
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence‘Need for speed: high throughput’ – mass spectrometry approaches for high-throughput directed evolution screening of natural product enzymes
Robert A.
Shepherd
 a,
Conrad A.
Fihn
a,
Alex J.
Tabag
ab,
Shaun M. K.
McKinnie
a and
Laura M.
Sanchez
*a
a,
Conrad A.
Fihn
a,
Alex J.
Tabag
ab,
Shaun M. K.
McKinnie
a and
Laura M.
Sanchez
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Cruz, Santa Cruz, CA 95064, USA +1 831-459-4676. E-mail: lmsanche@ucsc.edu
bPharmaceutical Sciences, University of Kentucky, Lexington, KY 40506, USA
First published on 27th February 2025
Abstract
Covering: 2015 to 2024
Mass spectrometry (MS)-based methods have been implemented extensively for enzyme engineering due to their label-free nature, making them suitable for screening a wide range of biochemical systems. Over the past decade, advancements in mass spectrometry, separation science, and the implementation of hyphenated methods have allowed for more streamlined analysis of large volumes of samples while maximizing the richness and dimensionality of the data collected. In this review we highlight recent advancements in mass spectrometry that have allowed for more efficient, robust, and rigorous enzyme engineering for various applications relating to natural products chemistry.
 Robert A. Shepherd | Robert earned his BSc in Chemistry at the University of North Carolina at Greensboro in 2022. He has since begun his PhD studies at the University of California, Santa Cruz under the mentorship of Dr Laura Sanchez as an NSF Graduate Research Fellow. His research interests include implementation of MALDI-trapped ion mobility spectrometry-mass spectrometry for high-throughput dereplication of microbial natural products and the directed evolution of enzymes. |
 Conrad A. Fihn | Dr Fihn earned their PhD in Medicinal Chemistry at the University of Minnesota under the guidance of Erin Carlson, where they studied histidine kinase inhibitors as virulence blockers in pathogenic bacteria. They have since joined Laura Sanchez's lab at the University of California, Santa Cruz, as an NIH IRACDA Fellow, where they are working to establish methods for MALDI-TOF-based screening methods for directed evolution and examining small molecule–protein interactions in the symbiosis of Vibrio fischeri and Euprymna scolopes. |
 Alex J. Tabag | Alex Tabag earned his BSc degree in Chemistry from the University of California, Santa Cruz in 2023 and was an NIH postbaccalaureate fellow. He began his PhD studies in Pharmaceutical Sciences at the University of Kentucky in Fall 2024. His research interests include natural products, medicinal chemistry, and neurotherapeutic chemistry. |
 Shaun M. K. McKinnie | Shaun McKinnie obtained his PhD in organic chemistry at the University of Alberta, Canada in 2015 under the supervision of Prof. John Vederas, focusing on the chemical syntheses of bioactive peptide analogues. He then completed his postdoctoral studies on the biosyntheses of a variety of marine natural products with Prof. Bradley Moore at the Scripps Institution of Oceanography. Dr McKinnie joined the Chemistry and Biochemistry Department at UC Santa Cruz in 2019 and his research involves identifying, characterizing, and employing natural product enzymes for biosynthetic and chemoenzymatic applications. |
 Laura M. Sanchez | Dr Sanchez earned a PhD in Chemistry at the University of California, Santa Cruz as an NSF graduate research fellow, followed by an NIH IRACDA postdoctoral fellowship at UC San Diego. She started her independent lab in the Fall of 2015 at the University of Illinois at Chicago in the Department of Pharmaceutical sciences. The lab relocated to the University of California, Santa Cruz Department of Chemistry and Biochemistry in January 2021. Her team specializes in using and adapting mass spectrometry imaging, tandem mass spectrometry, and ion mobility spectrometry for small molecule analyses in complex systems. |
1 Introduction
Natural products are invaluable entities to many industries, including cosmetics, food, and pharmaceuticals.1–3 Due to the broad applicability and structural diversity of natural products, engineering their biosynthesis has become increasingly important towards accessing the compounds themselves and creating new analogues. The use of naturally occurring enzymes in tandem with strain development and metabolic engineering strategies has enabled the routine synthesis of complex and/or notable natural products such as alkaloids, flavonoids, fatty acids, and various others.4–7 Advancements in the field of protein engineering have offered researchers a breadth of opportunities for expanding access to natural products and analogues thereof, and insight into their biosynthesis. However, a major bottleneck to engineering biosynthetic enzymes is the scale at which this can be performed and the analytical methodologies that are available and compatible with high-throughput enzymatic screening.8–10An increasingly popular approach to creating new chemical diversity from natural product enzymes is to employ directed evolution (DE). DE is a protein engineering method that mimics evolution in a laboratory setting over a short period of time to drive the activity of enzymes toward an intended, non-native biochemical function.11,12 For example, ‘new-to-nature’ chemistries enabled by heme protein DE efforts have led to development of efficient biocatalysts for performing complex chemistries, such as cyclopropanation, and formation of carbon–silicon bonds.12,13 DE entails performing iterative rounds of mutagenesis on a specific gene, screening of genetic mutants (enzyme variants), and selection of variants that better perform the desired function.8 These functional genes are then replicated and amplified, serving as the starting point for subsequent rounds of genetic diversification, screening, and selection. Methods such as random mutagenesis (i.e. error-prone PCR, tandem repeat insertion, rolling circle amplification, etc.) have enabled rapid generation of thousands-to-millions of genetic mutants (upwards of 1010 mutants), thus introducing adequate genetic diversity in short amounts of time.8,14,15 One major limitation to DE efforts is that screening large mutant libraries can be time-consuming and laborious, presenting a need for high throughput screening (HTS) methods that simultaneously increase efficiency while lowering costs.8,14,16
Many conventional DE screening efforts have relied on visual and/or optical methods for screening and selection of improved enzyme variants. An early example of this comes from Chen and Arnold, where subtilisin E, a serine protease isolated from Bacillus subtilis, was assessed for its activity in dimethylformamide by measuring the halo formed around mutant B. subtilis colonies resulting from the hydrolysis of agar-embedded casein by engineered subtilisin E variants.17 Additionally, colorimetric assays employing chromogenic substrates have been developed to obtain a visual readout of enzyme activity. A popular example is the use of X-galactose (X-Gal) as a substrate for measuring β-galactosidase activity on solid media. The prefix ‘X’ is an abbreviation for the 5-bromo-4-chloro-3-indoxyl moiety on the anomeric carbon of (X-)galactose, which dimerizes post-hydrolyzation, forming a blue precipitate.18 Colonies with a darker blue color are thus selected for subsequent rounds of DE.19 These visual screening methods are generally cost effective and easy to employ, but are often limited to specific enzymatic systems, rendering them inapplicable to a broader range of experimental scenarios.
To further increase the limited throughput offered by these manual screening and selection approaches, researchers have employed automated screening and selection technologies such as digital imaging (DI),20,21 microtiter plate workflows,22–26 and microfluidic (“lab-on-a-chip”) sorting devices in combination with various optical assays to measure the readout of individual enzyme mutants in an automated, high-throughput manner. Currently, microfluidic sorting devices, such as those used in fluorescence-activated droplet sorting (FADS),27,28 offer the highest throughput for mutant enzyme selection, with sorting rates as high as 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 droplets per second (1 droplet contains 1 enzyme variant). Multichannel microfluidic devices are used to suspend single cells expressing variant enzymes within aqueous microdroplets containing the respective reaction substrate and components. This allows for the enzyme reaction to occur within each droplet during a short incubation period. The microdroplets are emulsified within a steady flow of biocompatible oil and ultimately dielectrophoretically (DEP) sorted based on the fluorescence of the reaction products. Droplets that induce fluorescent signals that meet a specified fluorescence threshold are sorted, and the variant enzymes they harbor are prioritized for further rounds of DE.
000 droplets per second (1 droplet contains 1 enzyme variant). Multichannel microfluidic devices are used to suspend single cells expressing variant enzymes within aqueous microdroplets containing the respective reaction substrate and components. This allows for the enzyme reaction to occur within each droplet during a short incubation period. The microdroplets are emulsified within a steady flow of biocompatible oil and ultimately dielectrophoretically (DEP) sorted based on the fluorescence of the reaction products. Droplets that induce fluorescent signals that meet a specified fluorescence threshold are sorted, and the variant enzymes they harbor are prioritized for further rounds of DE.
While the discussed methods address the problem of throughput, they all require the use of chromogenic, absorbent, or fluorescent reaction components which severely limits the range of their applicability.14,29–31 Thus label-free approaches to DE screening have become a necessity in recent years. Over the last decade, mass spectrometry (MS)-based methods have gained significant traction for enzymatic screening due to their label-free nature, making them suitable for screening a wide range of biochemical systems without the need for extensive optimization.4
In this review we discuss different MS-based approaches that have been applied to screen the chemical products resulting from various DE screening campaigns. A discussion of the advantages, limitations, and throughput capabilities of MS-based DE screening workflows will be a major highlight throughout. Additionally, we briefly discuss alternative MS-based HTS techniques employed for enzymatic reaction screening in general, and highlight their potential for high-throughput natural product DE screening (Fig. 1 and Table 1).
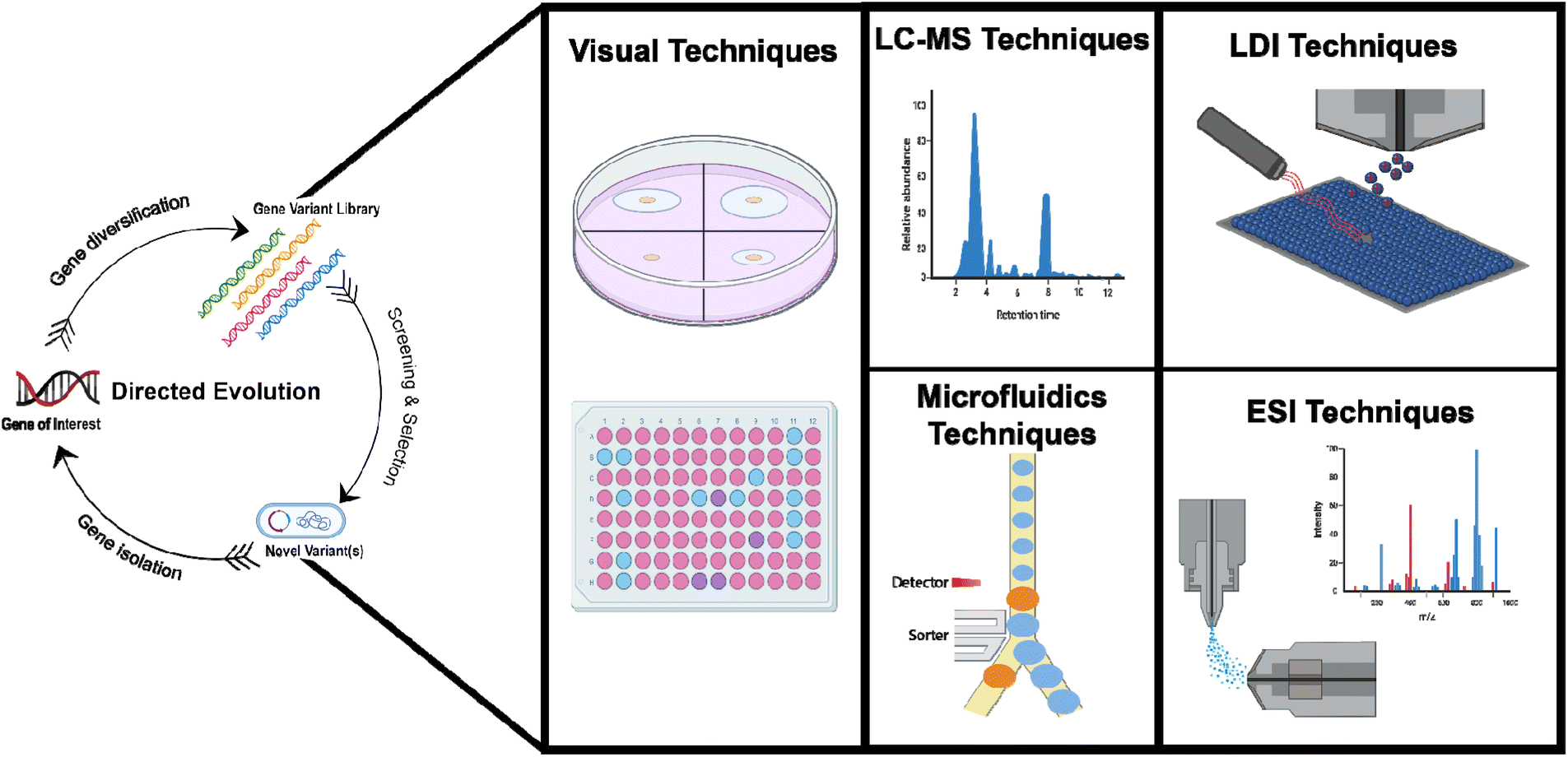 | ||
| Fig. 1 Summary of the techniques discussed in this review. | ||
| Technique | Speed (seconds per sample) | Advantages | Limitations | Application examples |
|---|---|---|---|---|
| References | ||||
| Visual techniques | Not reported | Inexpensive | Relies on human selection of ‘hits’ | 17 and 19 |
| Lower throughput compared to other methods mentioned below | ||||
| Data is easy to interpret | Not easily automated | |||
| Relies on a visibly observable change in phenotype | ||||
| Colorimetric microplates | ∼8 | Automated data collection and analysis | Reaction scope limited to those with fluorescent products | 84 |
| Minimal human intervention | Limited screening capacity | |||
| Requires access to a plate reader and sometimes robotics | ||||
| Digital imaging | ∼1.2 | Relatively inexpensive | Reaction scope limited to those with fluorescent products | 21 and 85 |
| Data is easy to interpret | X-gal methods where side product detection is employed have higher risk for false positives | |||
| Microfluidics sorting | ∼3.6 × 10−4 | Extremely fast. Well suited for extremely large mutant libraries | Reaction scope limited to those with fluorescent products | 9 |
| Screening devices must be adapted/customized for every biological system screened | ||||
| LC-MS | 600–1200 for standard LC-MS | Label-free | Slow. Often <10 variants per hour of screening | 86 |
| High sensitivity | The above raises challenges in shared instrument settings | |||
| Does not require chromogenic substrate or products | Access to expensive equipment (i.e. MS instrumentation) | |||
| Direct infusion ESI-MS | 10–20 | Label-free | No online separation of analytes | 30, 87 and 45 |
| High sensitivity | Sensitive to ion suppression from salts and buffers | |||
| Does not require chromogenic substrate or products | Access to expensive equipment (i.e. MS instrumentation) | |||
| LDI-MS | 1–5 | Label-free | Matrix effects | 14 and 60 |
| High sensitivity | Sample heterogeneity makes reliable quantitation more challenging | |||
| Addresses throughput limitation of LC-MS | No online separation of analytes | |||
| Does not require chromogenic substrate or products | Sensitive to ion suppression from salts and buffers | |||
| Access to expensive equipment (i.e. MS instrumentation) |
2 DE MS data interpretation
We will begin by addressing how mass spectrometry data is typically interpreted for DE and other enzyme engineering campaigns. The key driving component to these campaigns from a data analysis standpoint can be summarized as follows: with every change in genotype, there must exist an observable change in phenotype. Additionally, phenotypic changes must be detectable in a reliable manner, and directly traceable back to the corresponding change in genotype. The most common phenotypic change associated with engineered enzymes is their small molecule products (i.e. the ‘chemotype’). The desired change in chemotype can vary from case-to-case, but is typically monitored through means of a substrate/product ratio, or the production of m/z values correlating to new product species. The reaction substrate(s) and product(s) must have a resolvable mass difference in the case of mass spectrometry alone, or differing retention times in the case of LC-MS.After genetic perturbation (mutagenesis), mutant genes are transformed into a microbial host (typically E. coli or yeast) that subsequently acts as an ‘enzyme factory’ after protein expression is induced, producing high amounts of the enzyme of interest. Once a sufficient level of protein expression has been achieved, the enzyme reaction is performed either in whole-cell lysates, or in vitro with purified enzymes. Enzyme activity is then most commonly assessed using LC-MS, where the reaction substrate(s) and product(s) are monitored using extracted ion chromatograms (EICs), extracting solely m/z values of interest from complex reaction mixtures (Fig. 2A). If the change in the chemotype is desirable, such as an improved substrate/product ratio, the gene responsible for production of the engineered enzyme variant is then sequenced, identifying specific changes to the enzyme structure responsible for the favorable change in activity. The gene is then isolated, amplified, and reintroduced into the DE cycle, acting as the starting point (template) for further mutagenesis, screening, and selection (Fig. 2B). While this process is robust, it is time-consuming to perform lysate reactions on large mutant libraries, and even more so to perform in vitro reactions. The additional time required for chromatographic separation and MS analysis of hundreds-to-thousands of samples is also a notable challenge,14,15,32,33 however several methods are discussed in the following sections that address this.
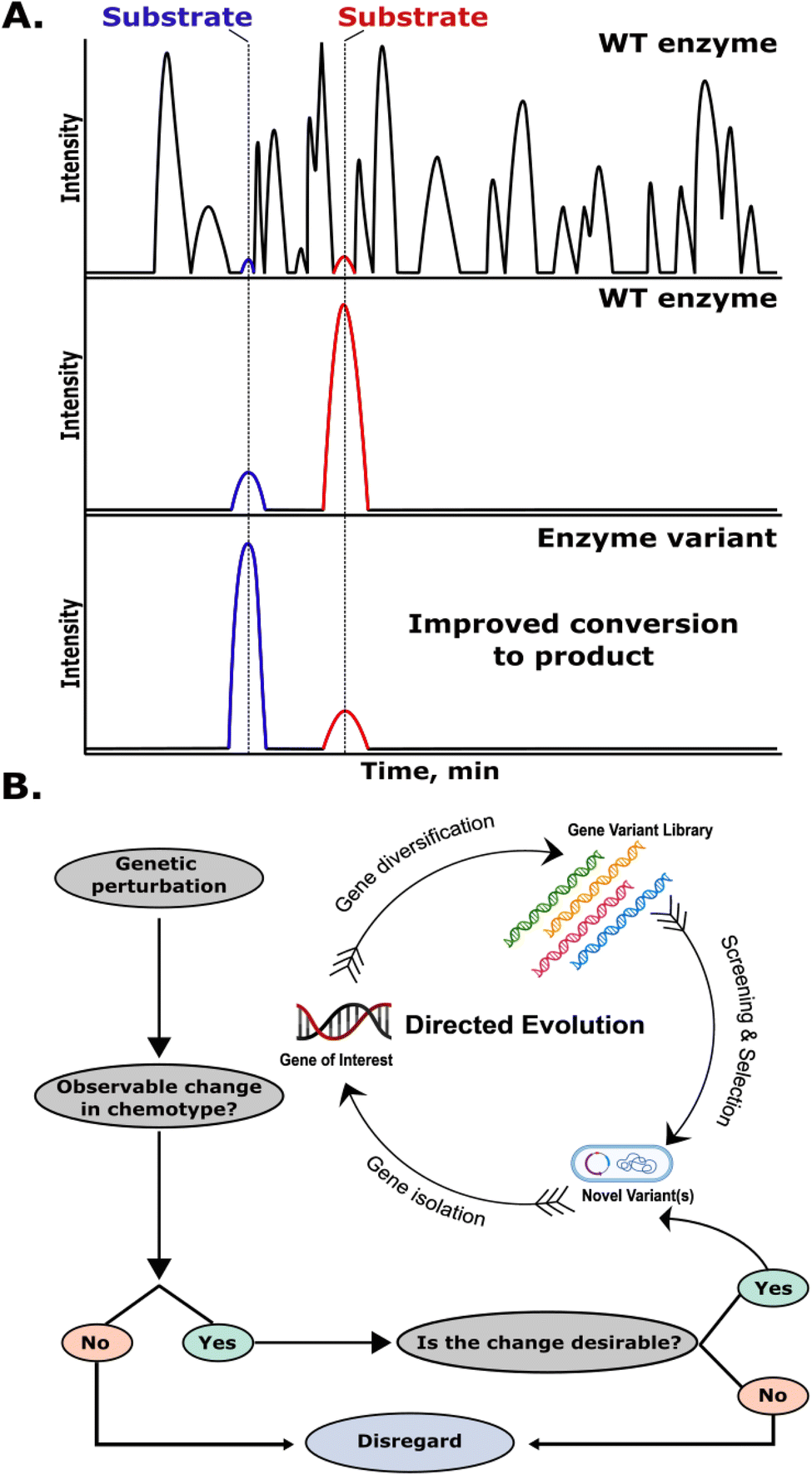 | ||
| Fig. 2 Data interpretation and variant selection for LC-MS-based DE assays. (A) Schematic depiction of a total ion chromatogram (TIC) of an enzymatic reaction mixture (top) followed by an EIC of the product and substrate from the wild type (WT) enzyme (middle) and from an enzyme variant with improved conversion to the product (bottom). (B) Flowchart depicting the selection process for improved enzyme variants. Variants that show a desirable change in chemotype are chosen for further rounds of DE. Variants without improved enzyme activity are typically ignored or discarded. | ||
It should be noted that Fig. 2 highlights the simplest scenario depicting the detection of a single expected product. A unique advantage of MS-based screening methods is the flexibility regarding the simultaneous monitoring of multiple unique molecular species within a single analysis, this sets MS apart from many of the screening methods discussed in Introduction, which are often limited to monitoring for a single analyte in a given experiment. As long as resolution of the analytes can be achieved (by m/z, retention time, and/or collisional cross section in the case of ion mobility spectrometry), multiple analytes can be monitored within a single MS experiment. One approach coined ‘Substrate Multiplexed Screening’ (SUMS) leverages this advantage of MS to select for catalytically promiscuous enzyme variants generated via DE. SUMS involves screening enzyme variants with a mixture of substrates to directly assess mutations that affect substrate preference and scope.34,35 DE campaigns utilizing SUMS have relied on LC-MS for analysis which impacts overall throughput. However, it is worth highlighting that the compatibility of MS with multidimensional, multiplexed analyses has allowed for significantly increased data richness from a single experiment (monitoring of multiple reaction products) when compared to conventional DE screening techniques, and has fostered the implementation of unique screening approaches that extend outside the conventional theme of improving enzyme activity for a single substrate. Finally, MS-based screening approaches lend themselves to monitoring the formation of unexpected side products, which represents a notable possibility when working with variant enzymes. In this scenario, relative abundance of side products could be factored into the selection criteria (i.e. a desirable chemotype), allowing for more informed decisions to be made related to the relative efficiency and/or promiscuity of variant enzymes for a specific (i.e. intended) transformation.
3 Direct infusion/ESI methods
Electrospray ionization (ESI) is the most widely used ionization technique in biochemical analyses.36–38 ESI is a soft ionization technique, meaning very little residual energy is retained by the analyte(s) and limited fragmentation occurs upon ionization in the source. This in turn allows for mass-to-charge (m/z) analysis of intact analyte(s) and its compatibility with aqueous solvent systems facilitates the study of biological macromolecules. The mechanism(s) and fundamentals of ESI technology have been discussed extensively in many notable review articles.39–41 In short, the basic principle of ESI can be summarized in three key steps: (1) production of charged droplets at the tip of the electrospray capillary; (2) evaporative droplet shrinkage and charge-repulsion-induced droplet dissociation; (3) the formation of gas phase ions from these droplets. The resulting gas phase ions then enter the mass spectrometer for analysis. One key advantage to ESI is its compatibility with liquid chromatography solvent systems, allowing for online separation of analytes prior to MS analysis. It is important to highlight that this advantage of ESI becomes its most notable disadvantage in the context of HTS efforts: the implementation of chromatography drastically reduces throughput. As such, there have been efforts to make ESI more amenable to higher throughput analyses by exploiting its compatibility with direct-infusion techniques. Pioneering work from Reetz et al. showcases the use of direct infusion ESI-MS to screen for enantioselective mutant lipases from Pseudomonas aeruginosa.42 Using a mixture of isotopically labeled pseudo-enantiomers, or pseudo-prochiral compounds bearing enantiotopic functional groups, the enantioselectivity (in the form of enantiomeric excess) for a given asymmetric transformation is determined by measuring the relative abundance of reactants and/or products via ESI-MS. As highlighted by Reetz et al., a variety of asymmetric transformations can be monitored using this method, providing isotopically-labeled substrates can be synthesized. While this can present a major challenge and time constraint for DE campaigns, especially with increased substrate complexity, Reetz et al. provide one of the earliest implementations of non-chromatographic ESI-MS for high-throughput screening of DE libraries, and highlights its accuracy and precision to conventional chromatographic techniques, namely chiral gas chromatography (GC). With the introduction of new ESI-MS technologies, researchers have developed DE screening approaches that further streamline sample preparation, eliminate the need for chemical perturbation (isotopic labeling or derivatization), and allow for increased rates of experiment acquisition, highlighted below.3.1 DiBT-MS
Many MS-based screening approaches for biotransformations in vitro or within microbial colonies require multiple liquid-handling steps, often including extraction, dilutions, and chromatography for analyte separation, increasing the probability of systematic error while decreasing throughput. The employment of ambient ionization techniques coupled to MS, namely desorption electrospray ionization (DESI) has overcome many of these obstacles, allowing for direct sampling of analytical targets under ambient conditions. DESI was originally popularized for mass spectrometry imaging (MSI) approaches to study the spatial distributions of chemical species in a variety of biological systems, including metabolic profiling of microbial colonies.43 Yan et al. employed DESI coupled with ion mobility spectrometry-mass spectrometry (IMS-MS) to study the activity of ammonia lyases and P450 monooxygenases. Since this workflow employed direct analysis of the products from living microbial colonies, the methodology was coined ‘direct biotransformation ion mobility mass spectrometry’ (DiBT-IMMS) (Fig. 3).30 | ||
| Fig. 3 Truncated workflow for screening biocatalytic reactions and identifying improved phenylalanine ammonia lyase (PAL) variants. Reprinted and adapted with permission from Kempa et al.44 Copyright © 2021 American Chemical Society. Created with http://BioRender.com. | ||
Kempa et al. later expanded the application of DiBT-IMMS to a HTS campaign of mutant phenylalanine ammonia lyases (PALs) to enhance their activity toward the conversion of electron-rich cinnamic acid derivatives to their noncanonical phenylalanine counterparts via the enantioselective addition of ammonia.44 A metagenomic PAL, AL-11 (GenBank accession number: MW026687), was subjected to DE based on its promiscuity toward a variety of substituted cinnamic acid derivatives, namely di- and trimethoxycinnamic acids, which are generally considered inactive with most PALs. Employment of DiBT-IMMS for biocatalytic screening enabled an analysis rate of ∼40 s per sample. While this is a longer acquisition time than other ESI-MS techniques discussed below, this is primarily due to the nature of the samples themselves and how they are presented to the mass spectrometer. For DiBT-MS, biotransformation reaction solutions are spotted onto membranes, and directly analyzed using DESI-MS in a spatially-resolved fashion omitting automated liquid handling and sample-cleanup steps. This decreases sample handling and preparation time pre-acquisition. Additionally, by acquiring data as a spatially-resolved MS ‘image’, data visualization as a heatmap of relative product abundance is effectively incorporated into the data acquisition itself, saving time post-screening while also providing adequate sample coverage. These factors shorten overall DE campaign length, despite the slightly increased sample acquisition time required for DiBT-MS. This facilitated rapid and comprehensive screening of three mutagenic PAL libraries to identify key amino acid residues necessary for both successful and enhanced conversion to the L-amino acid products. Engineering of AL-11 began with a point mutation of the large polar ligand Q84 to several amino acids of varying size and polarity. The mutation Q84V improved conversion of the substrate to the target amino acid product by nearly 7-fold compared to WT AL-11. To further expand the genetic diversity of this engineering approach, three additional combinatorial libraries were constructed using a degenerate codon set RBT (coding for T, S, I, G, A, V), iteratively targeting neighboring active site residues (N199, L196, L148, I400) in an activity-driven manner. The authors were ultimately successful at engineering AL-11 variants with enhanced activities, some capable of >99% conversion of multi-substituted cinnamic acid substrates to their corresponding L-phenylalanine derivative products. Most hits harbored the L196T mutation in combination with mutated residue N199I, N199G, or N199V, which ultimately gave >95% conversion of 2,3-dimethoxycinnamic acid to the respective enantioselective amino acids. Additionally, Kempa et al. highlight the use of IMS-MS for increasing confidence in their screening workflow using an example from a separate panel of mutagenized imine reductases (IREDs). While IMS can be used to determine an analyte's collisional cross section (CCS), further increasing confidence in the identification of specific analytes, it can alternatively be used as an additional mass filter when coupled to MS systems. If the IMS device is tuned to monitor within a specific mobility range, only ions that fall within this specified range will be detected. Ultimately, this allows for molecules that may have the same or similar m/z value (isomers vs. isobars, respectively), but different CCS values from the analyte of interest to be disregarded during the data acquisition process. Doing so prevents false positives resulting from the detection of m/z's that correspond to constitutional isomers or isobaric species. In this example, Kempa et al. used traveling wave ion mobility spectrometry (TWIMS) to filter a signal corresponding to an IRED substrate's second 13C isotopologue peak (m/z 208; [M + 2]+ of the substrate). This approach prevented false positives resulting from the detection of an isobaric m/z value that did not correspond to the target analyte.
3.2 RapidFire MS
Another notable direct infusion ESI technique is Agilent's RapidFire MS technology, which implements a solid phase extraction (SPE) and desalting step prior to direct infusion of the sample into the mass spectrometer. This removes unwanted contaminants and reduces the complexity of the sample solution. While having no chromatography means analyte separation is dependent entirely on mass resolution (or IMS separation if available), the acquisition time is reduced from 5–30 minutes to as low as 10–20 s per sample.45,46 Unlike DiBT-MS, RapidFire MS performs a sample-cleanup step, yet maintains rapid DI ESI-MS acquisition. Again, this is primarily due to the nature of the sample being analyzed. As explained in further detail below, the samples analyzed in the following example are solution-based and injected directly into the MS. As such, the solution itself provides an averaged landscape for MS-analysis, allowing for a robust assessment of enzyme activity from the collection of relatively few spectra (whereas DiBT-MS requires collection of more spectra to increase confidence in the measurement of enzyme activity due to the nature of the sample preparation).With the aid of RapidFire MS screening, Zetzsche et al. developed an engineered biocatalytic platform for the selective cross coupling of phenolic substrates through oxidative C–C bond formation.45 Biaryl compounds have many uses pertaining to drug design, materials science, and asymmetric catalysis, but there are many challenges in their synthesis. Specifically, metal-catalyzed cross-coupling offering tunable site-selectivity comes at the expense of extra synthetic steps and arduous optimization of reaction conditions, with the formation of tetra-ortho-substituted biaryl bonds remaining a notable synthetic challenge.45,47,48 Several classes of oxidative enzymes that mediate the dimerization of biaryl natural products have been observed from various natural sources including, but not limited to, bacteria and fungi.45,49,50 Notable examples fall within the cytochrome P450 enzyme superfamily, with many arbitrating highly selective oxidation reactions, and a smaller subset catalyzing site-selective and atroposelective dimerizations. Following optimization, Zetzsche et al.45 were successful at expressing an Aspergillus oxidative P450 enzyme, KtnC, in Pichia pastoris (now classified Komagataella phaffi). Using this optimized biosynthetic system, successful dimerization of the native coumarin substrate was observed with increased formation of the expected biaryl product (Fig. 4).
 | ||
| Fig. 4 (A) Biotransformation reactions with S. cerevisiae are performed in 96-well plate format for high-throughput RapidFire MS analysis. (B) Truncated schematic of RapidFire MS SPE cartridge system. Sample is loaded into the cartridge and the initial wash removes any salts and buffers from the sample mixture. A secondary wash can be performed to remove molecules similar to the analytes of interest (not shown). An elution solvent is then used to elute the analytes of interest for ESI-MS analysis. Finally, a re-equilibration step can be performed prior to the next injection (not shown). (C) Observed activity of wild-type KtnC when attempting cross-coupling of 7-hydroxy-5-methyl-2H-chromen-2-one with 2-naphthol. Site-selectivity is poor using the WT KtnC enzyme, resulting in high yields of isomeric products for both the biaryl coupling, and dimerization reactions. Notably, without tuning reaction stoichiometry, formation of the dimerized product is favored in WT KtnC compared to the target molecule and its associated isomeric products. | ||
The authors later expanded the scope of reactions toward unnatural (non-native) biaryl cross couplings between non-equivalent phenolic substrates with various patterns of substitution. It was observed that WT KtnC catalyzed the formation of various unnatural cross-coupled products with maximized yields over dimerized products, so long as the stoichiometry was tuned. KtnC was also shown to tolerate a range of coumarins substituted with various electron-rich and electron-deficient functional groups. However, as the substrates varied further in structure from that of the native coumarin scaffold, a notable decrease in cross-coupling activity and site-selectivity was observed.
Zetzsche et al. next sought to use DE to expand the activity of WT KtnC and address the limitations of chemo-, site-, and atroposelectivity associated with its biosynthetic capabilities. Using a semi-rational approach, thousands of KtnC enzyme variants were generated containing one or more substitutions within 12 Å of the active site. Following transformation of the mutant genes into S. cerevisiae (strain BY4742) on histidine dropout agar plates (2% glucose), the resulting colonies were inoculated into 96-well plates containing histidine dropout minimal media (4% glucose). Following overnight incubation, the cells were pelleted by centrifugation and the media removed, followed by resuspension into histidine dropout minimal media containing the reaction substrates. After incubation for 2–3 days, reactions were quenched with methanol and centrifuged. The resulting supernatants were subsequently screened for enzymatic product formation using RapidFire MS at a rate of ∼12 s per sample, not including blank injections performed between each new sample to minimize carryover. Peak areas of EIC's for internal standard, substrate, and cross-coupled products were collected for each sample and the relative percent conversions were calculated. The average conversion resulting from template reactions for each plate was set to 1.0, and the conversions for each variant were normalized to that of the template, providing a readout of relative fold improvement. Variants with increased conversion relative to WT KtnC were selected for further DE. Over five rounds of evolution, nine active site amino acid substitutions were performed (P142R, D322E, E329M, C331R, F336Y, G396W, R401Q, S513R, V516M), generating the engineered KtnC variant, LxC5, which was observed to have a 92-fold improvement in both activity and site-selectivity toward the target product. However, this did come at the expense of decreased atroposelectivity as determined by orthogonal chiral reversed phase chromatography. This limitation was addressed through two additional rounds of DE to create the KtnC variant LxC7, which catalyzed the cross-coupling to the target product in a 77:23 er, improving atroposelectivity relative to LxC5 (52:48 er), but decreasing overall reaction yield.
4 LDI-MS methods
Laser desorption ionization (LDI) MS based methods have recently gained traction for increased sample throughput while maintaining signal-to-noise levels, precision, reproducibility, and flexibility to monitor multiple products within a given sample. Of the LDI techniques, matrix assisted laser desorption ionization (MALDI) is by far the most common source used for MS-based assays. Briefly, in MALDI, the sample of interest is mixed with an organic matrix and spotted onto a conductive target plate. Upon laser irradiation, the matrix absorbs UV energy from the laser, converting it to electronically excited energy. This in turn transforms the solid co-crystallized matrix-analyte mixture into the gas phase. The analyte molecules become charged in the plume, and are accelerated into a mass spectrometer for detection.51 Notably, MALDI-MS lacks chromatographic dimensionality prior to ionization. The benefit to this is high-throughput generation of MS spectra, with the fastest applicable acquisition times at ∼1–2 seconds per sample. The main limitation to this lack of dimensionality however, is the presence of matrix and/or sample peaks that often overlap with signals of interest, particularly when analyzing ions in the 200–800 m/z range.52 This has recently been addressed with wider implementation of IM as a timescale-compatible method of ion resolution for MALDI-based analyses.53–58 Additionally, limitations concerning sample preparation have prevented widespread use of MALDI-MS for HTS of enzymatic reactions, since many enzymatic screening assays contain high levels of MS-incompatible components, including salts, buffers, and various other additives.59 Advancements in MALDI sample preparation, and optimization of enzymatic reaction conditions have largely been able to address these limitations. For example, liquid–liquid partitions60 and SPE61,62 have been adapted and implemented for high-throughput, MALDI-MS-compatible sample cleaning procedures. There are also various examples highlighted below that use MALDI-MS to screen enzymatic reactions performed in vivo, thus minimizing ion suppression resulting from common components present in enzymatic reaction mixtures (lysates or in vitro) altogether. In the following section we discuss a variety of both MALDI dried-droplet and MALDI-MSI approaches for HTS of natural product biocatalytic reactions.4.1 MALDI dried-droplet MS
There are several examples in the literature that utilize MALDI-TOF-based HTS for monitoring enzyme activity to uncover enzyme drug candidates in situ.59,63 While these examples set precedent for the use of MALDI-TOF MS for high-throughput drug discovery via enzyme reaction screening, they are employed as orthogonal approaches for measuring IC50 values of human protein inhibitors, and are not specifically relevant to natural products. However, the quantification approaches, internal standards, and use of positive and negative controls are notable for how these HTS assays that are commonly employed at pharmaceutical companies, might be extended to large mutant libraries for natural product based applications.Zhang et al. implemented an automated sample preparation workflow coupled to MALDI-TOF MS analysis to detect the biocatalytic reaction products at a rate of ∼5 seconds per sample.60 The generality of their sample preparation procedure suggests this HTS workflow could be applied to a variety of biocatalytic systems with diverse substrate scopes so long as a suitable matrix was chosen and the proper equipment was available. In their study, Zhang et al. aimed to use DE to engineer a cyclodipeptide synthase (CDPS) to produce novel diketopiperazine (DKP) products; CDPSs are recognized as challenging targets for rational engineering approaches, and DKPs can be useful pharmacophores.64–67 The CDPS AlbC was central to the assay and utilizes phenylalanyl-tRNAPhe (Phe-tRNAPhe) and leucyl-tRNALeu (Leu-tRNALeu) as substrates to synthesize cyclo(L-Phe-L-Leu) (cFL) in its native host Streptomyces noursei. They successfully screened a random mutagenesis library consisting of ∼4500 CDPS variants within a week using unlabeled MALDI-TOF MS analysis coupled to an integrated, robotic workcell (Fig. 5).
 | ||
| Fig. 5 (A) Biosynthetic scheme of diketopiperazines by cyclopeptide synthase (CDPS) (B) truncated workflow of the MALDI-MS-based HTS method for directed evolution of AlbC in E. coli by Zhang et al.60 Copyright © 2022 Royal Society of Chemistry (RSC). Created with http://BioRender.com. | ||
This ultimately led to the discovery of an AlbC mutant that produced a new cyclopeptide product (cFV), which revealed a previously unknown residue (F186) that had a substantial impact on the substrate specificity of AlbC. This result was confirmed by further LC-MS and computational analyses of AlbC clones harboring the same mutation (F186L). Additionally, by generating and screening site-saturation mutagenesis (SSM) libraries of AlbC, Zhang et al. were able to confirm the impact of known mutations and reveal new specificity-modulating mutations (T206F) in AlbC, further highlighting the utility of random engineering approaches like DE, and the importance of broadly applicable (unlabeled/untargeted) HTS methods for screening challenging biochemical systems (Fig. 6).
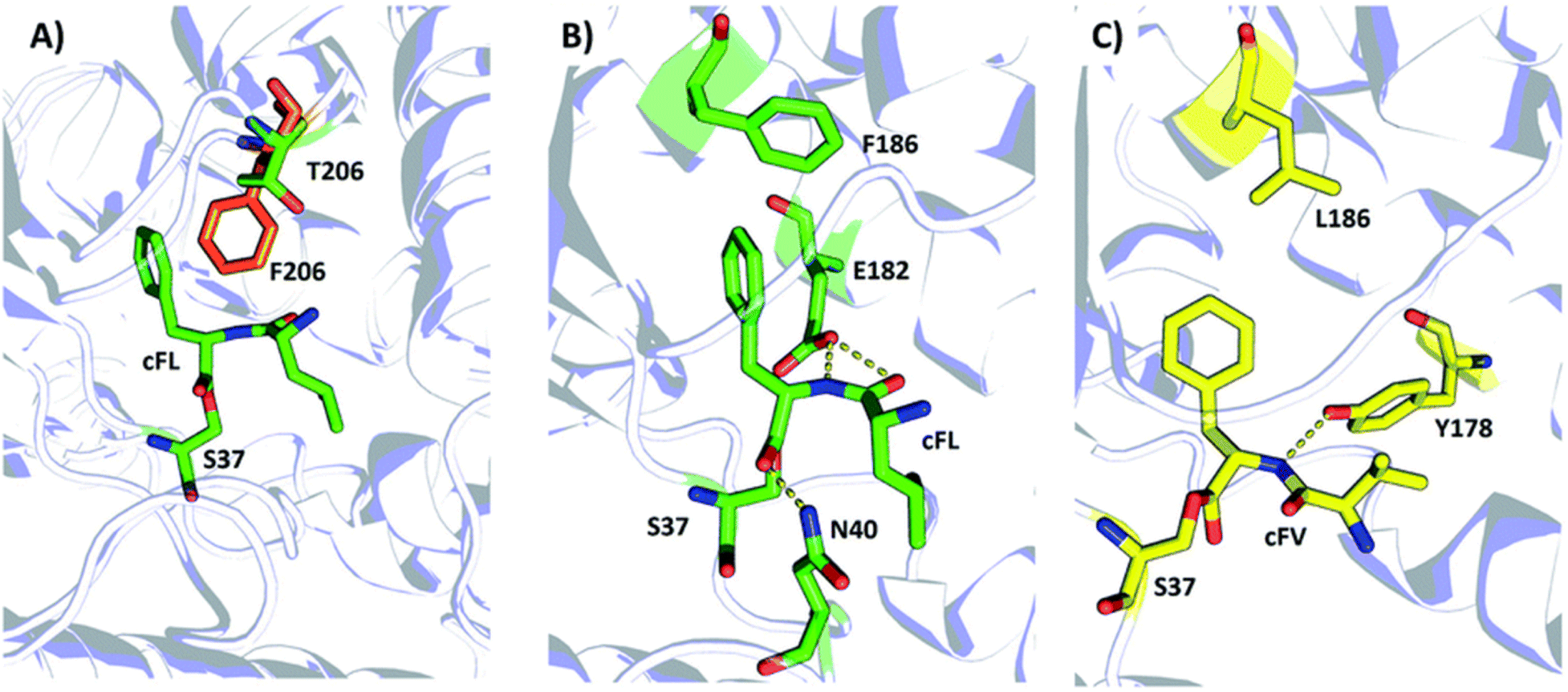 | ||
| Fig. 6 Structural illustration of the substrate binding pocket of the wildtype AlbC and the mutants F186L and T206. Note: Panel (A) superimposition of the WT with T206F. Panels (B and C) schematic representation of the hydrogen-bond and hydrophobic interaction between cFL WT (B) and cFV F186L (C). Dashed lines indicate hydrogen bond (yellow) interactions. The key amino acid residues of WT, F186L, and T206F are drawn as sticks in green, yellow and orange, respectively. Figure and caption reprinted from Zhang et al.60 Copyright © 2022 Royal Society of Chemistry (RSC). | ||
4.2 Self assembled monolayers for matrix assisted laser desorption/ionization (SAMDI)
Zhang et al. recently reported the evolution of a cytochrome P450BM3 variant from Bacillus megaterium, known as cytochrome P411, to perform selective alkylation of sp3 C–H bonds through carbene C–H insertion, presenting an efficient biocatalytic approach for a challenging and valuable chemical transformation.68 In the initial study, the reported reactions required GC-MS for the detection of the reaction products, as they do not contain suitable chromophores or fluorophores. Additionally, the substrates and products could not be reliably coupled to chromo- or fluorogenic co-factors without sacrificing enzyme efficiency or inhibiting downstream synthetic steps (Fig. 7). These points highlight the importance of MS-based techniques for the detection of specific enzymatic transformations that may be otherwise spectrophotometrically silent.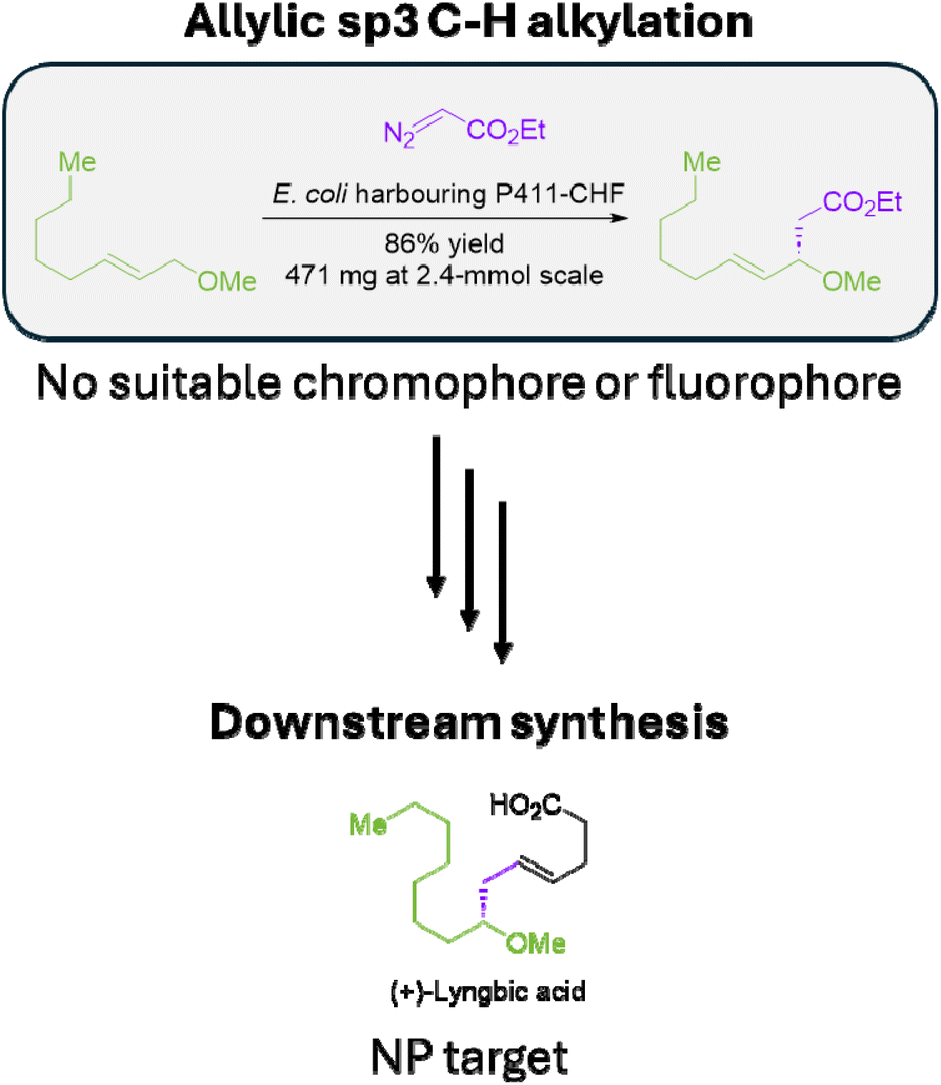 | ||
| Fig. 7 Allylic sp3 C–H alkylation by cytochrome P411 followed by subsequent synthesis of (+)-lyngbic acid. The enzymatic substrate and products do not contain a suitable chromo- or fluorophore. Attachment of a chromogenic moiety via the ethyl diazoacetate co-substrate risks disrupting the biotransformation, and inhibits downstream synthetic steps. | ||
With a goal of continuing to evolve this catalyst in a high-throughput manner, Pluchinsky et al. sought the use of an LDI-based methodology, ‘SAMDI’, to overcome the need for chromatography while monitoring this reaction.14 SAMDI works by applying MALDI to self-assembled monolayers (SAMs) that assemble on a surface (gold) by adsorption. When the surface is submerged in a solution of alkane thiolates, the sulfur atoms coordinate to the gold in a densely packed array. The alkane portion extends from the gold and can be functionalized to provide surfaces with defined chemical reactivities.69 Pluchinsky et al.14 utilized a gold-coated MALDI plate, which was soaked in a solution of disulfide alkanes functionalized with a mixture of maleimide and tri-(ethylene glycol) functional groups, chosen based on the chemistry available on the reaction substrate and products. This generated a MALDI plate that, when lysate reaction mixtures were spotted, selectively immobilized the reaction substrate and product on the surface via a conjugate/Michael addition. This allowed subsequent analysis of the analyte-alkane thiolate conjugates by MALDI-MS.70 The researchers identified the products by a corresponding change in mass of 86 Da and integrated the peaks for the substrate and product to obtain a reaction yield. For each of the libraries, heat maps were generated to exhibit the relative activities of the variants (Fig. 8). Promising library members were run at an analytical scale and their activities were validated using GC-MS. The variant exhibiting the highest conversion to the desired product was then chosen to be the template for the succeeding round of evolution.14
 | ||
| Fig. 8 SAMDI employed for HTS of cytochrome P411 variants. (A) Schematic representation of SAMDI. The thiol headgroup of functionalized alkane thiolates coordinates to the gold surface of the MALDI plate, forming a SAM. The reaction substrate and product are subsequently immobilized on the functionalized surface, followed by matrix application and MALDI-MS analysis. (B) Exposed thiols resulting from HCl induced deprotection of (E)-S-(7-methoxyhept-5-en-1-yl) ethanethioate (substrate) and ethyl (E)-9-(acetylthio)-3-methoxynon-4-enoate (product) are immobilized (covalently tethered) to the SAM via a Michael addition to the maleimide functional group. (C) MALDI analysis was performed, specifically monitoring for the respective m/z values corresponding to the thiolated substrate ([M + Na]+ = 1033.5861) and product ([M + Na]+ = 1119.6229). A mass shift of 86 Da is indicative of successful insertion of the ethyl acetate moiety onto the substrate via cytochrome P411. Reprinted (adapted) with permission from Pluchinsky et al.14 Copyright © 2020 American Chemical Society. (D) High throughput visualization of the MALDI-MS spectra represented as heat maps displaying mutants shaded and organized by relative fold improvement normalized by the average of template controls on each respective 96-well plate. Specifically, reaction yield was calculated (yield = [product area]/[substrate area + product area]) for each enzyme variant, and visualized relative to the average yield observed by the template control enzymes for each plate screened. Reprinted and adapted with permission from Pluchinsky et al. Copyright © 2020 American Chemical Society. | ||
To identify enzymes with increased activity, iterative rounds of random mutagenesis and screening were performed in E. coli. Over three rounds of evolution, data for ∼5000 variants were acquired. SAMDI screening was estimated to be ∼140-fold faster than using GC-MS as the primary screening method. The reproducibility of the technique was demonstrated by selecting and scaling up one variant from the final round of evolution to be repeatedly screened using SAMDI. Overall, Pluchinsky et al. showed that SAMDI-MS analysis can be applied as an effective and efficient DE screening technique.14 Given SAMDI's previous applications in HTS and optimization of both synthetic71 and enzymatic reactions,72–74 it is likely that SAMDI could be employed to monitor a diverse range of biochemical transformations relevant to NP enzyme engineering that are not limited to fluorescent probes or downstream signaling molecules. The caveat to this is that the transformation being observed must exhibit a corresponding change in mass between the substrate and the product(s), and must contain a selectively reactive functional group for covalent attachment to the reactive headgroup of the disulfide alkanes. Lastly, SAMDI does require the use of MALDI plates specifically modified for constructing SAMs, however these have become commercially available in recent years (Charles River Laboratories).
4.3 MALDI MSI
As introduced above, mass spectrometry imaging (MSI) is an advanced analytical technique that combines mass spectrometry with spatial imaging, allowing for the visualization and analysis of the distribution of molecules directly on tissue sections or other surfaces. MALDI MSI is a technique that has been applied to increase throughput and ease sample preparation requirements in the screening of enzymatic activities.75–77 MALDI MSI has enabled the screening of multistep enzyme reactions directly from microbial colonies without the need for lysis. Si et al. applied MALDI MSI to screen recombinant variants of a multi-enzymatic pathway for modified rhamnolipids (RLs).75,78 RLs are naturally occurring biosurfactants produced by certain bacteria, such as Pseudomonas aeruginosa. They are composed of rhamnose sugar molecules linked to hydroxy fatty acids, which give them amphiphilic properties, allowing them to reduce surface tension and act as an emulsifier. RLs have been extensively studied for their potential industrial applications. The transformants were plated on a filter membrane to allow the simultaneous manipulation of many colonies. Microbial cells were initially grown on PVDF (polyvinylidene fluoride or polyvinylidene difluoride) membrane filters on non-inducing agar media to obtain individual colonies. Then, the membrane was transferred to inducing plates to initiate enzyme expression and target molecule production. This separation of growth and induction phases allowed sufficient biomass accumulation before diverting resources to enzyme production. For MALDI-TOF MS analysis, colonies were imprinted onto conductive indium tin oxide (ITO)-coated glass slides. Imprinting the biomass onto a uniform target surface allowed better cell-associated compound profiling than directly mounting agar cultures. Whole slide images of the transparent MALDI targets were acquired using microscopy, and a python script was used to generate laser coordinates for colonies for automatic profiling (Fig. 9c). | ||
| Fig. 9 MALDI-MSI screening method (A) plantazolicin structure. Positions that were modified are highlighted in red. (B) Two-step biochemical pathway for biosynthesis of rhamnolipids (RLs). Different variants synthesized by RhlA (rhamnosyltransferase 1 chain A) using chain length and saturation variations of β-hydroxydecanoyl-ACP. RhlB (rhamnosyltransferase 1 chain B) catalyzes a condensation to form monorhamnolipids. (C) Recombinant variants of a multi-enzymatic pathway are constructed as plasmid DNA libraries and transformed into a production host like Escherichia coli. Colonies are grown on noninducing agar to accumulate biomass, then transferred to inducing plates to express enzymes and produce target molecules, minimizing uneven growth and fitness effects. Colonies are imprinted on indium tin oxide (ITO)-coated, conductive glass slides, yielding better ion signals for cell-associated compounds. (D) The slides can be imaged by bright-field and autofluorescence microscopy to generate laser target coordinates. This automated colony finding precedes the addition of matrix and the analysis by MALDI-TOF MS. Select colonies are recovered for further characterization, such as DNA sequencing or liquid fermentation. | ||
This method was first applied to examine the substrate tolerance of the biosynthesis for the antibiotic plantazolicin (PZN), a linear azol(in)e-containing peptide member of the ribosomally synthesized and post-translationally modified peptides (RiPPs) family of natural products produced by Bacillus velezensis. PZN is first synthesized as an unmodified peptide via translation at the ribosome. A trimeric heterocycle synthetase—composed of the enzymes PznB, PznC, and PznD—then converts select cysteine, serine, and threonine residues in the C-terminal (core) region of the precursor peptide into thiazol(in)e and (methyl)oxazol(in)e heterocycles through a series of cyclodehydration and dehydrogenation reactions. These heterocycles are then stabilized through a series of post-translational modifications, including additional oxidations catalyzed by the dehydrogenase subunit, which converts thiazoline and oxazoline intermediates into aromatic thiazole and oxazole rings. This step is followed by dimethylation by PznL and leader peptide cleavage mediated by the protease PznE, culminating in the production of the bioactive natural product.79 Analogues of PZN were assembled by site-saturation mutagenesis of the precursor peptide gene at two non-cyclized positions (Fig. 9a). After data collection, it was analyzed by t-distributed stochastic neighbor embedding (t-SNE) analyses for unsupervised clustering of spectra and manual examination, and the positions of mutants were mapped to the plate. t-SNE is a dimensionality reduction technique used to visualize high-dimensional data in two or three dimensions. This method captures the data structure by preserving local relationships, making exploring clusters or patterns in complex datasets practical. Thirteen PZN analogues, previously isolated by liquid cultivation and extraction, and ten previously unreported variants were detected.
After success with PZN, the screening method was aimed to quantify changes in the relative abundance of rhamnosyltransferases (RhLs) reaction products by modifying enzymatic specificities in the RhL biosynthesis pathway (Fig. 9b). Directed protein evolution of a two-enzyme pathway for the biosynthesis of RLs was carried out and two mutant strains, identified by MALDI-TOF analysis, were confirmed to produce significantly different ratios of RhL products than WT. The authors showed that this method can be both reproducible in multiple contexts as well as improve upon the rapidity and cost with which the mutants can be screened relative to traditional methods such as automated liquid handling (5 s and $0.0065 versus 15 s and $0.86, respectively).78
Another example of MALDI MSI for enzymatic screening can be highlighted by its use for metabolic biosensing to map enzyme activity of a G2PS1 type-3 polyketide synthase (PKS) from Gerbera hybrida.80 Through their unique screening approach, Xu et al. combine the scalability of microfluidics technology with the generalizability of MALDI MSI for assessing enzyme activity (Fig. 10). G2PS1 natively catalyzes the biosynthesis of triacetic acid lactone (TAL) via condensation of a starter acetyl-CoA unit with two malonyl-CoA units, followed by cyclization of the triketide chain. Importantly, it has been shown that active site mutations in type-3 PKSs can drastically alter the kinetics and scope of polyketide products, allowing access to potentially novel product species.80–82 Since the G2PS1 PKS enzyme of interest in this study catalyzes molecules associated with primary metabolism, the activity of the enzyme has a direct influence on the metabolite profile of the host, which can be leveraged to detect enzyme activity even if the target enzyme product is not directly observed. As such, this approach can also be used as a general method to characterize a range of products produced by engineered enzymes of interest.
 | ||
| Fig. 10 Truncated schematic of the printed droplet microfluidics (PDM) – MALDI MSI sample preparation and analysis workflow used for enzymatic screening of G2PS1 mutants by Xu et al.80 (A) Yeast cells harboring mutant G2PS1 genes are suspended in aqueous yeast nitrogen base minimal media solution. (B) Individual cells are encapsulated into aqueous droplets suspended in fluorocarbon oil. (C) The encapsulated single cells are collected into a 5 mL syringe and stored vertically in a shaking 30 °C incubator for 5 days to form isogenic colonies within each droplet. (D) Droplets containing isogenic colonies are scanned at multiple wavelengths and sorted based on cell density. (E) Droplets containing a uniform cell density are printed onto the custom glass MALDI slide into individual wells and allowed to dry. 2,5-dihydroxybenzoic acid (DHB) MALDI matrix is subsequently sprayed over the printed MALDI slide. (F) Once dry, the slide containing 10000 variant enzymes is subjected to MALDI-MSI analysis. | ||
First, a semi-rational G2PS1 type-3 PKS mutant library was constructed, varying four key amino positions within or near the substrate binding pocket (T199, L202, M259, L261). Ultimately, the library consisted of 1960 codon-shuffled members that were subsequently synthesized into a plasmid backbone and transformed into Yarrowia lipolytica. Single cells were encapsulated and cultured in 300 pL droplets to generate isogenic colonies. This cultivation produced additional material compared to a single cell, thus boosting MS signal and aiding in the collection of reliable metabolomic data. The resulting isogenic colonies were individually dispensed using PDM (∼30 minutes for 10000 colonies) onto custom glass slides etched with 10000 wells (80 μm in diameter) with rounded bottoms to concentrate the dispensed material to the center of each well (Fig. 10E). With PDM, the encapsulated colonies are individually scanned at multiple wavelengths, allowing only those within a narrow cell density range (corresponding to a specified optical threshold processed by a field-programmable gate array) to be dispensed onto the glass slide (Fig. 10C). Finally, the glass slide is dried and spray-coated with matrix, ultimately being subjected to MALDI MSI analysis between m/z 30–630 (Fig. 10F). The authors note that higher capacity slides with the dimensions of a standard MALDI target plate can also be constructed, allowing for up to 100000 wells.
Since MS is used for analysis, information on many molecules within the host cells can be obtained, enabling discovery of unexpected enzyme activities. To visualize and identify these varying enzyme activities, Xu et al. plot variations in the metabolome of the cells as a Uniform Manifold Approximation and Projection (UMAP), which visualizes high-dimensional data within a single plane while preserving clustering information. Through UMAP analysis, four clusters were observed, each representing groups of cells with distinct, yet related, metabolite profiles. Upon mapping the m/z for the target polyketide (TAL, m/z 127), it was observed that one large cluster represented productive G2PS1 mutants biosynthesizing varying amounts of the TAL product. Through further analysis of the generated UMAP, cells within a separate prominent cluster were shown to produce high amounts of another metabolite corresponding to m/z 169. Using LC-MS/MS, this mass was confirmed to correspond to another reported alternative product of G2PS1, 6-acetonyl-4-hydroxy-2-pyrone (AHP). Approximately 50 mutants from each cluster were selected for sequencing and results were validated by re-transforming and testing several mutants in bulk analyses using LC-MS. From their initial mutagenesis campaign, mutant TLMV (differing from WT G2PS1 by the mutation L261V) was observed to have high selectivity for the TAL product, with a 1.62-fold improvement in activity over WT G2PS1. Conversely, mutant TLIG (differing from WT G2PS1 by the mutations M259I and L261G) was observed to have high selectivity for the AHP product, with a 52.1-fold improvement in activity over WT G2PS1. Taking the observed mutations and their relative production of TAL and AHP into account, Xu et al.80 used a consensus design approach83 to engineer the TLLL mutant (with all key active site residues remaining the same relative to the WT, with the exception of the mutation M259L) which showed a 1.98-fold increase in production of the TAL product, and a 17.4-fold increase in production of the AHP product relative to the WT (Fig. 11). Ultimately, these results support that generation of specific enzymatic products can be inferred by clustering metabolomic profiles of the hosts, without directly clustering the m/z values of the products themselves. This also allows for a more generalized metabolomic analysis that could potentially be expanded for the assessment of promiscuous (engineered) enzyme activity without the use of time-consuming analytical screening technologies. It is important to note that this approach may not obtain the same results when analyzing enzymes that do not use endogenous substrates associated with the hosts' primary metabolism. Finally, it should be highlighted that this approach requires specialized equipment that extends beyond access to a MALDI-MS instrument alone. As stated previously, custom glass slides must be constructed, and specialized software for selection of droplets containing adequate cell density must be used. As with many microfluidic devices, the physical preparation of the device itself requires highly specific conditions and parameters for adequate sorting of droplets, which could present further challenges to both access and implementation.
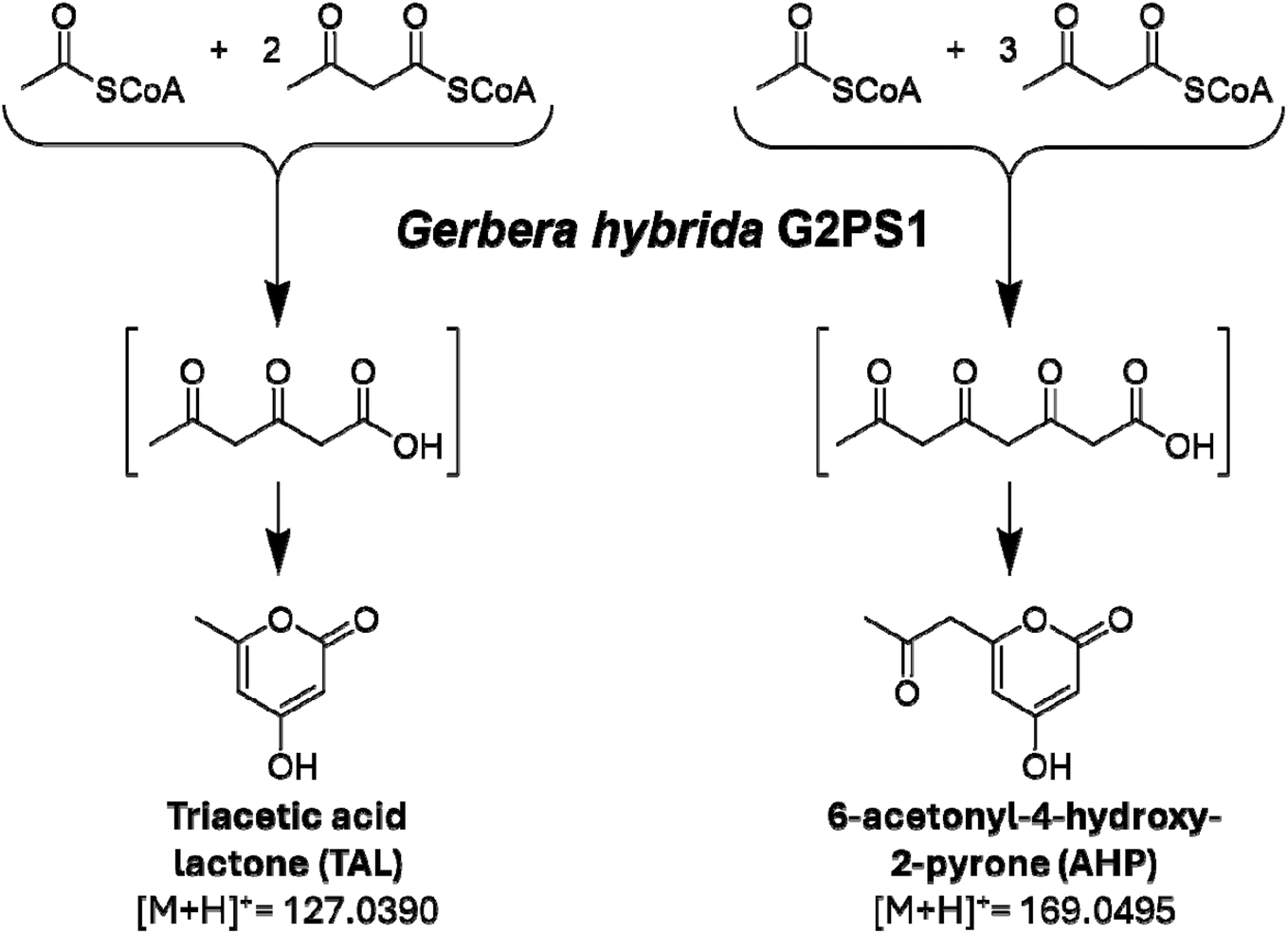 | ||
| Fig. 11 The smallest condensation/cyclization products typically produced by type III polyketide synthase include triacetic acid lactone (TAL, the native product of G2PS1), formed from one acetyl-CoA and two malonyl-CoA, and 6-acetonyl-4-hydroxy-2-pyrone (AHP), formed from one acetyl-CoA and three malonyl-CoA. Schema adapted from Xu et al.80 | ||
5 Forward-looking directions
With ongoing advancements in analytical technology occurring on multiple fronts, we posit that the field of enzyme engineering will see increased use of hyphenated analytical screening technologies in the coming years. Many of the limitations presented by one analytical technique can be addressed by the implementation of another in regards to both screening and selection of enzyme variants. Take microfluidic sorting as an example: microfluidic droplet sorting has enabled HTS of water-in-oil microreactions at unparalleled speeds and volumes compared to automated well-plate screening approaches.88,89 However, we belabor that many of these systems require specially designed fluorescent indicators that must be retained within the droplets and are inert relative to the reaction being investigated so as to provide assay-specific readouts. To address this limitation, Holland-Moritz et al. coupled ESI-MS to microfluidics dielectrophoretic (DEP) droplet sorting, aptly coining this methodology ‘mass activated droplet sorting’ (MADS).87,90 Droplets containing a specified m/z with increased intensity relative to droplets containing a WT or template enzyme are sorted and kept for subsequent experiments. By incorporating ESI-MS as the detection/screening method, the scope of reactions that can be monitored and conversely selected for is greatly increased. ESI-MS also eliminates extensive modification of the reaction conditions and screening system as a whole, making it easier to optimize conditions between different screening campaigns. MADS has been shown to allow analysis of 0.5 samples per seconds, but throughput is ultimately dependent on the scan rate of the mass spectrometer used, as sufficient data points must be collected across a single droplet for analysis.While MADS has not yet been reported for de novo DE screening and selection, Payne et al. assessed its application for the selection of a previously engineered variant of a 4-hydroxyhydrodipicolinate synthase (DapA E84T) producing increased amounts of lysine in vivo relative to WT DapA (Fig. 12).91 From their test dataset, MADS offered a 2.9% false positive rate (i.e. 2.9% of droplets sorted into the ‘winning’ pool contained cells expressing WT DapA), and an acceptably low false negative rate of 4.3% (i.e. 4.3% of droplets sorted into the ‘waste’ pool contained cells expressing DapA E84T). This level of accuracy is consistent with widely used fluorescence-based droplet sorting techniques, such as FADS.27,28,91
 | ||
| Fig. 12 Schematic representation of the MADS enzyme screening pipeline as outlined by Payne et al.91E. coli cells expressing either WT DapA or DapA E84T are suspended into water-in-oil droplets and subsequently incubated to allow the formation of droplet-encapsulated isogenic colonies. The droplets are then split, allowing one portion to be analyzed by ESI-MS. The other portion is sorted by DEP based on the corresponding MS signal intensity. Selected droplets (“winners”) are resuspended in fresh cell media for bulk culture, allowing subsequent phenotyping of the gene(s) and validation of activity by LC-MS/MS. Reprinted (adapted) with permission from Payne et al.91 Copyright © 2023 American Chemical Society. | ||
These results suggest that utilizing mass spectrometry as the detection/selection method in microfluidic sorting campaigns may be a viable option for screening challenging biocatalytic transformations in a label-free, high-throughput manner. Though just one example, MADS highlights the capabilities and importance of leveraging the combined strength(s) of existing analytical technologies to address the ever-evolving challenges associated with HTS and DE campaigns.
Previously, we discussed the applications of IMS-MS in DiBT-IMMS as a means of removing false positives.44 IMS-MS has potential further applications beyond this as an efficient dereplication tool, such as quickly differentiating isomeric compounds in DE screens. Prodiginines, for example, are a family of natural pigments produced by certain bacteria. These pigments have been widely studied, including their biosynthesis, for their promising wide range of biological activities, including antibacterial, antifungal, anticancer, and immunosuppressive properties.92 Prodiginines can exist in various isomeric and tautomeric forms due to their complex structure, which requires more lengthy liquid chromatographic methods to separate and fragmentation, as well as NMR for differentiation. Ramachandra et al. highlighted the differentiation of these products through these methods recently.93
Marshall et al. highlighted using IMS-MS as a rapid alternative for differentiating prodiginines' isomeric forms. Collisional cross-section (CCS) values were employed to differentiate isomeric cyclic prodiginines and confirm their identities. The measured CCS values for synthetic standards were matched to theoretical CCS calculations (<2% difference), which shows these types of differentiations could be made in the absence of standards.94 This work demonstrates ion mobility's power for rapid natural product identification, conformational analysis, and HTS capabilities compared to traditional techniques.
The increasing advantages of using native and engineered enzymes in chemical synthesis has made the field of biocatalysis a vibrant, exciting, and dynamic scientific subdiscipline.95,96 As the chemical industry continues to adapt to the growing presence of biocatalysis and how it can complement traditional chemical methods,97,98 this also highlights the importance of improving technologies to better identify and characterize biocatalysts and enable their downstream applications. Advancements in microfluidics,99 elegant multi-enzyme cascade assays,100,101 the fusion of biocatalysis with photocatalysis,102,103 and the de novo design of novel biocatalysts104 highlight a handful of the landmark technological achievements in protein engineering and biocatalysis within recent years. However, as emphasized throughout this article, advances in MS techniques and modalities can provide nearly universal support for DE campaigns while also considering their individual needs. The versatility of MS ionization, detection, and resolution modalities alongside compatibility across different biological and chemical matrices allows this technique to be broadly applicable across DE screening approaches. MS-based DE screens are most beneficial for biocatalytic transformations that generate obvious differences in m/z features or incorporate moieties with diagnostic isotope patterns (like halogenases). However, the balance between rapid screening and depth of sampling is more challenging when assessing closely related chemical structures, including regioisomers, atropoisomers, or enantiomers with identical m/z values. Orthogonal validation is frequently required when assessing these chemical products, showing that there is not a ‘one size fits all’ model for the application of MS-based techniques in DE campaigns. As MS technologies continue to evolve themselves, it is exciting to see how this will continue to enhance the discovery of novel biocatalysts and their reactions.
Finally, in surveying these literature associated with these different screening approaches, a noted bottleneck and challenge for many technologies is a lack of integrated data analysis capabilities for visualizing the output of these assays. This complicates how the data is processed and visualized with each individual campaign setting thresholds for hits. Creating a vendor agnostic analysis approach would be highly beneficial to the continued development and implementation of MS-based HTS DE assays. In surveying the literature and taking into account best practice examples from pharmaceutical HTS screening, use of internal standards to normalize the mass spectrometry based readouts is highly beneficial in the design of these assays. In our own efforts, we have found the addition of heavy internal standards to be highly critical in normalizing the analysis when comparing the reactant to product ratios as well as clearly defining positive and negative controls for each assay. Synthesis of heavy internal standards to use for natural products would still present a potential challenge in implementation at times. Overall, the number of high throughput approaches is growing and the incorporation of orthogonal separations, such as ion mobility, that add dimensionality compatible with the MS time scales will facilitate the measurement of these DE campaigns that keeps pace with the genetic based approaches.
6 Conflicts of interest
There are no conflicts of interest to declare.7 Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the NIH award R21GM148870 (LMS and SMKMc), K12GM139185 (CAF), and by National Science Foundation grants IOS-2220510 (LMS) and NSF graduate research fellowship program (RAS).8 References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, International Natural Product Sciences Taskforce and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- T. Ahmadu and K. Ahmad, in Bioactive Natural Products for Pharmaceutical Applications, ed. D. Pal and A. K. Nayak, Springer International Publishing, Cham, 2021, pp. 41–91 Search PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. J. Volk, V. G. Tran, S.-I. Tan, S. Mishra, Z. Fatma, A. Boob, H. Li, P. Xue, T. A. Martin and H. Zhao, Chem. Rev., 2023, 123, 5521–5570 CrossRef CAS PubMed.
- J. Montaño López, L. Duran and J. L. Avalos, Nat. Rev. Microbiol., 2022, 20, 35–48 CrossRef PubMed.
- S. Romanowski and A. S. Eustáquio, Curr. Opin. Chem. Biol., 2020, 58, 137–145 CrossRef CAS PubMed.
- S. Kunakom and A. S. Eustáquio, ACS Synth. Biol., 2020, 9, 241–248 CrossRef CAS PubMed.
- H. Xiao, Z. Bao and H. Zhao, Ind. Eng. Chem. Res., 2015, 54, 4011–4020 CrossRef CAS PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- U. Markel, K. D. Essani, V. Besirlioglu, J. Schiffels, W. R. Streit and U. Schwaneberg, Chem. Soc. Rev., 2020, 49, 233–262 RSC.
- Y. Wang, P. Xue, M. Cao, T. Yu, S. T. Lane and H. Zhao, Chem. Rev., 2021, 121, 12384–12444 CrossRef CAS PubMed.
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379–394 CrossRef CAS PubMed.
- Z. Liu and F. H. Arnold, Curr. Opin. Biotechnol., 2021, 69, 43–51 CrossRef CAS PubMed.
- A. J. Pluchinsky, D. J. Wackelin, X. Huang, F. H. Arnold and M. Mrksich, J. Am. Chem. Soc., 2020, 142, 19804–19808 CrossRef CAS PubMed.
- L. Sellés Vidal, M. Isalan, J. T. Heap and R. Ledesma-Amaro, RSC Chem. Biol., 2023, 4, 271–291 RSC.
- T. de Rond, J. Gao, A. Zargar, M. de Raad, J. Cunha, T. R. Northen and J. D. Keasling, Angew Chem. Int. Ed. Engl., 2019, 58, 10114–10119 CrossRef CAS PubMed.
- K. Q. Chen and F. H. Arnold, Biotechnology, 1991, 9, 1073–1077 CrossRef CAS PubMed.
- A. Shafi and Q. Husain, in Glycoside Hydrolases, ed. A. Goyal and K. Sharma, Academic Press, 2023, pp. 323–347 Search PubMed.
- J. H. Zhang, G. Dawes and W. P. Stemmer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4504–4509 CrossRef CAS PubMed.
- S. Delagrave, D. J. Murphy, J. L. Pruss, A. M. Maffia 3rd, B. L. Marrs, E. J. Bylina, W. J. Coleman, C. L. Grek, M. R. Dilworth, M. M. Yang and D. C. Youvan, Protein Eng., 2001, 14, 261–267 CrossRef CAS PubMed.
- F. M. T. Koné, M. Le Béchec, J.-P. Sine, M. Dion and C. Tellier, Protein Eng. Des. Sel., 2009, 22, 37–44 CrossRef PubMed.
- J. B. Behrendorff, C. E. Vickers, P. Chrysanthopoulos and L. K. Nielsen, Microb. Cell Fact., 2013, 12, 76 CrossRef PubMed.
- M. Mack, M. Burger, P. Pietschmann and B. Hock, Protein Expr. Purif., 2008, 61, 92–98 CrossRef CAS PubMed.
- Y.-C. He, C.-L. Ma, J.-H. Xu and L. Zhou, Appl. Microbiol. Biotechnol., 2011, 89, 817–823 CrossRef CAS PubMed.
- M. Samorski, G. Müller-Newen and J. Büchs, Biotechnol. Bioeng., 2005, 92, 61–68 CrossRef CAS PubMed.
- W. A. Duetz, Trends Microbiol., 2007, 15, 469–475 CrossRef CAS PubMed.
- O. Caen, S. Schütz, M. S. S. Jammalamadaka, J. Vrignon, P. Nizard, T. M. Schneider, J.-C. Baret and V. Taly, Microsyst. Nanoeng., 2018, 4, 33 CrossRef PubMed.
- D. Vallejo, A. Nikoomanzar, B. M. Paegel and J. C. Chaput, ACS Synth. Biol., 2019, 8, 1430–1440 CrossRef CAS PubMed.
- E. J. Watkins, P. J. Almhjell and F. H. Arnold, Chembiochem, 2020, 21, 80–83 CrossRef CAS PubMed.
- C. Yan, F. Parmeggiani, E. A. Jones, E. Claude, S. A. Hussain, N. J. Turner, S. L. Flitsch and P. E. Barran, J. Am. Chem. Soc., 2017, 139, 1408–1411 CrossRef CAS PubMed.
- V. Tarallo, K. Sudarshan, V. Nosek and J. Míšek, Chem. Commun., 2020, 56, 5386–5388 RSC.
- A. Knorrscheidt, P. Püllmann, E. Schell, D. Homann, E. Freier and M. J. Weissenborn, ChemCatChem, 2020, 12, 4788–4795 CrossRef CAS.
- Y. V. Sheludko and W.-D. Fessner, Curr. Opin. Struct. Biol., 2020, 63, 123–133 CrossRef CAS PubMed.
- A. D. McDonald, P. M. Higgins and A. R. Buller, Nat. Commun., 2022, 13, 5242 CrossRef CAS PubMed.
- A. D. McDonald, S. K. Bruffy, A. T. Kasat and A. R. Buller, Angew Chem. Int. Ed. Engl., 2022, 61, e202212637 CrossRef CAS PubMed.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem., 2013, 85, 2–9 CrossRef CAS PubMed.
- G. R. D. Prabhu, E. R. Williams, M. Wilm and P. L. Urban, Nat. Rev. Methods Primers, 2023, 3, 23 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS PubMed.
- J. E. Spraker, G. T. Luu and L. M. Sanchez, Nat. Prod. Rep., 2020, 37, 150–162 RSC.
- C. M. Grim, G. T. Luu and L. M. Sanchez, FEMS Microbiol. Lett., 2019, 366(11), fnz135 CrossRef CAS PubMed.
- N. B. Cech and C. G. Enke, Mass Spectrom. Rev., 2001, 20, 362–387 CrossRef CAS PubMed.
- M. T. Reetz, M. H. Becker, H.-W. Klein and D. Stöckigt, Angew Chem. Int. Ed. Engl., 1999, 38, 1758–1761 CrossRef CAS PubMed.
- J. Watrous, P. Roach, T. Alexandrov, B. S. Heath, J. Y. Yang, R. D. Kersten, M. van der Voort, K. Pogliano, H. Gross, J. M. Raaijmakers, B. S. Moore, J. Laskin, N. Bandeira and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1743–E1752 CrossRef CAS PubMed.
- E. E. Kempa, J. L. Galman, F. Parmeggiani, J. R. Marshall, J. Malassis, C. Q. Fontenelle, J.-B. Vendeville, B. Linclau, S. J. Charnock, S. L. Flitsch, N. J. Turner and P. E. Barran, JACS Au, 2021, 1, 508–516 CrossRef CAS PubMed.
- L. E. Zetzsche, J. A. Yazarians, S. Chakrabarty, M. E. Hinze, L. A. M. Murray, A. L. Lukowski, L. A. Joyce and A. R. H. Narayan, Nature, 2022, 603, 79–85 CrossRef CAS PubMed.
- B. T. Veach, T. K. Mudalige and P. Rye, Anal. Chem., 2017, 89, 3256–3260 CrossRef CAS PubMed.
- M. C. Kozlowski, Acc. Chem. Res., 2017, 50, 638–643 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- W. Hüttel and M. Müller, Nat. Prod. Rep., 2021, 38, 1011–1043 RSC.
- H. Aldemir, R. Richarz and T. A. M. Gulder, Angew Chem. Int. Ed. Engl., 2014, 53, 8286–8293 CrossRef CAS PubMed.
- D. Li, J. Yi, G. Han and L. Qiao, ACS Meas. Sci. Au, 2022, 2, 385–404 CrossRef CAS PubMed.
- A. Treu and A. Römpp, Rapid Commun. Mass Spectrom., 2021, 35, e9110 CrossRef CAS PubMed.
- J. Stauber, L. MacAleese, J. Franck, E. Claude, M. Snel, B. K. Kaletas, I. M. V. D. Wiel, M. Wisztorski, I. Fournier and R. M. A. Heeren, J. Am. Soc. Mass Spectrom., 2010, 21, 338–347 CrossRef CAS PubMed.
- S. Heuckeroth, A. Behrens, C. Wolf, A. Fütterer, I. D. Nordhorn, K. Kronenberg, C. Brungs, A. Korf, H. Richter, A. Jeibmann, U. Karst and R. Schmid, Nat. Commun., 2023, 14, 7495 CrossRef CAS PubMed.
- K. V. Djambazova, K. N. Gibson-Corley, J. A. Freiberg, R. M. Caprioli, E. P. Skaar and J. M. Spraggins, J. Am. Soc. Mass Spectrom., 2024, 35, 1692–1701 CrossRef CAS PubMed.
- J. M. Spraggins, K. V. Djambazova, E. S. Rivera, L. G. Migas, E. K. Neumann, A. Fuetterer, J. Suetering, N. Goedecke, A. Ly, R. Van de Plas and R. M. Caprioli, Anal. Chem., 2019, 91, 14552–14560 CrossRef CAS PubMed.
- K. V. Djambazova, M. Dufresne, L. G. Migas, A. R. S. Kruse, R. Van de Plas, R. M. Caprioli and J. M. Spraggins, Anal. Chem., 2023, 95, 1176–1183 CAS.
- K. V. Djambazova, D. R. Klein, L. G. Migas, E. K. Neumann, E. S. Rivera, R. Van de Plas, R. M. Caprioli and J. M. Spraggins, Anal. Chem., 2020, 92, 13290–13297 CrossRef CAS PubMed.
- K. Beeman, J. Baumgärtner, M. Laubenheimer, K. Hergesell, M. Hoffmann, U. Pehl, F. Fischer and J.-C. Pieck, SLAS Discovery, 2017, 22, 1203–1210 CrossRef CAS PubMed.
- S. Zhang, J. Zhu, S. Fan, W. Xie, Z. Yang and T. Si, Chem. Sci., 2022, 13, 7581–7586 RSC.
- S. Ekström, L. Wallman, D. Hök, G. Marko-Varga and T. Laurell, J. Proteome Res., 2006, 5, 1071–1081 CrossRef PubMed.
- L. Zhang and R. Orlando, Anal. Chem., 1999, 71, 4753–4757 CrossRef CAS PubMed.
- M. Winter, T. Bretschneider, C. Kleiner, R. Ries, J. P. Hehn, N. Redemann, A. H. Luippold, D. Bischoff and F. H. Büttner, SLAS Discovery, 2018, 23, 561–573 CrossRef CAS PubMed.
- M. B. Martins and I. Carvalho, Tetrahedron, 2007, 63, 9923–9932 CrossRef CAS.
- J. Bojarska, A. Mieczkowski, Z. M. Ziora, M. Skwarczynski, I. Toth, A. O. Shalash, K. Parang, S. A. El-Mowafi, E. H. M. Mohammed, S. Elnagdy, M. AlKhazindar and W. M. Wolf, Biomolecules, 2021, 11, 1515 CrossRef CAS PubMed.
- C. Romero-Diaz, S. M. Campos, M. A. Herrmann, K. N. Lewis, D. R. Williams, H. A. Soini, M. V. Novotny, D. K. Hews and E. P. Martins, Sci. Rep., 2020, 10, 4303 CrossRef PubMed.
- Y.-M. Ma, X.-A. Liang, Y. Kong and B. Jia, J. Agric. Food Chem., 2016, 64, 6659–6671 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed.
- C. Nicosia and J. Huskens, Mater. Horiz., 2013, 1, 32–45 RSC.
- M. Mrksich, ACS Nano, 2008, 2, 7–18 CrossRef CAS PubMed.
- A. B. Diagne, S. Li, G. A. Perkowski, M. Mrksich and R. J. Thomson, ACS Comb. Sci., 2015, 17, 658–662 CrossRef CAS PubMed.
- M. D. Scholle, D. McLaughlin and Z. A. Gurard-Levin, SLAS Discov., 2021, 26, 974–983 CrossRef CAS PubMed.
- L. C. Szymczak, C.-F. Huang, E. J. Berns and M. Mrksich, Methods Enzymol., 2018, 607, 389–403 CAS.
- S. E. Anderson, N. S. Fahey, J. Park, P. T. O'Kane, C. A. Mirkin and M. Mrksich, Analyst, 2020, 145, 3899–3908 RSC.
- P. Xue, T. Si and H. Zhao, Methods Enzymol., 2020, 644, 255–273 CAS.
- L. Ban, N. Pettit, L. Li, A. D. Stuparu, L. Cai, W. Chen, W. Guan, W. Han, P. G. Wang and M. Mrksich, Nat. Chem. Biol., 2012, 8, 769–773 CrossRef CAS PubMed.
- R. A. Heins, X. Cheng, S. Nath, K. Deng, B. P. Bowen, D. C. Chivian, S. Datta, G. D. Friedland, P. D'Haeseleer, D. Wu, M. Tran-Gyamfi, C. S. Scullin, S. Singh, W. Shi, M. G. Hamilton, M. L. Bendall, A. Sczyrba, J. Thompson, T. Feldman, J. M. Guenther, J. M. Gladden, J.-F. Cheng, P. D. Adams, E. M. Rubin, B. A. Simmons, K. L. Sale, T. R. Northen and S. Deutsch, ACS Chem. Biol., 2014, 9, 2082–2091 CrossRef CAS PubMed.
- T. Si, B. Li, T. J. Comi, Y. Wu, P. Hu, Y. Wu, Y. Min, D. A. Mitchell, H. Zhao and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 12466–12473 CrossRef CAS PubMed.
- R. Scholz, K. J. Molohon, J. Nachtigall, J. Vater, A. L. Markley, R. D. Süssmuth, D. A. Mitchell and R. Borriss, J. Bacteriol., 2011, 193, 215–224 CrossRef CAS PubMed.
- L. Xu, K.-C. Chang, E. M. Payne, C. Modavi, L. Liu, C. M. Palmer, N. Tao, H. S. Alper, R. T. Kennedy, D. S. Cornett and A. R. Abate, Nat. Commun., 2021, 12, 6803 CrossRef CAS PubMed.
- J. M. Jez, M. B. Austin, J. Ferrer, M. E. Bowman, J. Schröder and J. P. Noel, Chem. Biol., 2000, 7, 919–930 CrossRef CAS PubMed.
- I. Abe, S. Oguro, Y. Utsumi, Y. Sano and H. Noguchi, J. Am. Chem. Soc., 2005, 127, 12709–12716 CrossRef CAS PubMed.
- B. T. Porebski and A. M. Buckle, Protein Eng. Des. Sel., 2016, 29, 245–251 CrossRef CAS PubMed.
- J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5869–5874 CrossRef CAS PubMed.
- H. Joo, A. Arisawa, Z. Lin and F. H. Arnold, Chem. Biol., 1999, 6, 699–706 CrossRef CAS PubMed.
- D. T. Tran, V. J. Cavett, V. Q. Dang, H. L. Torres and B. M. Paegel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14686–14691 CrossRef CAS PubMed.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun and R. T. Kennedy, Angew Chem. Int. Ed. Engl., 2020, 59, 4470–4477 CrossRef CAS PubMed.
- A. Sciambi and A. R. Abate, Lab Chip, 2015, 15, 47–51 RSC.
- G. Sun, L. Qu, F. Azi, Y. Liu, J. Li, X. Lv, G. Du, J. Chen, C.-H. Chen and L. Liu, Biosens. Bioelectron., 2023, 225, 115107 CrossRef CAS PubMed.
- E. E. Kempa, C. A. Smith, X. Li, B. Bellina, K. Richardson, S. Pringle, J. L. Galman, N. J. Turner and P. E. Barran, Anal. Chem., 2020, 92, 12605–12612 CrossRef CAS PubMed.
- E. M. Payne, B. E. Murray, L. I. Penabad, E. Abbate and R. T. Kennedy, Anal. Chem., 2023, 95, 15716–15724 CrossRef CAS PubMed.
- T. de Rond, P. Stow, I. Eigl, R. E. Johnson, L. J. G. Chan, G. Goyal, E. E. K. Baidoo, N. J. Hillson, C. J. Petzold, R. Sarpong and J. D. Keasling, Nat. Chem. Biol., 2017, 13, 1155–1157 CrossRef CAS PubMed.
- M. Ramachandra, J. L. M. Innis, J. Yu, G. W. Howe, F. Sauriol, R. D. Oleschuk and A. C. Ross, J. Am. Chem. Soc., 2025, 147(5), 3937–3942 CrossRef CAS PubMed.
- A. P. Marshall, A. R. Johnson, M. M. Vega, R. J. Thomson and E. E. Carlson, J. Nat. Prod., 2020, 83, 159–163 CrossRef CAS PubMed.
- J. B. Pyser, S. Chakrabarty, E. O. Romero and A. R. H. Narayan, ACS Cent. Sci., 2021, 7, 1105–1116 CrossRef CAS PubMed.
- E. N. Kissman, M. B. Sosa, D. C. Millar, E. J. Koleski, K. Thevasundaram and M. C. Y. Chang, Nature, 2024, 631, 37–48 CrossRef CAS PubMed.
- S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715–735 CrossRef CAS PubMed.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, eadh8615 CrossRef CAS PubMed.
- C. J. Markin, D. A. Mokhtari, F. Sunden, M. J. Appel, E. Akiva, S. A. Longwell, C. Sabatti, D. Herschlag and P. M. Fordyce, Science, 2021, 373, eabf8761 CrossRef CAS PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- M. E. Neugebauer, E. N. Kissman, J. A. Marchand, J. G. Pelton, N. A. Sambold, D. C. Millar and M. C. Y. Chang, Nat. Chem. Biol., 2022, 18, 171–179 CrossRef CAS PubMed.
- H. Fu, J. Cao, T. Qiao, Y. Qi, S. J. Charnock, S. Garfinkle and T. K. Hyster, Nature, 2022, 610, 302–307 CrossRef CAS PubMed.
- L. Cheng, D. Li, B. K. Mai, Z. Bo, L. Cheng, P. Liu and Y. Yang, Science, 2023, 381, 444–451 CrossRef CAS PubMed.
- A. H.-W. Yeh, C. Norn, Y. Kipnis, D. Tischer, S. J. Pellock, D. Evans, P. Ma, G. R. Lee, J. Z. Zhang, I. Anishchenko, B. Coventry, L. Cao, J. Dauparas, S. Halabiya, M. DeWitt, L. Carter, K. N. Houk and D. Baker, Nature, 2023, 614, 774–780 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |