
Synergizing superwetting and architected electrodes for high-rate water splitting
Qiu
Ren
a,
Cassidy
Tran
a,
Kangkang
Zhang
b,
Cheng
Zhu
b and
Yat
Li
 *a
*a
aDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California, 95064, USA. E-mail: yatli@ucsc.edu
bLawrence Livermore National Laboratory, 7000 East Avenue, Livermore, California 94550, USA
First published on 30th December 2024
Abstract
Water splitting is one of the most promising technologies for generating green hydrogen. To meet industrial demand, it is essential to boost the operation current density to industrial levels, typically in the hundreds of mA cm−2. However, operating at these high current densities presents significant challenges, with bubble formation being one of the most critical issues. Efficient bubble management is crucial as it directly impacts the performance and stability of the water splitting process. Superwetting electrodes, which can enhance aerophobicity, are particularly favorable for facilitating bubble detachment and transport. By reducing bubble contact time and minimizing the size of detached bubbles, these electrodes help prevent blockage and maintain high catalytic efficiency. In this review, we aim to provide an overview of recent advancements in tackling bubble-related issues through the design and implementation of superwetting electrodes, including surface modification techniques and structural optimizations. We will also share our insights into the principles and mechanisms behind the design of superwetting electrodes, highlighting the key factors that influence their performance. Our review aims to guide future research directions and provides a solid foundation for developing more efficient and durable superwetting electrodes for high-rate water splitting.
1. Introduction
Hydrogen is a clean, renewable fuel with high energy density, making it an ideal replacement for fossil fuels.1,2 However, over 95% of hydrogen is currently produced through unsustainable petrochemical processes. Due to the ongoing energy crisis and environmental concerns, there is a growing need to explore alternative hydrogen generation methods.3,4 Water splitting is one of the most promising technologies for producing green hydrogen.5–7 The main methods of water splitting include polymer electrolyte membrane (PEM), alkaline water splitting (AWS), and solid oxide electrolysis cell (SOEC) technology.8–10 These methods share a common principle: the hydrogen evolution reaction (HER) occurs at the cathode, while the oxygen evolution reaction (OER) takes place at the anode. AWS and PEM electrolysis operate at temperatures between 60 and 90 °C, while SOEC operates at much higher temperatures, ranging from 600 to 900 °C. Each technology has its own set of challenges. PEM utilizes a proton-conducting solid membrane, which provides superior higher ionic conductivity, reduces ohmic losses, and supports high current density operation. Therefore, high current density is not a limiting factor for PEM-based water splitting. However, this system relies on noble metal-based catalysts (e.g., IrO2, Pt) to operate effectively in the acidic environment, which increases costs and limits large scale adoption.11,12 In contrast, AWS offers a more cost-effective alternative by employing non-precious metal (non-PGM) electrodes, such as Ni-based catalysts, and more affordable diaphragms in alkaline electrolytes, significantly reducing overall system costs. Nevertheless, AWS is limited in achieving high current densities at low voltages due to the lower catalytic activity of the electrodes and the higher ionic resistivity of the diaphragm, resulting in moderate energy efficiency.13–17 SOEC technology faces difficulties due to its significantly shorter lifespan compared to PEM and AWS.18,19The efficiency and durability of any water-splitting technology largely depends on the performance of its electrode materials. To meet industrial needs, it is essential to develop electrode materials that can maintain high performance under demanding conditions, such as high current density, extended operation, and specific pressure and temperature requirements.20–22 The primary difference between low-rate (tens of mA cm−2) and high-rate (hundreds of mA cm−2) water splitting is the amount of gas bubbles generated during the electrochemical process.23–25 This significantly impacts the two main types of overpotentials, activation (ηact) and ohmic overpotentials (ηohm), during water splitting.26–28 Electrochemical gas bubble evolution on the electrodes occurs in three stages: nucleation, growth, and detachment.29,30 As shown in Fig. 1, during low-rate water splitting, the concentration of gas bubbles is lower than the gas solubility limit of the bulk electrolyte, therefore allowing for efficient bubble dissolution and transport. However, in high-rate water splitting, the rate of gas production exceeds the rate of gas diffusion. This leads to the accumulation of bubbles on the electrode surface, which blocks active sites, increases activation energy, and results in higher ηact.31–33ηohm is influenced by the various resistances throughout the electrochemical process, including those of the electrode, electrolyte, membrane, wire connection and gas bubbles. At low current densities, the number of bubbles is minimal, so their contribution to ohmic resistance is small. However, as current density increases, the amount of gas bubbles rises significantly. This extensive bubble coverage on the electrodes creates a barrier between the electrolyte and electrode, resulting in increased ionic diffusion resistance and, consequently, higher ηohm.34–36 Additionally, gas bubbles that adhere to the catalyst surface continue to grow until their buoyancy overcomes the adhesion forces with the catalyst. When larger bubbles eventually detach from the catalyst surface, the forces exerted at the bubble-electrode interface can damage the catalyst layer, potentially leading to structural failure or loss of catalyst material.37
 | ||
| Fig. 1 (a) Gas bubble generation during low-rate water splitting, where minimal bubbles form on the electrode surface, and (b) gas bubble generation during high-rate water splitting, where a layer of bubbles forms on the electrode surface, obstructing actives sites and impeding ion diffusion. | ||
Numerous strategies have been developed to address bubble-related issues during water splitting.38–40 For instance, passive methods like adjusting the electrolyte composition to modify surface tension can significantly influence bubble behavior in electrochemical systems.41 Research has shown that bubble detachment radii decrease with increasing pH values.42 Additionally, the addition of surfactants like sodium dodecyl sulfate,43 hexadecyltrimethylammonium bromide44 or potassium perfluorobutyl sulfonate45 can reduce bubbles detachment radii by improving surface hydrophilicity, promoting better wetting behavior. They facilitate bubble detachment by reducing the contact angles between gas bubbles and the electrode surface. Surfactants also reduce surface tension (e.g., liquid/vapor interfacial tension) and modify the interfacial tension at the solid/liquid interface. This reduction enhances the mass transfer rate within the gas diffusion layer during water splitting. However, while surfactants can help reduce energy losses caused by bubbles, they also complicate the system by potentially participating in unwanted electrode reactions. To further enhance bubble detachment and transport, various active methods utilizing external fields, such as magnetic and acoustic fields, have been developed.46–48 These methods aim to reduce the impact of bubbles on the system's overpotential. For example, applying magnetic fields can create Lorentz forces in the electrolyte, which induce convection. This convective flow improves mass transfer, reducing both ohmic and concentration overpotentials.49 Matsushima and coworkers found that the HER performance of a platinum electrode in an acidic electrolyte (0.5 M H2SO4) improved with increasing magnetic flux intensity.50 Furthermore, acoustic fields explore the use of ultrasonic radiation in conjunction with electrochemical process. The goal is to enhance mass transfer in the liquid electrolyte and facilitates the removal of bubbles from electrode surfaces during reactions.51 For instance, Li and coworkers demonstrated that applying acoustic fields improved hydrogen generation efficiency by 5–18% on a RuO2 and IrO2-plated Ti electrode in alkaline electrolytes.52 However, the practical application of these active methods is often limited by the need for external field generators and the potential for these fields to degrade the catalyst layer.53
Furthermore, biomimetic materials offer inspiration for developing innovative methods to engineer functional surfaces with unique wetting properties.54–57 Recent studies have demonstrated that by tailoring the compositions and micro/nanoarchitectures of electrode surfaces, it is possible to effectively control the adhesion between gas bubbles and the electrode surface underwater.58,59 This approach has created new opportunities to enhance electrochemical performance by manipulating surface properties related to superwettability.60 Nanostructured superaerophobic electrodes, for instance, create discontinuous three-phase contact lines (solid–liquid–gas, TPCL), which minimize gas bubble adhesion and accelerate gas evolution.61–63 As a result, there is a growing interest in designing superwetting electrodes based on this principle.
In this review, we aim to provide a thorough overview of superwetting electrodes for high-rate water splitting. We start by highlighting the significance of operating water splitting processes at high current densities. Next, we introduce the concept of superaerophobicity in aqueous environments. Then, we summarize recent advancements in the use of superwetting electrodes for high-rate water splitting, dividing them into two-dimensional (2D) substrates (such as foil-based electrodes) and three-dimensional (3D) substrates (such as Ni foam and 3D-printed electrodes). Finally, we discuss future directions for the development of superwetting electrodes and the challenges they face in practical applications.
2. High-rate water splitting
For industrial applications, it is essential to develop electrode materials that can perform effectively under industry-relevant conditions, such as high current density, extended operational periods, and specific pressure and temperature requirements. High current density is particularly important because it increases the rate of hydrogen production, which can reduce capital costs and improve the profitability of hydrogen production.64–66The ability to produce hydrogen at high rates is vital to effectively meet the growing demand for hydrogen in various applications, including hydrogen-powered fuel cell vehicles, ammonia production, and fuel cell related energy storage systems.67,68 Various governments and organizations have established technical targets for high-rate water splitting to address different application needs. For example, the U.S. Department of Energy (DOE) has set ambitious targets for PEM water splitting.69 By 2026, the DOE aims to achieve a current density of 3000 mA cm−2 at a cell voltage of 1.8 V, with a long-term goal of reaching 3000 mA cm−2 at 1.6 V. Similarly, the Fuel Cells and Hydrogen Joint Undertaking (FCH JU) in Europe has set broader targets,70 aiming for a current density of 800 mA cm−2 for AWS and 2500 mA cm−2 for PEM water splitting by 2030. These targets are part of a broader strategy to enhance performance and reduce the costs of hydrogen production technologies, making them more accessible and practical for widespread use. However, achieving high current densities can accelerate the degradation of electrode materials, highlighting the need for more robust and durable catalysts and electrodes. Research efforts have been focused on developing advanced catalysts, innovating electrode architectures to improve mass transport and reduce overpotentials, and integrating electrolysis systems with renewable energy sources to ensure sustainable hydrogen production. Table 1 summarizes some of the future technical performance targets, which are driving research into electrochemical water splitting under high current density conditions.
| Source | Technique | Year | Current density (mA cm−2) | Voltage (V) | Energy efficiency (%) | Durability (h) |
|---|---|---|---|---|---|---|
| DOE | PEM | 2026 | 3000 | 1.8 | 69 | 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| DOE | PEM | Ultimate | 3000 | 1.6 | 77 | 80000 |
| FC HJU | PEM | 2030 | 2500 | — | — | Degraded by 0.12% per 1000 h |
| FC HJU | AWS | 2030 | 800 | — | — | Degraded by 0.1% per 1000 h |
3. Principle of superwetting electrode
Achieving high current densities can lead to increased gas bubble formation and faster degradation of electrode materials, highlighting the need for more efficient gas-removal electrodes. The development of superwetting electrodes has emerged as a promising approach. These electrodes are engineered to optimize the interaction between the solid electrode surface and gas bubbles, reducing bubble-induced resistance and therefore enhancing overall performance. The relationship between water contact angle (WCA) and a solid surface is described by Young's equation (eqn (1)), which explains hydrophilicity or hydrophobicity under ideal conditions by balancing forces at the TPCL, where the liquid, solid, and gas bubbles meet.71–73 Essentially, Young's equation quantifies how a liquid droplet interacts with a solid surface, determining whether the surface will repel or attract water molecule. Similarly, Young's equation can also be applied as a theoretical model for analyzing the aerophilicity and aerophobicity properties of a solid surface, as shown in eqn (2). Using the same assumptions as those applied for liquid interactions, this equation helps explain how gas bubbles interact with the solid surface, providing insights into the surface's behavior in gaseous environments.74,75 | (1) |
 | (2) |
These wettability characteristics are crucial for various applications, particularly in gas evolution reactions like water splitting. The Cassie–Baxter equation (eqn (3)) was introduced to explore how electrode surface design can achieve superaerophobicity underwater.78,79 This equation accounts for the heterogeneous composite surface within the rough texture of the electrode, which reduces the contact area between gas bubbles and the solid surface. As a result, this promotes the release of gas bubbles and enhances the overall efficiency of the electrochemical process.
| cosα* = fs(cosα + 1) − 1 | (3) |
 | ||
| Fig. 2 Schematic illustration showing the effect of surface roughness on the bubble contact angle and bubble detachment size: (a) on a flat surface, and (b) on a rough surface, as predicted by the Cassie-Baxter equation. Reproduced from ref. 81 with permission from Wiley, copyright 2023. | ||
In high-rate water splitting, gas bubbles formed at active sites follow a direct injection model, where the surrounding dissolved gas flows into the bubble, causing its radius to increase in proportion to the cube root of the growth time. According to this model, the dynamics of bubble growth are influenced by the local concentration of dissolved gases and the properties of the electrode surface. As shown in the eqn (4), the radius of the detached gas bubble  is proportional to the contact area, surface tension, and the sine function of BCA raised to the power of 1/3.82
is proportional to the contact area, surface tension, and the sine function of BCA raised to the power of 1/3.82
 | (4) |
4. Implementation of superwetting electrodes on different substrates
Superwetting electrodes can be fabricated on a variety of substrates using a range of methods, including electrodeposition and hydrothermal reactions. As outlined in Table 2, they include two-dimensional (2D) substrates, mainly flat foils, and three-dimensional (3D) substrates, which offer more complex and structured architectures. For example, 3D-printed electrodes, which leverage additive manufacturing techniques to create customized electrode structures with periodic pores or channels. By exploring and utilizing these diverse substrate geometries, researchers can optimize electrode/device design and functionality to meet specific requirements and improve the performance of high-rate water splitting.| Substrate/electrode | Electrode architecture | Electrolyte | Reaction | Current density (mA cm−2) | Overpotential (mV) | Ref. | |
|---|---|---|---|---|---|---|---|
| 2D | Pt foil | Superaerophilic/superaerophobic cooperative electrode | 0.5 M H2SO4 | HER | 471 | 300 | 85 |
| NiFe LDHs on Ni plate | Superaerophilic/superaerophobic cooperative electrode | 1.0 M KOH | AWE | 500 | — | 86 | |
| Co–Ni phosphide/spinel oxide on Ti plate | Nanowires and nanosheets | 6.0 M KOH | Zero-gap AWE | 3500 | 1020 | 77 | |
| 3D Ni foam | CoMoSx | Hierarchical micro-/nanostructures | 1.0 M KOH | AWE | 500 | 660 | 87 |
| NiMo alloy film and NiFe-LDH film | Hemispherical structure (NiMo) and nanoplates (NiFe-LDH) | 6.0 M KOH | AWE | >400 | 670 | 58 | |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF)-Ni3S2 | Heterodimensional nanostructure | 1.0 M KOH | AWE | 1000 | 563 | 88 | |
| CoMoSx on Ni foam | Nanosheet | 1.0 M KOH | AWE | 500 | 660 | 89 | |
| 3D porous CoxP | Ordered nanoarray | 1.0 M KOH | HER | 200 | 272 | 90 | |
| NiP2@MoO2/Co(Ni)MoO4 | Bouquet-like core–shell cuboid array | 1.0 M KOH | HER | 1000 | 297 | 91 | |
| NiMoO4@NiFeP | Nanoarray structure | 1.0 M KOH | AWE | 500 | 700 | 92 | |
| Polyethyleneimine hydrogels | Polyethyleneimine hydrogels coating | 1.0 M KOH | HER | 500 | 608 | 93 | |
| Ni2P nanoarray | Nanowires | 1.0 M KOH | HER | 1500 | 368 | 94 | |
| 3D-printed | MoNi4-MoO2/3DP Ni||NiFe LDH/3DP Ni | Hierarchical porous structure | 1.0 M KOH | AWE | 500 | 400 | 95 |
| Anisotropic porous NiMo | Anisotropic porous structure | 1.0 M KOH | HER | 500 | ∼150 | 96 | |
| Carbon-doped NiO | Nanorods on periodic lattice structure | 1.0 M KOH | AWE | 850 | 970 | 37 | |
| Ni nanocones | Nanocones modified lattice structure | 1.0 M KOH | AWE | 910 | 1220 | 81 | |
| Porous nickel-based alloy | Porous structure | 1.0 M KOH | AWE | 500 | — | 97 | |
4.1. 2D substrates
The Yan group developed a superaerophobic heterostructured catalyst composed of Ni2P-CoP phosphide and NiCo2O4 spinel oxide on a nickel sheet.77 The phosphide provided hydrophilicity and activity for both OER and HER, while the spinel oxide, after oxidation, contributed superaerophilicity and OER activity. The morphology of the hybrid catalyst was optimized to form low density nanosheet bundles (BS-1), which showed high wettability with a water contact angle of 0°. Compared to NF, the hybrid catalyst showed reduced bubble adhesion at various HER current densities (Fig. 3a). The catalyst was then incorporated into a zero-gap alkaline water electrolyzer (AWE) using 6.0 M KOH. At 85 °C, as shown in Fig. 3b, the cell achieved a current density of 3500 mA cm−2 at 2.25 V and remained stable after 330 hours at 85 °C at a current density of 2000 mA cm−2. The cell also exhibited a less significant increase in ohmic resistance with rising current density compared to the cell with NF electrodes. The enhancement was attributed to the reduced bubble adhesion, which dominates ohmic overpotential at high current densities. | ||
| Fig. 3 (a) Comparison of surface bubbles on the NF and BS-1 electrodes at various HER current densities. (b) Overall water splitting performance of NF and BS-1 electrodes using a zero-gap AWE at 85 °C. Reproduced from ref. 98 with permission from Wiley, copyright 2022. (c–f) Optical images and schematic illustrations of H2 bubble behavior on the four different electrodes. (g) Linear sweep voltammograms (LSV) of the four HER electrodes. Reproduced from ref. 85 with permission from Science, copyright 2023. | ||
The Jiang group developed a strategy of using superaerophobic/superaerophilic (SAB/SAL) electrode to enhance mass transfer for HER.85 As shown in Fig. 3c–f, the electrode features alternating stripes of Pt decorated with superaerophilic SiO2 nanoparticles and superaerophobic nanostructured pine-shaped Pt on a titanium sheet (SAL/SAB Pt). During HER, the superaerophilic stripes form a gas cushion that acts as a diffusion channel for H2 bubbles, promoting rapid bubble transport through asymmetric Laplace pressure. Additionally, these gas cushions, connected to ambient air, provide a shorter diffusion path for H2 gas generated in solution, leading to more efficient diffusion and lower H2 concentration near the electrode. As shown in Fig. 3g, this enhanced H2 bubble transfer improves the HER performance of SAL/SAB Pt in an acidic electrolyte. The Jiang group further developed a similar superaerophobic/superaerophilic patterned electrode for overall water splitting using NiFe layered-double-hydroxides (LDHs).86 The electrode, consisting of alternating stripes of superaerophilic SiO2 nanoparticles and superaerophobic NiFe-LDHs on a nickel plate, functions as both the anode and the cathode in overall water splitting, achieving a current density of 500 mA cm−2 at around 2.4 V in 1.0 M KOH.
