafficking Induced by Anti-HER2 Biparatopic Antibody, Antibodies, 2020, 9, 49, DOI:10.3390/antib9030049.
- F. M. Yakes, W. Chinratanalab and C. A. Ritter, et al., Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action, Cancer Res., 2002, 62, 4132–4141 CAS.
- L. Arnould, M. Gelly and F. Penault-Llorca, et al., Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism?, Br. J. Cancer, 2006, 94, 259–267, DOI:10.1038/sj.bjc.6602930.
- J. V. Leyton, Improving Receptor-Mediated Intracellular Access and Accumulation of Antibody Therapeutics-The Tale of HER2, Antibodies, 2020, 9, 32, DOI:10.3390/antib9030032.
- P. R. Pohlmann, I. A. Mayer and R. Mernaugh, Resistance to Trastuzumab in Breast Cancer, Clin. Cancer Res., 2009, 15, 7479–7491, DOI:10.1158/1078-0432.CCR-09-0636.
- D. Tural, E. Akar and H. Mutlu, et al., P95 HER2 fragments and breast cancer outcome, Expert Rev. Anticancer Ther., 2014, 14, 1089–1096, DOI:10.1586/14737140.2014.929946.
- T. Vu and F. X. Claret, Trastuzumab: updated mechanisms of action and resistance in breast cancer, Front. Oncol., 2012, 2, 62, DOI:10.3389/fonc.2012.00062.
- L. Guo, H. Zhang and P. Liu, et al., Preclinical Assessment of Paclitaxel- and Trastuzumab-Delivering Magnetic Nanoparticles Fe3O4 for Treatment and Imaging of HER2-Positive Breast Cancer, Front. Med., 2021, 8, 738775, DOI:10.3389/fmed.2021.
- N. Wathoni, L. E. Puluhulawa and I. M. Joni, et al., Monoclonal antibody as a targeting mediator for nanoparticle targeted delivery system for lung cancer, Drug Delivery, 2022, 29, 2959–2970, DOI:10.1080/10717544.2022.2120566.
- A. Kumar Mehata, S. Bharti and P. Singh, et al., Trastuzumab decorated TPGS-g-chitosan nanoparticles for targeted breast cancer therapy, Colloids Surf., B, 2019, 173, 366–377, DOI:10.1016/j.colsurfb.2018.10.007.
- A. Juan, F. J. Cimas and I. Bravo, et al., Antibody Conjugation of Nanoparticles as Therapeutics for Breast Cancer Treatment, Int. J. Mol. Sci., 2020, 21, 6018, DOI:10.3390/ijms21176018.
- A. Badkas, E. Frank and Z. Zhou, et al., Modulation of in vitro phagocytic uptake and immunogenicity potential of modified Herceptin®-conjugated PLGA-PEG nanoparticles for drug delivery, Colloids Surf., B, 2018, 162, 271–278, DOI:10.1016/j.colsurfb.2017.12.001.
- B. Colzani, L. Pandolfi and A. Hoti, et al., Investigation of antitumor activities of trastuzumab delivered by PLGA nanoparticles, Int. J. Nanomed., 2018, 13, 957–973, DOI:10.2147/IJN.S152742.
- H. Spreen, C. Barth and L. Keuter, et al., Tuning the protein corona of PLGA nanoparticles: Characterization of trastuzumab adsorption behavior and its cellular interaction with breast cancer cell lines, J. Drug Delivery Sci. Technol., 2022, 74, 03543, DOI:10.1016/j.jddst.2022.103543.
- R. K. Dhritlahre and A. Saneja, Recent advances in HER2-targeted delivery for cancer therapy, Drug Discovery Today., 2021, 26(5), 1319–1329, DOI:10.1016/j.drudis.2020.12.014.
- T. M. Caputo, A. M. Cusano and S. Principe, et al., Sorafenib-Loaded PLGA Carriers for Enhanced Drug Delivery and Cellular Uptake in Liver Cancer Cells, Int. J. Nanomed., 2023, 18, 4121–4142, DOI:10.2147/IJN.S415968.
