
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of 3D Cr2C through solid state reaction-mediated Al extraction within Cr2AlC/Cu thin films†
Clio
Azina
 *a,
Justinas
Palisaitis
b,
Dimitri
Bogdanovski
a,
Tim
Bartsch
a,
Rajib
Sahu
ac,
Christina
Scheu
c,
Per O. Å.
Persson
b,
Per
Eklund
bd and
Jochen M.
Schneider
a
*a,
Justinas
Palisaitis
b,
Dimitri
Bogdanovski
a,
Tim
Bartsch
a,
Rajib
Sahu
ac,
Christina
Scheu
c,
Per O. Å.
Persson
b,
Per
Eklund
bd and
Jochen M.
Schneider
a
aMaterials Chemistry, RWTH Aachen University, Kopernikusstr. 10, 52074 Aachen, Germany. E-mail: azina@mch.rwth-aachen.de
bThin Film Physics Division, Department of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83 Linköping, Sweden
cMax-Planck-Institute for Sustainable Materials, Max-Planck-Str. 1, 40237 Düsseldorf, Germany
dInorganic Chemistry, Department of Chemistry - Ångström Laboratory, Uppsala University, Box 538, SE-751 21 Uppsala, Sweden
First published on 21st January 2025
Abstract
We report on the formation of the Cr2C compound using chemical etching-free methodology to extract Al from a Cr2AlC MAX phase thin film. Cr2AlC/Cu assemblies were deposited on sapphire substrates, using magnetron sputtering, and were subsequently annealed in vacuum. The Al from the MAX phase was shown to diffuse into Cu resulting in the formation of Al4Cu9 and causing the MAX phase to collapse into Cr2C grains. These carbide grains were characterized by transmission electron microscopy and the interatomic distances extracted were in good agreement with ab initio calculations predicting the equilibrium volume of the Cr2C phase.
1. Introduction
MAX phases constitute a family of ternary nanolaminated compounds given by the formula Mn+1AXn where M is a transition metal, A is a group A element, X is either carbon or nitrogen, and n = 1, 2, or 3.1 This family of ceramics has attracted significant attention due to their exceptional properties which bridge the gap between metals and ceramics.2,3 The unusual set of properties that MAX phases possess is a result of their hexagonal crystal structure and bonding, specifically their charge density modulation.4 Indeed, MAX phases are characterized by strong metallic-covalent M–X bonds and weaker M–A bonds.5 In the case of Al-containing MAX phases, such as Cr2AlC or Ti2AlC, the weakly bonded Al has been shown to diffuse out of the MAX phase when placed in oxidizing environments at high temperatures.6,7 The weak nature of the M–A bonds has also led to the discovery of the MAX phases’ 2D derivatives, MXenes.8,9MXenes are 2D materials which are mostly obtained by chemical etching of the A-layers.10–14 As the A-elements are etched away, the 2D M–X slabs are functionalized with terminations which are dependent on the etchant.15 The resulting 2D materials can then be extracted and characterized further. MXenes have shown potential for a variety of applications, in particular electromagnetic shielding,16 and energy-related ones, such as catalysis,17 supercapacitors,18,19 and batteries.18,20
While there are currently more than 100 ternary MAX phases reported,21 not all MAX phases are found to form MXenes.22 In fact, Cr-based MAX phases such as Cr2AlC23 and Cr2GaC24 were proven to be difficult to etch, when compared to Ti-based MAX phases such as Ti2AlC,25 Ti3AlC2,26,27 and Ti3SiC2.14 A number of studies, based on ab initio calculations, have discussed the potential of Cr2C MXenes with particular focus on their electronic and magnetic properties,28–30 their catalytic activity,31,32 and on their use for batteries.33 Attempts at synthesizing Cr2C have been reported by Tran et al. starting from Cr2GaC by chemical etching using hydrofluoric acid (HF),24 and by Zou et al. starting from Cr2AlC also using chemical etching (H2SO4 and LiF).34 While etching Cr2GaC was not successful, etching Cr2AlC led to the formation of Cr2CO2, in which O replaced Al. Therefore, etching in aqueous environments is not optimal for the formation of non-oxidized Cr2C MXenes.
Several studies have investigated alternative methods to produce different MXenes without the use of F-based substances (HF, LiF). Li et al. reported the synthesis of Ti3C2Cl2 MXenes by prolonged reaction of Ti3AlC2 with ZnCl2 which was further extended to different A-elements and molten salts.14 Another option reported by Ding et al. involved the intercalation of species into the layered structure to increase the distance between slabs which can facilitate delamination into single layers.12 In all these cases, the 2D MXenes were accompanied by surface terminations. Sahu et al. reported the formation of MBene regions during magnetron sputtering of MoAlB which is a structurally similar material to MAX phases.35 It was shown that the MoB MBene formed close to AlOx phases by Al deintercalation because of the Al affinity with O. This method enables direct MBene synthesis and does not involve etchants or intercalants, which leads to termination-free MBenes. Therefore, using a getter material to attract Al can allow for Al removal from MAB and/or MAX phases in order to produce MBenes and/or MXenes.
Herein, we report, for the first time, the formation of 3-dimensional Cr2C by Al extraction from a Cr2AlC thin film using Cu as a getter material. The formation of Cr2C was enabled by vacuum annealing and therefore, did not involve any chemical etching step, and was confirmed by a combination of microscopy and density functional theory approaches.
2. Methods
Cr2AlC thin films of ∼400 nm in thickness were deposited by magnetron sputtering in a lab-scale high vacuum deposition chamber (base pressure <1 × 10−4 Pa). A powder metallurgical composite target (50 mm ∅) with a stoichiometry of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 provided by Plansee Composite Materials GmbH, Germany, was operated in direct current magnetron sputtering (DCMS). 10 × 10 mm2 Al2O3(0001) substrates were rotated and kept at 650 °C during deposition to promote crystallization of the MAX phase. The working pressure was set to 0.55 Pa in presence of Ar and the power used on the Cr–Al–C target was set to 200 W. The thickness of ∼400 nm was achieved after 20 min of deposition. After deposition the samples were left to cool overnight. Then, Cu was deposited from an elemental target (Gemmel Metalle, ≥99.95%) which was operated at 200 W in DCMS for 3 min to achieve a thickness of ∼190 nm. Cu was deposited at a working pressure of 0.55 Pa, at room temperature to avoid diffusion.
:1:1 provided by Plansee Composite Materials GmbH, Germany, was operated in direct current magnetron sputtering (DCMS). 10 × 10 mm2 Al2O3(0001) substrates were rotated and kept at 650 °C during deposition to promote crystallization of the MAX phase. The working pressure was set to 0.55 Pa in presence of Ar and the power used on the Cr–Al–C target was set to 200 W. The thickness of ∼400 nm was achieved after 20 min of deposition. After deposition the samples were left to cool overnight. Then, Cu was deposited from an elemental target (Gemmel Metalle, ≥99.95%) which was operated at 200 W in DCMS for 3 min to achieve a thickness of ∼190 nm. Cu was deposited at a working pressure of 0.55 Pa, at room temperature to avoid diffusion.
In order to track changes in the assemblies with temperature, in situ X-ray diffraction (XRD) during heating was carried out in a Bruker AXS D8 Discover X-ray diffractometer with an integrated General Area Detector Diffraction System (GADDS). The diffractometer was equipped with an Anton Paar DHS 1100 heating stage. The measurements were carried out using Cu Kα radiation, in vacuum (<1 Pa). The incident angle was 15° and the measurements were carried out over the 2θ range of 30–50°, using 30° frames, and a collection time of 5 min per frame. The samples were heated up to 700 °C in 50 °C steps and the temperature was left to stabilize for 5 min before each measurement.
The structure of the annealed film was evaluated by XRD using a Panalytical Empyrean diffractometer with Cu Kα radiation in Bragg–Brentano geometry. Data were collected in the 10–70° 2θ range with a step size of 0.03° and a collection time of 96 s per step. An omega offset of −2° was used to remove the contribution of the substrate. Cr2AlC/Cu samples were vacuum-annealed in a Nabertherm tube furnace. The annealing program consisted of heating the sample to 620 °C for 10 h followed by cooling to room temperature. The heating and cooling rates were set to 10 °C min−1 and the average pressure during annealing was <1 × 10−4 Pa.
For scanning transmission electron microscopy (STEM) analysis, cross-sectional lamellae were extracted from as-deposited and annealed samples using a FEI Helios NanoLab dual-beam focused ion beam (FIB) microscope. The sample microstructure and elemental distribution were investigated at the atomic level using high-angle annular dark field (HAADF) STEM imaging, energy-dispersive X-ray spectroscopy (EDX) and electron energy-loss spectroscopy (EELS) mapping. Investigations were performed using the Linköping double Cs-corrected FEI Titan3 60-300, operated at 300 kV, equipped with a Super-X EDX system and Gatan GIF Quantum ERS post-column imaging filter. Atomically resolved HAADF-STEM imaging was acquired by using a 21.5 mrad convergence semi-angle providing sub-Ångström resolution probes with ∼100 pA beam current. The HAADF-STEM angular detection was set to 46–200 mrad range. EDX spectrum images of 256 × 256 pixels were recorded in 3 min using 0.3 nA probe current in cumulative mode. Elemental Cr-Kα, Al-Kα, and Cu-Kα distribution maps were extracted from the STEM-EDX spectrum images. EELS spectra were acquired for 1 min using a 0.5 eV per channel energy dispersion, 5 s pixel dwell time, and a 55 mrad collection semi-angle. Characteristic C-K and Cr-L edges were obtained by background subtraction using a power law.36
The calculations were performed using a density-functional theory approach,37,38 as implemented in the Vienna ab initio Simulation Package (VASP, version 5.4.4, University of Vienna),39–41 with the resulting output post-processed by other programs, as will be described below. The projector-augmented wave (PAW) method42,43 with a plane-wave cutoff energy of 500 eV was used for basis set construction, while electronic exchange and correlation were treated by employing the standard parametrization of the generalized gradient approximation by Perdew, Burke and Ernzerhof (PBE).44 The valence electron configuration utilized for the individual atomic potentials was 4s23d43p6 for Cr, 3s23p1 for Al and 2s22p2 for C, thus incorporating the 3p “semi-core” states of Cr, as specified in the respective atomic potential files. The dimensions of the k-mesh spanning the Brillouin zone (BZ) and constructed with the Monkhorst–Pack approach45 were specified as 13 × 13 × 2, which ensured both energetic and force convergence within 5 meV and 0.6 meV per atom, respectively, as verified by convergence tests. Integration over the BZ was performed via the Methfessel-Paxton method.46
The initial structural model for Cr2AlC was obtained from literature,47 with removal of the Al atoms resulting in the initial models for the MXene-like Cr2AlC systems. All systems underwent full structural optimization, and the total energy of the optimized system was used to calculate the energy of formation at the ground state vs. the constituent elements per eqn (1):
 | (1) |
It must be noted that Cr2AlC has been reported to be antiferromagnetic in the ground state;49 however, our calculations of the total energy show a difference between the non-spin polarized case and the energetically more favorable antiferromagnetic case of <50 meV, or approx. 6 meV per atom. Considering this small difference and the significantly higher computational cost of spin-polarized calculations particularly for lattice dynamics simulations (outlined below), which would have been prohibitive, the non-spin polarized approach was used throughout for consistency across the different simulation runs.
To obtain Gibbs’ free energies of formation as a function of temperature, quasi-harmonic lattice dynamics simulations employing VASP as the main quantum-mechanical program and phonopy (version 2.11.0, University of Kyoto)50 as a postprocessor were performed. To this end, a set of supercells of roughly cubic shape with a ≈ b ≈ c ≈ 10 Å was automatically generated, introducing displacements on the order of 0.02 Å from the equilibrium position of each symmetry-inequivalent atom. The exact number of displacements strongly depends on the space group of the underlying system. For each supercell, a static VASP calculation was performed to obtain the interatomic force constants, which were then processed by phonopy across all simulation runs (for each displacement) to yield a force constant matrix, from which the phonon frequencies can be obtained. From these, the lattice vibration contribution to the Helmholtz free energy, Aph was calculated, which together with the ground-state energy Etot given by VASP provides the dominant part of A(V,T). The aforementioned set of simulation runs for distinct supercells with unique displacements was repeated for a set of different volumes besides the equilibrium volume, accounting for the volume dependence of A(V,T). Fitting this data to the Birch–Murnaghan equation of state51,52 then yields the temperature dependence as well, so that the Gibbs free energy for a given system was obtained. The Gibbs free energies of formation for a given reaction, ΔG(T) were then calculated as per eqn (1), with G replacing Etot. We refer to prior literature for further methodological details on this approach.53,54
Finally, bonding analysis based on the Crystal Orbital Hamilton Population (COHP) technique55 using the LOBSTER package (version 4.0.0., Institute of Inorganic Chemistry, RWTH Aachen University)56–58 was performed to quantify the interatomic interactions in the examined systems. To this end, static VASP simulations of the optimized systems provided the PAW-based wavefunctions of the individual systems, which were then projected onto local atomic basis sets, allowing for the treatment of localized interatomic interactions (i.e., chemical bonds). This results in integrated crystal orbital Hamilton population (ICOHP) values, describing the strength of an interaction up to the Fermi level, with more negative values signifying stronger bonds. While not equal to the “true” bond strength, which is hard to quantify, the ICOHP value is strongly correlated with the latter and is routinely used as a descriptor for it.59–61
3. Results and discussion
The XRD patterns of the as-deposited Cr2AlC (400 nm)/Cu (190 nm) thin film assembly as well as the patterns collected during in situ heating are shown in Fig. 1 (extended step size in Fig. S1†). As the temperature was progressively increased, the structural changes occurring in the sample could be identified. The as-deposited sample exhibited characteristic peaks of the MAX phase (given by the blue dashed lines) at 36–37° and ∼42° corresponding to the (100)/(101) and (103)/(006) reflections, respectively. Additionally, the (111) Cu reflection was identified at ∼43°. As the temperature increased the signals of the MAX phase and Cu began to decrease in intensity and shift to lower 2θ angles, either because of the formation of solid solutions or because of the thermal expansion of the lattice upon heating. At 200 °C, the Cu reflection disappeared, suggesting that the Cu had been consumed. At 300 °C, a peak and a broad band appeared at approx. 35° and 38°, respectively, suggesting crystallization of a new phase, which could be indexed as Cr3C2. Their intensity increased with increasing temperature, while their full width at half maximum decreased. Based on identification carried out on the sample post-annealing, the peak was indexed as (310) of Al4Cu9. Another peak which could be indexed as (332) of Al4Cu9 appeared between 350–450 °C. The MAX phase peaks at ∼42° were shifted to lower 2θ angles during heating and returned to their original peak position post-annealing, confirming that the shift was due to thermal expansion. | ||
| Fig. 1 XRD patterns obtained during, and after, in situ XRD annealing of as-deposited Cr2AlC/Cu assemblies, up to 700 °C with 100 °C steps. | ||
To enhance the diffusion of species, the Cr2AlC/Cu assemblies were annealed in a vacuum furnace for 10 h at 620 °C. The appearance of the assemblies before and after vacuum annealing are shown in Fig. 2. As can be seen, the appearance changed drastically from metallic copper to a darker color. The XRD pattern of the annealed sample is also shown in Fig. 2. The MAX phase and Al4Cu9 phases were indexed, in addition to Cr3C2, which is in good agreement with observations made during the in situ XRD, in Fig. 1.
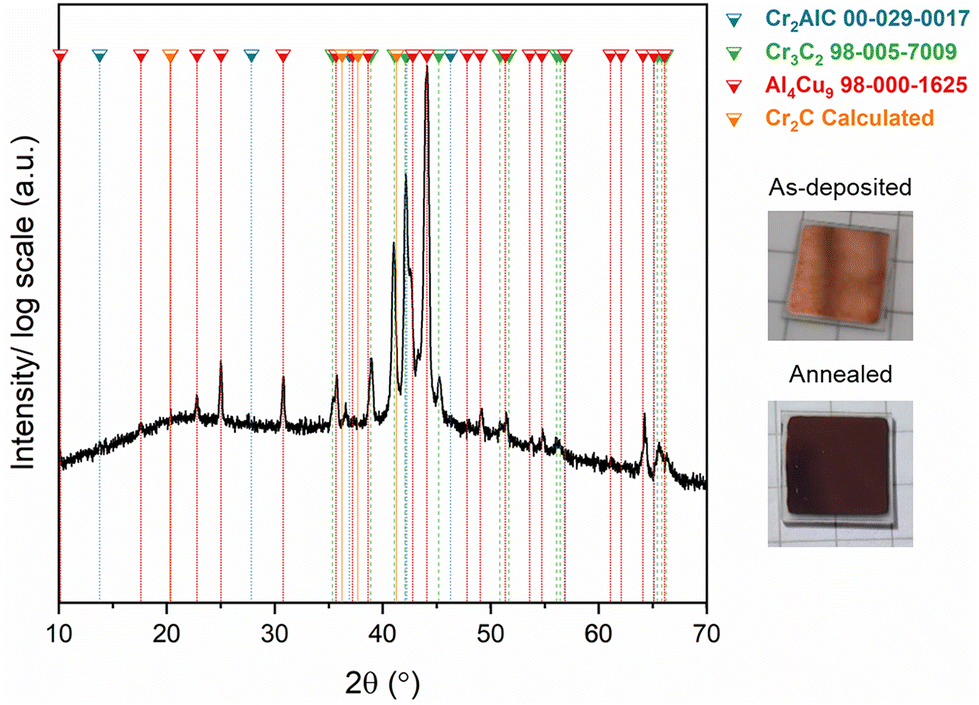 | ||
| Fig. 2 XRD pattern of the Cr2AlC/Cu assembly post-annealing at 620 °C for 10 h, in vacuum. Photographs of the Cr2AlC/Cu assembly surface before and after annealing. | ||
It is well known from oxidation experiments that Al is mobile and tends to diffuse out of Cr2AlC when subjected to high-temperature oxidizing environments.6,7,62 In fact, one of the major challenges when oxidizing Cr2AlC at high temperatures is the decomposition of the MAX phase into Cr-carbides as Al leaves the MAX phase.6,7,63,64 Similarly, as Al interacts with Cu to either form a solid solution or the intermetallic phase (Al4Cu9), the MAX phase is expected to be Al-depleted and therefore decompose into Cr-carbides. Evidence for the formation of Cr2C based on X-ray diffraction data (Fig. 2 and S2†) could not be obtained.
Fig. 3 shows a STEM image and corresponding EDX maps obtained from a lamella extracted from the vacuum annealed sample. STEM images and corresponding EDX maps of the as-deposited Cr2AlC can be seen in Fig. S3.† The as-deposited films exhibited a homogeneous distribution of Cr and Al throughout the thickness of the film, as well as a clear interface between the MAX phase film and the top Cu layer, indicating that no intermixing occurred during deposition. From the STEM image of the annealed sample in Fig. 3(a) one can notice that the Cu layer deposited on top of the MAX film is still present, therefore, confirming that the MAX phase has access to a continuous supply of Cu.65 It is evident from the maps in Fig. 3(b)–(d) that a phase separation occurred since Cr-rich particles are separated from Cu–Al-rich particles, as there is no indication for the formation of Cu–Al alloys within MAX phase grains. In fact, the Cu–Al alloys are found in the vicinity of Cr-rich grains (Cr2C, Cr3C2 or Cr2AlC) as evidenced on the EDX maps in Fig. 3. It can also be seen that Cr-rich grains exhibit little to no Al presence, consistent with the formation of Cr2C or Cr3C2. The formation of Cr2C is supported by the EELS spectrum, shown in Fig. S3,† where Cr-rich areas/grains contain Cr and C in a 2:1 ratio which corresponds to the stoichiometry of a Cr2C carbide. However, based on the XRD patterns in Fig. 1 and 2, no clear indication of the presence of the Cr2C phase could be deduced while Cr3C2 was indexed in both the in situ and ex situ measurements.
 | ||
| Fig. 3 (a) HAADF-STEM micrograph of the annealed Cr2AlC/Cu assembly, and (b)–(d) corresponding EDX maps. | ||
To confirm the phase that has formed after Al removal from Cr2AlC, atomically resolved STEM images were acquired from Cr-rich grains (Fig. S4†) where little to no Al is present, and are presented in Fig. 4. Two perpendicular zone axes are shown and the corresponding calculated fast Fourier transform (FFT) patterns are provided as insets. In Fig. 4(a) one can observe the typical honeycomb structure characteristic of a hexagonal structure, with n = 1. The lack of chemical contrast in addition to the EELS spectrum (Fig. S3†) suggests that the bright regions correspond to Cr and that the structure corresponds to Cr2C. Indeed, the Cr to C ratio of 2:1 could suggest the formation of carbon-deficient Cr3C2, however, the STEM images and corresponding FFTs do not correspond to the Cr3C2 structure but rather the Cr2C one. Along the [11−20] zone axis, one can observe the Cr2C slabs which remain intact after Al removal from the starting MAX phase. The spacing between Cr2C slabs is smaller by nearly half than in the MAX phase due to Al removal. This suggests, that part of the MAX phase structure has collapsed into Cr2C. To compare the measured projected bond distances with predictions, a Cr2C cell was simulated by DFT by removing Al from a Cr2AlC cell and allowing the configuration to relax. The resulting structure is depicted in Fig. 4 along (c) [0001] and (d) [11−20].
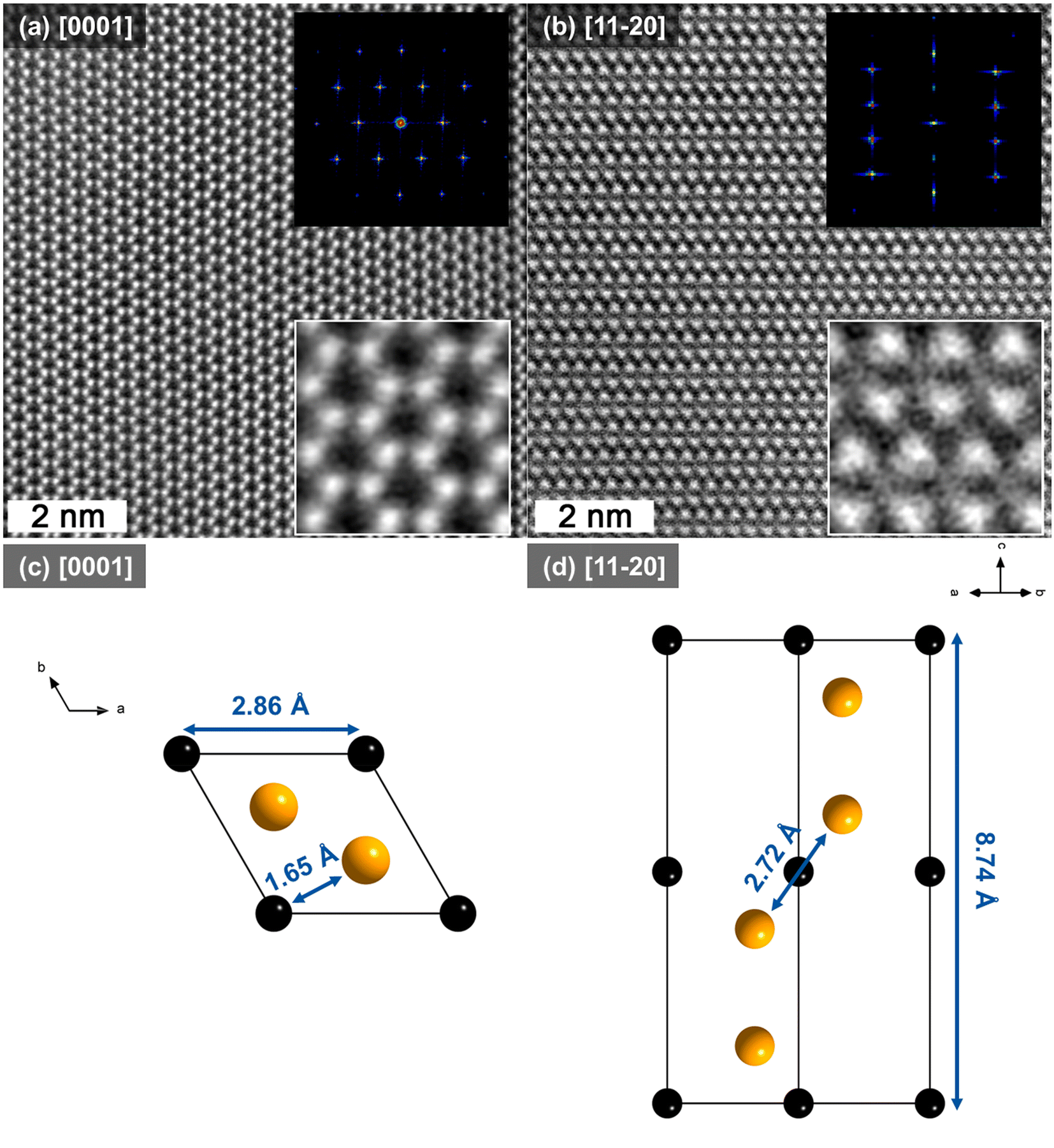 | ||
| Fig. 4 HAADF-STEM images along (a) [0001] and (b) [11−20] zone axes and corresponding FFT patterns. (c) and (d) correspond to the representation of the cell along [0001] and [11−20]. Black spheres represent C and orange spheres represent Cr atoms. | ||
The interatomic distances were extracted from the simulated cell (Fig. 4(c) and (d)) and compared with those extracted from the corresponding FFTs from the STEM data in Fig. 4. The resulting projected distances are provided in Table 1. One can notice that the experimentally deduced values are in good agreement with the ones predicted by theory. In fact, the deviations for projected bond distances deduced from calculations and TEM is 0.6% for Cr–Cr. Interestingly, the largest difference is measured for intra-slab Cr–Cr bonds where the experimentally deduced distance is ∼2.2% smaller than the theoretical one. According to Paier et al., this deviation has to be expected considering the exchange correlation functionals employed here.66
| Distances | Cr2C (STEM) (Å) | Cr2C (DFT) (Å) | Relative deviation (%) |
|---|---|---|---|
| Cr–Cr(0001) | 1.66 ± 0.05 | 1.65 | 0.6 |
| Cr–Cr(11−20) | 2.66 ± 0.05 | 2.72 | 2.2 |
| a parameter | 2.7 ± 0.1 | 2.86 | 5.6 |
| c parameter | 9.5 ± 0.1 | 8.74 | 8.7 |
A complete bonding analysis was carried out to assess whether large differences would be observed between Cr2C slabs in the case of a free standing MXene and in the case of the MAX phase. Fig. 5 gathers the actual (i.e. unprojected) interatomic distances, the average ICOHP per bond and the sum of all ICOHP values in the unit cell up to and including the second coordination shell, thus totaling all bonds, for Cr2AlC and Cr2C. First of all, one may notice that the interatomic distances (Fig. 5(a)) in both Cr2AlC and Cr2C are only slightly different, which is to be expected considering the electronic structure has been rearranged because of Al removal. Then, the average ICOHP values per bond, in Fig. 5(b) show that Cr–Cr and Cr–C bonds in the Cr2C slabs have weakened compared to those in Cr2AlC, while the C–C bonds were slightly strengthened. Strong Cr–Cr inter-slab bonds (see inset in Fig. 5 for visualization) are also observed compared to Cr–Al bonds in the MAX phase which is because the Cr–Cr bonds are shorter. The sum over all interactions are shown in Fig. 5(c) and show a similar trend as for the individual interactions, despite a weaker interaction for the Cr–Cr inter-slabs in Cr2C compared to the Cr–Al and Al–Al interactions in the MAX phase.