4.2. 3D Ni foam substrates
2D electrodes, characterized by their flat and planar structures, offer simplicity in design and ease of fabrication, making them ideal for fundamental studies and applications. However, their limited surface area constrains the number of active sites, reducing efficiency at higher current densities. Furthermore, gas bubbles formed during electrolysis tend to adhere to the flat surface, increasing mass transport resistance and diminishing performance under practical conditions. 2D electrodes are simple in design and easy to fabricate, making them ideal for fundamental studies and some applications. However, their limited surface area restricts the number of active sites, reducing efficiency at higher current densities. Additionally, gas bubbles formed during electrolysis tend to adhere to their flat surfaces, increasing mass transport resistance and diminishing performance under practical conditions.In contrast, 3D electrodes, with their high surface area, provide a significantly greater number of active sites. Porous nickel foam (NF) is a widely used commercial electrode for alkaline water splitting due to its low cost, stability, and scalability.99,100 The addition of superaerophobic catalysts to the NF surface provides an effective pathway for developing effective superwetting electrodes. For example, the Jiang group developed a superhydrophilic/superaerophobic CoMoSx chalcogel on NF for overall water splitting.87 They attributed the electrode's superaerophobicity to its hierarchical micro/nano structure, which created a discontinuous TPCL between bubbles and the electrode surface. This reduced contact area allowed for faster bubble release (Fig. 4a–c). Additionally, the amorphous morphology of CoMoSx provided abundant active sites and defects, enhancing catalytic activity. The electrode achieved a current density of 500 mA cm−2 at 1.89 V in 1.0 M KOH and maintained this performance for over 100 hours (Fig. 4d). Similarly, the Yu group developed a Ni2P nanoarray catalyst on NF (Ni2P/NF) for HER.94 This catalyst's morphology, consisting of Ni2P nanosheets uniformly distributed on Ni2P nanowire arrays, turned the electrode into a superaerophobic surface, which facilitated rapid bubble release (Fig. 4e). The Ni2P/NF electrode achieved high HER current densities of 1000 and 1500 mA cm−2 at overpotentials of 306 and 368 mV, respectively. When paired with a NiFe-LDH anode for overall water splitting, the device outperformed the combination of traditional noble metal catalysts Pt/C and Ir/C at high current densities (Fig. 4f). The catalyst remained stable at 1200 mA cm−2 for 160 hours, with no changes in structure or phase observed after testing.
 | ||
| Fig. 4 Digital images showing bubbles released from (a) NF, (b) Pt/C, and (c) CoMoSx/NF during HER at a current density of 200 mA cm−2. Scale bars are 0.5 mm. (d) LSV for CoMoSx/NF, MoSx/NF, IrO2-Pt/C, and NF electrodes at a scan rate of 5 mV s−1. Reproduced from ref. 87 with permission from Wiley, copyright 2019. (e) Digital images of hydrogen bubbles on NF and Ni2P/NF. (f) LSV of the catalysts at a scan rate of 10 mV s−1, with the inset showing EIS Nyquist plots of the catalysts at 1.7 V. Reproduced from ref. 101 with permission from ACS, copyright 2019. | ||
The Park group developed a bifunctional superaerophobic/superhydrophilic heterostructured catalyst on NF designed for high-rate water splitting.88 The catalyst is composed of one-dimensional (1D) Ni3S2 nanorods, which are coated by zero-dimensional (0D) PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) nanoparticles, as shown in Fig. 5a. This unique morphology enhances the availability of active sites at the exposed regions of the heterointerface regions, enabling a high electrochemical active surface area (ECSA). Additionally, this increased exposure of active sites provides an effective pathway for mass transfer at the TPCL. As a result, the catalyst promotes enhanced bubble release, with bubbles detaching from the PBSCF-Ni3S2 electrode with a smaller average diameter of 83.6 μm, compared to 392.6 μm for NF and 187.4 μm for Ni3S2 (Fig. 5b–g). The PBSCF-Ni3S2 catalyst achieved a current density of 1000 mA cm−2 at 1.793 V (Fig. 5h) and demonstrated stability for 500 hours at a current density of 500 mA cm−2 (Fig. 5i), with no noticeable degradation or change in morphology, chemical composition, crystal structure or bonding. In a different approach, the Ryu group introduced a method to impart superaerophobicity to various electrodes by coating them with a porous polymeric hydrogel.93 Substrates, either NF or flat Pt, were first functionalized with amine groups, and then coated by crosslinking with polyethyleneimine (PEI). The coating's pore size, porosity and superaerophobicity were controlled by varying concentrations of PEI. Concentrations between 2.0% and 7.3% PEI resulted in a porous network structure, with pore size and porosity decreasing as PEI concentration increased (Fig. 5j–m). The 2.0% PEI electrode exhibited the highest aerophobicity, with a bubble contact angle of 153.7° (Fig. 5n), likely due to the agglomeration of PEI and the covering of hydrophilic amine groups at higher PEI concentrations. This electrode also demonstrated the best HER performance, outperforming other HER electrodes at high current densities (Fig. 5o). The structural stability of the hydrogel-modified electrodes was confirmed with repeated CV cycles and chronopotentiometry tests (Fig. 5p). The 2.0% PEI hydrogel maintained its superaerophobicity and porous structure after testing at 500 mA cm−2 for 20 hours (Fig. 5q).
 | ||
| Fig. 5 (a) SEM image of PBSCF-Ni3S2. Images of the generated bubbles on (b) NF, (c) Ni3S2, and (d) PBSCF-Ni3S2. Dashed circles represent the gas bubbles produced. Bubbles size distributions in (e) NF, (f) Ni3S2, and (g) PBSCF-Ni3S2. (h) LSV of PBSCF-Ni3S2||PBSCF-Ni3S2 and Pt/C||IrO2 coupled electrodes for overall water electrolysis in 1.0 M KOH. Dashed lines indicate cell voltages at 100 and 500 mA cm−2. (i) Stability test of PBSCF-Ni3S2||PBSCF-Ni3S2 and Pt/C||IrO2 at a constant current density of 500 mA cm−2. Reproduced from ref. 88 with permission from ACS, copyright 2024. SEM images of (j) Bare NF, (k) PEI-0.5%, (l) PEI-2.0%, and (m) PEI-7.3%. (n) Air contact angles of the NF-based electrodes. (o) LSV of various HER electrodes (without iR correction). (p) Chronopotentiometry of bare and hydrogel-modified NF electrodes at 500 mA cm−2 for 20 h without external convection. (q) SEM images of the hydrogel-modified NF electrodes after the stability test, with the inset showing air contact angles. Reproduced from ref. 93 with permission from Wiley, copyright 2022. | ||
4.3. 3D-printed substrates
Additive manufacturing, commonly known as 3D printing, enables the creation of advanced electrode architectures, enhancing the performance of electrochemical devices.102–106 The complex architecture of NF, with disordered pore sizes and irregular spatial distribution, can hinder efficient gas removal at high current densities, thereby limiting performance. 3D printing techniques present a promising solution to these challenges by enabling the fabrication of electrodes with well-defined, ordered architectures. This approach allows precise control over channel design, optimizing reactant flow and gas bubble transport to enhance mass transport and overall performance. Moreover, the flexibility of 3D printing makes it possible to customize 3D electrode structures for specific operational requirements. Almost any electrode geometries can be realized through computer programming.107 Additionally, 3D printing technology supports a wide range of materials, including polymers, metals and metal oxides, carbon, and ceramics.108,109 The highly automated manufacturing process and straightforward synthesis routes also help reduce the cost of production compared to traditional methods. Various 3D-printing techniques are currently available, including direct ink writing (DIW),110 selective laser sintering (SLS),111 fused deposition modeling (FDM),112 selective laser melting (SLM),112 digital light processing (DLP),113 and binder jetting (BJ).114Kou et al. developed catalyst-decorated 3D-printed Ni lattice-based electrodes (3DPNi) using the DIW technique for AWS. These electrodes feature periodic channels and open structures that effectively suppress gas bubble coalescence, jamming, and trapping, resulting in more efficient gas bubble transport and release compared to NF-based electrodes with randomly distributed pores (Fig. 6a and b).37 Simulation studies (Fig. 6c–e) reveal that the critical bubble size (dc) at which bubbles become trapped in the porous medium is larger in the periodic 3DPNi electrode (dc = 29) compared to the NF structure (dc = 20). As shown in Fig. 6h and i, the 3D-printed Ni lattice-based electrode exhibits superior HER and OER performance, particularly at high current densities. This enhanced performance was attributed to its periodic channels that enable smoother bubble transport and release.
 | ||
| Fig. 6 High-speed camera images of (a) C-Ni1−xO/NF and (b) C-Ni1−xO/3DPNi electrodes during overall AWS. Scale bars are 2 mm. Structure model of (c) 3DPNi and (d) NF. (e) Relative bubble migration time through 3DPNi and NF as a function of bubble diameter, determined by time to cross the central plane of the structure (units are dimensionless). (f and g) Simulation frames showing the shape of a bubble (d = 20) during transport in (f) 3DPNi and (g) NF. The arrow in the inset of (g) highlights an interaction with the NF surface, which causes bubble deformation. LSV of C-Ni1−xO/NF and C-Ni1−xO/3DPNi collected in 1.0 M KOH at a scan rate of 0.1 mV s−1 for (h) HER and (i) OER. Reproduced from ref. 37 with permission from Wiley, copyright 2020. (j) Examples of triply periodic minimal surfaces (TPMS) geometries, all with a lattice size of 8 mm and a t parameter of 0.26. Arrows indicate the direction of forced electrolytic flow. (k) Velocity streamlines for structures with the same theoretical surface area (32 cm2) and porosity (61%) in Schwarz: L = 4 and t = 0.18; Gyroid: L = 3.94 and t = 0.34; and Fischer–Koch: L = 6.5 and t = 0.25 structure. (l) Flow sensitivity at a projected current density of 0.4 A cm−2 for structures with a measured ECSA around 100 cm2 (Gyroid: L = 4 mm, t = 1.2; Fischer–Koch: L = 8 mm, t = 0.7; Schwarz: L = 4 mm, t = 0.26). Reproduced from ref. 115 with permission from Wiley, copyright 2023. | ||
Building on this, recent innovations have further explored the role of 3D-printed electrode geometry in optimizing gas removal and overall efficiency. For instance, the Proost group developed tailored 3D-printed electrodes with various geometries, such as Gyroid, Fischer–Koch, and Schwarz CLP structures (Fig. 6j).115 These geometries, characterized by their zero-mean curvature, are ideal for enhancing gas removal due to their three-dimensional periodicity. A key advantage of these designs is the ability to independently adjust porosity, which affects the available surface area, and pore size or flow channel dimensions, which influence bubble entrapment. Computational models reveal that different electrode geometries create distinct flow patterns. For example, as displayed in Fig. 6k, Gyroid and Fischer–Koch structures induce lateral flow mixing, leading to increased pressure drop at a constant vertical velocity, which may encourage bubble coalescence. In contrast, the Schwarz structure, with its unique symmetry along one axis, directs flow vertically, reducing pressure drop. This characteristic suggests that the Schwarz geometry may be the most effective among the analyzed structures, given its inherent high permeability and notable flow sensitivity, offering the highest electrocatalytic efficiency for AWS (Fig. 6l).
The Huang group utilized DLP technology to construct a 3D porous nickel structure (3DP Ni) with a precisely designed periodic arrangement and unique surface chemistry.95 As shown in Fig. 7a–d, the average diameter of bubbles formed on the 3DP Ni surface is significantly smaller (d(average) = 13 μm) compared to those on NF (d(average) = 500 μm), with a reduced number of bubbles as well. The macroscopic ordered pores in the structure facilitate rapid bubble evolution and release (Fig. 7e–l), while the microporosity contributes to a high ECSA. As previously discussed, lateral mixing can promote bubble coalescence, so structures like gyroids that can direct flow are preferred. This design enhances the efficiency of continuous gas evolution reactions in industrial production. When further loaded with MoNi4 and NiFe LDH active materials, the 3DP Ni structure achieves low overpotentials of 104 mV for the HER and 310 mV for the OER at a current density of 500 mA cm−2. Additionally, they demonstrated a fully 3D-printed electrolyzer using 3DP Ni composite electrodes (Fig. 7m). The device achieved a low overpotential of 1.63 V and maintained performance at a high current density of 500 mA cm−2 for 1000 hours (Fig. 7n).
 | ||
| Fig. 7 SEM images of (a) a 3DP Ni and (c) a NF electrode. Optical microscope images showing the bubble evolution behavior at the same voltage for the (b) 3DP Ni and (d) NF electrodes. Camera images of bubble release from (e–h) 3DP Ni and (i–l) NF structures at different time intervals. Red and blue dotted lines highlight the first and second bubbles, respectively. (m) Digital images of the fully 3D-printed electrolyzer, showing the membrane separating hydrogen from oxygen during water splitting. (n) LSV of MoNi4-MoO2/3DP Ni||NiFe LDH/3DP Ni and MoNi4-MoO2/NF||NiFe LDH/NF devices for overall water splitting. Reproduced from ref. 95 with permission from ACS, copyright 2023. (o and p) Schematic diagrams illustrating the preparation of 3DP NiMoAS and 3DP NiMoIS, respectively. (q and r) Digital images of 3DP NiMoAS and 3DP NiMoIS electrodes captured during the HER process. The marked potentials are versus RHE. (s) LSV for HER of 3DP NiMoAS and 3DP NiMoIS. Reproduced from ref. 96 with permission from Elsevier, copyright 2023. | ||
Architected electrodes have also been used in seawater electrolysis to promote catalyst-electrolyte interaction and gas bubble releasing. The Lu group used DIW technology to print two 3D electrodes with different internal porous channel structures, utilizing distinct NiMo-based nanostructures: nanorods and nanospheres. They printed both anisotropic (3DP NiMoAS) and isotropic structures (3DP NiMoIS) (Fig. 7o and p).96 The 3DP NiMoAS maintained a long-range ordered structure under well-controlled processing parameters. This unique structure allowed nearly all electrocatalysts within the internal space to participate in the reaction driven by capillary pressure, while also facilitating rapid electrolyte-hydrogen phase conversion during electrochemical reactions. When the 3DP NiMoIS was used as the cathode, hydrogen gas bubbles were released only from the electrode surface (Fig. 7r). In contrast, for the 3DP NiMoAS electrode, a larger number of bubbles emerged from the tip region (Fig. 7q). The obtained 3DP NiMoAS exhibited superior electrocatalytic performance and excellent long-term operational stability, with an extremely low overpotential of 150 mV at a current density of 500 mA cm−2 in 1.0 M KOH seawater (Fig. 7s).
While the 3D-printed electrodes promote gas bubbles releasing from porous structures, they are not helping on the bubble nucleation and detachment. To address the issues of bubble detachment during AWS, Ren et al. successfully enhance the wettability of a 3D-printed Ni lattice electrode by decorating it with Ni nanocone (NC) structures (Fig. 8a–d), thereby facilitating gas bubble detachment.81 As shown in Fig. 8e and f, BCA measurements reveal that, compared to a flat Ni electrode (134.6°), the nanocone-modified Ni electrode exhibits superaerophobic properties with a contact angle of 151.8°. The rough structures significantly reduce the hydrogen gas bubble detachment time on the NC-decorated electrode to 3.05 ± 6 s, compared to 56.90 ± 26.18 s on the flat surface. Additionally, the gas bubble detachment size on the NC-modified electrode (97 ± 42 μm) is significantly smaller than that on the flat Ni electrode (379 ± 121 μm), with a similar trend observed for oxygen gas bubbles (Fig. 8g–j). Significantly, the NC surface modification approach was also successfully applied on the 3D-printed nickel lattice (Lattice_NC). As shown in Fig. 8k and l, the Lattice_NC shows substantially reduced HER and OER overpotentials at 1000 mA cm−2 compared to the pristine lattice (Lattice). The electrolyzer, assembled with two Lattice_NC electrodes, retains over 95% of its performance after testing at approximately 900 mA cm−2 for 100 hours, demonstrating excellent electrochemical durability.
 | ||
| Fig. 8 Schematic illustrations show bubble contact angle, area and evolution on (a) a smooth Ni surface and (b) a NC-modified Ni surface. SEM images of (c) Foil and (d) Foil_NC. Scale bars are 1 μm. Digital images showing air contact angles on (e) Foil and (f) Foil_NC. Histograms illustrate the distribution of (g and h) bubble detachment time (Td) and (i and j) detachment size (Dd) on Foil and Foil_NC substrates. LSV of Lattice and Lattice_NC electrodes collected in 1.0 M KOH at a scan rate of 0.1 mV s−1 for (k) HER and (l) OER. Reproduced from ref. 81 with permission from Wiley, copyright 2023. | ||
In a membrane electrolyzer, the gas diffusion layer (GDL) position between the catalyst layer (CL) and the flow field plate plays a vital role in removing gas bubbles. Efficient bubble escape within the GDL significantly reduces the dead volume of gas products and decreases gas saturation at the GDL/CL interface, thereby retaining bubble formation in the CL.116,117 Thanks to advanced 3D printing techniques, the geometry of the GDL can be precisely tailored to optimize mass transport.
The Zhuang group developed a Ni GDL (GDL3D print) with straight-through pores and a three-dimensional periodic structure using the DIW technique (Fig. 9a).118 In a pure-water-fed anion exchange membrane water electrolysis (AEMWE), the GDL3D print achieved an industrial current density of 1000 mA cm−2 at 1.8 V, compared to 2.0 V required for a NF electrode to reach the same current density (Fig. 9b). Using an in situ high-speed camera (Fig. 9c), it was observed that the average size of bubbles released from the GDL3D print surface (450 μm) was considerably smaller than those from the NF surface (600 μm on average) at 1000 mA cm−2. The GDL3D print also improved bubble release dynamics, allowing oxygen bubbles to fully detach from its surface within 0.05 s, while NF required more time. Adhesion measurements showed that the bubble adhesion force on NF was 19.7 μN, approximately three times higher than that on the GDL3D print (6.3 μN). The higher hydrophilicity and aerophobicity of the GDL3D print reduce the contact area between bubbles and the GDL surface, resulting in a lower adhesion force (Fig. 9d–g), which explains the smaller bubble detachment diameter on the GDL3D print compared to NF. Furthermore, the Zhuang group used DIW technology to 3D print Ni GDLs with various grid sizes: 40, 100, 240, 380, and 440 μm (Fig. 9h).119 Among these, the grid size of 240 μm (Ni-240) demonstrated the best AEMWE cell performance (Fig. 9i–k). High-speed camera observations, shown in Fig. 9l, provided statistical results of bubble detachment diameter and bubble residence time for different GDLs with various grid sizes. For example, the diameter of bubbles released from Ni-440 was 304 ± 77 μm, while it was 788 ± 152 μm for Ni-40 at a current density of 1000 mA cm−2. The bubbles residence time was 57 ± 24 ms on Ni-40 but mostly only 10 ms on Ni-440. Additionally, bubbles release from the electrode with the 440 μm grid size without contacting the grids, indicating that the contact area can be zero when the grid is sufficiently large, resulting in almost zero adhesion. However, this also leads to lower catalyst utilization due to higher in-plane electrical resistance, which explains why Ni-240 provides the best cell efficiency by balancing mass transport and catalyst utilization.
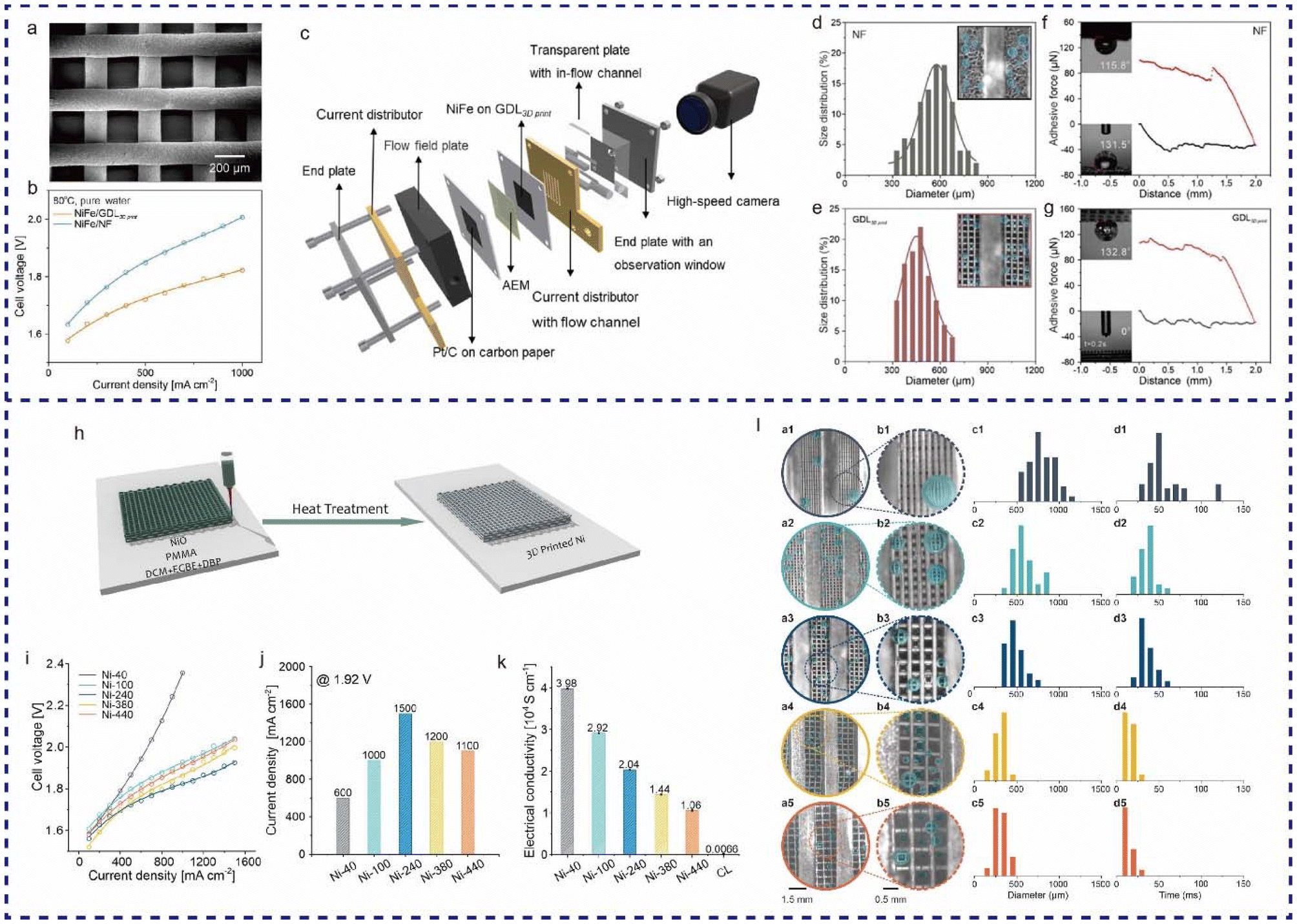 | ||
| Fig. 9 (a) Optical microscope images of GDL3D print. (b) performance comparison curves of pure-water-fed AEMWE between NiFe/GDL3D print and NiFe/NF at 80 °C. (c) Schematic of an AEMWE with a high-speed camera and an in situ observation system. Size distribution statistics of oxygen bubbles released form (d) NF and (e) GDL3D print in an operating AEMWE at 1000 mA cm−2. Insets show the corresponding images of bubbles from NF and GDL3D print. Adhesion force measurement of oxygen bubbles on (f) NF and (g) GDL3D print. Insets show the corresponding images of bubble contact angles and liquid contact angles. Reproduced from ref. 118 with permission from Elsevier, copyright 2023. (h) Schematic of the preparation process for 3D printed Ni GDLs. (i) LSV and (j) performance comparison of 3D printed GDLs with different grid sizes in pure water at 80 °C. (k) Conductivity comparison of the catalyst layer and 3D printed GDLs with different grid sizes. (l) Photos of bubbles released from 3D printed Ni GDLs with different grid sizes, observed in a circular pore of an operating AEMWE at 1000 mA cm−2, (a1 and b1) Ni-40, (a2 and b2) Ni-100, (a3 and b3) Ni-240, (a4 and b4) Ni-380, and (a5 and b5) Ni-440. (c1–c5) Size distribution statistics of oxygen bubbles released form 3D printed Ni GDLs with different grid sizes. (d1–d5) Time distribution statistics of bubbles growth and detachment on the surface of 3D printed Ni GDLs with different grid sizes. Reproduced from ref. 119 with permission from Wiley, copyright 2023. | ||
Furthermore, by leveraging 3D printing techniques, the water electrolyzer device can be optimized to improve efficiency through enhanced mass transport and ion diffusion. Ren et al. recently developed a novel device featuring 3D interpenetrating gyroid electrodes using DLP technology, specifically designed to improve mass transport and ion diffusion in overall AWS.120 This innovative configuration includes two intertwined bicontinuous struts with smoothly curved surfaces, promoting efficient bubble evacuation (Fig. 10a–c). The use of two lattices with opposite chirality enables the creation of an interpenetrating topology. The open structure of each single electrode in this compact design ensures rapid bubble release. The 3D interpenetrating gyroid device (3DIG) achieves an ultrahigh current density of 1000 mA cm−2 at 2.85 V, outperforming a control device with two separate 3D gyroid electrodes (3DG), which only reaches 225 mA cm−2 at the same voltage (Fig. 10d). This performance enhancement is attributed to the reduced distance between the electrodes in the 3DIG device. Electrochemical Impedance Spectroscopy (EIS) analysis confirms that the 3DIG exhibits lower solution (Rs) and charge transfer resistance (RCT), facilitating ion diffusion and enhancing reaction efficiency (Fig. 10e and f).
 | ||
| Fig. 10 Schematic illustration of (a) a 3DIG device and (b) a 3DG device. (c) SEM image of the 3DIG device, showing its thickness and the pore size. (d) LSVs of 3DIG and 3DG, collected in 1.0 M KOH at a scan rate of 1 mV s−1. Histograms display the (e) RS and (f) RCT values for both 3DIG and 3DG at voltages corresponding to a current density of 10 mA cm−2. Reproduced from ref. 120 with permission from ACS, copyright 2024. | ||
5. Conclusion and outlook
This review highlights the significant advantages of integrating superwetting strategies with architected electrodes to tackle the issues of bubble formation, detachment and transport for high-rate water splitting. We have summarized recent works to showcase various approaches and their effectiveness. These electrodes possess unique properties that enhance both the efficiency and stability of the water-splitting process, making them highly promising for industrial applications. Despite these advancements, there remain several promising directions for further exploration.First, stability is a crucial factor when evaluating electrode materials for high-rate water splitting. These systems must reliably operate over extended periods – often thousands of hours – under harsh conditions such as elevated temperatures, pressure, strong acidic or alkaline electrolytes. Maintaining stability, both electrochemical and mechanical, is critical. Electrode materials must be robust and resilient to withstand these demanding conditions without significant degradation in electrochemical performance. Advanced electrode materials, such as 3d transition metal oxides and hydroxides, have shown promise due to their abundance, great stability, and tunable structure.121–123 Moreover, superwetting properties are typically related to electrodes’ nanostructured surface features such as nanoflakes and nanocones. While these nanostructures enhance the electrode/water interaction and facilitate gas bubbles detachment, they often undergo surface reconstruction during water splitting process.124–126 Electrode wettability can degrade over time due to structural deterioration under harsh electrolytic conditions. Prolonged exposure to reactive intermediates and gas bubbles can alter surface properties, leading to reduced electrolysis and bubble mitigation efficiency. Advanced electrode designs have been investigated to improve wettability retention during extended operation. For example, superwetting electrodes with nanostructures have demonstrated enhanced stability and sustained superaerophobic properties.25 Additionally, durable surface coatings, such as corrosion-resistant hydrophilic polymers, can minimize degradation, while adaptive wettability features that dynamically adjust to operational conditions offer promising solutions.93 Among these advancements, 3D-printed electrodes stand out for their unique benefits. Their ordered channels and customizable surface properties enhance electrolyte flow, promote efficient gas bubble releasing, and maintain wettability during prolonged operation.37 For superwetting electrodes, ensuring that these materials retain their superwetting properties and structural integrity throughout the water splitting process is vital for efficient gas bubble removal and overall system performance.
Second, the use of 3D printing technology to optimize electrode structure for gas bubble detachment and release is highly desirable. The 3D-printed Schwarz structure, for example, shows great potential for efficient gas diffusion. Nanostructuring to enhance aerophobicity further aids in controlling bubble dynamics. Combining 3D printed geometries with superwetting surface design can significantly improve the efficiency of high-rate water splitting by facilitating both gas bubble detachment and transport. Advancements in 3D printing and computational simulations can accelerate the development of optimized superwetting electrode materials and customized electrolyzer devices, enhancing overall performance and durability.
Third, observing bubble behavior under industrial operation conditions using in situ techniques is crucial for understanding the mechanisms governing bubble evolution and interface chemistry. First, it is essential to observe bubble formation and dynamics under varying current densities. Besides, industrial water splitting typically occurs at higher temperatures (80 °C) and pressures (several hundred psi),127–131 which are significantly different from the experimental settings commonly reported in literature. Elevated temperatures enhance reaction kinetics and reduce the overpotentials required for HER and OER, thereby improving catalytic activity. However, high temperatures also present challenges, including material degradation through corrosion and loss of structural integrity. To address these issues, superwetting electrodes must be fabricated using thermally stable materials capable of maintaining performance under such conditions. Similarly, elevated pressures introduce mechanical stress, increasing the risk of deformation or cracking. Materials with strong mechanical properties are crucial to ensuring electrode stability. While higher pressure reduces bubble size, it can also increase bubble adherence to the electrode surface, potentially hindering gas release. Additionally, porous or layered electrode architectures can balance mass transport, mechanical robustness, and thermal expansion. These designs help accommodate the stresses induced by high temperatures and pressures, ensuring sustained performance under industrial operating conditions.
Fourth, seawater, constituting over 96% of Earth's water, is an abundant resource, making it a promising candidate for large-scale water electrolysis. The principles of electrode design are similar for seawater electrolysis; however, seawater electrolysis poses additional challenges compared to freshwater electrolysis. Seawater contains high concentrations of salts and impurities, particularly chloride ions (Cl−), which drive the chlorine evolution reaction at the anode. This reaction produces chlorine gas, compromising the stability of both the catalyst and the electrolytic cell. Additionally, cations such as Ca2+ and Mg2+, along with microbial impurities, can accumulate on the electrode surface, forming fouling layers that diminish efficiency. To address these issues, it is crucial to develop superwetting electrodes and catalysts with high selectivity for the OER and long-term stability to mitigate the effects of Cl− at industrially relevant current densities.
Finally, beyond scientific and technological challenges, economic considerations are crucial for developing superwetting electrodes. It is essential to evaluate the costs associated with the production processes, including raw materials, electrode fabrication including the architected electrode and superwetting surface features, and energy consumption during electrochemical reactions. Efforts to develop inexpensive and efficient fabrication processes is critical to ensure large-scale and sustainable implementation of superwetting electrodes for water splitting technologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Y. L. thanks the financial support from the faculty research grant and the Center for Coastal Climate Resilience of the University of California, Santa Cruz. This work was supported by Lawrence Livermore National Laboratory (LLNL) under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344. The LLNL information management release number is LLNL-JRNL-869412.References
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33(31), 2007100 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137(7), 2688–2694 CrossRef CAS PubMed.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2024, 36(4), e2306108 CrossRef PubMed.
- H. Xu, J. Zhang, M. Eikerling and J. Huang, J. Am. Chem. Soc., 2024, 146(29), 19720–19727 CrossRef CAS PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28(2), 215–230 CrossRef CAS PubMed.
- T. Naito, T. Shinagawa, T. Nishimoto and K. Takanabe, ChemSusChem, 2020, 13(22), 5921–5933 CrossRef CAS PubMed.
- X. M. Xu, Z. P. Shao and S. P. Jiang, Energy Technol., 2022, 10(11), 2200573 CrossRef CAS.
- R. T. Liu, Z. L. Xu, F. M. Li, F. Y. Chen, J. Y. Yu, Y. Yan, Y. Chen and B. Y. Xia, Chem. Soc. Rev., 2023, 52(16), 5652–5683 RSC.
- N. Thissen, J. Hoffmann, S. Tigges, D. A. M. Vogel, J. J. Thoede, S. Khan, N. Schmitt, S. Heumann, B. J. M. Etzold and A. K. Mechler, ChemElectroChem, 2023, 11(1), e202300432 CrossRef.
- J. W. Cao, Y. F. Li, Y. Zheng, S. B. Wang, W. Q. Zhang, X. F. Qin, G. Geng and B. Yu, Adv. Energy Mater., 2022, 12(28), 2200899 CrossRef CAS.
- P. Sun, Z. Qiao, X. Dong, R. Jiang, Z.-T. Hu, J. Yun and D. Cao, J. Am. Chem. Soc., 2024, 146(22), 15515–15524 CrossRef CAS PubMed.
- Z. Shi, J. Li, Y. Wang, S. Liu, J. Zhu, J. Yang, X. Wang, J. Ni, Z. Jiang, L. Zhang, Y. Wang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2023, 14(1), 843 CrossRef CAS PubMed.
- H. Y. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11(39), 2102074 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38(12), 4901–4934 CrossRef CAS.
- P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukauloo and C. Etiévant, Int. J. Hydrogen Energy, 2011, 36(6), 4134–4142 CrossRef CAS.
- H. Tuysuz, Alkaline Water Electrolysis for Green Hydrogen Production, Acc. Chem. Res., 2024, 57(4), 558–567 Search PubMed.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- Z. X. Wang, Y. Wang, N. Z. Li, Y. C. Tong, Y. Teng, D. Wang, C. S. Chen and Z. L. Zhan, Int. J. Hydrogen Energy, 2023, 48(35), 12949–12957 CrossRef CAS.
- Y. Wang, W. Y. Li, L. Ma, W. Li and X. B. Liu, J. Mater. Sci. Nanotechnol., 2020, 55, 35–55 CrossRef CAS.
- T. Holm, T. Borsboom-Hanson, O. E. Herrera and W. Mérida, Energy Convers. Manage., 2021, 237, 114106 CrossRef CAS.
- Y. Li, Y. Peng, W. Dong, X. Jiang, L. Lu, D. Yang, L. C. Hsu, W. Li, B. Su and A. Lei, J. Am. Chem. Soc., 2024, 146(20), 14194–14202 CrossRef CAS PubMed.
- A. Badgett, M. Ruth, B. James and B. Pivovar, Curr. Opin. Chem. Eng., 2021, 33, 100714 CrossRef.
- S. Yuan, C. F. Zhao, X. Y. Cai, L. An, S. Y. Shen, X. H. Yan and J. L. Zhang, Prog. Energy Combust. Sci., 2023, 96, 101075 CrossRef.
- R. Wu, Z. Hu, H. Zhang, J. Wang, C. Qin and Y. Zhou, Langmuir, 2024, 40(1), 721–733 CrossRef CAS PubMed.
- Q. Ren, L. S. Feng, C. W. Ye, X. Z. Xue, D. Lin, S. Eisenberg, T. Y. Kou, E. B. Duoss, C. Zhu and Y. T. Li, Adv. Energy Mater., 2023, 13(39), 2302073 CrossRef CAS.
- K. Zouhri and S. Y. Lee, Int. J. Hydrogen Energy, 2016, 41(18), 7253–7263 CrossRef CAS.
- C. Immerz, B. Bensmann, P. Trinke, M. Suermann and R. Hanke-Rauschenbach, J. Electrochem. Soc., 2019, 166(15), F1200–F1208 CrossRef CAS.
- G. Schmidt, M. Suermann, B. Bensmann, R. Hanke-Rauschenbach and I. Neuweiler, J. Electrochem. Soc., 2020, 167(11), 114511 CrossRef CAS.
- G. Sakuma, Y. Fukunaka and H. Matsushima, Int. J. Hydrogen Energy, 2014, 39(15), 7638–7645 CrossRef CAS.
- H. Matsushima, D. Kiuchi, Y. Fukunaka and K. Kuribayashi, Electrochem. Commun., 2009, 11(8), 1721–1723 CrossRef CAS.
- Y. L. Xu, C. Wang, Y. H. Huang and J. Fu, Nano Energy, 2021, 80, 105545 CrossRef CAS.
- D. Kitajima, R. Misumi, Y. Kuroda and S. Mitsushima, Electrochim. Acta, 2024, 144772 CrossRef CAS.
- S. Yuan, C. F. Zhao, X. H. Mei, S. Y. Shen, Q. Wang, X. H. Yan and J. L. Zhang, Int. J. Heat Mass Transfer, 2023, 212, 124249 CrossRef CAS.
- P. Mandin, Z. Derhoumi, H. Roustan and W. Rolf, Electrochim. Acta, 2014, 128, 248–258 CrossRef CAS.
- J. T. Davis, D. E. Brown, X. Pang and D. V. Esposito, J. Electrochem. Soc., 2019, 166(4), F312–F321 CrossRef CAS.
- D. K. Zhang and K. Zeng, Ind. Eng. Chem. Res., 2012, 51(42), 13825–13832 CrossRef CAS.
- T. Y. Kou, S. W. Wang, R. P. Shi, T. Zhang, S. Chiovoloni, J. Q. Lu, W. Chen, M. A. Worsley, B. C. Wood, S. E. Baker, E. B. Duoss, R. Wu, C. Zhu and Y. Li, Adv. Energy Mater., 2020, 10(46), 2002955 CrossRef CAS.
- M. M. Bakker and D. A. Vermaas, Electrochim. Acta, 2019, 319, 148–157 CrossRef CAS.
- G. F. Swiegers, R. N. L. Terrett, G. Tsekouras, T. Tsuzuki, R. J. Pace and R. Stranger, Sustainable Energy Fuels, 2021, 5(5), 1280–1310 RSC.
- G. B. Darband, M. Aliofkhazraei and S. Shanmugam, Renewable Sustainable Energy Rev., 2019, 114, 109300 CrossRef CAS.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Rivas, Joule, 2020, 4(3), 555–579 CrossRef CAS.
- N. P. Brandon and G. H. Kelsall, J. Appl. Electrochem., 1985, 15(4), 475–484 CrossRef CAS.
- X. L. Lu, D. Yadav, B. C. Ma, L. J. Ma and D. W. Jing, J. Power Sources, 2024, 599, 234217 CrossRef CAS.
- Z. D. Wei, M. B. Ji, S. G. Chen, Y. Liu, C. X. Sun, G. Z. Yin, P. K. Shen and S. H. Chan, Electrochim. Acta, 2007, 52(9), 3323–3329 CrossRef CAS.
- H. P. Wang, Z. X. Xu, W. Lin, X. Yang, X. R. Gu, W. Zhu and Z. B. Zhuang, Nano Res., 2023, 16(1), 420–426 CrossRef CAS.
- M. Y. Wang, Z. Wang, X. Z. Gong and Z. C. Guo, Renewable Sustainable Energy Rev., 2014, 29, 573–588 CrossRef CAS.
- T. Iida, H. Matsushima and Y. Fukunaka, J. Electrochem. Soc., 2007, 154(8), E112–E115 CrossRef CAS.
- K. M. Cho, P. R. Deshmukh and W. G. Shin, Ultrason. Sonochem., 2021, 80, 105796 CrossRef CAS PubMed.
- M. Y. Lin, L. W. Hourng and C. W. Kuo, Int. J. Hydrogen Energy, 2012, 37(2), 1311–1320 CrossRef CAS.
- H. Matsushima, D. Kiuchi and Y. Fukunaka, Electrochim. Acta, 2009, 54(24), 5858–5862 CrossRef CAS.
- X. Duan, J. Xiao, W. Lin, S. Wang and J. Wen, Int. J. Hydrogen Energy, 2024, 49, 404–416 CrossRef CAS.
- S. D. Li, C. C. Wang and C. Y. Chen, Electrochim. Acta, 2009, 54(15), 3877–3883 CrossRef CAS.
- W. Li, H. C. Tian, L. Ma, Y. Wang, X. B. Liu and X. F. Gao, Mater. Adv., 2022, 3(14), 5598–5644 RSC.
- L. Ionov, Biomimetic Hydrogel-Based Actuating Systems, Adv. Funct. Mater., 2013, 23(36), 4555–4570 CrossRef CAS.
- A. R. Parker and H. E. Townley, Nat. Nanotechnol., 2007, 2(6), 347–353 CrossRef CAS PubMed.
- H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24(24), 4353–4364 CrossRef CAS PubMed.
- M. Kuang, J. Wang and L. Jiang, Chem. Soc. Rev., 2016, 45(24), 6833–6854 RSC.
- W. Xu, Z. Lu, P. Wan, Y. Kuang and X. Sun, Small, 2016, 12(18), 2492–2498 CrossRef CAS PubMed.
- M. Li, P. Xie, L. Yu, L. Luo and X. Sun, ACS Nano, 2023, 17(23), 23299–23316 CrossRef CAS PubMed.
- K. Ando, Y. Uchimoto and T. Nakajima, J. Phys. Chem. C, 2022, 126(44), 18988–18993 CrossRef CAS.
- G. B. Darband, M. Aliofkhazraei and S. Shanmugam, Renewable Sustainable Energy Rev., 2019, 114, 109300 CrossRef CAS.
- J. S. Qin, T. H. Xie, D. J. Zhou, L. Luo, Z. Y. Zhang, Z. C. Shang, J. W. Li, L. Mohapatra, J. W. Yu, H. J. Xu and X. M. Sun, Nano Res., 2021, 14(7), 2154–2159 CrossRef CAS.
- Y. L. Hu, Y. F. Yang, Z. Zeng, T. X. Zhou, F. Yang, W. Sun and L. L. He, Int. J. Hydrogen Energy, 2024, 61, 754–761 CrossRef CAS.
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2022, 34(16), e2108133 CrossRef PubMed.
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. T. Chen, G. T. K. K. Gunasooriya, I. McCrum, R. Mom, N. López and M. T. M. Koper, Water electrolysis, Nat. Rev. Methods Primers, 2022, 2(1), 84 CrossRef CAS.
- Y. C. Wan, L. X. Zhou and R. T. Lv, Mater. Chem. Front., 2023, 7(23), 6035–6060 RSC.
- Y. X. Zhao, Q. Y. Sun, C. F. Zhang, F. S. Liu, L. Wang and G. R. Xu, J. Alloys Compd., 2023, 968, 172286 CrossRef CAS.
- X. Zhang, M. T. Jin, F. F. Jia, J. Q. Huang, A. Amini, S. X. Song, H. Yi and C. Cheng, Energy Environ. Mater., 2023, 6(5), e12457 CrossRef CAS.
- Technical Targets for Proton Exchange Membrane Electrolysis. https://www.energy.gov/eere/fuelcells/technical-targets-proton-exchange-membraneelectrolysis.
- Annual Work Plan 2014–2020. https://www.fch.europa.eu/page/multi-annual-work-plan.
- N. K. Adam, Nature, 1957, 180(4590), 809–810 CrossRef.
- H. J. Butt, D. S. Golovko and E. Bonaccurso, J. Phys. Chem. B, 2007, 111(19), 5277–5283 CrossRef CAS PubMed.
- P. Letellier, A. Mayaffre and M. Turmine, J. Colloid Interface Sci., 2007, 314(2), 604–614 CrossRef CAS PubMed.
- J. C. Garcia-Navarro, M. Schulze and K. A. Friedrich, Int. J. Hydrogen Energy, 2019, 44(50), 27190–27203 CrossRef CAS.
- P. Tiwari, G. Tselzouras, K. Wagner, G. F. Swiegers and G. G. Wallace, Int. J. Hydrogen Energy, 2019, 44(42), 23568–23579 CrossRef CAS.
- S. Jeong, U. Kim, S. Lee, Y. Zhang, E. Son, K.-J. Choi, Y.-K. Han, J. M. Baik and H. Park, ACS Nano, 2024, 18(10), 7558–7569 CrossRef CAS PubMed.
- L. Li, P. C. M. Laan, X. Yan, X. Cao, M. J. Mekkering, K. Zhao, L. Ke, X. Jiang, X. Wu, L. Li, L. Xue, Z. Wang, G. Rothenberg and N. Yan, Adv. Sci., 2023, 10(4), 2206180 CrossRef CAS PubMed.
- J. Das, S. Mandal, A. Borbora, S. Rani, M. Tenjimbayashi and U. Manna, Adv. Funct. Mater., 2023, 34(6), 2311648 CrossRef.
- R. C. Iwata, L. N. Zhang, K. L. Wilke, S. Gong, M. F. He, B. M. Gallant and E. N. Wang, Joule, 2021, 5(4), 887–900 CrossRef CAS.
- J. Li, S. Gong, L. Zhang, P. Cheng, X. Ma and F. Hong, Langmuir, 2022, 38(10), 3180–3188 CrossRef CAS PubMed.
- Q. Ren, L. Feng, C. Ye, X. Xue, D. Lin, S. Eisenberg, T. Kou, E. B. Duoss, C. Zhu and Y. Li, Adv. Energy Mater., 2023, 13(39), 2302073 CrossRef CAS.
- G. Duhar and C. Colin, Phys. Fluids, 2006, 18(7), 077101 CrossRef.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang, J. Luo, X. Lei and L. Jiang, Adv. Mater., 2014, 26(17), 2683–2687 CrossRef CAS PubMed , 2615.
- Y. J. Li, H. C. Zhang, T. H. Xu, Z. Y. Lu, X. C. Wu, P. B. Wan, X. M. Sun and L. Jiang, Adv. Funct. Mater., 2015, 25(11), 1737–1744 CrossRef CAS.
- C. Zhang, Z. Xu, N. Han, Y. Tian, T. Kallio, C. Yu and L. Jiang, Sci. Adv., 2023, 9(3), eadd6978 CrossRef PubMed.
- C. Zhang, C. Teng, S. Guo, Y. Sun, C. Yu, X. Jin, K. Liu and L. Jiang, Nano Lett., 2024, 24(6), 1959–1966 CrossRef CAS PubMed.
- X. Shan, J. Liu, H. Mu, Y. Xiao, B. Mei, W. Liu, G. Lin, Z. Jiang, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2020, 59(4), 1659–1665 CrossRef CAS PubMed.
- S. Jeong, U. Kim, S. Lee, Y. Zhang, E. Son, K. J. Choi, Y. K. Han, J. M. Baik and H. Park, ACS Nano, 2024, 18(10), 7558–7569 CrossRef CAS PubMed.
- H. R. Mu, G. Lin, Y. Y. Zhang, Y. Xiao and J. Liu, Colloids Surf., A, 2021, 623, 126734 CrossRef CAS.
- X. B. Chen, L. Sheng, S. X. Li, Y. Cui, T. R. Lin, X. Y. Que, Z. H. Du, Z. Y. Zhang, J. Peng, H. L. Ma, J. Q. Li, J. Y. Qiu and M. L. Zhai, Chem. Eng. J., 2021, 426, 131029 CrossRef CAS.
- Q. Xu, P. Wang, L. Wan, Z. Xu, M. Z. Sultana and B. Wang, ACS Appl. Mater. Interfaces, 2022, 14(17), 19448–19458 CrossRef CAS PubMed.
- D. X. Guo, Z. Zhao, M. Y. Zong, C. Z. Fan, W. J. Zheng and D. H. Wang, ACS Sustainable Chem. Eng., 2023, 11(22), 8362–8373 CrossRef CAS.
- M. Bae, Y. Kang, D. W. Lee, D. Jeon and J. Ryu, Adv. Energy Mater., 2022, 12(29), 2201452 CrossRef CAS.
- X. Yu, Z. Y. Yu, X. L. Zhang, Y. R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M. R. Gao, G. Wang and S. H. Yu, J. Am. Chem. Soc., 2019, 141(18), 7537–7543 CrossRef CAS PubMed.
- X. Xu, G. Fu, Y. Wang, Q. Cao, Y. Xun, C. Li, C. Guan and W. Huang, Nano Lett., 2023, 23(2), 629–636 CrossRef CAS PubMed.
- X. Bu, Z. Mao, Y. Bu, Q. Quan, Y. Meng, Z. Lai, D. Chen, P. Xie, H. Li, C. Liu, X. Wang, S. Yip, J. Lu and J. C. Ho, Appl. Catal., B, 2023, 320, 121995 CrossRef CAS.
- Z. Duan, Y. Liu, Y. Wang, M. K. Kim, Y. Fang, Q. Yuan, Y. Zhang, P. Xiong and J. Suhr, Nano Lett., 2024, 24(28), 8558–8566 CrossRef CAS PubMed.
- L. Li, P. C. M. Laan, X. Yan, X. Cao, M. J. Mekkering, K. Zhao, L. Ke, X. Jiang, X. Wu, L. Li, L. Xue, Z. Wang, G. Rothenberg and N. Yan, Adv. Sci., 2023, 10(4), e2206180 CrossRef PubMed.
- F. Rocha, R. Delmelle, C. Georgiadis and J. Proost, J. Environ. Chem. Eng., 2022, 10(3), 107648 CrossRef CAS.
- S. E. S. Olesen, A. W. Jensen, M. Klove, F. Fenini, J. Nissen, B. B. Iversen, A. Bentien and L. P. Nielsen, ACS Catal., 2024, 11931–11940 CrossRef CAS.
- X. Yu, Z.-Y. Yu, X.-L. Zhang, Y.-R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M.-R. Gao, G. Wang and S.-H. Yu, J. Am. Chem. Soc., 2019, 141(18), 7537–7543 CrossRef CAS PubMed.
- L. Y. Zhou, J. Z. Fu and Y. He, Adv. Funct. Mater., 2020, 30(28), 2000187 CrossRef CAS.
- J. Zhu, P. W. Wu, Y. H. Chao, J. T. Yu, W. S. Zhu, Z. C. Liu and C. M. Xu, Chem. Eng. J., 2022, 433, 134341 CrossRef CAS.
- E. Sachyani Keneth, A. Kamyshny, M. Totaro, L. Beccai and S. Magdassi, Adv. Mater., 2021, 33(19), e2003387 CrossRef PubMed.
- R. A. Márquez, K. Kawashima, Y. J. Son, R. Rose, L. A. Smith, N. Miller, O. A. Carrasco Jaim, H. Celio and C. B. Mullins, ACS Appl. Mater. Interfaces, 2022, 14(37), 42153–42170 CrossRef PubMed.
- T. Zeng, B. Guo, Z. Xu, F. Mo, X. Chen, L. Wang, Y. Ding and J. Bai, Adv. Sci., 2023, 10(13), 2207495 CrossRef CAS PubMed.
- X. Z. Xue, D. Lin and Y. Li, Small Struct., 2022, 3(12), 2200159 CrossRef CAS.
- K. Zub, S. Hoeppener and U. S. Schubert, Adv. Mater., 2022, 34(31), e2105015 CrossRef PubMed.
- M. A. Ali, C. Hu, E. A. Yttri and R. Panat, Adv. Funct. Mater., 2022, 32(9), 2107671 CrossRef CAS PubMed.
- M. Saadi, A. Maguire, N. T. Pottackal, M. S. H. Thakur, M. M. Ikram, A. J. Hart, P. M. Ajayan and M. M. Rahman, Adv. Mater., 2022, 34(28), e2108855 CrossRef CAS PubMed.
- Z. H. Wu, D. Sun, C. C. Shi, S. Chen, S. H. Tang, Y. K. Li, C. Z. Yan, Y. S. Shi and B. Su, Adv. Funct. Mater., 2023, 33(45), 2304897 CrossRef CAS.
- E. Kargar and A. Ghasemi-Ghalebahman, Sci. Rep., 2023, 13(1), 18194 CrossRef CAS PubMed.
- Z. Zhao, X. X. Tian and X. Y. Song, J. Mater. Chem. C, 2020, 8(40), 13896–13917 RSC.
- S. Barui, H. Ding, Z. Wang, H. Zhao, S. Marathe, W. Mirihanage, B. Basu and B. Derby, ACS Appl. Mater. Interfaces, 2020, 12(30), 34254–34264 CrossRef CAS PubMed.
- F. Rocha, R. Delmelle, C. Georgiadis and J. Proost, Adv. Energy Mater., 2022, 13(1), 2203087 CrossRef.
- S. De Angelis, T. Schuler, M. A. Charalambous, F. Marone, T. J. Schmidt and F. N. Büchi, J. Mater. Chem. A, 2021, 9(38), 22102–22113 RSC.
- F. Arbabi, A. Kalantarian, R. Abouatallah, R. Wang, J. S. Wallace and A. Bazylak, J. Power Sources, 2014, 258, 142–149 CrossRef CAS.
- B. R. Huang, C. Lei, X. M. Sun, L. Luo, G. W. Wang, L. Zhuang and L. Xiao, Int. J. Hydrogen Energy, 2023, 48(91), 35453–35462 CrossRef CAS.
- B. Huang, X. Wang, W. Li, W. Tian, L. Luo, X. Sun, G. Wang, L. Zhuang and L. Xiao, Angew. Chem., Int. Ed., 2023, 62(33), e202304230 CrossRef CAS PubMed.
- Q. Ren, L. Feng, C. Tran, S. Eisenberg, X. Xue, A. Pinongcos, E. B. Duoss, C. Zhu and Y. Li, ACS Mater. Lett., 2024, 6(8), 3705–3712 CrossRef CAS.
- W. F. Zhai, Y. Y. Ma, D. Chen, J. C. Ho, Z. F. Dai and Y. Q. Qu, InfoMat, 2022, 4(9), e12357 CrossRef CAS.
- T. Liang, A. Wang, D. Ma, Z. Mao, J. Wang and J. Xie, Nanoscale, 2022, 14(48), 17841–17861 RSC.
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13(30), 12788–12817 RSC.
- X. Liu, R. Guo, K. Ni, F. Xia, C. Niu, B. Wen, J. Meng, P. Wu, J. Wu, X. Wu and L. Mai, Adv. Mater., 2020, 32(40), e2001136 CrossRef PubMed.
- Y. Zeng, M. T. Zhao, Z. H. Huang, W. J. Zhu, J. X. Zheng, Q. Jiang, Z. C. Wang and H. F. Liang, Adv. Energy Mater., 2022, 12(33), 2201713 CrossRef CAS.
- T. Liang, A. Wang, D. Ma, Z. Mao, J. Wang and J. Xie, Nanoscale, 2022, 14, 17841 RSC.
- M. R. Domalanta, J. N. Bamba, D. D. Matienzo, J. A. Del Rosario-Paraggua and J. Ocon, ChemSusChem, 2023, 16(13), e202300310 CrossRef PubMed.
- A. Villagra and P. Millet, Int. J. Hydrogen Energy, 2019, 44(20), 9708–9717 CrossRef CAS.
- M. Suermann, T. J. Schmidt and F. N. Büchi, Electrochim. Acta, 2016, 211, 989–997 CrossRef CAS.
- X. Wang, Y. Fei, J. Chen, Y. Pan, W. Yuan, L. Y. Zhang, C. X. Guo and C. M. Li, Small, 2022, 18, 2103866 CrossRef CAS PubMed.
- W. Hu, Q. Yan, X. Wang, J. Lu, Q. He, Q. Zhang and W. Yuan, Adv. Funct. Mater., 2024, 2411904 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |