
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of DYRK1b as a novel regulator of small extracellular vesicle release using a high throughput nanoscale flow cytometry screening platform†
Sina
Halvaei
,
Nikki
Salmond
 and
Karla C.
Williams
*
and
Karla C.
Williams
*
Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, Canada. E-mail: karla.williams@ubc.ca
First published on 10th March 2025
Abstract
Extracellular vesicles (EVs) are important mediators of intercellular communication and have various roles in physiological and pathological processes. Discovery of regulators of EV biogenesis and release has led to significant improvements in our understanding of EV biology and has highlighted disease-specific pathways. Large scale discovery studies of EV regulators are limited by conventional methods of EV analysis with limited throughput and sensitivity. To address this, this study presents a high-throughput flow cytometry-based platform for the quantification of EVs released from cells. Here, a system was developed using the MDA–MB-231 cell line stably expressing ZsGreen, which passively loads ZsGreen proteins into EVs, and nanoscale flow cytometry. EV detection and quantitation was optimized and validated for a 96-well format. The high-throughput flow cytometry screening platform quantified the effect of 156 kinase inhibitors on EV number and identified AZ191 – a DYRK1b inhibitor – as a potent EV inhibitor. DYRK1b inhibition and knockdown confirmed a significant reduction in total EV number, with small EVs demonstrating the largest reduction. DYRK1b knockdown altered the intracellular distribution of EV marker CD63, suggesting a role for DYRK1b in EV trafficking. In conclusion, our study establishes a platform for high-throughput analysis of EV dynamics and introduces DYRK1b kinase as a novel EV-regulator.
1. Introduction
Extracellular vesicles (EVs) are nano-sized lipid bi-layer enclosed particles released by all cell types into the extracellular space and biofluids.1 EVs contain nucleic acids,2 proteins3 and lipids,4 and their contents can reflect the cell of origin.5 There are several EV subtypes, with ectosomes and exosomes being the most extensively studied. Ectosomes bud directly from the plasma membrane or from membrane protrusions such as filipodia and microvilli.6 Ectosomes can be formed and released using lipid membrane re-organization,7 ESCRT machinery,8 and actin–myosin bleb forming mechanisms.9,10 Exosomes are formed in the endosomal system. Exosome biogenesis starts with the inward budding of the endosomal membrane into the endosomal lumen; this forms intraluminal vesicles (ILV) and results in a multivesicular body.11 Trafficking and fusion of the multivesicular body with the plasma membrane releases the ILVs into the extracellular environment/biofluids as exosomes.11 Because exosomes and ectosomes can be difficult to separate and distinguish from one another using common EV isolation methods, MISEV 2023 (the latest guideline for EV studies), recommends describing purified EVs <200 nm in size as small EVs, and >200 nm in size as large EVs.1EVs carry a diverse array of biologically active molecules, including oncogenic proteins,12 RNA,13 miRNA,13 lipids,4 genomic14 and mitochondrial DNA.2 EVs are taken up by neighbouring cells at local and distant sites.15 The transfer of EV-associated biologically active molecules to recipient cells can modify downstream signaling and gene regulation pathways. In the context of cancer, EV education of cells within the tumor microenvironment can result in more aggressive characteristics and phenotypes of neighbouring cells,16 endothelial cell tubule formation and facilitation of angiogenesis,17 activation of fibroblasts to promote the pre-metastatic niche formation,18 and immune cell suppression19 and apoptosis.20 Recent progress highlights the function of breast cancer EVs in shaping a tumor microenvironment conducive to cancer cell growth, metastasis,21 immune system evasion,22 and the developing resistance to therapy.23
Given the role of EVs in cell–cell communication and promotion of pathological process such as cancer progression and metastasis,18,24 modulating EV release could represent a potential therapeutic strategy.25 Targeting EV biogenesis and release could be achieved through manipulating lipid reorganization, sphingomyelinase activity, translocation of phosphatidylserine, ATP-binding cassette transporters, and cytoskeletal organization.26 Kinases also participate in EV biogenesis and release at multiple points of the pathway. For example, phosphorylation of synaptosome-associated protein 23 (SNAP-23) is necessary for SNARE complex formation which facilitates exosome release.27 In tumor cells, SNAP-23 phosphorylation at Ser95 is mediated by pyruvate kinase type M2 (PKM2) which further upregulates exosome release from tumor cells.27 Also, SNAP-23 phosphorylation at Ser110 is mediated by diacylglycerol/protein Kinase C (DAG/PKC), which contributes to multivesicular bodies’ fusion with the plasma membrane.28 On the other hand, Rho family small G protein and Rho-associated protein kinase (ROCK) are essential for actin–myosin-based contraction and ectosome release.29 ARF6 or ARF1 proteins activate RhoA and result in actin–myosin contraction and ectosome budding.24 These studies suggest that altering kinase enzyme activity could be a promising approach to modulate EV biogenesis and release.
Current methods for EV isolation and analysis are not scalable, expensive, and time-consuming,30 making EVs difficult to study. This has impeded the implementation of high throughput discovery studies investigating EV biogenesis and release. Nanoscale flow cytometry is a promising platform that could be used to develop a high-throughput screening platform. We have previously shown that nanoscale flow cytometry can be used to quantitatively analyze EVs at the single particle resolution level in complex biological fluids using a 96-well plate format.31–33 In this study, we further advance nanoscale flow cytometry technology by developing it as a high-throughput compound screening platform. We optimized and validated a robust system to analyze the effect of 156 kinase inhibitors on EV production. This method does not require any EV isolation steps and is performed directly in a 96-well plate format. Using this approach, we identify a previously unknown EV modulator− DYRK1b.
2. Results
2.1. Quantitative analysis of ZsGreen-EV release dynamics by nanoscale flow cytometry
MDA–MB-231 cells stably expressing ZsGreen proteins were used to develop a nanoscale flow cytometry screening platform (Fig. 1A). ZsGreen was used as it is significantly brighter compared to enhanced GFP and, thus, would support the detection of EVs above background noise. To demonstrate this, conditioned media from MDA–MB-231–ZsGreen cells seeded in a 96-well plate was analysed by nanoscale flow cytometry. ZsGreen EVs were readily detected and quantified by nanoscale flow cytometry (Fig. 1B–E). To ensure that the EVs detected were not protein aggregates or cell debris, 1% Triton-X was added into the conditioned media prior to analysis by nanoscale flow cytometry to lyse EVs. Particle signal was completely lost after detergent treatment demonstrating that the system is detecting ZsGreen-positive EVs (Fig. 1B). Additionally, large EVs (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g) and small EVs (100000g) isolated by differential centrifugation were readily detected by nanoscale flow cytometry and an observed shift in size was noted from the small EV population compared to the large EV population (Fig. 1C). Isolated EVs were characterized by western blot, nanoparticle tracking analysis and scanning transmission electron microscope (STEM) (Fig. S1A–S1C†). Western blot showed that EV-associated proteins CD9, CD63, and CD81 were abundant in the EV pellets (Fig. S1A†). Comparing the small EVs and large EVs, the expression level of the EV markers was higher in the small EVs (Fig. S1A†). Moreover, both the small and large EVs contained little calnexin compared to cell lysate, indicating minimal EV preparation contamination with protein or cell debris (Fig. S1A†). Nanoparticle tracking analysis data showed that the small EV fraction was smaller in size compared to the large EV fraction (Fig. S1B†). STEM confirmed that cup shaped particles, consistent with EV morphology, were present in the small and large EV fractions (Fig. S1C†). In addition, size analysis of STEM images showed that the mean particle size for large EV and small EV isolated fractions was approximately 203.69 nm and 99.73 nm, respectively (Fig. S1D†).
000g) and small EVs (100000g) isolated by differential centrifugation were readily detected by nanoscale flow cytometry and an observed shift in size was noted from the small EV population compared to the large EV population (Fig. 1C). Isolated EVs were characterized by western blot, nanoparticle tracking analysis and scanning transmission electron microscope (STEM) (Fig. S1A–S1C†). Western blot showed that EV-associated proteins CD9, CD63, and CD81 were abundant in the EV pellets (Fig. S1A†). Comparing the small EVs and large EVs, the expression level of the EV markers was higher in the small EVs (Fig. S1A†). Moreover, both the small and large EVs contained little calnexin compared to cell lysate, indicating minimal EV preparation contamination with protein or cell debris (Fig. S1A†). Nanoparticle tracking analysis data showed that the small EV fraction was smaller in size compared to the large EV fraction (Fig. S1B†). STEM confirmed that cup shaped particles, consistent with EV morphology, were present in the small and large EV fractions (Fig. S1C†). In addition, size analysis of STEM images showed that the mean particle size for large EV and small EV isolated fractions was approximately 203.69 nm and 99.73 nm, respectively (Fig. S1D†).
 | ||
| Fig. 1 Optimization of nanoscale flow cytometry for high-throughput screening of EVs in 96-well plate format. (A) A schematic diagram detailing the set-up and testing for development of a high-throughput screen platform to accurately quantify EV release dynamics. For this, (i) ZsGreen cells were tested for EV release, (ii and iii) plated in a 96-well format with small molecule EV-modulators, and (iv) changes to EV number assessed by nanoscale flow cytometry. (B) Representative flow cytometer dot plots of ZsGreen EVs in MDA–MB-231–ZsGreen cell conditioned media (diluted 1:1 in PBS). Upon addition of 1:1 1% Triton-X, EV lysis was confirmed by reduction of ZsGreen-EV signal on the flow cytometer. Red frames represent the gate for FITC-H signal threshold. (C) After isolating large EVs (10000g) and small EVs (100000g) by differential ultracentrifugation, the ZsGreen-EV were detected on the flow cytometer in a 30 second measurement. (D) MDA–MB-231 cells were seeded into a 96-well plate in increasing cell numbers: 2.5 × 103, 5 × 103, 10 × 103, 15 × 103, 20 × 103. After 24 h of conditioning the media, the ZsGreen-EVs were analyzed using nanoscale flow cytometry. Red frames represent the gate for FITC-H signal threshold. (E) The data acquired in (D) was quantified and presented in graphical format. ±SEM, n = 3. (F) MDA–MB-231 cells were seeded into a 96-well plate in increasing cell numbers: 2.5 × 103, 5 × 103, 10 × 103, 15 × 103, 20 × 103 and imaged. The end-point confluence was calculated using IncuCyte imaging. ±SEM, n = 3. (G) MDA–MB-231 cells were seeded into a 96-well plate at 15 × 103 seeding density, and cells were treated with 99.2 nM bafilomycin or 4 μM GW4869 for 24 h before conditioned media was harvested and analyzed on the flow cytometer for 30 seconds. Red frames represent the gate for FITC-H signal threshold. GW4869 and bafilomycin were dissolved in DMSO; different volumes of DMSO were used for each compound. GW4869 and Bafilomycin Vehicle Control (DMSO alone) were matched acccordingly. (H) Data acquired in (G) was quantified and presented in graphical format. One-way ANOVA test with post-hoc uncorrected Dunnett's test was perform for statistical analysis. ±SEM, n = 3. ns: not significant. **p < 0.01. ****p < 0.0001. | ||
To test whether our system could detect changes in the number of EVs released by cells, increasing numbers of MDA–MB-231 cells were plated in a 96-well format (ranging between 2 × 103–20 × 103). The number of EVs detected in the conditioned media by nanoscale flow cytometry increased accordingly with increasing cell number (Fig. 1D and E) until cell confluence reached 100% (Fig. 1F), at which point EV release decreased potentially due to changes in cell behaviour at this confluence. To validate that the system can detect changes in EV release due to small molecule manipulation of EV biogenesis and release dynamics, MDA–MB-231–ZsGreen cells were treated for 24 h with bafilomycin and GW4869, which increase and decrease EV number, respectively. The concentration of GW4869 to decrease EV release was optimized compared to vehicle control dimethyl sulfoxide (DMSO) (Fig. S2A†). Analysis of the conditioned media by nanoscale flow cytometry showed that the number of EVs released from cells treated with EV stimulator bafilomycin was significantly higher compared to the vehicle-treated control cells (Fig. 1G and H). Treatment with EV biogenesis inhibitor GW4869 was found to significantly reduce the number of EVs detected by nanoscale flow cytometry compared to the vehicle-treated control (Fig. 1G and H). Importantly, the treatment of MDA–MB-231–ZsGreen cells with bafilomycin or GW4869 did not change cell proliferation or viability, indicating that the change in EV number was solely due to the effect of the compounds upon EV regulation (Fig. S2B and S2C†).
2.2. High-throughput screen of the effect that 156 kinase inhibitors have upon the biogenesis and release of EVs by MDA–MB-231–ZsGreen cells
Next, using the nanoscale flow cytometry platform, in a 96-well plate format, a kinase inhibitor library was selected to screen for changes in EV quantity. In a 96-well plate format, 15 × 103 MDA–MB-231–ZsGreen cells were treated with a library of 156 non-selective and selective kinase inhibitors (500 nM) and DMSO as the vehicle control for 24 h. Cytotoxicity assay were performed to identify compounds that >10% increased cell toxicity (Fig. S3†). Compounds causing >10% cytotoxicity compared to vehicle control were removed from analysis as cell death could affect EV number irrespective of the kinase mechanism of action. Seven compound were removed due to increased cytotoxicity. Compared to the untreated control, the vehicle control, did not show any significant change differences in EV release and cytotoxicity assay. The remaining 149 compounds were used for the screen. MDA–MB-231–ZsGreen cells were incubated with each of the 149 compounds and conditioned media was analyzed by nanoscale flow cytometry. The number of EV events detected by nanoscale flow cytometry were normalized to end-point cell confluence to ensure that the EV number accounted for any differences in cell number from well-to-well (Fig. S4†). The kinase inhibitor screen identified 16 compounds that significantly reduced the number of the EVs released by MDA–MB-231–ZsGreen cells into the conditioned media (Fig. 2). Among these 16 compounds, 7 compounds (KN-93, H-9, JNK inhibitor XVI, BIBF1120, AZ191, VE-822, LCK inhibitor, MLCK Inhibitor Peptide 18; Fig. 2, Red Dots) were selected for validation (Fig. 3). MB-231–ZsGreen cells were treated for 24 hours with 500 nm of each compound, DMSO as vehicle control, or left untreated (untreated control). EVs were isolated using differential ultracentrifugation and isolated EVs were analyzed and quantified by nanoscale flow cytometry. This confirmed a reduction in EV number for six of the seven compounds (Fig. 3A and B). AZ191 was the only compound that consistently, and significantly, reduced the number of large and small EVs. Compared to vehicle control, AZ191 reduced the number of small EVs and large EVs by 60.9% and 40.9%, respectively. AZ919 was chosen as the lead compound for further validation.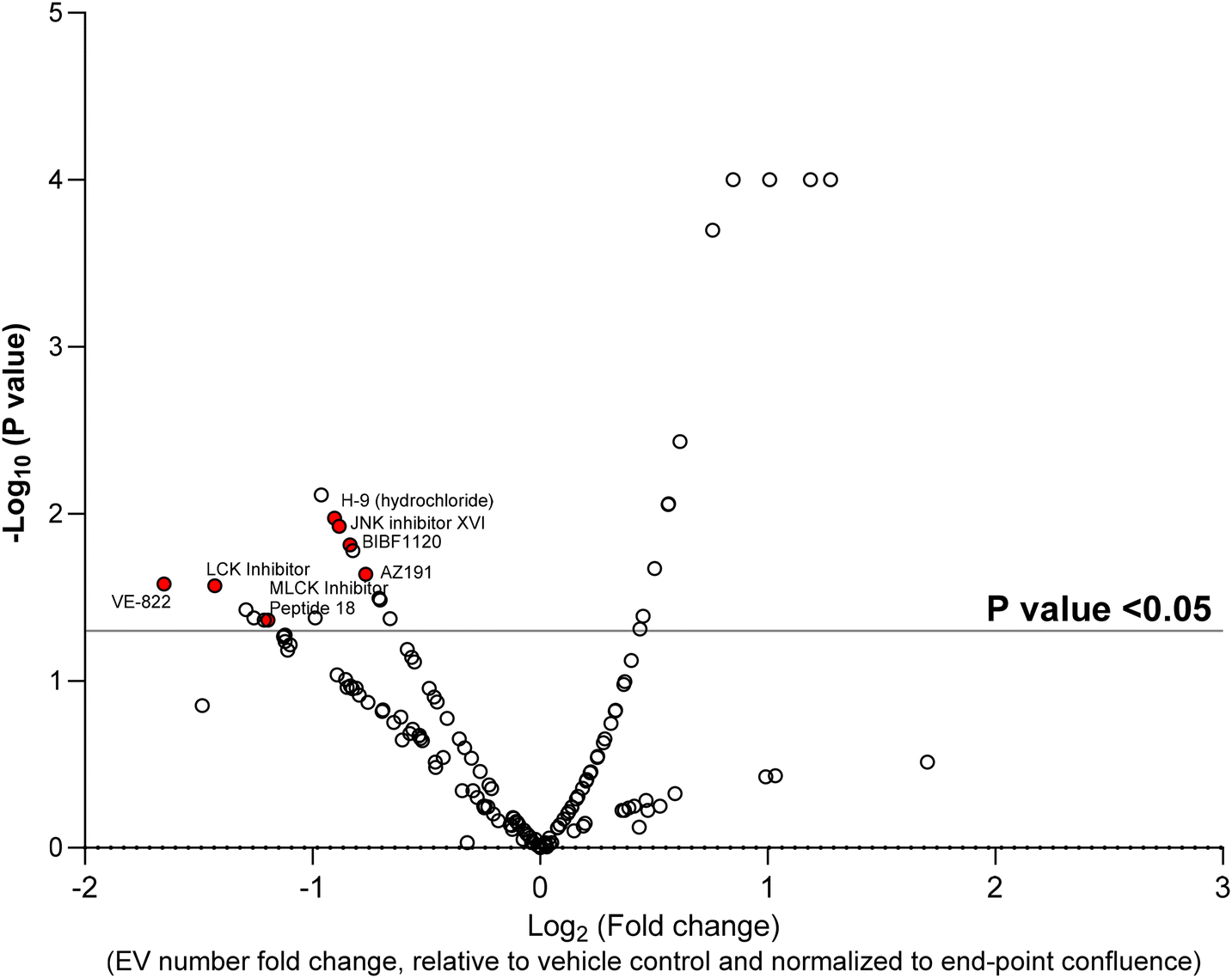 | ||
| Fig. 2 High-throughput screen to determine the effect of 149 kinase inhibitors on EV release. 15 × 103 MDA–MB-231 cells were seeded into each well of a 96-well plate. The cells were treated with 500 nM final concentration of each compound for 24 h and conditioned media harvested, diluted 1:1 with PBS and run on the flow cytometer for 30 seconds. The number of EVs detected in each condition was quantified and normalized to end-point confluence, and fold change of EV release compared to vehicle was calculated in excel. P-Value was calculated using Kruskal–Wallis's test in GraphPad Prism comparing each compound to vehicle. The log10 (P value) and log2 (fold change) were used for the volcano plot. n = 2. | ||
 | ||
| Fig. 3 Validation of EV-compound screen by large scale EV isolation using differential ultracentrifugation. MDA–MB-231 cells were seeded into 15 cm plates (5 × 106 per plate) in EV-depleted media and treated with 500 nM of each compound for 24 h. After 24 h, conditioned media was collected and large EVs (10 K pellet) and small EVs (100 K pellet) were isolated via differential ultracentrifugation. Cell confluency for each condition was quantified at end-point. (A) Large EVs and (B) small EVs were analyzed and quantified using nanoscale flow cytometry. The number of events quantified by flow cytometry (particle counts per 30 s run) were normalized to end-point cell confluence. One-way ANOVA with Uncorrected Fisher's LSD test. ±SEM, n = 3. ns: not significant. *p < 0.05. **p < 0.01. ***p < 0.001. | ||
AZ191 is a small molecule selective DYRK1b inhibitor with 10-fold selectivity toward DYRK1b over DYRK1a.34 To validate the role of AZ191 in decreasing EV release, EVs were harvested from MDA–MB-231–ZsGreen cells following treatment with 500 nm AZ191 for 24 h. EVs were isolated using size-exclusion chromatography (SEC) whereby five fractions were collected representing EVs of decreasing size (1 (largest) to 5 (smallest)). SEC isolated EVs were analyzed and quantified using nanoscale flow cytometry. Similar to the results obtained by differential ultracentrifugation, AZ191 reduced the number of total EVs with the largest reductions found for the smaller EV fractions. Analysis of SEC EV fractions by nanoscale flow cytometry demonstrated that compared to vehicle control, AZ191 decreased EV release by 28.7%, 51.4%, 54.9%, 63.2% in fractions 1, 2, 3, and, 4/5, respectively (Fig. 4A). While a reduction in EV number was noted for fraction 1, which contains the largest EV sizes,31,33 no significant difference was found compared to vehicle control.
 | ||
| Fig. 4 Validation of AZ191 as an EV release modulator. (A) MDA–MB-231 cells were seeded into 15 cm plates and treated with AZ191 for 24 h. EVs were isolated using size exclusion chromatography. After 3 ml of flow through, five 500 μl EV fractions ranging from 1 (larger) to 5 (smaller) were collected. The number of EVs in each fraction was analyzed by nanoscale flow cytometry. Fraction 4 and 5 were pooled. Two-way ANOVA test with uncorrected Dunn's test. ±SEM, n = 3, ns: not significant, *p < 0.05, ****p < 0.0001. (B and C) MDA–MB-231 cells were transfected with siRNA against (B) DYRK1b or (C) DYRK1a. Untreated: no treatment. Non-targeting: non-targeting siRNA control. The protein expression of each kinase was assessed by western blot 24 h and 48 h post-transfection. (D) MDA–MB-231 cells were left untreated or transfected with siRNA against DYRK1b, DYRK1a, or non-targeting. 24 h post-transfection, cells were re-plated and allowed to condition media for 48 h. Conditioned media was harvested from each condition, and EVs were isolated by ultracentrifugation. Isolated EVs were analysed by nanoscale flow cytometry. One-way ANOVA test with uncorrected Dunn's test. ±SEM, n = 3, ns: not significant, *p < 0.05. | ||
To validate that the target of AZ191 –DYRK1b− has a role in EV regulation we performed DYRK1b knockdown studies in MDA–MB-231–ZsGreen cells. Knockdown was confirmed by western blot (Fig. 4B). To show that the effect of AZ191 in reducing the EV quantity was selective toward DYRK1b and not due to any off-target effects on DYRK1a, DYRK1a knockdown was also performed. DYRK1a knockdown was confirmed with western blot (Fig. 4C) Flow cytometry analysis of conditioned media from non-transfected cells (untreated), non-targeting siRNA control cells (si-Control), DYRK1a knockdown (si-DYRK1a), and DYRK1b knockdown (si-DYRK1b), found a significant reduction (36.6% reduction) in EV number with loss of DYRK1b (Fig. 4D). Knockdown of DYRK1a did not show any effect on EV quantity (Fig. 4D). This demonstrates that inhibition of DYRK1b function reduces EV number and suggests that the DYRK1b may play an important role in EV biogenesis or release.
2.3. DYRK1b modulates CD63 localization in the cytosol
To further investigate the role of DYRK1b in EV biogenesis or release we assessed CD63 expression and localization. CD63 is a marker of the multivesicular body and found to be enriched in exosomes. Immunofluorescence staining showed that CD63 localization changed with loss of DYRK1b (Fig. 5A). Upon loss of DYRK1b, CD63 signal was less dispersed and found to cluster around the nucleus whereas control cells had a distinct punctate distribution throughout the cytoplasm (Fig. 5A, white arrows). There was no difference in CD63 expression between control and DYRK1b knockdown cell lines (Fig. 5B). Quantification of CD63 total area per cell demonstrated that that CD63 was less distributed throughout the cell in DYRK1b knockdown cells compared to non-targeting siRNA control cells (Fig. 5C and E, white arrows). This lack of distribution of CD63 vesicular structures resulted in an observed increased intensity of a perinuclear signal in DYRK1b knockdown cells (Fig. 5E, white arrows). Quantification of CD63 signal intensity found an increase in CD63 signal intensity in DYRK1b knockdown cells compared to non-targeting siRNA control cells (Fig. 5D). Overall, the results indicate that in the absence of DYRK1b, CD63-positive structures accumulate rather than dispersing suggesting a defect in multivesicular trafficking or maturation. | ||
| Fig. 5 DYRK1b crosstalk with CD63. (A) MDA–MB-231 cells transfected with either non-targeting siRNA or DYRK1b siRNA were fixed and fluorescently immunostained for CD63 (red) followed by staining with DAPI (blue) and actin (Alexa488 Phallodin, green). Imaging of cells was done using 20× objective (top row). A higher magnification image shows CD63 punctate (control and siControl, white arrows) and CD63 perinuclear accumulation (siDYRK1b, white arrow) (bottom row). (B) A representative western blot of CD63 expression in MDA–MB-231 non-transfected control, siRNA non-targeting control, siRNA against DYRK1b. Scale bar = 100 μm or 20 μm (zoom). (C & D) The images acquired in (B) were analyzed using ImageJ, and two parameters were measured: (C) the total area within each cell that CD63 occupied and (D) the fluorescent intensity of each CD63 pixel within each cell. (E) Representative images of MDA–MB-231 cells used for analysis in (D). CD63 (red) and DAPI (blue); arrows point to distributed CD63 punctae (top two rows) and aggregated CD63 perinuclear clusters (bottom row). Images were taken using a 40× oil emersion objective. One-way ANOVA test with uncorrected Dunn's test. ±SEM, n = 3, ns: not significant, *p < 0.05, ***p < 0.001. | ||
3. Discussion
EVs have been repeatedly shown to have an important role in healthy physiology and in the pathophysiology of many diseases, including neurological disorders,35 autoimmune diseases,36 cardiovascular diseases,37 and cancer.38 Identifying modulators of EV biogenesis, trafficking, and secretion is important for an improved understanding of EV biology. In addition, the study of modulators of EV biogenesis and release in the context of disease could identify disease specific EV-pathways, which may represent new therapeutic options. Discovery of modulators of EV biogenesis, trafficking, and secretion has been limited due the inherent challenges in studying EVs. Current EV isolation methods are time-demanding and often require a high volume of sample;39 they are not practical for running high-throughput drug screens. Hence, there is a need for the development of novel techniques to quantify EV release in a large-scale screening format. Nanoscale flow cytometry is an emerging technology useful for the detection and quantification of nanoparticles, including sEVs and lEVs. Relying on its sensitivity and resolution,33 nanoscale flow cytometry provides a unique advantage by detecting EVs without requiring EV isolation and purification steps. In addition, its 96-well plate format offers a practical and feasible platform for high-throughput drug screenings.This study aimed to optimize and validate nanoscale flow cytometry for use in a high-throughput kinase screen to identify novel kinase inhibitors that modulate EV biogenesis and release. We showed that nanoscale flow cytometry provides a direct, fast, and quantitative measurement of ZsGreen-positive EVs in the conditioned media of MDA–MB-231–ZsGreen cells, without the need to pre-isolate EVs. The 96-well plate format of the nanoscale flow cytometer allowed rapid and high-throughput analysis of the inhibitory or inducing effects of 149 small molecule kinase inhibitors on EV release using only 50 μl of conditioned media.
While other studies have carried out high-throughput analysis of EV release modulators, they have used in-direct measurement of EV release, whereas our study is one of the first to directly quantify EVs in a high throughput screen. For example, Datta et al., used a CD63-GFP transfected cell line as their model to study prostate cancer EVs. They developed an imaging assay on a microplate-based laser scanning imaging cytometer and quantified the GFP signal from cells as an indirect representation of EV dynamics and conducted a high throughput study on more than 4000 compounds.40 Another tetraspanin-oriented high throughput approach was to make a CD63-Tluc-CD9-EmGFP reporter cell line and study the effect of 27895 compounds on monocyte leukemia cells. After treating the cells with the compounds, the conditioned media was harvested, and a chemiluminescent assay was employed to measure luciferase activity, which acts as an indirect readout of EV release.41 To our knowledge, all the other high throughput studies that have been conducted by other research groups looked at the indirect readout of the EV levels. What sets our study apart from the previous studies is that our system directly measures and quantifies the EV numbers released by cells under each condition and performs this measurement in a short 30 second run. Overall, the field of EV research has shown a keen interest in developing novel systems or optimizing existing technologies for high throughput screening of EV dynamics. Various approaches, ranging from emerging novel fluidic devices36 to adjustments in conventional flow cytometry,42 have been utilized to detect EV dynamics. Our work offers an insightful approach in quantitative high-throughput screening to assess the effect of small molecules on the release of EVs using nanoscale flow cytometry.
Despite the numerous advantages of our high-throughput methodology—such as its speed (30 s), simplicity, cost-effectiveness (minimal plastic/media), and direct EV detection via flow cytometry—it has limitations. Autofluorescent compounds, for instance, cannot be easily used in this platform. Also, suspension cells, such as lymphocytes, may require further optimization steps. Also, our method depends heavily on fluorescence expression by cell lines for the generation of fluorescent EVs, making it unsuitable for hard-to-transfect cells like primary or stem cells. Moreover, the flow cytometer used has a detection limit around 80 nm,33,43 which limits detection of smaller EV populations. While this method provides a platform to study EV dynamics further validation for reliable results is required using larger scale EV preparations. Nonetheless, for cell lines amenable to our platform, the potential for discovery of new EV regulators is substantial; indeed, this supported our identification of DYRK1b as an EV-regulatory kinase.
To our knowledge, this is the first report on the role of DYRK1b in the modulation of EV dynamics. We showed that DYRK1b knockdown affected CD63 localization in cells indicating that DYRK1b could play a role in CD63-exosome biogenesis or trafficking pathway. It has been shown that CD63 is localized in multivesicular bodies and traffics towards the plasma membranes.44 Impairing regulators of CD63-positive multivesicular bodies has been shown to lead to an accumulation of CD63-positive bodies in the cytoplasm, consequently reducing the number of secreted EVs.45 Potentially, DYRK1 regulates either directly, or in-directly, trafficking of the multivesicular body and thus, impairing its activity or expression results in a reduction in EV trafficking and subsequent secretion.
Vesicle trafficking is crucial in endocytosis and exocytosis46 as well as in EV docking and release. In some mammals DYRK1 has two homologs, DYRK1a and DYRK1b, but in Ascidian Ciona it has one homolog making it an interesting model to study DYRK1 function.46 In has been shown in the Ascidian Ciona model that DYRK1 regulates the phosphorylation of endocytic and exocytic components such as: dynamin1 at Ser728, amphiphysin1 at Ser408, endophilin at Ser263, and VAMP2 at Ser112.46 VAMP2 (v-SNARE) interacts with t-SNAREs and plays a role in SNARE complex assembly for vesicle docking.47 Therefore, VAMP2 may potentially be involved in docking and release of multivesicular bodies as well. In addition, DYRK1b directly binds and interacts with STAT3 which leads to phosphorylation and accumulation of p-STAT3 in the nucleus.48 Upregulation and downregulation of p-STAT3 increases and decreases EV protein markers and EV release in cancer cells.49 p-STAT3 promotes transcription of SHMT2 mRNA in the nucleolus which positively correlates with PKM2 transition from tetrameric to phosphorylated dimeric form.49 p-PKM2 then acts as kinase and phosphorylates SNAP23 (t-SNARE) protein and p-SNAP23 engages in forming SNARE complex and controls EV release.27 In conclusion, DYRK1b may control EV release by directly phosphorylating VAMP2 or indirectly leading to SNAP23 phosphorylation. Further studies are warranted to investigate the precise molecular mechanism of DYRK1b-mediated EV regulation.
4. Conclusion
In conclusion, this study presents a platform to permit high-throughput screening of EV modulators. It proposes a straightforward, fast, and cost-effective protocol for conducting screens on EV biogenesis and release that is applicable to both pharmaceutical and academic settings. This study describes a direct platform for accurately measuring and quantifying the number of EVs released by cells and eliminates the need for tedious EV isolation steps. It also introduces DYRK1b kinase as an EV modulator for the first time. Future work needed to expand on these studies would involve exploring alternative EV labelling methods, such as fluorescently labelled subcellular compartment markers, or incorporating antibodies for sub-population analysis. Using our platform enabled the identification of the kinase, DYRK1b, as an EV modulator; the possibility for using this platform to screen other regulators is tremendous and could support studies assessing EV-regulation by lipids, metabolism, and other small molecule libraries.5. Experimental section
5.1. Cell culture
Triple-negative breast cancer cell line, MDA–MB-231, was purchased from ATCC (ATCC HTB-26). MDA–MB-231 cell line was cultivated in DMEM with 10% fetal bovine serum (FBS; Gibco Life Technologies, Grand Island, NY, USA). All cell lines were cultured at 37 °C in a humidified environment, supplemented with 5% CO2. Cells used in all experiments were between 70% to 75% confluent at the time of experiment initiation and had a greater than 90% viability. pLC-ZsGreen-P2A-Hygro vector was purchased from Addgene (Plasmid# 124301) and used to transfect MDA–MB-231 cells using lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturers protocol to generate a stably transfected MDA–MB-231–ZsGreen cell line.5.2. Electron microscopy
Isolated EVs were fixed by dilution 1:1 in 4% paraformaldehyde and adsorbed onto formvar/carbon-coated copper 200 mesh grids for 2 minutes. EVs were negatively stained for 30 seconds using 1% pre-filtered Uranyl acetate pH 4.6 (Fisher Scientific). After the grids were blotted with filter paper and air dried, they were imaged using the scanning transmission mode of a Helios NanoLab 650, fitted with a STEM detector, (Thermofisher, Systems for Research, Kanata, ON, Canada) at 30 kV. Then images obtained from STEM were analyzed via ImageJ and ten images per grid per sample were used to measure the size of particles.
5.3. Nanoparticle tracking analysis
EVs were diluted 1:200 in 0.02 μm filtered PBS. The concentration and size of diluted EVs were analyzed using the NanoSight LM10 equipped with a blue 488 nm laser (Malvern Panalytical) and sCMOS camera. The laser gain was set on 366 and 200 tracks per video were considered as valid measurements. Three repeats of three 30 second measurements with 749 video frames were acquired while the syringe pump sustained EV movement at speed 40 through the fluidic compartment. For all the samples the detection threshold for analysis was set at 5. NTA software version 3.4 was used to analyze data.
5.4. Differential ultracentrifugation
For EV isolation via ultracentrifugation, 1.75 × 106 cells were seeded into a 10 cm plate in 17 ml complete media for each condition. After 24 h, the plate was washed with PBS and the media was refreshed with 17 ml 10% EV-depleted FBS supplemented DMEM media. The cells were treated with 500 nM final concentration of the compound of interest. 24 h post-treatment, conditioned media was harvested and spun at 300g for 10 minutes, at 4 °C to remove dead and floating cells. Then, the supernatant was transferred into a new tube and it was spun at 2500g for 15 minutes, 4 °C, to remove cell debris and apoptotic bodies. Next, the supernatant was transferred to polyallomer 38.5 ml open-top ultracentrifuge tubes (Beckman Coulter) and centrifuged at 10000g for 30 min, 4 °C, to isolate large EVs. The supernatant was transferred to new ultracentrifuge tubes and small EVs were pelleted at 100000g for 70 min, 4 °C. The resulting large and small EV pellets were washed in 1.5 ml PBS and transferred into an Eppendorf tube. The Eppendorf tubes were placed into open-top ultracentrifuge tubes and re-pelleted at 100000g for 70 min, 4 °C. EV pellets were re-suspended in 30 μl PBS. The SW32Ti Rotor (Beckman Coulter) was used in an Optima XPN-100 ultracentrifuge (Beckman Coulter) for this procedure.
5.5. EV isolation via size-exclusion chromatography
Cells were prepared for EV isolation via size exclusion chromatography (SEC) as described in the differential centrifugation protocol. After 24 h of conditioning, media was harvested and spun at 300g for 10 minutes, at 4 °C then, at 2500g for 15 minutes, at 4 °C. The conditioned media was then concentrated to 500 μl using a 10 kDa molecular weight cut-off regenerated cellulose membrane Amicon® Ultra-4 centrifugal concentrator (Millipore, Sigma). A room temperature IZON qEVoriginal/70 nm size-exclusion chromatography column (IZON Sciences Ltd) was equilibrated with two-column volumes (20 ml) of 0.2 μm filtered PBS. Subsequently, 500 μl of the concentrated sample was applied to the column fret and 3 ml flow-through was collected, followed by five 500 μl EV fractions (1–5 larger EVs to smaller). The flow of the sample through the column was sustained by continuously adding 1 ml 0.02 μm filtered PBS to prevent the column from drying.5.6. Nanoscale flow cytometry
The commercially available kinase screen library (#10505, Cayman Chemicals, USA) consisted of 156 kinase inhibitors and were tested for their effects on EV biogenesis and release. In a 96-well plate, 15 × 103 cells were seeded in 100 μl complete media. After 24 h, the plate was washed with PBS and the media was refreshed with 100 μl 10% EV-depleted FBS supplemented DMEM media. The cells were treated with 500 nM of each kinase inhibitor for 24 h. The 500 nM concentration was a concentration suggested by the manufacturing company as it was the concentration near and above the IC50 for most of the compounds. All the compounds were dissolved in DMSO and since the stock concentration and final concentration were similar for all the compounds one vehicle condition was included and reported in this experiment. After treatment, 50 μl conditioned media was harvested, diluted 1:1 in 50 μl 0.02 μm filtered PBS (MultiCell), and transferred into a flat-bottomed 96-well plate. Alternatively, upon analysis of the large and small EV samples isolated using ultracentrifugation or SEC, samples were diluted 1:10 in 90 μl 0.02 μm filtered PBS. Samples were analyzed using the CytoFLEX S (Beckman). Instead of sheath fluid, water obtained from the MilliQ water purification system was used. Samples were triggered with 405 nm violet side scatter (VSSC), with a 1027 detection threshold, on slow speed setting, and gain settings as follows: VSSC = 100. FITC = 500. Data was acquired for 30 seconds. The population of interest was gated with reference to complete media. For the two compounds, SU 6668 and 17β-hydroxy Wortmannin, which showed autofluorescence, manual gating was performed on populations of interest with reference to complete media plus compounds. All data was exported into an Excel file, and analysis was performed on the total number of events within the manually gated areas during the 30 second acquisition.
5.7. Lysis of EVs
EVs were lysed before running on the CytoFLEX S by mixing samples in 1:1 1% Triton-X 100 (Fisher Bioreagents) in PBS. Samples were analyzed via nanoscale flow cytometry as mentioned above.
5.8. Kinase screen
A commercial library of 156 selective and non-selective kinase inhibitors was purchased from Cayman Chemical (#10505). The compounds were applied at the 500 nM final concentration according to the manufacturer's recommendation. To see the full list of compounds and their target kinase, refer to ESI Table 1.†5.9. Cell lysate preparation
After collecting the conditioned media, 200 μl cold RIPA lysing buffer (Thermo Scientific #PI89900) was added to each plate on ice. A cell scraper was used to harvest the cells into a 1.5 mL tube and then rotated for 10 min at 4 °C. The tube was then centrifuged at 16 × 103g for 10 min, at 4 °C. The pellet was discarded, and the supernatant was collected in a new 1.5 mL tube and kept on ice for protein concentration quantification.5.10. Protein concentration quantification
The protein concentration of isolated EVs and cell lysates were determined using Micro BCA™ Protein Assay Kit and Pierce™ Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific) respectively, according to the manufacturer's instructions.5.11. Western blot
After protein isolation and concentration quantification, 30–80 μg protein was prepared for western blot by adding 25% loading buffer (Novex Life Technologies). Protein samples were boiled at 95 °C for 9 minutes. Protein samples were loaded onto 4–12% gradient gels (Thermo Fisher Scientific) and separated by molecular weight using electrophoresis at 200 V for 30 minutes in MES running buffer (50 mM MES (Sigma), 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.7). Proteins were transferred (190 mM glycine, 25 mM Tris Base) from the gel onto a nitrocellulose 0.45 μm membrane (BioRad) at 30 V for 90 minutes. The membrane was blocked in 5% milk in TBS-T (20 mM Tris base, 160 mM NaCl, 0.1% Tween) for 1 hour while shaking at room temperature. Primary antibodies were incubated overnight while shaking at 4 °C in 1% milk-TBS-T. Then, blots were washed with three (10 minutes) washes in TBS-T. Secondary Licor IRDye® 680RD antibody was applied in 1% milk-TBS-T for 1 hour in the dark while shaking at room temperature. Blots were washed with three (10 minutes) washes in TBS-T and imaged on the Licor Odyssey® CLx using Image Studio Lite software (5.2.5). All reagents were sourced from Thermo Fisher unless otherwise specified. For all antibody information, please see ESI Table 1.†5.12. MTT assay
In each well of a 96-well plate, 15 × 103 MDA–MB-231 cells were seeded. After overnight culture, cells were treated with bafilomycin (99.2 nM) and GW4689 (4 μM) for 24 h. The conditioned media was removed and replaced with serum free media containing 0.01% thiazolyl blue tetrazolium bromide (T-030-1; GOLDBIO). Cells were incubated at 37 °C for 4 h before the media was removed, and 100 μL of DMSO was added to each well to dissolve the purple formazan crystals. After incubating the plate at 37 °C for 10 min and shaking the plate for 5 min on a plate shaker in the dark, absorbance was measured at a wavelength of 570 nm to determine the viability.5.13. Cytotoxicity assay
Lactose dehydrogenase (LDH) release was measured using the CytoTox 96® Non-Radioactive Cytotoxicity Assay kit (Promega Corporation, Madison, USA) based on the manufacturer's protocol. The conversion of a tetrazolium salt into a formazan crystal determines LDH release. Briefly, 15 × 103 MDA–MB-231 cells were seeded in each well of a 96-well plate. After overnight culture, cells were treated with 500 nM kinase inhibitors for 24 h. All the compounds were dissolved in DMSO and since the stock concentration and final concentration were similar for all the compounds one vehicle condition was included and reported in this experiment. Conditioned media were collected and used to measure the amount of LDH. To determine the level of LDH the absorbance was measured at 490 nm.5.14. Confluence analysis
For experiments done in flat bottom 96-well plates, 15 × 103 MDA–MB-231 cells per well were seeded in 100 μl complete media. After 24 h, the plate was washed with PBS, and the media was refreshed with 100 μl DMEM media supplemented with 10% EV-depleted FBS. The cells were treated with 500 nM final concentration of the compounds of interest. All the compounds were dissolved in DMSO and since the stock concentration and final concentration were the same for all compounds, only one vehicle control condition was included and reported in this experiment. Cells were left to settle for at least 30 minutes, and the plate was imaged at 0 h and 24 h on the IncuCyte Zoom HD72CLR (Essen BioScience, USA), 9 phase contrast images per well using 20× magnification. The confluence of cells was analyzed via IncuCyte Zoom 2018A software according to the manufacturer's instructions for a label-free proliferation assay. For experiments done in a 15 cm plates, 5 × 106 MDA–MB-231 cells per plate were seeded in EV-depleted media and treated with 500 nM of each compound for 24 h. After 24 h, conditioned media was collected for EV isolation via differential ultracentrifugation or SEC. The remaining cells were trypsinized with 0.53 mM EDTA/0.05% trypsin (Wisent), stained 1:1 with 0.4% trypan blue solution (Gibco) and counted via Luna-II automated cell counter. Isolated EVs were then analyzed and quantified using nanoscale flow cytometry. The number of events obtained via flow cytometry were normalized to end-point cell confluence of 1 × 106 cells.
5.15. siRNA knockdown
In a 6-well plate, 250 × 103 MDA–MB-231 cells were seeded in 2 ml complete media. After 24 h, when cells were 70% confluent, siRNA against DYRK1b or a non-targeting siRNA control (Dharmacon Research) was mixed with 7.5 μl Lipofectamine 3000 (Thermo Fisher) in 250 μl Gibco OptiMEM media (Fisher Scientific) according to the manufacturer's protocol. After a 6 h incubation, the lipoplexes were removed, and the media was refreshed. The plate was imaged at 48 h post-transfection on the IncuCyte Zoom HD72CLR (Essen BioScience, USA), 9 phase contrast images per well using 20× magnification. The confluence of cells was analyzed via IncuCyte Zoom 2018A software according to the manufacturer's instructions for a label-free proliferation assay. The cell lysates were harvested at 48 h post-transfection. The siRNA was used at a final concentration of 25 nM.5.16. Immunocytochemistry
MDA–MB-231 cells were seeded onto 13 mm diameter coverslips with a seeding density of 0.05 × 106 cells. After 6 h of siRNA transfection, cells were fixed with 4% paraformaldehyde, pH 7.4, for 5 min at room temperature. The coverslips were washed with 1% Glycine for 10 min and permeabilized with 0.1% Triton X-100 for 3 min. Then, the coverslips were blocked with 5% milk in PBS for 90 min at room temperature. Cells were washed with PBS and incubated with the primary antibody (CD63) for 90 minutes in PBS using the concentration shown in Table S1.† The coverslips were washed in PBS using three 5 minute washes. The secondary anti-mouse antibody was diluted in 1% milk in PBS and incubated for 60 min. Finally, the coverslips were mounted onto slides with DAPI+ mounting media (Invitrogen #S36920). The slides were allowed to dry overnight at room temperature. Samples were protected from light throughout the process until imaging.5.17. Software, statistical analysis, and data acquisition
All experiments, were performed as three independent experiments unless stated otherwise in the figure legend. Adobe Illustrator 27.7 was used for figure preparation. ImageJ2 2.14.0/1.54f was used for analyzing STEM images. CytExpert 2.3 software was used to export nanoscale flow cytometry images and data. Western blot visualization was performed on Image Studio 2.5.2 via the Li-COR Odyssey CLx. Data was handled in Microsoft Excel 16.90.2 and analyzed using GraphPad Prism 10.1.1. The normal distribution of the data was analyzed by Kolmogorov–Smirnov test. All normally distributed data were analyzed using parametric one-way ANOVA and two-way ANOVA with Dunnett's post-hoc test. The kinase screen data was analyzed using Kruskal–Wallis's test to calculate the P-value in GraphPad Prism comparing each compound to vehicle control (DMSO) and the fold change of EV release compared to vehicle was calculated in excel. The log10 (P Value) and log2 (fold change) were used for the volcano plot. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.Abbreviation
| EVs | Extracellular vesicles |
| sEVs | Small extracellular vesicles |
| lEVs | Large extracellular vesicles |
| SEC | Size-exclusion chromatography |
| ESCRT | Endosomal sorting complexes required for transport machinery |
| SNAP-23 | Synaptosome associated protein 23 |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide |
| DYRK | Dual-specificity tyrosine-regulated kinases |
| ATCC | American type culture collection |
| PBS | Phosphate buffer saline |
| BCA | Bicinchoninic acid |
| H | Hour |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular-signal-regulated kinase |
Author contributions
SH and NS conducted experiments. SH, NS, and KCW analyzed the data. KCW conceptualized the manuscript. KCW, SH, and NS assembled the manuscript. SH drafted a manuscript and NS, SH, and KCW revised the manuscript. All authors reviewed and approved the manuscript.Data availability
Any raw data not included in the article and ESI† in this current study is available from the corresponding author on reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants Program awarded to K. C. W. K. C. W. holds a Tier 2 Canada Research Chair. The authors would like to acknowledge the Centre for High-Throughput Phenogenomics group in the Faculty of Dentistry for assistance in acquiring electron microscopy images during this study. The authors also thank Dr Russ Algar for use of their NanoSight instrument for nanoparticle tracking analysis of extracellular vesicles.References
- J. A. Welsh, D. C. I. Goberdhan, L. O'Driscoll, E. I. Buzas, C. Blenkiron, B. Bussolati, H. Cai, D. Di Vizio, T. A. P. Driedonks, U. Erdbrügger, J. M. Falcon-Perez, Q. Fu, A. F. Hill, M. Lenassi, S. K. Lim, M. G. Mahoney, S. Mohanty, A. Möller, R. Nieuwland, T. Ochiya, S. Sahoo, A. C. Torrecilhas, L. Zheng, A. Zijlstra, S. Abuelreich, R. Bagabas, P. Bergese, E. M. Bridges, M. Brucale, D. Burger, R. P. Carney, E. Cocucci, R. Crescitelli, E. Hanser, A. L. Harris, N. J. Haughey, A. Hendrix, A. R. Ivanov, T. Jovanovic-Talisman, N. A. Kruh-Garcia, V. Ku'ulei-Lyn Faustino, D. Kyburz, C. Lässer, K. M. Lennon, J. Lötvall, A. L. Maddox, E. S. Martens-Uzunova, R. R. Mizenko, L. A. Newman, A. Ridolfi, E. Rohde, T. Rojalin, A. Rowland, A. Saftics, U. S. Sandau, J. A. Saugstad, F. Shekari, S. Swift, D. Ter-Ovanesyan, J. P. Tosar, Z. Useckaite, F. Valle, Z. Varga, P. Van Der Pol, M. J. C. Van Herwijnen, M. H. M. Wauben, A. M. Wehman, S. Williams, A. Zendrini, A. J. Zimmerman, MISEV Consortium, C. Théry and K. W. Witwer, Minimal Information for Studies of Extracellular Vesicles (MISEV2023): From Basic to Advanced Approaches, J. Extracell. Vesicles, 2024, 13(2), e12404, DOI:10.1002/jev2.12404.
- P. Sansone, C. Savini, I. Kurelac, Q. Chang, L. B. Amato, A. Strillacci, A. Stepanova, L. Iommarini, C. Mastroleo and L. Daly, Packaging and Transfer of Mitochondrial DNA via Exosomes Regulate Escape from Dormancy in Hormonal Therapy-Resistant Breast Cancer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(43), E9066–E9075 CrossRef CAS PubMed.
- Y. Vinik, F. G. Ortega, G. B. Mills, Y. Lu, M. Jurkowicz, S. Halperin, M. Aharoni, M. Gutman and S. Lev, Proteomic Analysis of Circulating Extracellular Vesicles Identifies Potential Markers of Breast Cancer Progression, Recurrence, and Response, Sci. Adv., 2020, 6(40), eaba5714 CrossRef CAS PubMed.
- N. Nishida-Aoki, Y. Izumi, H. Takeda, M. Takahashi, T. Ochiya and T. Bamba, Lipidomic Analysis of Cells and Extracellular Vesicles from High-and Low-Metastatic Triple-Negative Breast Cancer, Metabolites, 2020, 10(2), 67 CrossRef CAS PubMed.
- S. Kehrloesser, O. Cast, T. S. Elliott, R. J. Ernst, A. C. Machel, J.-X. Chen, J. W. Chin and M. L. Miller, Cell-of-Origin–Specific Proteomics of Extracellular Vesicles, PNAS Nexus, 2023, 2(4), pgad107 CrossRef PubMed.
- F. Teng and M. Fussenegger, Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering, Adv. Sci., 2021, 8(1), 2003505 Search PubMed.
- J. Mariscal, T. Vagner, M. Kim, B. Zhou, A. Chin, M. Zandian, M. R. Freeman, S. You, A. Zijlstra and W. Yang, Comprehensive Palmitoyl-Proteomic Analysis Identifies Distinct Protein Signatures for Large and Small Cancer-Derived Extracellular Vesicles, J. Extracell. Vesicles, 2020, 9(1), 1764192 Search PubMed.
- C. E. Jackson, B. S. Scruggs, J. E. Schaffer and P. I. Hanson, Effects of Inhibiting VPS4 Support a General Role for ESCRTs in Extracellular Vesicle Biogenesis, Biophys. J., 2017, 113(6), 1342–1352, DOI:10.1016/j.bpj.2017.05.032.
- V. Muralidharan-Chari, J. Clancy, C. Plou, M. Romao, P. Chavrier, G. Raposo and C. D'Souza-Schorey, ARF6-Regulated Shedding of Tumor Cell-Derived Plasma Membrane Microvesicles, Curr. Biol., 2009, 19(22), 1875–1885 Search PubMed.
- D. Di Vizio, J. Kim, M. H. Hager, M. Morello, W. Yang, C. J. Lafargue, L. D. True, M. A. Rubin, R. M. Adam and R. Beroukhim, Oncosome Formation in Prostate Cancer: Association with a Region of Frequent Chromosomal Deletion in Metastatic Disease, Cancer Res., 2009, 69(13), 5601–5609 Search PubMed.
- G. Van Niel, G. D'Angelo and G. Raposo, Shedding Light on the Cell Biology of Extracellular Vesicles, Nat. Rev. Mol. Cell Biol., 2018, 19(4), 213–228, DOI:10.1038/nrm.2017.125.
- S. Kruger, Z. Y. A. Elmageed, D. H. Hawke, P. M. Wörner, D. A. Jansen, A. B. Abdel-Mageed, E. U. Alt and R. Izadpanah, Molecular Characterization of Exosome-like Vesicles from Breast Cancer Cells, BMC Cancer, 2014, 14, 1–10 CrossRef PubMed.
- P. Jenjaroenpun, Y. Kremenska, V. M. Nair, M. Kremenskoy, B. Joseph and I. V. Kurochkin, Characterization of RNA in Exosomes Secreted by Human Breast Cancer Cell Lines Using Next-Generation Sequencing, PeerJ, 2013, 1, e201 CrossRef PubMed.
- J. M. Diamond, C. Vanpouille-Box, S. Spada, N.-P. Rudqvist, J. R. Chapman, B. M. Ueberheide, K. A. Pilones, Y. Sarfraz, S. C. Formenti and S. Demaria, Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs, Cancer Immunol. Res., 2018, 6(8), 910–920 CrossRef CAS PubMed.
- A. Zomer, C. Maynard, F. J. Verweij, A. Kamermans, R. Schäfer, E. Beerling, R. M. Schiffelers, E. de Wit, J. Berenguer, S. I. J. Ellenbroek, T. Wurdinger, D. M. Pegtel and J. van Rheenen, In Vivo Imaging Reveals Extracellular Vesicle-Mediated Phenocopying of Metastatic Behavior, Cell, 2015, 161(5), 1046–1057, DOI:10.1016/j.cell.2015.04.042.
- T. H. Lee, S. Chennakrishnaiah, B. Meehan, L. Montermini, D. Garnier, E. D'Asti, W. Hou, N. Magnus, T. Gayden and N. Jabado, Barriers to Horizontal Cell Transformation by Extracellular Vesicles Containing Oncogenic H-Ras, Oncotarget, 2016, 7(32), 51991 CrossRef PubMed.
- B. Han, H. Zhang, R. Tian, H. Liu, Z. Wang, Z. Wang, J. Tian, Y. Cui, S. Ren and X. Zuo, Exosomal EPHA2 Derived from Highly Metastatic Breast Cancer Cells Promotes Angiogenesis by Activating the AMPK Signaling Pathway through Ephrin A1-EPHA2 Forward Signaling, Theranostics, 2022, 12(9), 4127 CrossRef CAS PubMed.
- S.-S. Yang, S. Ma, H. Dou, F. Liu, S.-Y. Zhang, C. Jiang, M. Xiao and Y.-X. Huang, Breast Cancer-Derived Exosomes Regulate Cell Invasion and Metastasis in Breast Cancer via miR-146a to Activate Cancer Associated Fibroblasts in Tumor Microenvironment, Exp. Cell Res., 2020, 391(2), 111983 CrossRef CAS PubMed.
- A. Clayton, S. Al-Taei, J. Webber, M. D. Mason and Z. Tabi, Cancer Exosomes Express CD39 and CD73, Which Suppress T Cells through Adenosine Production, J. Immunol., 2011, 187(2), 676–683, DOI:10.4049/jimmunol.1003884.
- S. K. Mondal, D. Haas, J. Han and T. L. Whiteside, Small EV in Plasma of Triple Negative Breast Cancer Patients Induce Intrinsic Apoptosis in Activated T Cells, Commun. Biol., 2023, 6(1), 815 CrossRef CAS PubMed.
- P. González-Callejo, P. Gener, Z. V. Díaz-Riascos, S. Conti, P. Cámara-Sánchez, R. Riera, S. Mancilla, M. García-Gabilondo, V. Peg and D. Arango, Extracellular Vesicles Secreted by Triple-Negative Breast Cancer Stem Cells Trigger Premetastatic Niche Remodeling and Metastatic Growth in the Lungs, Int. J. Cancer, 2023, 152(10), 2153–2165 Search PubMed.
- D. Huang, J. Chen, L. Yang, Q. Ouyang, J. Li, L. Lao, J. Zhao, J. Liu, Y. Lu and Y. Xing, NKILA lncRNA Promotes Tumor Immune Evasion by Sensitizing T Cells to Activation-Induced Cell Death, Nat. Immunol., 2018, 19(10), 1112–1125 CrossRef CAS PubMed.
- V. Ciravolo, V. Huber, G. C. Ghedini, E. Venturelli, F. Bianchi, M. Campiglio, D. Morelli, A. Villa, P. D. Mina and S. Menard, Potential Role of HER2-overexpressing Exosomes in Countering Trastuzumab-based Therapy, J. Cell. Physiol., 2012, 227(2), 658–667 CrossRef CAS PubMed.
- S. Schlienger, S. Campbell and A. Claing, ARF1 Regulates the Rho/MLC Pathway to Control EGF-Dependent Breast Cancer Cell Invasion, Mol. Biol. Cell, 2014, 25(1), 17–29, DOI:10.1091/mbc.e13-06-0335.
- N. McNamee, M. Catalano, A. Mukhopadhya and L. O'Driscoll, An Extensive Study of Potential Inhibitors of Extracellular Vesicles Release in Triple-Negative Breast Cancer, BMC Cancer, 2023, 23(1), 654 CrossRef CAS PubMed.
- R. Hayatudin, Z. Fong, L. C. Ming, B.-H. Goh, W.-L. Lee and N. Kifli, Overcoming Chemoresistance via Extracellular Vesicle Inhibition, Front. Mol. Biosci., 2021, 8, 629874 CrossRef CAS PubMed.
- Y. Wei, D. Wang, F. Jin, Z. Bian, L. Li, H. Liang, M. Li, L. Shi, C. Pan, D. Zhu, X. Chen, G. Hu, Y. Liu, C.-Y. Zhang and K. Zen, Pyruvate Kinase Type M2 Promotes Tumour Cell Exosome Release via Phosphorylating Synaptosome-Associated Protein 23, Nat. Commun., 2017, 8(1), 14041, DOI:10.1038/ncomms14041.
- F. J. Verweij, M. P. Bebelman, C. R. Jimenez, J. J. Garcia-Vallejo, H. Janssen, J. Neefjes, J. C. Knol, R. De Goeij-de Haas, S. R. Piersma, S. R. Baglio, M. Verhage, J. M. Middeldorp, A. Zomer, J. Van Rheenen, M. G. Coppolino, I. Hurbain, G. Raposo, M. J. Smit, R. F. G. Toonen, G. Van Niel and D. M. Pegtel, Quantifying Exosome Secretion from Single Cells Reveals a Modulatory Role for GPCR Signaling, J. Cell Biol., 2018, 217(3), 1129–1142, DOI:10.1083/jcb.201703206.
- B. Li, M. A. Antonyak, J. Zhang and R. A. Cerione, RhoA Triggers a Specific Signaling Pathway That Generates Transforming Microvesicles in Cancer Cells, Oncogene, 2012, 31(45), 4740–4749, DOI:10.1038/onc.2011.636.
- N. Salmond and K. C. Williams, Isolation and Characterization of Extracellular Vesicles for Clinical Applications in Cancer–Time for Standardization?, Nanoscale Adv., 2021, 3(7), 1830–1852 Search PubMed.
- K. Khanna, N. Salmond, S. Halvaei, A. Johnson and K. C. Williams, Separation and Isolation of CD9-Positive Extracellular Vesicles from Plasma Using Flow Cytometry, Nanoscale Adv., 2023, 5(17), 4435–4446 Search PubMed.
- K. Khanna, N. Salmond, K. S. Lynn, H. S. Leong and K. C. Williams, Clinical Significance of STEAP1 Extracellular Vesicles in Prostate Cancer, Prostate Cancer Prostatic Dis., 2021, 24(3), 802–811 Search PubMed.
- N. Salmond, K. Khanna, G. R. Owen and K. C. Williams, Nanoscale Flow Cytometry for Immunophenotyping and Quantitating Extracellular Vesicles in Blood Plasma, Nanoscale, 2021, 13(3), 2012–2025 Search PubMed.
- A. L. Ashford, D. Oxley, J. Kettle, K. Hudson, S. Guichard, S. J. Cook and P. A. Lochhead, A Novel DYRK1B Inhibitor AZ191 Demonstrates That DYRK1B Acts Independently of GSK3β to Phosphorylate Cyclin D1 at Thr286, Not Thr288, Biochem. J., 2014, 457(1), 43–56, DOI:10.1042/BJ20130461.
- R. H. Takahashi, T. A. Milner, F. Li, E. E. Nam, M. A. Edgar, H. Yamaguchi, M. F. Beal, H. Xu, P. Greengard and G. K. Gouras, Intraneuronal Alzheimer Aβ42 Accumulates in Multivesicular Bodies and Is Associated with Synaptic Pathology, Am. J. Pathol., 2002, 161(5), 1869–1879, DOI:10.1016/S0002-9440(10)64463-X.
- H.-G. Zhang, C. Liu, K. Su, S. Yu, L. Zhang, S. Zhang, J. Wang, X. Cao, W. Grizzle and R. P. Kimberly, A Membrane Form of TNF-α Presented by Exosomes Delays T Cell Activation-Induced Cell Death, J. Immunol., 2006, 176(12), 7385–7393, DOI:10.4049/jimmunol.176.12.7385.
- F. Wang, F. Chen, Y. Shang, Y. Li, Z. Wang, L. Han, Y. Li, L. Zhang, Y. Ti, W. Zhang and M. Zhong, Insulin Resistance Adipocyte-Derived Exosomes Aggravate Atherosclerosis by Increasing Vasa Vasorum Angiogenesis in Diabetic ApoE −/− Mice, Int. J. Cardiol., 2018, 265, 181–187, DOI:10.1016/j.ijcard.2018.04.028.
- G. La Camera, L. Gelsomino, R. Malivindi, I. Barone, S. Panza, D. De Rose, F. Giordano, V. D'Esposito, P. Formisano, D. Bonofiglio, S. Andò, C. Giordano and S. Catalano, Adipocyte-Derived Extracellular Vesicles Promote Breast Cancer Cell Malignancy through HIF-1α Activity, Cancer Lett., 2021, 521, 155–168, DOI:10.1016/j.canlet.2021.08.021.
- T. Baranyai, K. Herczeg, Z. Onódi, I. Voszka, K. Módos, N. Marton, G. Nagy, I. Mäger, M. J. Wood, S. El Andaloussi, Z. Pálinkás, V. Kumar, P. Nagy, Á. Kittel, E. I. Buzás, P. Ferdinandy and Z. Giricz, Isolation of Exosomes from Blood Plasma: Qualitative and Quantitative Comparison of Ultracentrifugation and Size Exclusion Chromatography Methods, PLoS One, 2015, 10(12), e0145686, DOI:10.1371/journal.pone.0145686.
- A. Datta, H. Kim, L. McGee, A. E. Johnson, S. Talwar, J. Marugan, N. Southall, X. Hu, M. Lal and D. Mondal, High-Throughput Screening Identified Selective Inhibitors of Exosome Biogenesis and Secretion: A Drug Repurposing Strategy for Advanced Cancer, Sci. Rep., 2018, 8(1), 8161 Search PubMed.
- N. M. Shukla, F. Sato-Kaneko, S. Yao, M. Pu, M. Chan, F. S. Lao, Y. Sako, T. Saito, K. Messer, T. Hayashi, H. B. Cottam, M. Corr and D. A. Carson, A Triple High Throughput Screening for Extracellular Vesicle Inducing Agents With Immunostimulatory Activity, Front. Pharmacol., 2022, 13, 869649, DOI:10.3389/fphar.2022.869649.
- V. Pospichalova, J. Svoboda, Z. Dave, A. Kotrbova, K. Kaiser, D. Klemova, L. Ilkovics, A. Hampl, I. Crha, E. Jandakova, L. Minar, V. Weinberger and V. Bryja, Simplified Protocol for Flow Cytometry Analysis of Fluorescently Labeled Exosomes and Microvesicles Using Dedicated Flow Cytometer, J. Extracell. Vesicles, 2015, 4(1), 25530, DOI:10.3402/jev.v4.25530.
- G. C. Brittain IV, Y. Q. Chen, E. Martinez, V. A. Tang, T. M. Renner, M.-A. Langlois and S. Gulnik, A Novel Semiconductor-Based Flow Cytometer with Enhanced Light-Scatter Sensitivity for the Analysis of Biological Nanoparticles, Sci. Rep., 2019, 9(1), 16039 Search PubMed.
- M. Mathieu, N. Névo, M. Jouve, J. I. Valenzuela, M. Maurin, F. J. Verweij, R. Palmulli, D. Lankar, F. Dingli, D. Loew, E. Rubinstein, G. Boncompain, F. Perez and C. Théry, Specificities of Exosome versus Small Ectosome Secretion Revealed by Live Intracellular Tracking of CD63 and CD9, Nat. Commun., 2021, 12(1), 4389, DOI:10.1038/s41467-021-24384-2.
- M. Ostrowski, N. B. Carmo, S. Krumeich, I. Fanget, G. Raposo, A. Savina, C. F. Moita, K. Schauer, A. N. Hume and R. P. Freitas, Rab27a and Rab27b Control Different Steps of the Exosome Secretion Pathway, Nat. Cell Biol., 2010, 12(1), 19–30 Search PubMed.
- X. Ouyang, B. Wu, H. Yu and B. Dong, DYRK1-Mediated Phosphorylation of Endocytic Components Is Required for Extracellular Lumen Expansion in Ascidian Notochord, Biol. Res., 2023, 56(1), 10 Search PubMed.
- C. Wang, J. Tu, S. Zhang, B. Cai, Z. Liu, S. Hou, Q. Zhong, X. Hu, W. Liu and G. Li, Different Regions of Synaptic Vesicle Membrane Regulate VAMP2 Conformation for the SNARE Assembly, Nat. Commun., 2020, 11(1), 1531 Search PubMed.
- L. Zhuang, K. Jia, C. Chen, Z. Li, J. Zhao, J. Hu, H. Zhang, Q. Fan, C. Huang and H. Xie, DYRK1B-STAT3 Drives Cardiac Hypertrophy and Heart Failure by Impairing Mitochondrial Bioenergetics, Circulation, 2022, 145(11), 829–846 Search PubMed.
- M. Fan, W. Sun, X. Gu, S. Lu, Q. Shen, X. Liu and X. Zhang, The Critical Role of STAT3 in Biogenesis of Tumor-Derived Exosomes with Potency of Inducing Cancer Cachexia in Vitro and in Vivo, Oncogene, 2022, 41(7), 1050–1062 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02510e |
| This journal is © The Royal Society of Chemistry 2025 |