 | ||
| Fig. 5 (a) Interatomic distances, (b) average ICOHP per bond, and (c) sum of ICOHP in the 1st and 2nd coordination spheres of the cell for Cr2AlC and Cr2C. | ||
As stated above, the oxidation resistance of Cr2AlC has been widely investigated and it is common knowledge that upon Al depletion, Cr2AlC decomposes into stable Cr-carbides, namely Cr7C3 and Cr3C2.6 While the mechanism of Al extraction is similar to that which occurs during oxidation, the local formation of Cr2C is observed here. The temperature-dependent Gibbs free energies of formation of different Cr-carbides upon Al removal from Cr2AlC are provided in Fig. 6. One can see that the most stable carbide formed is expected to be Cr3C2, which is in good agreement with the XRD (Fig. 1, S1, and 2†). Cr2C formation, however, is not favorable averaging at ∼100 kJ per mol at 900 K (∼1 eV per atom), which suggests that the compound is metastable. While the transformation of Cr2C into a more stable carbide such as Cr3C2 was not investigated here, based on the Gibb's free energies presented herein, it is likely that this transformation occurs.
 | ||
| Fig. 6 Gibbs’ free energy of formation curves of decomposition routes of Cr2AlC derived from lattice dynamics simulations. | ||
The main difference between the process presented herein and oxidation are the temperature at which the samples are annealed (620 vs. ≥1000 °C), the environment (air vs. vacuum), and the duration of the process (10 h vs. 1–2 h). Considering that oxidation occurs at temperatures ≥1000 °C, the Al diffusion is much more rapid than at 620 °C. The rapid depletion in Al is expected to destabilize the MAX phase structure abruptly causing it to transform into a stable carbide (e.g. Cr3C2 and/or Cr7C3, as both have been reported experimentally6,7). Depleting the Al progressively, however, was shown here to result in the formation of Cr2C by collapse of the MAX phase. Another important difference between the two cases is the getter element, Cu for this work and O during oxidation. The resulting compound will either be a ductile and expandable Cu alloy or solid solution for the former, while a brittle Al2O3 will be the resulting compound for the latter. These compounds can also affect the final product after Al-extraction.
The metastable Cr2C phase coexists with Cr2AlC, Cr3C2, Al4Cu9, and a Cu–Al solid solution. As observed on the HAADF-STEM micrographs, upon Al removal, part of the MAX phase had collapsed as the Cr–Cr distance between slabs was much smaller compared to when Al was still present. Furthermore, we observed that Cr-rich grains are directly adjacent to a Cu-containing phase. By combining HAADF-STEM observations and calculations we are suggesting the following mechanism for the formation of the metastable Cr2C phase, see Fig. 7. Al diffuses out of the MAX phase into the Cu layer to either form a solid solution or the intermetallic Al4Cu9 phase. As Al is extracted from the MAX phase, the Cr2C slabs collapse as is evidenced by the smaller Cr–Cr inter-slab distances.
 | ||
| Fig. 7 Schematic of (a) starting Cr2AlC MAX phase, (b) Al-depleted Cr2AlC, and (c) collapsed MAX phase structure resulting in Cr2C. | ||
4. Conclusion
We reported the formation of metastable Cr2C obtained by Al extraction within a Cr2AlC/Cu thin film, without the use of chemical etching. While oxidation studies have reported the decomposition of the MAX phase into Cr7C3 and Cr3C2 upon Al depletion, the observations made herein show that the metastable Cr2C can also form upon Al removal. Indeed, vacuum annealing of Cr2AlC/Cu assemblies resulted in a multiphase film composed of Cr2C, Cr2AlC, Cr3C2, Al4Cu9 and a solid solution of Al in Cu. HAADF-HRSTEM allowed determining the interatomic distances in the newly formed Cr2C which were in good agreement with distances extracted from ab initio calculations. The bonding analysis suggests that the interactions between Cr–Cr intra-slabs in Cr2C and Cr–Cr in Cr2AlC are similar as the sum of the ICOHP values varies between −2.1 and −2.6 eV. However, the distance between slabs has significantly shrunk (2.2 Å in Cr2C instead of 2.6 Å for the Cr–Al distance in the MAX phase) because of the Al removal which has caused the MAX phase to collapse into Cr2C. While the final product is a multiphase film, optimization of the process conditions (use of powder instead of thin films, annealing temperature and duration, Cu volume fraction, etc.), may result in a higher fraction of Cr2C enabled by efficient Al extraction.Author contributions
C. A.: conceptualization, data curation, formal analysis, investigation, methodology, funding acquisition, project, administration, visualization, writing original draft. J. P.: formal analysis, data curation, investigation, methodology, writing – review & editing. D. B.: formal analysis, investigation, methodology, writing – review & editing. T. B.: formal analysis, investigation, writing – review & editing. R. S.: formal analysis, writing – review & editing. C. S.: supervision, writing – review & editing. P. O. Å. P.: resources, supervision, funding acquisition, writing – review & editing. P. E.: resources, funding acquisition, writing – review & editing. J. M. S.: resources, supervision, writing – review & editing.Data availability
The data supporting this study are available within the article and the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
C. A. acknowledges funding from the European Union's H2020-MSCA-IF-2019 research and innovation programme under the Marie Curie grant agreement no. 892501 (REALMAX). The Swedish Research Council (VR) and Swedish Foundation for Strategic Research (SSF) are acknowledged for access to ARTEMI, the Swedish National Infrastructure in Advanced Electron Microscopy (2021-00171 and RIF21-0026). The support of the IT Center of RWTH Aachen University for providing computational resources within the framework of the NHR4CES initiative and the jara0221 project of the Jülich-Aachen Research Alliance (JARA) is most gratefully acknowledged. P. E. also acknowledges the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009 00971) and the Knut and Alice Wallenberg foundation through the Wallenberg Academy Fellows program (KAW-2020.0196).References
- M. W. Barsoum, Prog. Solid State Chem., 2000, 28, 201 Search PubMed.
- M. Radovic and M. W. Barsoum, Am. Ceram. Soc. Bull., 2013, 92, 20 Search PubMed.
- J. Gonzalez-Julian, J. Am. Ceram. Soc., 2021, 104, 659 Search PubMed.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, Trends Chem., 2019, 1, 210 Search PubMed.
- M. Magnuson and M. Mattesini, Thin Solid Films, 2017, 621, 108 Search PubMed.
- D. E. Hajas, M. To Baben, B. Hallstedt, R. Iskandar, J. Mayer and J. M. Schneider, Surf. Coat. Technol., 2011, 206, 591 Search PubMed.
- C. Azina, M. Poll, D. M. Holzapfel, E. Tailleur, A. Zuber, S. Dubois, P. Eklund and J. Gonzalez-Julian, J. Eur. Ceram. Soc., 2024, 44, 4895–4904 Search PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248 Search PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992 Search PubMed.
- M. Naguib, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2021, 33, 2103393 Search PubMed.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Adv. Mater., 2021, 33, 2103148 Search PubMed.
- H. Ding, Y. Li, M. Li, K. Chen, K. Liang, G. Chen, J. Lu, J. Palisaitis, P. O. Å. Persson, P. Eklund, L. Hultman, S. Du, Z. Chai, Y. Gogotsi and Q. Huang, Science, 2023, 379, 1130 Search PubMed.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman, P. Eklund, P. O. Å. Persson, S. Du, Z. Chai, Z. Huang and Q. Huang, J. Am. Chem. Soc., 2019, 141, 4730 Search PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Å. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X.-H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Piñero, P.-L. Taberna, P. Simon and Q. Huang, Nat. Mater., 2020, 19, 894 Search PubMed.
- V. Natu and M. W. Barsoum, J. Phys. Chem. C, 2023, 127, 20197–20206 Search PubMed.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. M. Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 Search PubMed.
- I. M. Chirica, A. G. Mirea, Ş. Neaţu, M. Florea, M. W. Barsoum and F. Neaţu, J. Mater. Chem. A, 2021, 9, 19589 Search PubMed.
- Y. Sun, D. Chen and Z. Liang, Mater. Today Energy, 2017, 5, 22 Search PubMed.
- Q. Zhu, J. Li, P. Simon and B. Xu, Energy Storage Mater., 2021, 35, 630 Search PubMed.
- Z. Chen, Z. Chang, Z. Liu and N. Zhou, Appl. Surf. Sci., 2022, 602, 154375 Search PubMed.
- M. Dahlqvist, M. W. Barsoum and J. Rosen, Mater. Today, 2024, 72, 1 Search PubMed.
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491 Search PubMed.
- O. Akinola, I. Chakraborty, H. Celio, D. Akinwande and J. A. C. Incorvia, J. Mater. Res., 2021, 36, 1980 Search PubMed.
- M. H. Tran, A. M. Malik, M. Dürrschnabel, A. Regoutz, P. Thakur, T.-L. Lee, D. Perera, L. Molina-Luna, K. Albe, J. Rohrer and C. S. Birkel, Dalton Trans., 2020, 49, 12215 Search PubMed.
- L. Liu, H. Zschiesche, M. Antonietti, M. Gibilaro, P. Chamelot, L. Massot, P. Rozier, P.-L. Taberna and P. Simon, Adv. Energy Mater., 2023, 13, 2203805 Search PubMed.
- L. Liu, M. Orbay, S. Luo, S. Duluard, H. Shao, J. Harmel, P. Rozier, P.-L. Taberna and P. Simon, ACS Nano, 2022, 16, 111 Search PubMed.
- N. Goossens, K. Lambrinou, B. Tunca, V. Kotasthane, M. C. Rodríguez González, A. Bazylevska, P. O. Å. Persson, S. De Feyter, M. Radovic, F. Molina-Lopez and J. Vleugels, Small Methods, 2024, 8, 2300776 Search PubMed.
- B. Akgenc, B. Akgenc, E. Vatansever and F. Ersan, Phys. Rev. Mater., 2021, 5, 083403 Search PubMed.
- C. Si, J. Zhou and Z. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 17510 Search PubMed.
- Q. Sun, Z. Fu and Z. Yang, J. Magn. Magn. Mater., 2020, 514, 167141 Search PubMed.
- Y. Cheng, J. Dai, Y. Zhang, Y. Zhang, Y. Song and Y. Song, J. Mater. Chem., 2018, 6, 20956 Search PubMed.
- I. A. M. Ibrahim, S. Abdel-Azeim, A. M. El-Nahas, O. Kühn, C.-Y. Chung, A. El-Zatahry and M. F. Shibl, J. Phys. Chem. C, 2022, 126, 14886 Search PubMed.
- Y. Li, L. Bai, N. Ma and L. Niu, Comput. Theor. Chem., 2022, 1217, 113892 Search PubMed.
- X. Zou, H. Liu, H. Xu, X. Wu, X. Han, J. Kang and K. M. Reddy, Mater. Today Energy, 2021, 20, 100668 Search PubMed.
- R. Sahu, D. Bogdanovski, J.-O. Achenbach, S. Zhang, M. Hans, D. Primetzhofer, J. M. Schneider and C. Scheu, Nanoscale, 2021, 13, 18077 Search PubMed.
- R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope, 2011 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558 Search PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 Search PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 Search PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B:Condens. Matter Mater. Phys., 1989, 40, 3616 Search PubMed.
- J. C. Schuster, H. Nowotny and C. Vaccaro, J. Solid State Chem., 1980, 32, 213 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- M. Dahlqvist, B. Alling and J. Rosén, J. Appl. Phys., 2013, 113, 216103 Search PubMed.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1 Search PubMed.
- F. Birch, Phys. Rev., 1947, 71, 809 Search PubMed.
- F. D. Murnaghan, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244 Search PubMed.
- R. P. Stoffel, C. Wessel, M.-W. Lumey and R. Dronskowski, Angew. Chem., Int. Ed., 2010, 49, 5242 Search PubMed.
- D. Bogdanovski, P. J. Pöllmann and J. M. Schneider, Nanoscale, 2022, 14, 12866 Search PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617 Search PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34, 2557 Search PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030 Search PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, J. Comput. Chem., 2020, 41, 1931 Search PubMed.
- G. A. Landrum and R. Dronskowski, Angew. Chem., Int. Ed., 2000, 39, 1560 Search PubMed.
- A. Saksena, D. Bogdanovski, H. Sahasrabuddhe, D. Music and J. M. Schneider, Materials, 2020, 13, 2298 Search PubMed.
- S. Amano, D. Bogdanovski, H. Yamane, M. Terauchi and R. Dronskowski, Angew. Chem., Int. Ed., 2016, 55, 1652 Search PubMed.
- X. Chen, B. Stelzer, M. Hans, R. Iskandar, J. Mayer and J. M. Schneider, Mater. Res. Lett., 2021, 9, 127 Search PubMed.
- J. Gonzalez-Julian, T. Go, D. E. Mack and R. Vaßen, J. Am. Ceram. Soc., 2018, 101, 1841 Search PubMed.
- A. Zuber, V. Gauthier-Brunet, J. Roger, J. Gonzalez-Julian, T. Ouisse and S. Dubois, J. Eur. Ceram. Soc., 2022, 42, 2089 Search PubMed.
- H. Fashandi, M. Dahlqvist, J. Lu, J. Palisaitis, S. I. Simak, I. A. Abrikosov, J. Rosen, L. Hultman, M. Andersson, A. Lloyd Spetz and P. Eklund, Nat. Mater., 2017, 16, 814 Search PubMed.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03664f |
| This journal is © The Royal Society of Chemistry 2025 |