- C. Liverani, L. Mercatali and L. Cristofolini, et al., Investigating the Mechanobiology of Cancer Cell-ECM Interaction Through Collagen-Based 3D Scaffolds, Cell. Mol. Bioeng., 2017, 10, 223–234, DOI:10.1007/s12195-017-0483-x.
- C. Liverani, A. De Vita and C. Spadazzi, et al., Lineage-specific mechanisms and drivers of breast cancer chemoresistance revealed by 3D biomimetic culture, Mol. Oncol., 2022, 16, 921–939, DOI:10.1002/1878-0261.13037.
- T. M. Caputo, et al., Development of High-Loading Trastuzumab PLGA Nanoparticles: A Powerful Tool Against HER2 Positive Breast Cancer Cells, Int. J. Nanomed., 2023, 6999–7020 CrossRef CAS PubMed.
- M. Iqbal, N. Zafar and H. Fessi, et al., Double emulsion solvent evaporation techniques used for drug encapsulation, Int. J. Pharm., 2015, 496, 173–190, DOI:10.1016/j.ijpharm.2015.10.057.
- E. Piñón-Segundo, M. G. Nava-Arzaluz and D. Lechuga-Ballesteros, Pharmaceutical polymeric nanoparticles prepared by the double emulsion- solvent evaporation technique, Recent Pat. Drug Delivery Formulation, 2012, 6, 224–235, DOI:10.2174/187221112802652606.
- C. Liverani, A. De Vita and S. Minardi, et al., A biomimetic 3D model of hypoxia-driven cancer progression, Sci. Rep., 2019, 9, 12263, DOI:10.1038/s41598-019-48701-4.
- A. De Vita, F. Recine and G. Miserocchi, et al., The potential role of the extracellular matrix in the activity of trabectedin in UPS and L-sarcoma: evidences from a patient-derived primary culture case series in tridimensional and zebrafish models, J. Exp. Clin. Cancer Res., 2021, 40, 165, DOI:10.1186/s13046-021-01963-1.
- A. De Vita, S. Vanni and G. Miserocchi, et al., A Rationale for the Activity of Bone Target Therapy and Tyrosine Kinase Inhibitor Combination in Giant Cell Tumor of Bone and Desmoplastic Fibroma: Translational Evidences, Biomedicines, 2022, 10, 372, DOI:10.3390/biomedicines10020372.
- Herceptin: summary of product characteristics, https://www.ema.europa.eu/en/documents/product-information/herceptin-epar-product-information_it.pdf, (accessed September 2024).
- C. E. Holy, S. M. Dang and J. E. Davies, et al., In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam, Biomaterials, 1999, 20, 1177–1185, DOI:10.1016/s0142-9612(98)00256-7.
- B. S. Zolnik and D. J. Burgess, Effect of acidic pH on PLGA microsphere degradation and release., J. Controlled Release, 2007, 122, 338–344, DOI:10.1016/j.jconrel.2007.05.034.
- H. K. Makadia and S. J. Siegel, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers, 2011, 3, 1377–1397, DOI:10.3390/polym3031377.
- R. A. Gatenby, E. T. Gawlinski and A. F. Gmitro, et al., Acid-mediated tumor invasion: a multidisciplinary study., Cancer Res., 2006, 66, 5216–5223, DOI:10.1158/0008-5472.CAN-05-4193.
- R.J Gillies, Z. Liu and Z. Bhujwalla, 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate., Am. J. Physiol., 1994, 267, C195–C203, DOI:10.1152/ajpcell.1994.267.1.C195.
- A. Kumar Mehata, S. Bharti and P. Singh, et al., Trastuzumab decorated TPGS-g-chitosan nanoparticles for targeted breast cancer therapy, Colloids Surf., B, 2019, 173, 366–377, DOI:10.1016/j.colsurfb.2018.10.007.
- J. Hamzehalipour Almaki, R. Nasiri and A. Idris, et al., Trastuzumab-decorated nanoparticles for in vitro and in vivo tumor-targeting hyperthermia of HER2 + breast cancer, J. Mater. Chem. B, 2017, 5, 7369–7383, 10.1039/c7tb01305a.
- G. Brockhoff, B. Heckel and E. Schmidt-Bruecken, et al., Differential impact of Cetuximab, Pertuzumab and Trastuzumab on BT474 and SK-BR-3 breast cancer cell proliferation, Cell Proliferation, 2007, 40, 488–507, DOI:10.1111/j.1365-2184.2007.00449.x.
- B. Carotenuto, A. Ricciardi and A. Micco, et al., Smart Optical Catheters for Epidurals, Sensors, 2018, 18, 2101, DOI:10.3390/s18072101.
- C. Monat, P. Domachuk and B. J. Eggleton, Integrated optofluidics: a new river of light, Nat. Photonics, 2007, 1, 106–114, DOI:10.1038/nphoton.2006.96.
- K. Sui, M. Meneghetti and J. Kaur, et al., Microstructured soft fiber-based neural device for drug delivery and optical neuromodulation, Biophotonics Congr. Biomed. Opt., 2022, 2022, BW4C.3, DOI:10.1364/BRAIN.2022.BW4C.3.
- M. Nazari, M. Rubio-Martinez and G. Tobias, et al., Metal-organic-framework-coated optical fibers as light-triggered drug delivery vehicles, Adv. Funct. Mater., 2016, 26, 3244–3249, DOI:10.1002/adfm.201505260.
- M. Pisco and A. Cusano, Lab-On-Fiber Technology: A Roadmap toward Multifunctional Plug and Play Platforms, Sensors, 2020, 20, 4705, DOI:10.3390/s20174705.
- P. Caldas, P. A. Jorge and G. Rego, et al., Fiber optic hot-wire flowmeter based on a metallic coated hybrid long period grating/fiber Bragg grating structure, Appl. Opt., 2011, 50, 2738–2743, DOI:10.1364/AO.50.002738.
- R. Gao and D. Lu, Temperature compensated fiber optic anemometer based on graphene-coated elliptical core micro-fiber Bragg grating, Opt. Express, 2019, 27, 34011–34021, DOI:10.1364/OE.27.034011.
- Z. Liu, F. Wang and Y. Zhang, et al., Low-power-consumption fiber-optic anemometer based on long-period grating with SWCNT coating, IEEE Sens. J., 2019, 19, 2592–2597, DOI:10.1109/JSEN.2019.2891044.
- F. Wang, Y. Duan and M. Lu, et al., Linear-response and simple hot-wire fiber-optic anemometer using high-order cladding mode, Opt. Express, 2020, 28, 27028–27036, DOI:10.1364/OE.399774.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2020, CA Cancer J. Clin., 2020, 70, 7–30, DOI:10.3322/caac.21590.
- S. M. Swain, D. Miles and S. B. Kim, et al., Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study, Lancet Oncol., 2020, 21, 519–530, DOI:10.1016/S1470-2045(19)30863-0.
- A. Matusz-Fisher and A. R. Tan, Combination of HER2-targeted agents with immune checkpoint inhibitors in the treatment of HER2-positive breast cancer, Expert Opin. Biol. Ther., 2022, 22, 385–395, DOI:10.1080/14712598.2021.1981284.
- T. Yang, L. Kang and D. Li, et al., Immunotherapy for HER-2 positive breast cancer, Front. Oncol., 2023, 13, 1097983, DOI:10.3389/fonc.2023.1097983.
- D. Gajria and S. Chandarlapaty, HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies, Expert Rev. Anticancer Ther., 2011, 11, 263–275, DOI:10.1586/era.10.226.
- H. Tonbul, A. Sahin and E. Tavukcuoglu, et al., Combination drug delivery with actively-targeted PLGA nanoparticles to overcome multidrug resistance in breast cancer, J. Drug Delivery Sci. Technol., 2019, 54, 101380, DOI:10.1016/j.jddst.2019.101380.
- N. Jafarzadeh, M. Nadafan and R. Malekfar, et al., Structural, optical and dielectric studies of Ag nanoparticles decorated by herceptin, Phys. E, 2019, 114, 113562, DOI:10.1016/j.physe.2019.113562.
- L. Guo, H. Zhang and P. Liu, et al., Preclinical Assessment of Paclitaxel- and Trastuzumab-Delivering Magnetic Nanoparticles Fe3O4 for Treatment and Imaging of HER2-Positive Breast Cancer, Front. Med., 2021, 8, 738775, DOI:10.3389/fmed.2021.738775.
- A. Catala, M. Dzieciatkowska and G. Wang, et al., Targeted Intracellular Delivery of Trastuzumab Using Designer Phage Lambda Nanoparticles Alters Cellular Programs in Human Breast Cancer Cells, ACS Nano, 2021, 15, 11789–11805, DOI:10.1021/acsnano.1c02864.
- R. K. Dhritlahre and A. Saneja, Recent advances in HER2-targeted delivery for cancer therapy, Drug Discovery Today, 2021, 26, 1319–1329, DOI:10.1016/j.drudis.2020.12.01.
- X. Xu, W. Ho and X. Zhang, et al., Cancer nanomedicine: from targeted delivery to combination therapy, Trends Mol. Med., 2015, 21(4), 223–232, DOI:10.1016/j.molmed.2015.01.001.
- J. G. Dancy, A. S. Wadajkar and N. P. Connolly, et al., Decreased nonspecific adhesivity, receptor-targeted therapeutic nanoparticles for primary and metastatic breast cancer, Sci. Adv., 2020, 6(3), eaax3931, DOI:10.1126/sciadv.aax3931.
- R. Vivek, R. Thangam and V. NipunBabu, et al., Multifunctional HER2-antibody conjugated polymeric nanocarrier-based drug delivery system for multi-drug-resistant breast cancer therapy, ACS Appl. Mater. Interfaces, 2014, 6, 6469–6480, DOI:10.1021/am406012g.
- H. Chen, L. Wang and Q. Yu, et al., Anti-HER2 antibody and ScFvEGFR-conjugated antifouling magnetic iron oxide nanoparticles for targeting and magnetic resonance imaging of breast cancer, Int. J. Nanomed., 2013, 8, 3781–3794, DOI:10.2147/IJN.S49069.
- S. Zalba, A. M. Contreras and A. Haeri, et al., Cetuximab-oxaliplatin-liposomes for epidermal growth factor receptor targeted chemotherapy of colorectal cancer, J. Controlled Release, 2015, 210, 26–38, DOI:10.1016/j.jconrel.2015.05.271.
- N. Li, Q. Zhao and C. Shu, et al., Targeted killing of cancer cells in vivo and in vitro with IGF-IR antibody-directed carbon nanohorns based drug delivery, Int. J. Pharm., 2015, 478, 644–654, DOI:10.1016/j.ijpharm.2014.12.015.
- M. K. Greene, D. A. Richards and J. C. F. Nogueira, et al., Forming next-generation antibody-nanoparticle conjugates through the oriented installation of non-engineered antibody fragments, Chem. Sci., 2017, 9, 79–87, 10.1039/c7sc02747h.
- Z. Zhou, A. Badkas and M. Stevenson, et al., Herceptin conjugated PLGA-PHis-PEG pH sensitive nanoparticles for targeted and controlled drug delivery, Int. J. Pharm., 2015, 487, 81–90, DOI:10.1016/j.ijpharm.2015.03.081.
- C. Nieto, A. Centa and J. A. Rodríguez-Rodríguez, et al., Paclitaxel-Trastuzumab Mixed Nanovehicle to Target HER2-Overexpressing Tumors, Nanomaterials, 2019, 9, 948, DOI:10.3390/nano9070948.
- R. Domínguez-Ríos, D. R. Sánchez-Ramírez and K. Ruiz-Saray, et al., Cisplatin-loaded PLGA nanoparticles for HER2 targeted ovarian cancer therapy, Colloids Surf., B, 2019, 178, 199–207, DOI:10.1016/j.colsurfb.2019.03.011.
- V. O. Shipunova, A. S. Sogomonyan and I. V. Zelepukin, et al., PLGA Nanoparticles Decorated with Anti-HER2 Affibody for Targeted Delivery and Photoinduced Cell Death, Molecules, 2021, 26, 3955, DOI:10.3390/molecules26133955.
- T. K. D. Doan, M. Umezawa and K. Ikeda, et al., Influence of Carboxyl Group Ratios on the Design of Breast Cancer Targeting Bimodal MR/NIR-II Imaging Probe from PLGA@Gd-DOTA@PEG Micelles Conjugating Herceptin, ACS Appl. Bio. Mater., 2023, 6, 2644–2650, DOI:10.1021/acsabm.3c00260.
- Y. Liu, K. Li and B. Liu, et al., A strategy for precision engineering of nanoparticles of biodegradable copolymers for quantitative control of targeted drug delivery, Biomaterials, 2010, 31, 9145–9155, DOI:10.1016/j.biomaterials.2010.08.053.
- I. Steinhauser, B. Spänkuch and K. Strebhardt, et al., Trastuzumab-modified nanoparticles: optimisation of preparation and uptake in cancer cells, Biomaterials, 2006, 27, 4975–4983, DOI:10.1016/j.biomaterials.2006.05.016.
- A. Juan, F. J. Cimas and I. Bravo, et al., Antibody Conjugation of Nanoparticles as Therapeutics for Breast Cancer Treatment, Int. J. Mol. Sci., 2020, 21, 6018, DOI:10.3390/ijms21176018.
- M. Sakhi, A. Khan and Z. Iqbal, et al., Design and Characterization of Paclitaxel-Loaded Polymeric Nanoparticles Decorated With Trastuzumab for the Effective Treatment of Breast Cancer, Front. Pharmacol., 2022, 13, 855294, DOI:10.3389/fphar.2022.855294.
- P. Kocbek, N. Obermajer and M. Cegnar, et al., Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody, J. Controlled Release, 2007, 120, 18–26, DOI:10.1016/j.jconrel.2007.03.012.
- L. Bregoli, D. Movia and J. D. Gavigan-Imedio, et al., Nanomedicine applied to translational oncology: A future perspective on cancer treatment, Nanomedicine, 2016, 12, 81–103, DOI:10.1016/j.nano.2015.08.006.
- C. A. Hudis, Trastuzumab–mechanism of action and use in clinical practice, N. Engl. J. Med., 2007, 357, 39–51, DOI:10.1056/NEJMra043186.
- F. Corsi, L. Fiandra and C. De Palma, et al., HER2 expression in breast cancer cells is downregulated upon active targeting by antibody-engineered multifunctional nanoparticles in mice, ACS Nano, 2011, 5, 6383–6393, DOI:10.1021/nn201570n.
- H. Tian, T. Zhang and S. Qin, et al., Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies, J. Hematol. Oncol., 2022, 15, 132, DOI:10.1186/s13045-022-01320-5.
- M. Overchuk and G. Zheng, Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics, Biomaterials, 2018, 156, 217–237, DOI:10.1016/j.biomaterials.2017.10.024.
- K. M. Song, S. Lee and C. Ban, Aptamers and their biological applications, Sensors, 2012, 12, 612–631, DOI:10.3390/s120100612.
- M. Fazio, J. Ablain and Y. Chuan, et al., Zebrafish patient avatars in cancer biology and precision cancer therapy, Nat. Rev. Cancer, 2020, 20, 263–273, DOI:10.1038/s41568-020-0252-3.
- B. Costa, M. F. Estrada and R. V. Mendes, et al., Zebrafish Avatars towards Personalized Medicine-A Comparative Review between Avatar Models, Cells, 2020, 9, 293, DOI:10.3390/cells9020293.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 Specific HER2-TZ binding documented in SPR sensorgrams and relative RU. Fig. S2 Cumulative release profile of TZ. Fig. S3 Efficacy of free TZ on cell proliferation inhibition in BT-474 cell line after 72 hours or 7 days of treatment in 2D and 3D culture models. Fig. S4 Effect of naked PLGA (N-PLGA) and trehalose on cell proliferation inhibition in BT-474 cell line after 72 hours or 7 days of treatment in 2D and 3D culture models. Fig. S5 Flow cytometry analysis of HER2 expression in BT-474 cell line untreated (CTRL), treated with different PLGA + TZ nanoparticles or treated with free TZ after 72 hours or 7 days. See DOI: https://doi.org/10.1039/d4nr03907f |
| ‡ These authors share first authorship. |
| § These authors share last authorship. |
| This journal is © The Royal Society of Chemistry 2025 |