DOI:
10.1039/D5NJ01451D
(Paper)
New J. Chem., 2025,
49, 11870-11884
Synthesis and evaluation of edaravone-1,3,4-oxadiazole derivatives as potential anti-cancer inhibitors†
Received
2nd April 2025
, Accepted 6th June 2025
First published on 9th June 2025
Abstract
Edaravone, a pyrazole-based compound, exhibits inherent antitumor activity. Furthermore, its combination with compounds like 1,3,4-oxadiazoles may have significant potential in pharmaceutical chemistry, due to their diverse biological activities—including antiviral, antibacterial, anti-inflammatory, and anticancer effects. In this study, we synthesized edaravone-1,3,4-oxadiazole derivatives through a multistep route starting from phenyl-hydrazine and ethyl acetoacetate, involving pyrazole formation, ether synthesis, hydrazinolysis, cyclization, and final amine coupling. All the synthesized derivatives were thoroughly characterized using NMR, LC-MS and IR to confirm their chemical structures and assess their purity and molecular integrity. Through an in silico approach we identified two of the derivatives exhibited inhibition activity comparable with that of Erlotinib, an approved drug for inhibiting Epidermal Growth Factor Receptor (EGFR) kinase. Further in vitro analysis with the two derivatives showed significant results, they exhibited cytotoxicity toward MDA-MB-231 cells at low concentrations with the IC50 values of the two derivatives being 1 μM and 1.9 μM, and these results were compared with the approved Erlotinib drug. This in vitro analysis corroborated the in silico approach. These results suggest that edaravone-1,3,4-oxadiazole derivatives are promising candidates for EGFR-targeted anticancer therapies.
1. Introduction
Compounds with heterocyclic rings are prevalent in metabolic systems and nature, playing crucial roles in sustaining life. Heterocyclic compounds also significantly contribute to the synthesis of new molecules in pharmaceutical chemistry. Substituted 1,3,4-oxadiazoles, a class of heterocyclic compounds, have been utilized across various fields, including pharmaceutical chemistry and material sciences, and as organic catalysts. These compounds exhibit diverse biological activities, such as antiviral, antibacterial, and anti-inflammatory effects. The low lipophilicity and thermal stability of 1,3,4-oxadiazole derivatives make them well-suited for drug design and discovery, whether mono- or di-substituted. Substituted 1,3,4-oxadiazoles have demonstrated different modes of action, which have been harnessed in the development of anticancer drugs.1–7 Other drugs, including antivirals, antimicrobials, antitussives, and antihypertensives, have also been developed.8–13 Recently, our group published a review on the chemistry of such oxazole analogues, discussing various drugs and inhibitors based on the oxazoles.14 Another interesting entity is pyrazole, a well-known five-membered electron-rich heterocycle with two nitrogen atoms in adjacent positions. Pyrazoles have fascinating pharmacological activities such as anticancer,15,16 antimicrobial,17 anti-inflammatory,18 and antioxidant19 activities.
Edaravone is an extension of pyrazole and a well-known drug recognized for its anti-inflammatory and antioxidant properties, and has sparked interest in its potential applications in cancer treatment. Research has focused on its cytoprotective and anti-proliferative effects, which may help mitigate the side effects of conventional cancer therapies. Additionally, edaravone also shows promise as a stand-alone antitumor agent or when used in combination with other drugs.20 Suzuki et al. suggested that edaravone inhibits tumor growth in vitro via modulating EGFR signalling and inducing cell cycle arrest, at least partially.21 Naveen et al. designed and synthesized a new series of edaravone derivatives that underwent characterization using various spectroscopic techniques. Edaravone derivatives were also screened for their in vitro anti-cancer and antioxidant activities. Out of all the screening tests, 5l showed strong anti-PC3 cancer cell activity. Strong activity has been proven by compounds 5i, 5l, and 7c against A549 cancer cells.22 Marković et al. previously conducted in vitro analyses using 4-aminomethylidene derivatives of edaravone and reported impressive anticancer activity against human breast cancer MDA-MB 361 and human cancer MDA-MB 453 cell lines.23
In this article, we have synthesized derivatives of edaravone combined with 1,3,4-oxadiazoles to check the enhancement in their anticancer activity.
The edaravone-1,3,4-oxadiazole derivatives were synthesized via a multistep synthetic route beginning with phenyl-hydrazine and ethyl acetoacetate. The strategy involved the formation of a pyrazolone core as the key intermediate, followed by a series of functional transformations including etherification, hydrazinolysis, cyclization to construct the 1,3,4-oxadiazole ring, and subsequent derivatization with selected amines at room temperature. This synthetic approach was designed to generate a targeted library of hybrid molecules that integrate both pyrazole and oxadiazole pharmacophores, known for their diverse and promising biological activities. Ten different derivatives were synthesized based on the amines given in Scheme 1. All these derivatives were characterised by FTIR, LCMS and NMR. The derivatives based on the amine groups were chosen to have acyclic as well as cyclic rings with different substitutions to enhance the interaction affinity towards the target site. We have taken two acyclic hydrocarbons with different chain lengths; four four-membered rings with different substitutions, and four six-membered heterocyclic rings.
 |
| | Scheme 1 The different amine groups based on which the derivatives of the edaravone have been prepared in this work. | |
Furthermore, in silico molecular docking studies were performed to assess the potential efficacy of the synthesized derivatives against epidermal growth factor receptor (EGFR) kinase—a membrane-bound protein of the receptor tyrosine kinase family that plays a pivotal role in regulating cell proliferation, survival, and differentiation. Overexpression or mutation of EGFR is commonly associated with the development and progression of various cancers. Due to its central role in oncogenic signalling and its involvement in mammary gland development, EGFR has become a critical target in the development of anticancer therapies. The docking simulations were employed to predict the binding interactions of the derivatives within the active site of EGFR, offering valuable insights into their potential as kinase inhibitors.24–27 The pharmacokinetic properties of each synthesized heterocyclic derivative were evaluated to identify the most promising candidates with strong EGFR inhibitory potential, and to compare their performance with the clinically approved EGFR inhibitor, Erlotinib. Molecular docking studies were employed to estimate binding affinities, followed by molecular dynamics (MD) simulations for the top-performing compounds to assess the stability and dynamics of their interactions with EGFR over time. The most potent derivatives, F-9 and F-10, were subsequently subjected to in vitro evaluation using the MTT assay. These compounds demonstrated anticancer activity comparable to that of Erlotinib, with similar IC50 values, thereby corroborating the computational predictions and supporting their potential as effective EGFR-targeted anticancer agents.
2. Results and discussions
2.1. Synthesis
Scheme 2 illustrates the synthesis of the F-series compounds. Initially, phenyl-hydrazine (A) was refluxed with ethyl acetoacetate in the presence of acetic acid for 3 h to afford 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (B) via condensation and cyclization. The R-NH2 shown in (Scheme 2) are described in (Scheme 1). The reaction proceeds through nucleophilic attack of the hydrazine nitrogen on the carbonyl carbon, followed by cyclization to form the pyrazolone ring. The synthetic process was the same as one found in the literature.28
 |
| | Scheme 2 Schematic representations of the reaction steps for the preparation of the ederavone derivatives (F-1 to F-10). | |
To a solution of compound B in 50 mL of DMF, potassium carbonate (11.0 g) was added at 0 °C, followed by the dropwise addition of bromoethyl acetate (7 mL) under the same conditions. The reaction mixture was stirred at room temperature for 6 h, leading to the formation of ethyl 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetate (C) through an SN2-type alkylation of the pyrazolone enolate intermediate. The product was isolated as a pale-yellow sticky oil. Subsequently, compound C was dissolved in methanol (40 mL), and hydrazine hydrate (1.3 mL) was added. The reaction mixture was stirred for 2 h at room temperature, during which hydrazinolysis of the ester occurred, yielding 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetohydrazide (D). A clear phase separation was observed, with the organic DCM layer containing the desired product as a white solid.
Compound D was then dissolved in DCM (20 mL) containing DIPEA (81.25 mL) and cooled to 0 °C. To this mixture, ethyl oxalyl chloride was added dropwise. After stirring for 1 h, tosyl chloride was added, and the reaction was stirred for an additional 18 h at room temperature. These conditions facilitated cyclodehydration to form the 1,3,4-oxadiazole ring, affording ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E).
In the final step, compound E was reacted with a series of selective amines (Scheme 1) in THF at room temperature to furnish the corresponding F-series derivatives via nucleophilic substitution at the ester moiety. Reaction parameters, including temperature, time, base, and isolated yields for each derivative, are summarized in Table 1. These F-series compounds represent ten novel edaravone-based analogues. Structural confirmation was achieved by 1H NMR, 13C NMR, mass spectrometry and IR, as described in the Experimental methods section. Scheme 3 shows the geometries of the novel synthesized derivatives F-1 to F-10 and are provided in the ESI.†
Table 1 Formation of the ederavone derivatives of the F series from E and suitable choice of amines, temperature, base, and time; the yield in % is also given here
| Entry |
Amine |
Base |
Temp. [°C] |
Time [h] |
Yield % |
|
F-1
|
Propane-1-amine (R1) |
— |
RT |
4 |
89 |
|
F-2
|
Butan-1-amine (R2) |
— |
RT |
6 |
83 |
|
F-3
|
1-Methylpiperazine (R3) |
— |
RT |
16 |
77 |
|
F-4
|
Azetidine (R4) |
— |
RT |
6 |
91 |
|
F-5
|
3-Fluoro-3-methylazetidine hydrochloride (R5) |
DIPEA |
RT |
16 |
79 |
|
F-6
|
3,3-Difluoroazetidine hydrochloride (R6) |
DIPEA |
RT |
10 |
88 |
|
F-7
|
3-Fluoroazetidine hydrochloride (R7) |
DIPEA |
RT |
16 |
77 |
|
F-8
|
Morpholine (R8) |
— |
50 |
16 |
82 |
|
F-9
|
Piperidine (R9) |
— |
50 |
16 |
73 |
|
F-10
|
Piperazine (R10) |
— |
50 |
16 |
82 |
 |
| | Scheme 3 Schematic structural representations of the different derivatives F-1 to F-10. | |
2.2. Mechanistic insight
The synthetic route outlined in Scheme 2 follows a series of well-known organic transformations that together allow for the efficient and selective synthesis of the F-series compounds. Initially the formation of intermediate B involves nucleophilic attack of phenyl-hydrazine on the β-ketoester carbonyl group of ethyl acetoacetate, followed by cyclization through enolization to yield the pyrazolone core similar to the work performed by Fuse et al.28 The subsequent SN2 reaction between the deprotonated pyrazolone and bromoethyl acetate generates compound C, facilitated by potassium carbonate. Furthermore, C undergoes hydrazinolysis by hydrazine hydrate leading to the corresponding hydrazide Dvia nucleophilic acyl substitution. The next step involves the formation of the 1,3,4-oxadiazole ring in compound E, which occurs through intramolecular cyclization after reacting with ethyl oxalyl chloride, followed by tosyl chloride to promote ring closure under mild conditions. In the final step, the ester group in compound E is converted into various amide derivatives through aminolysis with selected amines as given in Scheme 1, producing the F-series compounds (Scheme 3). This stepwise strategy enables late-stage diversification for exploring structure–activity relationships.
2.3. Computational analysis
The primary objective of the article is to evaluate the effectiveness of the designed compounds as potential anticancer drugs. To achieve this, in silico studies were initially conducted to identify the best inhibitors for the EGFR protein, a well-known cell–surface receptor from the tyrosine kinase family that regulates cell growth. Mutations leading to the overexpression of EGFR are associated with various types of cancer. Given its critical role in mammary gland development and its involvement in tumor growth and progression when overexpressed, EGFR has become a key target for anti-cancer therapies. Initially, all the 10 compounds F-1 to F-10 are optimized at the M062x/aug-cc-pVDZ level of theory29,30 using the Gaussian software package.31 These Minnesota functionals based on the meta-GGA approximation are highly parameterized approximate exchange–correlation functionals and give a high level of accuracy on the geometrical parameters. The optimized geometries were subjected to pharmacological studies to understand its drug likeliness, followed by docking analysis through Autodock, which narrows down to the high binding affinity of the EGFR with two synthetic compounds, F-9 and F-10, while comparing with the reference drug Erlotinib. The molecular docking was followed by molecular dynamics simulations compared with the same drug to understand the interaction patterns, H-bond analysis, and binding energy during the real-time motion and later was followed by an experimental assay to verify the theoretical results.
2.4. Pharmacological studies
Evaluating the drug-likeness of a compound is a key step in determining its potential suitability for human administration. Table 2 summarizes the predicted pharmacokinetic parameters of the synthesized edaravone derivatives (F-1 to F-10). The molecular weights of all the compounds fall within the range of 340–385 g mol−1, which is slightly lower than that of the reference EGFR inhibitor, Erlotinib (393.44 g mol−1). This suggests that the derivatives may possess favourable characteristics for cellular transport, distribution, and absorption. Notably, none of the compounds violated Lipinski's rule of five, implying a strong potential for good oral bioavailability. In addition, all derivatives exhibited topological polar surface area (TPSA) values below 100 Å2 and were predicted to have high gastrointestinal (GI) absorption. These properties further support their potential as orally active agents that can be efficiently absorbed at therapeutic doses. Based on these favourable parameters, molecular docking studies were subsequently carried out to investigate the binding affinity of each compound (F-1 to F-10) toward the EGFR receptor.
Table 2 Pharmacological parameters of the reference drug Erlotinib and phenyl-3-methyl-5-pyrazolone (edaravone) derivatives
| Compounds |
Molecular weight (g mol−1) |
HBD |
HBA |
TPSA |
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p p |
Bio availability |
Lipinski rule |
GI |
|
Erlotinib
|
393.44 |
1 |
6 |
74.73 |
1.89 |
0.55 |
Yes, 0 violation |
High |
|
F-1
|
341.36 |
1 |
6 |
95.07 |
1.63 |
0.55 |
Yes, 0 violation |
High |
|
F-2
|
355.39 |
1 |
6 |
95.07 |
1.87 |
0.55 |
Yes, 0 violation |
High |
|
F-3
|
382.42 |
0 |
7 |
89.52 |
1.32 |
0.55 |
Yes, 0 violation |
High |
|
F-4
|
341.36 |
0 |
6 |
84.06 |
1.47 |
0.55 |
Yes, 0 violation |
High |
|
F-5
|
373.38 |
0 |
7 |
84.06 |
1.83 |
0.55 |
Yes, 0 violation |
High |
|
F-6
|
377.35 |
0 |
8 |
84.06 |
1.71 |
0.55 |
Yes, 0 violation |
High |
|
F-7
|
359.35 |
0 |
7 |
84.06 |
1.59 |
0.55 |
Yes, 0 violation |
High |
|
F-8
|
369.37 |
0 |
7 |
95.51 |
1.09 |
0.55 |
Yes, 0 violation |
High |
|
F-9
|
367.40 |
0 |
6 |
86.28 |
2.10 |
0.55 |
Yes, 0 violation |
High |
|
F-10
|
368.39 |
1 |
7 |
98.31 |
1.09 |
0.55 |
Yes, 0 violation |
High |
2.5. Virtual screening through molecular docking
Table 3 shows the molecular docking analyses of the Erlotinib drug and the compounds F-1 to F-10 with EGFR kinase. Compound F-9 exhibited the highest binding affinity, −8.37 kcal mol−1, with an inhibition constant of 0.73 μM among all the docked compounds, followed by compound F-10, which exhibited the second highest binding affinity of −8.19 kcal mol−1 with an inhibition constant of 0.99 μM. Similarly, the compounds F-1, F-3, F-4 & F-8 exhibited binding affinities in the range of −7.93 kcal mol−1 to −8.08 kcal mol−1. The remaining derivatives, F-2, F-5, F-6 & F-7 compounds, showed lower binding affinities in the range of −7.23 kcal mol−1 to −7.65 kcal mol−1, though all the derivatives showed higher binding affinity than the reference drug Erlotinib, which exhibits a binding affinity of −6.37 kcal mol−1 with a higher inhibition constant of 21.54 μM. The residues involved in the hydrogen bonding with the protein are also given in Table 3. F-10 showed better hydrogen bonding with the EGFR, showing its better capability for interaction. All other derivatives, except the reference drug, F-1, and F-4, showed single hydrogen bonds. The best two interacting inhibitors (F-9 and F-10) were further used for the MD simulations. The interactions of the Erlotinib drug and the edaravone derivatives (F-9 and F-10) are given in Fig. 1, and all others are given in Fig. S53 in the ESI.†
Table 3 Molecular docking studies results of binding energy, Ki values, and several hydrogen bond interactions with the residues of the compounds (F-1 to F-10) against the epidermal growth factor receptor (PDB ID-4HJO)
| Compound |
Binding energy (kcal mol−1) |
Inhibition constant (μM) |
No. of hydrogen bonds |
Residues involved in hydrogen bonding (in Å) |
|
Erlotinib
|
−6.37 |
21.54 |
0 |
— |
|
F-1
|
−8.07 |
1.22 |
0 |
— |
|
F-2
|
−7.64 |
2.52 |
1 |
LYS721(2.18) |
|
F-3
|
−8.08 |
1.20 |
1 |
LYS721(2.68) |
|
F-4
|
−7.93 |
1.54 |
0 |
— |
|
F-5
|
−7.23 |
5.03 |
1 |
ARG817(2.01) |
|
F-6
|
−7.11 |
6.14 |
1 |
LYS721(2.17) |
|
F-7
|
−7.65 |
2.48 |
1 |
LYS721(2.32) |
|
F-8
|
−8.08 |
1.20 |
1 |
ASP831(2.52) |
|
F-9
|
−8.37 |
0.73 |
1 |
ASP831(2.52) |
|
F-10
|
−8.19 |
0.99 |
2 |
LYS721(2.10), ASP831(1.76) |
 |
| | Fig. 1 The binding interaction of the Erlotinib, F-9, and F-10 with the EGFR protein is given here. | |
2.6. Molecular dynamics simulation studies
Molecular dynamics (MD) simulations were conducted to gain insights into the structural stability, conformational adaptability, and binding energetics of the ligand–EGFR receptor complexes. Simulations were performed for 100 ns for the EGFR complexes with compounds F-9, F-10, and the reference drug Erlotinib. All systems were subjected to energy minimization and equilibration prior to the production runs, as described in the Computational methods section. Key parameters analysed included root mean square deviation (RMSD) to evaluate the overall stability of the complexes, root mean square fluctuation (RMSF) to examine residue-level flexibility, and radius of gyration (Rg) to assess the compactness of the receptor–ligand assemblies. Additionally, hydrogen bonding interactions and solvent-accessible surface area (SASA) were monitored to understand solvent exposure and intermolecular stability.
As shown in Fig. 2A, the RMSD values of the EGFR–Erlotinib complex remained relatively stable, ranging from ∼0.15 to 0.21 nm during the initial 2–18 ns, followed by a gradual increase to ∼0.28 nm around 43 ns, before stabilizing between ∼0.21 and 0.25 nm for the rest of the simulation. In contrast, the EGFR–F-9 and EGFR–F-10 complexes showed higher initial RMSD values, with moderate fluctuations during the first 20 ns, indicating a period of structural adjustment before reaching equilibrium. From 20 ns onward, the RMSD values for both the EGFR–F-9 and EGFR–F-10 complexes remained relatively stable, fluctuating within the range of ∼0.25–0.35 nm throughout the rest of the 100 ns simulation. The average RMSD values for F-9 and F-10 were calculated to be approximately 0.27 nm and 0.25 nm, respectively, which are slightly higher than that of the reference ligand Erlotinib (∼0.19 nm). Despite these differences, all three complexes displayed low RMSD values overall, suggesting that the binding of F-series ligands did not induce major conformational deviations in the receptor structure.
 |
| | Fig. 2 The structural stability of the inhibitor–receptor interactions was evaluated using the following indicators: (A) RMSD, (B) RMSF, (C) Rg, (D) SASA, and (E) number of HB (Erlotinib-ERL). | |
To further evaluate the dynamic behaviour of the protein in response to ligand binding, root mean square fluctuation (RMSF) analyses were performed to assess residue-level flexibility over the simulation trajectory. The RMSF profiles for Erlotinib, F-9, and F-10 are presented in Fig. 2B. Regions showing higher peak values correspond to flexible loop domains, where ligand-induced mobility is most pronounced. Notably, F-10 exhibited a fluctuation pattern similar to Erlotinib, with average RMSF values in the range of ∼0.1–0.2 nm. In contrast, the F-9 complex showed increased residue mobility, with fluctuations reaching up to ∼0.7 nm compared to ∼0.4 nm for Erlotinib, indicating greater flexibility in certain protein regions.
To assess the structural compactness of the EGFR–ligand complexes, the radius of gyration (Rg) of the Cα atoms was calculated for the reference compounds Erlotinib, F-9 and F-10 (Fig. 2C). The Rg values for F-10 were comparable to those of Erlotinib, fluctuating in the range of ∼1.90–1.95 nm, suggesting similar overall compactness of the complex. In contrast, the EGFR–F-9 complex exhibited slightly elevated Rg values (∼1.95–2.00 nm), indicating a marginally less compact structure.
Solvent-accessible surface area (SASA) analysis was performed to examine the exposure of hydrophobic residues upon ligand binding, which reflects conformational shifts in the receptor. As shown in Fig. 2D, the average SASA values for the EGFR complexes with F-9, F-10, and Erlotinib were 151.73 nm2, 147.90 nm2, and 149.03 nm2, respectively. The similarity in these values suggests that all three ligands maintain comparable solvent exposure and occupy the active site in a similar fashion.
Hydrogen bonding interactions were also monitored over the course of the simulation to evaluate ligand–receptor stability (Fig. 2E). The EGFR–Erlotinib complex formed up to five hydrogen bonds, while F-9 and F-10 formed up to four and three hydrogen bonds, respectively. Although slightly fewer, these interactions indicate that both F-9 and F-10 bind with good affinity and contribute to stabilizing the receptor–ligand complex, closely resembling the behaviour of Erlotinib. Overall, the results from RMSD, RMSF, Rg, SASA, and hydrogen bonding analyses suggest that the F-9 and F-10 derivatives have similar structural stability, flexibility, compactness, and solvent exposure with the reference drug Erlotinib and forms stable complexes with EGFR, suggesting that they could be promising alternatives with potential therapeutic value.
2.7. G_MMPBSA calculations for the binding energy
The binding free energies of the ligand–receptor complexes were calculated over the final 30 ns of the MD simulation trajectories, and the results are summarized in Table 4. The EGFR–F-9 complex exhibited a binding energy of −29.32 ± 0.08 kcal mol−1, indicating a stronger binding affinity compared to the reference drug Erlotinib (−25.64 ± 0.06 kcal mol−1). Compound F-10 also showed a comparable affinity, with a binding energy of −24.58 ± 0.06 kcal mol−1. These findings suggest that both F-9 and F-10 interact favourably with the EGFR receptor, with F-9 in particular demonstrating a potentially higher inhibitory capability than Erlotinib.
Table 4 The binding free energies (ΔGBind) of ligand–receptor in kcal mol−1. Van der Waals energy, electrostatic energy, polar solvation energy, and nonpolar-solvation energy are represented by ΔEvdW, ΔEele, ΔGpol, and ΔGn-pol, in kcal mol−1
| Sl. no. |
Compound |
ΔEvdW |
ΔEele |
ΔGpol |
ΔGn-pol |
ΔGbind |
| 1 |
F-9
|
−35.24 ± 0.07 |
−6.46 ± 0.05 |
16.79 ± 0.06 |
−4.41 ± 0.00 |
−29.32 ± 0.08 |
| 2 |
F-10
|
−32.78 ± 0.05 |
−4.99 ± 0.05 |
17.18 ± 0.05 |
−3.98 ± 0.00 |
−24.58 ± 0.06 |
| 3 |
ERL
|
−42.89 ± 0.05 |
−2.65 ± 0.02 |
25.20 ± 0.05 |
−5.11 ± 0.00 |
−25.64 ± 0.06 |
To gain deeper insight into the binding interactions, the energy components contributing to the ligand–receptor complexes were analyzed, focusing on electrostatic and non-bonded interactions. The electrostatic interaction energy (ΔEele) and non-polar solvation energy (ΔGn-pol) for F-9, F-10, and Erlotinib ranged from −2.65 to −6.46 kcal mol−1, indicating their significant role in stabilizing the complexes. Both edaravone derivatives exhibited stronger electrostatic interactions with EGFR compared to Erlotinib, suggesting enhanced charge-based complementarity with the binding site. Furthermore, F-9 and F-10 displayed lower polar solvation energies, which contributed positively to their overall binding affinities, while their non-polar solvation energy values were comparable to those observed for Erlotinib. Although van der Waals contributions were modest, the reduction in polar solvation energy notably improved the binding efficiency of F-9 and F-10, further supporting their potential as effective EGFR inhibitors.
To further elucidate the molecular basis of ligand binding, per-residue free energy decomposition analysis was performed to identify the key amino acid residues within the EGFR receptor involved in interactions with the edaravone derivatives. The residue-wise binding energy contributions for F-9, F-10, and the reference compound Erlotinib are shown in Fig. 3. Three major binding regions were identified as critical contributors to ligand interaction: region I (residues 694–721), region II (residues 764–779), and region III (residues 817–856).
 |
| | Fig. 3 The energetic contribution (kcal mol−1) of residue binding in complexes containing F-9, F-10, and Erlotinib (ERL) with the EGFR receptor. | |
In region I, F-9 and F-10 showed enhanced interactions with residues such as Leu694, Gly695, Ser696, Gly697, and Lys721, which were minimally involved in binding with Erlotinib, except for Val702, Ala719, and Ile720. Specifically, F-10 displayed stronger contributions from Leu694, Gly695, and Lys721, while F-9 showed higher binding interactions with Gly697 and Ala698. Region II appeared to favor Erlotinib, which engaged significantly with residues including Leu764, Ile765, Thr766, Met769, Gly772, and Arg779. In contrast, F-10 showed notable contributions from Leu768 and Cys773, suggesting a slightly altered binding orientation. Stronger interactions were observed for F-9 and F-10 in region III, with Arg817, Asn818, Lys822, and Lys828, in contrast to Erlotinib, which displayed limited binding within this region, interacting predominantly with Thr830 and Leu834. Notably, Trp856 contributed significantly only in the F-9 complex, indicating a unique interaction profile. These findings reveal that F-9 and F-10 engage a broader range of key EGFR residues compared to Erlotinib, supporting their potential as effective inhibitors with distinct yet complementary binding patterns.
2.8. Global reactivity parameters
The global reactivity parameters have been calculated for the F-9 and F-10 compared to the Erlotinib drug to understand the relationship between the structure, stability, and global chemical reactivity using the conceptual DFT method. The HOMO and LUMO energies have been calculated for the three systems at the same level of theory to analyse the different additional properties like differences in energy (ΔE), ionization potential (IP), electron affinity (EA), chemical potentials (μ), hardness of chemicals (η), electrophilicity index (ω) and softness of chemicals (σ) depicted in Table 5. The calculation of the parameters is performed following Koopman's theorem, where IE = −EHOMO and EA = −ELUMO. μ, η, ω, and σ are given as the following equations:| | 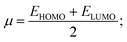 | (1) |
| |  | (2) |
| |  | (3) |
and| |  | (4) |
Table 5 Global reactivity indicators of edaravone derivatives (F-9 & F-10) and the reference drug. Energies of the frontier molecular orbitals (EHOMO & ELUMO), energy difference of the frontier molecular orbitals (ΔE), electron affinity (EA), ionization potential (IP), chemical potentials (μ), chemical hardness (η), and electrophilicity index (ω) in kcal mol−1 are given here. The chemical softness (σ) in (kcal mol−1)−1 is also given here
| Sl. no. |
Compounds |
E
HOMO in kcal mol−1 |
E
LUMO in kcal mol−1 |
ΔE |
IP |
EA |
η
|
σ
|
μ
|
ω
|
| 1 |
Erlotinib
|
−168.66 |
−21.91 |
146.74 |
168.66 |
21.91 |
73.38 |
0.013 |
−95.29 |
61.87 |
| 2 |
F-9
|
−172.25 |
−13.96 |
158.29 |
172.25 |
13.96 |
79.14 |
0.012 |
−93.11 |
54.77 |
| 3 |
F-10
|
−172.46 |
−15.33 |
157.12 |
172.46 |
15.33 |
78.56 |
0.012 |
−93.90 |
56.12 |
The wider HOMO–LUMO gap (Table 5) for the derivatives compared to the reference indicates that the edaravone derivatives have greater kinetic stability and lower chemical reactivity, leading to the adoption of the lowest energy conformation. Furthermore, the derivatives showed higher ionization potential and lower electron affinity than the reference drug, which indicates that there is a lower probability of electron loss and also a lower probability of electron gain for the derivatives (Table 5). The edaravone derivatives also showed higher chemical hardness, which is also directly related to stability. The chemical softness for all the drugs was comparable, indicating good reactivity for all of them. The chemical potential, which is proportional to Gibbs free energy, correlates with spontaneity and decreases with increasing chemical reactivity, showing lower values for the derivatives than the reference drug, indicating higher reactivity for both of them, with F-10 slightly higher (Table 5). The derivatives showed a lower electrophilicity index, which indicates that they are more susceptible to electrophilic attack than the reference drug. Thus, such results indicate good reactivity and also more stability for the edaravone derivatives than the reference drug.
3.
In vitro analysis
Erlotinib is a tyrosine kinase inhibitor targeting the epidermal growth factor receptor (EGFR), primarily used to inhibit the proliferation of cancer cells. Its main mechanism involves blocking EGFR signalling pathways, thereby reducing cancer cell survival.32,33 Cytotoxicity was induced in MDA-MB-231 cells by Erlotinib and edaravone derivatives (F-9 and F-10) compared to the untreated controls after 72 hours. However, no significant difference was observed between Erlotinib and edaravone derivatives, suggesting that the edaravone derivatives largely mimic the cytotoxic effects of Erlotinib. The IC50 values of Erlotinib (2 μM), F-9 (1 μM), and F-10 (1.9 μM), as given in Fig. 4 and Table 6, suggest that all three inhibitors were cytotoxic at low concentrations. The results suggest that all three compounds may be effective in inducing cytotoxicity in MDA-MB-231 cells. Future studies aimed at uncovering the effects of these compounds against normal cells will help understand if the cytotoxicity is specific to tumor cells or may also target other normal cells. This will lead to identifying the most potent compound with effective cytotoxicity against cancer cells and limited side effects.
 |
| | Fig. 4 Effect of Erlotinib and edaravone derivatives on MDA-MB-231 cells. The graph was plotted between the different concentrations of Erlotinib and edaravone derivatives and the cell viability percentage. | |
Table 6 The calculated IC50 values for the Erlotinib drug and the edaravone derivatives
| Inhibitors |
ERL
|
F-9
|
F-10
|
| IC50 (μM) |
2.0 |
1.0 |
1.9 |
4. Conclusion
In this study, we explored the potential of edaravone, a pyrazole derivative, combined with 1,3,4-oxadiazoles, a class of heterocyclic compounds known for their broad spectrum of biological activities. We have synthesized 10 derivatives with acyclic hydrocarbons, four-membered, and six-membered rings with different substitutions that interacted with edaravone and 1,3,4-oxadiazoles. Our in silico analysis identified two compounds with inhibition activity comparable to Erlotinib, a well-established EGFR kinase inhibitor. Further in vitro testing confirmed that these derivatives exhibited anticancer activity similar to Erlotinib, validating our computational predictions proven through the cytotoxicity towards the MDA-MB-231 cells. These promising findings suggest that edaravone-1,3,4-oxadiazole derivatives could serve as potential candidates for developing EGFR-targeted anticancer therapies, offering a novel approach to cancer treatment.
5. Experimental and computational methods
5.1. Reagents and materials
All the reagents and materials used in the work were procured with high purity. Phenyl hydrazine (97%), DMF (99%), chloroform-d (99.8%), propane-1-amine (99%) and morpholine (99%) were procured from Sigma Aldrich. Ethyl acetoacetate (98%), potassium carbonate (98%), bromo ethyl acetate (95%), sodium sulphate, p-toluenesulfonyl chloride (95%), piperidine (99%) and piperazine (98%) were procured from Avra synthesis Pvt Ltd. Ethyl acetate (99%), methanol (99%), hydrazine hydrate (99–100%) were procured from Finar (actylic lab solution). 1-Methylpiperazine (98%), azetidine (98%) and 3,3-difluoroazetidine hydrochloride (97%) were procured from BLD-pharma. Butan-1-amine (98%), 3-fluoro-3-methylazetidine hydrochloride (97%) and 3-fluoroazetidine hydrochloride (97%) were procured from Combi-blocks. DCM (99%) and DIPEA (98%) were procured from RCP Chemicals and Vinsa Pharmaceuticals respectively.
5.2. Material characterization
The melting points were determined by the open capillary method and were uncorrected. The IR spectra (in KBr pellets) were recorded on a JASCO FT/IR-4100 spectrophotometer. 1H NMR and 13C NMR spectra were recorded (chloroform-d) on a Bruker (400 MHz) spectrometer using TMS as the internal standard. Chemical shift values are given in d (ppm) scales. Liquid chromatography–mass spectrometry was carried out on a Waters-Acquity UPLC I class and detector SQ Water. The completion of the reaction was checked by thin layer chromatography (TLC) on silica gel-coated aluminium sheets (silica gel 60 F254) obtained from Merck. Compounds were purified using Biotage-Isolera One.
5.3. Materials and methods
5.3.1. 5-Methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (B).
To a solution of phenyl hydrazine (A) (20 g, 0.18 mole) in glacial acetic acid (80 mL) was added ethyl acetoacetate (24 g, 0.18 mol) at rt. The reaction mixture was refluxed for 3 h. The reaction mixture was concentrated under reduced pressure to obtain the residue, which was partitioned between water and EtOAc. The organic layer was washed with brine, dried over sodium sulphate and concentrated in a vacuum to obtain the crude product. The crude compound was purified by flash column chromatography eluted with 50% EtOAC/hexane to afford 29 g (90.6% of yield) of 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one as a white solid.28
5.3.2. Ethyl 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetate (C).
To a solution of 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (B) (10 g, 57.44 mmol) in DMF (50 mL) was added potassium carbonate (11.9 g, 86.16 mmol) at 0 °C. Then, bromo ethyl acetate (7.0 mL, 63.18 mmol) was added dropwise at 0 °C and the reaction mixture was stirred for 6 h at room temperature. The reaction mixture was poured into roughly shedded ice and the product was extracted with EtOAc. The organic layer was washed with brine, dried over sodium sulphate and concentrated in a vacuum to obtain the crude product. The crude compound was purified by flash column chromatography eluted with 5% EtOAC/hexane to afford 11.2 g (75% of yield) of ethyl 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl) oxy) acetate as a pale yellow sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.75–7.72 (m, 2H), 7.44–7.38 (m, 2H), 7.27–7.22 (m, 1H), 5.46 (s, 1H), 4.64 (s, 1H), 4.27 (q, J = 7.2 Hz, 2H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 167.44, 153.63, 148.68, 138.14, 128.75, 126.24, 122.24, 86.60, 68.11, 61.61, 14.32, 14.01. LC-MS, [M + H]+ = 261.16.
5.3.3. 2-((3-Methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetohydrazide (D).
To a solution of ethyl 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetate (C) (7 g, 26.9 mmol) in methanol (40 mL) was added hydrazine hydrate (1.3 mL, 40.36 mmol) at rt. The reaction mixture was stirred for 2 h at room temperature. After stirring for 2 h, the reaction mixture was partition between water and DCM. The organic layer was dried over sodium sulphate and concentrated in a vacuum to afford 6.2 g (93.6% of yield) of 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetohydrazide as an off-white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.83 (br s, 1H), 7.59–7.55 (m, 2H), 7.44–7.39 (m, 2H), 7.3–7.25 (m, 1H), 5.49 (s, 1H), 4.56 (s, 2H), 3.85 (br s, 2H), 2.25 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 167.02, 152.71, 148.82, 138.00, 128.94, 126.55, 122.19, 86.92, 69.92, 14.35. IR (KBr) 3278, 3165, 2922, 1656, 1595, 1560, 1512, 1257, 1163, 972, 752 cm1. LC-MS, [M + H]+ = 247.05, melting point 121–130 °C.
5.3.4. Ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E).
To a solution of 2-((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)acetohydrazide (D) (4 g, 16.25 mmol) in DCM (20 mL) and DIPEA (14.15 mL, 81.25 mmol) ethyl oxalyl chloride (2.36 mL, 21.12 mmol) was added dropwise at 0 °C. After 1 h of stirring, tosyl chloride (4.34 g, 22.75 mmol) was added and the reaction mixture was stirred for an additional 18 h at room temperature. The reaction mixture was diluted with DCM and washed with water. The organic layer was dried over sodium sulphate and concentrated in a vacuum to obtain the crude product. The crude compound was purified by flash column chromatography eluted with 50% EtOAC/hexane to afford 3.9 g (73% of yield) of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl) oxy) methyl)-1,3,4-oxadiazole-2-carboxylate as an off-white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.64–7.62 (m, 2H), 7.43–7.39 (m, 2H), 7.29–7.26 (m, 1H), 5.69 (s, 1H), 5.37 (s, 2H), 4.52 (q, J = 7.2 Hz, 2H), 2.26 (s, 3H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 163.04, 157.60, 153.74, 152.63, 148.75, 138.01, 128.85, 126.43, 122.23, 87.55, 63.80, 62.54, 14.44, 13.94. IR (KBr) 3140, 2980, 1745, 1597, 1564, 1506, 1442, 1186, 1145, 912, 846, 763 cm1. LC-MS, [M + H]+ = 329.29, melting point 76.8–85.3 °C.
5.3.5. 5-(((3-Methyl-1-phenyl-1H-pyrazol-5-yl) oxy) methyl)-N-propyl-1,3,4-oxadiazole-2-carboxamide (F-1).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl) oxy) methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added propan-1-amine (R1) (11 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 4 h at room temperature. The reaction mixture was diluted with ethyl acetate and washed with water. The organic layer was dried over sodium sulphate and concentrated in a vacuum to obtain the crude product. The crude compound was purified by flash column chromatography eluted with 50% EtOAC/hexane to afford 46 mg (89% of yield) of 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl) oxy) methyl)-N-propyl-1,3,4-oxadiazole-2-carboxamide31 (F-1) as an off-white solid: 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.64–7.61 (m, 2H), 7.42–7.38 (m, 2H), 7.29–7.25 (m, 1H), 7.15 (br s, 1H), 5.69 (s, 1H), 5.34 (s, 2H), 3.47–3.42 (m, 2H), 2.27 (s, 3H), 1.66 (sxt, J = 7.3 Hz, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 163.04, 159.68, 152.72, 152.64, 148.73, 138.04, 128.90, 126.47, 122.31, 87.49, 62.49, 41.63, 22.51, 14.50, 11.25. IR (KBr) 3066, 2966, 2927, 2872, 1691, 1595, 1560, 1512, 1448, 1149, 1062, 912, 758 cm1. LC-MS, [M + H]+ = 342.3, melting point 99–104 °C.
5.3.6.
N-Butyl-5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxamide (F-2).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added butan-1-amine (R2) (13.3 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 16 h at room temperature. The reaction mixture was worked up as above and resulted in 45 mg (83% of yield) of the purified product as an off-white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.64–7.60 (m, 2H), 7.43–7.38 (m, 2H), 7.29–7.25 (m, 1H), 7.12 (br s, 1H), 5.69 (s, 1H), 5.34 (s, 2H), 3.51–3.45 (m, 2H), 2.27 (s, 3H), 1.67–1.57 (m, 2H), 1.48–1.38 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 163.04, 159.69, 152.73, 152.60, 148.74, 138.04, 128.90, 126.49, 122.33, 87.50, 62.49, 39.71, 31.19, 19.93, 14.50, 13.60. IR (KBr) 2995, 2924, 2854, 1685, 1595, 1560, 1510, 1396, 1367, 1192, 1076, 759 cm1. LC-MS, [M + H]+ = 356.33, melting point 129–132 °C.
5.3.7. (5-(((3-Methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)(4-methylpiperazin-1-yl)methanone (F-3).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added 1-methylpiperazine (R3) (18.24 mg, 0.182 mmol) at room temperature. The reaction mixture was heated to 50 °C and stirred for 16 h. The reaction mixture was worked up as above and resulted in 45 mg (77% of yield) of the purified product as a pale yellow sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.65–7.62 (m, 2H), 7.41 (t, J = 7.9 Hz, 2H), 7.29–7.25 (m, 1H), 5.71 (s, 1H), 5.36 (s, 2H), 4.15 (br s, 2H), 3.84 (br t, J = 4.4 Hz, 2H), 2.53 (m, 4H), 2.43–2.30 (m, 4H), 2.27 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 162.05, 159.17, 152.80, 152.60, 148.77, 138.10, 128.91, 126.46, 122.28, 87.49, 62.47, 55.09, 54.37, 46.80, 45.80, 42.93, 14.52. IR (KBr) 2926, 2854, 2798, 1658, 1595, 1560, 1510, 1448, 1394, 1141, 1029, 763, 692 cm1. LC-MS, [M + H]+ = 383.34.
5.3.8. Azetidin-1-yl(5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)methanone (F-4).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added azetidine (R4) (10.5 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 6 h at room temperature. The reaction mixture was worked up as above and resulted in 47 mg (91% of yield) of the purified product as an off-white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.68–7.65 (m, 2H), 7.45–7.39 (m, 2H), 7.30–7.25 (m, 1H), 5.53 (s, 1H), 4.60 (s, 2H), 4.11 (td, J = 7.7, 19.0 Hz, 4H), 2.33–2.21 (m, 5H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 166.21, 153.66, 148.87, 138.34, 128.83, 126.29, 122.22, 86.78, 70.09, 51.15, 48.72, 16.38, 14.51. IR (KBr) 3134, 3062, 2960, 2920, 2881, 1668, 1595, 1560, 1512, 1436, 1346, 1161, 1080, 761 cm1. LC-MS, [M + H]+ = 340.23, melting point 90–98 °C.
5.3.9. (3-Fluoro-3-methylazetidin-1-yl)(5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)methanone (F-5).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added DIPEA (0.08 mL, 0.456 mmol) followed by 3-fluoro-3-methylazetidine hydrochloride (R5) (23 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 16 h at room temperature. The reaction mixture was worked up as above and resulted in 44 mg (79% of yield) of the purified product as a pale yellow sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.65–7.61 (m, 2H), 7.45–7.40 (m, 2H), 7.32–7.30–7.26 (m, 1H), 5.53 (s, 1H), 4.64 (s, 2H), 4.27–4.17 (m, 2H), 4.06–3.97 (m, 2H), 2.27 (s, 3H), 1.59–1.50 (d, J = 20 Hz, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 166.70, 153.37, 148.96, 138.21, 128.91, 126.59, 122.49, 91.36, 89.33, 86.78, 70.44, 63.01, 62.87, 60.79, 60.26, 22.73, 14.52. IR (KBr) 3136, 3061, 2931, 2873, 1674, 1597, 1560, 1512, 1446, 1392, 1253, 887, 761, 696 cm1. LC-MS, [M + H]+ = 372.33.
5.3.10. (3,3-Difluoroazetidin-1-yl)(5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)methanone (F-6).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added DIPEA (0.08 mL, 0.456 mmol) followed by 3,3-difluoroazetidine hydrochloride (R6·HCl) (23.5 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 10 h at room temperature. The reaction mixture was worked up as above and resulted in 50 mg (88% of yield) of the purified product as an off-white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.63–7.61 (m, 2H), 7.46–7.36 (m, 2H), 7.29–7.25 (m, 1H), 5.69 (s, 1H), 5.36 (s, 2H), 5.00 (t, J = 10.8 Hz, 2H), 4.51 (t, J = 11.8 Hz, 2H), 2.27 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 162.78, 158.55, 152.80, 148.91, 138.19, 129.06, 126.66, 122.46, 115.11 (t, J = 1085.4 Hz, 1C), 87.67, 65.39 (t, J = 29.8 Hz, 1C), 62.57, 61.15 (t, J = 29.8 Hz, 1C), 14.66. IR (KBr) 3138, 3059, 2958, 2924, 1674, 1595, 1560, 1506, 1444, 1382, 1273, 1246, 1103, 908, 756 cm1. LC-MS, [M + H]+ = 376.28, melting point 107–115 °C.
5.3.11. (3-Fluoroazetidin-1-yl)(5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)methanone (F-7).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added N, DIPEA (0.08 mL, 0.456 mmol) followed by 3-fluoroazetidine hydrochloride (R7·HCl) (20.8 mg, 0.182 mmol) at room temperature. The reaction mixture was stirred for 16 h at room temperature. The reaction mixture was worked up as above and resulted in 42 mg (77% of yield) of the purified product as a pale yellow solid. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.63–7.61 (m, 2H), 7.42–7.38 (m, 2H), 7.29–7.25 (m, 1H), 5.69 (s, 1H), 5.49–5.36 (m, 1H), 5.33 (s, 1H), 5.03–4.88 (m, 1H), 4.84–4.70 (m, 1H), 4.60–4.47 (m, 1H), 4.42–4.28 (m, 1H), 2.26 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 162.31, 158.58, 152.68, 152.40, 148.72, 138.03, 128.89, 126.45, 122.27, 87.46, 83.03, 80.98, 62.39, 61.36 (d, J = 26.9 Hz, 1C), 56.78 (d, J = 26.9 Hz, 1C), 14.48. IR (KBr) 3138, 3059, 2854, 1672, 1595, 1558, 1508, 1452, 1390, 1267, 1066, 999, 761 cm1. LC-MS, [M + H]+ = 358.3, melting point 108–114 °C.
5.3.12. (5-(((3-Methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)(morpholino)methanone (F-8).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added morpholine (R8) (15.85 mg, 0.182 mmol) at room temperature. The reaction mixture was heated to 50 °C and stirred for 16 h. The reaction mixture was worked up as above and resulted in 46 mg (82% of yield) of the purified product as a pale yellow sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.65–7.62 (m, 2H), 7.43–7.39 (m, 2H), 7.28–7.24 (m, 1H), 5.71 (s, 1H), 5.36 (s, 2H), 4.21–4.18 (m, 2H), 3.83–3.77 (m, 6H), 2.27 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 162.16, 159.03, 152.76, 152.60, 148.76, 138.08, 128.90, 126.46, 122.27, 87.47, 66.88, 66.54, 62.43, 47.44, 43.37, 14.52. IR (KBr) 3136, 2922, 2854, 1658, 1595, 1560, 1510, 1446, 1392, 1271, 1114, 1028, 763 cm1. LC-MS, [M + H]+ = 370.3.
5.3.13. (5-(((3-Methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)(piperidin-1-yl)methanone (F-9).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added piperidine (R9) (15.5 mg, 0.182 mmol) at room temperature. The reaction mixture was heated to 50 °C and stirred for 16 h. The reaction mixture was worked up as above and resulted in 41 mg (73% of yield) of the purified product as a colourless sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.65–7.63 (m, 2H), 7.43–7.39 (m, 2H), 7.29–7.24 (m, 1H), 5.71 (s, 1H), 5.35 (s, 2H), 3.96 (br d, J = 5.5 Hz, 2H), 3.75 (br d, J = 5.5 Hz, 2H), 2.27 (s, 3H), 1.72–1.69 (m, 6H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 161.86, 159.36, 152.85, 152.70, 148.76, 138.13, 128.90, 126.42, 122.27, 87.48, 62.52, 48.10, 44.17, 26.56, 25.53, 24.27, 14.51. IR (KBr) 2936, 2858, 1656, 1597, 1560, 1448, 1382, 1265, 1217, 1114, 1010, 769 cm1. LC-MS, [M + H]+ = 368.3.
5.3.14. (5-(((3-Methyl-1-phenyl-1H-pyrazol-5-yl) oxy) methyl)-1,3,4-oxadiazol-2-yl) (piperazin-1-yl)methanone (F-10).
To a solution of ethyl 5-(((3-methyl-1-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1,3,4-oxadiazole-2-carboxylate (E) (50 mg, 0.15 mmol) in THF (1 mL) was added piperazine (R10) (15.7 mg, 0.182 mmol) at room temperature. The reaction mixture was heated to 50 °C and stirred for 16 h. The reaction mixture was worked up as above and resulted in 46 mg (82% of yield) of the purified product as a pale yellow sticky oil. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.65–7.62 (m, 2H), 7.43–7.39 (m, 2H), 7.30–7.25 (m, 1H), 5.71 (s, 1H), 5.36 (s, 2H), 4.12 (t, J = 5.2 Hz, 2H), 3.81 (t, J = 5.2 Hz, 2H), 2.98 (br d, J = 3.7 Hz, 4H) 2.27 (s, 3H). 13C NMR (100 MHz, CHLOROFORM-d) δ = 162.03, 159.16, 152.81, 152.68, 148.77, 138.10, 128.91, 126.46, 122.28, 87.50, 62.48, 48.06, 46.36, 45.64, 44.00, 14.51. IR (KBr) 3072, 2931, 2858, 1658, 1593, 1560, 1510, 1452, 1381, 1263, 1219, 1112, 1008, 821, 704 cm1. LC-MS, [M + H]+ = 368.3. LC-MS, [M + H]+ = 369.2.
5.4. Computational methods
5.4.1. Preparation and optimization of the derivative compounds.
1-Phenyl-3-methyl-5-pyrazolone (edaravone) derivative compounds were designed as per Scheme 1 as 3D geometrical structures to make an input file for optimization utilizing the Gaussian 16 software package.31 Each 1-phenyl-3-methyl-5-pyrazolone derivative compound abbreviated as F-1 to F-10 was optimized at the M062x/aug-cc-pVDZ29,30 level of theory. No imaginary vibrational frequency shows that each optimized geometrical structure of F-1 to F-10 compounds relates to a stable state of the molecule.
5.4.2. Pharmacological properties.
Compounds (F-1 to F-10) derived from 1-phenyl-3-methyl-5-pyrazolone (edaravone) and the reference inhibitor Erlotinib were submitted to the Swiss ADME website (https://www.swissadme.ch/) to identify potential candidates for drug development.34,35 We have checked for each derivative under Lipinski's rule of five,36 which states that the molecular weight of each derivative cannot exceed 500 g mol−1, the presence of hydrogen bond donors and acceptors cannot be more than 5 and 10, respectively, and the lipophilicity value (logp) cannot go beyond 5. Polar surface area, bioavailability score, and gastrointestinal absorption potential were also assessed for each derivative to identify which compound is suitable for further analysis and to better understand the drug-likeness qualities of each compound.
5.4.3. Molecular docking.
Molecular docking studies were conducted using Auto Dock4 tools,37 with the epidermal growth factor receptor (EGFR) as the receptor target. The receptor was downloaded in pdb format (4HJO) from the Rcsb (https://www.rcsb.org/) protein data bank.38 Gaussview 6 software generated the pdb format from the optimal geometrical structures of 1-phenyl-3-methyl-5-pyrazolone derivative (F-1 to F-10) compounds before docking. We added the missing amino acid residues using SwissPDB-Viewer software39 to prepare the receptor structure for docking and also removed the heteroatoms, water molecules, and inhibitors from the crystallographic pdb structure. Auto Dock Tools is used to add the polar hydrogen and Kollman charges to the receptor. Before docking, a grid map with the docking number of grid points was set up with 60 × 60 × 60 dimensions, with a grid spacing of 0.375 Å, and was prepared. Docking analysis was performed using the Lamarckian genetic algorithm,40 and a higher binding affinity with a lower rmsd value was considered for each compound.
5.4.4. Molecular dynamics simulations.
As further studies to verify both the interaction analysis and the binding capability with the epidermal growth factor receptor, molecular dynamics (MD) simulations were carried out using the GROMACS 2022 package version41 for EGFR-F9 & EGFR-F10 complexes as well as EGFR-Erlotinib as reference complexes. For each MD simulation, GROMACS was utilized along with the charmm36 force field42 and TIP3P water model for the solvation. The ligand structure was extracted from the pdb file of the molecular docking results, and their topologies were retrieved from the CGenFF web server (https://cgenff.com/), and GROMACS created the EGFR receptor topology file. The protein–ligand complex was kept at the center of an 8.4 nm edge cubic box containing approximately 17832 molecules of solvent. Four chlorides as counter ions were then injected to neutralize the simulation system. The entire system experienced 50000 steps of energy minimization using the steepest descent approach, and the subsequent NVT and NPT equilibration was adopted for 50 ps each. The Berendsen thermostat43 and isotropic Berendsen barostat44 algorithm maintained the temperature at 300 K and the pressure at 1 bar, respectively. For Newtonian motion, the leap-frog integrator was used with a timestep of 2 fs. Using the Lincs algorithm,45 the bond length of the hydrogen atoms within the EGFR-ligated complex is constrained. While the particle mesh Ewald algorithm46 calculates long-range electrostatic correction interactions with a cutoff distance of 1.2 nm and a Fourier grid spacing of 0.16, respectively, the short-range interactions of van der Waals are computed with a minimum of 1.2 nm. The production MD run was subjected to 100 ns with 2 fs timestep production dynamics involving the Berendsen thermostat43 and Parrinello–Rahman algorithm47 to maintain the temperature at 300 K and pressure at 1 bar, respectively.
5.4.5. MM-PBSA methods.
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA), an implicit solvent model, was employed to estimate the free binding energies (ΔGbind) of ligands F-9, F-10, and with the reference drug Erlotinib opposition to the EGFR receptor. The estimation of free binding energies can be assembled as represented in eqn (5) by consisting of three energetic terms: receptor–ligand interaction energy (ΔGmm), effect of desolvation energetic terms (ΔGsol), and energetic part of the entropic contribution (TΔS).| | | ΔGbind = ΔGmm + ΔGsol − TΔS | (5) |
ΔGele (electrostatic interaction energy) and ΔGvdW (van der Waals energy) can be separated from ΔGmm energetic terms. ΔGbind has been determined without considering the entropy component (TΔS). Polar (ΔGpol) and non-polar solvation energy (ΔGnpol) are separated out from ΔGsol energetic terms. The GROMACS-based g_mmpbsa tool48 is employed to estimate the energy contributions of receptor–ligand interaction energy (ΔGmm), as well as the effect of the desolvation energetic component (ΔGsol) upon calculating the total binding energy of F-9, F-10, and Erlotinib complexed with the EGFR receptor. The MmPbSaStat.py Python script computes the average binding energy of the receptor–ligand complexes by recording snapshots of the last 30 ns extracted from the MD trajectories. Furthermore, the residual contribution of essential residues in binding free energy is calculated using the MmPbSaDecomp.py Python script.48
5.5. Cell culture
MDA-MB-231 human breast carcinoma cells were cultured in DMEM-high glucose medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin under standard conditions (37 °C, 5% CO2).
5.5.1. MTT assay.
MDA-MB-231 cells were seeded into 96-well plates at a density of 2 × 104 cells per well and incubated for 24 hours to allow attachment. After adherence, cells were treated with Erlotinib and edaravone derivatives at different concentrations (0.1 μM, 1 μM, 5 μM, 10 μM, and 20 μM) for 72 hours. The cytotoxic effects of Erlotinib and edaravone derivatives (F-9 and F-10) were assessed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. After the treatment period, 10 μL of MTT solution (5 mg mL−1 in PBS) was added to each well. Plates were incubated for 4 hours at 37 °C in the dark to allow formazan crystal formation. The medium was carefully removed, and 100 μL of DMSO was added to each well to dissolve the formazan crystals. The absorbance was measured at 570 nm using a microplate reader. The experiment was repeated thrice with triplicates each time, and the IC50 value was calculated. The percentage of viable cells was calculated using the formula:
| Cell viability (%) = (absorbance of treated cells/absorbance of control cells) × 100 |
Author contributions
MV Reddy: synthesis and characterization. P. Shyamala: conceptualisation and helped with the synthesis part. AK Sharma: conceptualisation and supervision of the synthesis part, and the initial draft of the manuscript. PN Sahu: in silico studies and drafting the manuscript. K. Subramaniyam: performed in vitro analysis. S. Harihar: conceptualisation and supervision in the in vitro analysis, and writing of the initial draft. A. Sen: conceptualisation, in silico studies, supervision, writing draft and revision, and collaborative discussions.
Data availability
The structure of the epidermal growth factor receptor (EGFR) was downloaded in pdb format (4HJO) from the Rcsb (https://www.rcsb.org/) protein data bank. Additional data generated during the research are available in the ESI.†
Conflicts of interest
There is no conflicts of interest.
Acknowledgements
MVR, PS, AKS are thankful to Aragen Life Sciences (formerly known as GVK Biosciences Pvt. Ltd) for the financial support and for the laboratory facilities. The assistance from the analytical department, Aragen Life Sciences, is greatly acknowledged. PNS, KS, SH, and AS acknowledge GITAM Deemed to be University for the support. KS and SH also acknowledge a SERB Grant (CRG/2022/005685) for funding.
References
- S. Garg and N. Raghav, 2,5-Diaryloxadiazoles and their precursors as novel inhibitors of cathepsins B, H and L, Bioorg. Chem., 2016, 67, 64–74, DOI:10.1016/j.bioorg.2016.05.003.
- J. T. Palmer, B. L. Hirschbein, H. Cheung, J. McCarter, J. W. Janc, Z. W. Yu and G. Wesolowski, Keto-1,3,4-oxadiazoles as cathepsin K inhibitors, Bioorg. Med. Chem. Lett., 2006, 16, 2909–2914, DOI:10.1016/j.bmcl.2006.03.001.
- T. J. Lukas, G. E. Schiltz, H. Arrat, K. Scheidt and T. Siddique, Discovery of 1,3,4-oxidiazole scaffold compounds as inhibitors of superoxide dismutase expression, Bioorg. Med. Chem. Lett., 2014, 24, 1532–1537, DOI:10.1016/j.bmcl.2014.01.078.
- P. Bhatt, A. Sen and A. Jha, Design and Ultrasound Assisted Synthesis of Novel 1,3,4-Oxadiazole Drugs for Anti-Cancer Activity, ChemistrySelect, 2020, 5, 3347–3354, DOI:10.1002/slct.201904412.
- M. Albratty, K. A. El-Sharkawy and H. A. Alhazmi, Synthesis and evaluation of some new 1,3,4-oxadiazoles bearing thiophene, thiazole, coumarin, pyridine and pyridazine derivatives as antiviral agents, Acta Pharm., 2019, 69, 261–276, DOI:10.2478/acph-2019-0015.
- E. F. Ewies, M. El-Hussieny, N. F. El-Sayed and M. A. Fouad, Design, synthesis and biological evaluation of novel α-aminophosphonate oxadiazoles via optimized iron triflate catalyzed reaction as apoptotic inducers, Eur. J. Med. Chem., 2019, 180, 310–320, DOI:10.1016/j.ejmech.2019.07.029.
- G. Verma, M. F. Khan, W. Akhtar, M. M. Alam, M. Akhter and M. Shaquiquzzaman, A Review Exploring Therapeutic Worth of 1,3,4-Oxadiazole Tailored Compounds, Mini-Rev. Med. Chem., 2019, 19, 477–509, DOI:10.2174/1389557518666181015152433.
- K. Paruch, Ł. Popiołek and M. Wujec, Antimicrobial and antiprotozoal activity of 3-acetyl-2,5-disubstituted-1,3,4-oxadiazolines: a review, Med. Chem. Res., 2020, 29, 1–16, DOI:10.1007/s00044-019-02463-w.
- S. S. De, M. P. Khambete and M. S. Degani, Oxadiazole scaffolds in anti-tuberculosis drug discovery, Bioorg. Med. Chem. Lett., 2019, 29, 1999–2007, DOI:10.1016/j.bmcl.2019.06.054.
- R. Bhutani, D. P. Pathak, G. Kapoor, A. Husain and Md. A. Iqbal, Novel hybrids of benzothiazole-1,3,4-oxadiazole-4-thiazolidinone: Synthesis, in silico ADME study, molecular docking and in vivo anti-diabetic assessment, Bioorg. Chem., 2019, 83, 6–19, DOI:10.1016/j.bioorg.2018.10.025.
- M. J. Ahsan, A. Choupra, R. K. Sharma, S. S. Jadav, P. Padmaja, M. Z. Hassan, A. B. S. Al-Tamimi, M. H. Geesi and M. A. Bakht, Rationale Design, Synthesis, Cytotoxicity Evaluation, and Molecular Docking Studies of 1,3,4-oxadiazole Analogues, Anti-Cancer Agents Med. Chem., 2018, 18, 121–138, DOI:10.2174/1871520617666170419124702.
- M. Agarwal, V. Singh, S. K. Sharma, P. Sharma, M. Y. Ansari, S. S. Jadav, S. Yasmin, R. Sreenivasulu, M. Z. Hassan, V. Saini and M. J. Ahsan, Design and synthesis of new 2,5-disubstituted-1,3,4-oxadiazole analogues as anticancer agents, Med. Chem. Res., 2016, 25, 2289–2303, DOI:10.1007/s00044-016-1672-1.
-
Y. K. Gorbunov and L. L. Fershtat, Recent chemistry and applications of 1,3,4-oxadiazoles, Advances in Heterocyclic Chemistry, Elsevier, 2024, pp. 1–26 DOI:10.1016/bs.aihch.2023.11.001.
- A. Jha, A. Sen and R. R. Malla, Chemistry of Oxadiazole Analogues: Current Status and Applications, Russ. J. Bioorg. Chem., 2021, 47, 670–680, DOI:10.1134/S1068162021030092.
- B. Sever, M. D. Altıntop, M. O. Radwan, A. Özdemir, M. Otsuka, M. Fujita and H. I. Ciftci, Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors, Eur. J. Med. Chem., 2019, 182, 111648, DOI:10.1016/j.ejmech.2019.111648.
- S. Zhou, L. Xu, M. Cao, Z. Wang, D. Xiao, S. Xu, J. Deng, X. Hu, C. He, T. Tao, W. Wang, A. Guan and X. Yang, Anticancer properties of novel pyrazole-containing biguanide derivatives with activating the adenosine monophosphate-activated protein kinase signaling pathway, Arch. Pharm., 2019, 352, 1900075, DOI:10.1002/ardp.201900075.
- M. I. Ansari and S. A. Khan, Synthesis and antimicrobial activity of some novel quinoline-pyrazoline-based coumarinyl thiazole derivatives, Med. Chem. Res., 2017, 26, 1481–1496, DOI:10.1007/s00044-017-1855-4.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. Ansar, Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review, Molecules, 2018, 23, 134, DOI:10.3390/molecules23010134.
- N. A. Khalil, E. M. Ahmed, H. B. El-Nassan, O. K. Ahmed and A. M. Al-Abd, Synthesis and biological evaluation of novel pyrazoline derivatives as anti-inflammatory and antioxidant agents, Arch. Pharm. Res., 2012, 35, 995–1002, DOI:10.1007/s12272-012-0606-9.
- E. Duranti, N. Cordani and C. Villa, Edaravone: A Novel Possible Drug for Cancer Treatment?, Int. J. Mol. Sci., 2024, 25, 1633, DOI:10.3390/ijms25031633.
- R. Suzuki, R. K. Gopalrao, H. Maeda, P. Rao, M. Yamamoto, Y. Xing, S. Mizobuchi and S. Sasaguri, MCI-186 inhibits tumor growth through suppression of EGFR phosphorylation and cell cycle arrest, Anticancer Res., 2005, 25, 1131–1138 CAS.
- N. Polkam, V. R. Ramaswamy, P. Rayam, T. R. Allaka, H. S. Anantaraju, S. Dharmarajan, Y. Perumal, D. Gandamalla, N. R. Yellu, S. Balasubramanian and J. S. Anireddy, Synthesis, molecular properties prediction and anticancer, antioxidant evaluation of new edaravone derivatives, Bioorg. Med. Chem. Lett., 2016, 26, 2562–2568, DOI:10.1016/j.bmcl.2016.03.024.
- V. Marković, S. Erić, Z. D. Juranić, T. Stanojković, L. Joksović, B. Ranković, M. Kosanić and M. D. Joksović, Synthesis, antitumor activity and QSAR studies of some 4-aminomethylidene derivatives of edaravone, Bioorg. Chem., 2011, 39, 18–27, DOI:10.1016/j.bioorg.2010.10.003.
- H. Zhang, A. Berezov, Q. Wang, G. Zhang, J. Drebin, R. Murali and M. I. Greene, ErbB receptors: from oncogenes to targeted cancer therapies, J. Clin. Invest., 2007, 117, 2051–2058, DOI:10.1172/JCI32278.
- J. McBryan, J. Howlin, S. Napoletano and F. Martin, Amphiregulin: Role in Mammary Gland Development and Breast Cancer, J. Mammary Gland Biol. Neoplasia, 2008, 13, 159–169, DOI:10.1007/s10911-008-9075-7.
- R. Roskoski, The ErbB/HER family of protein-tyrosine kinases and cancer, Pharmacol. Res., 2014, 79, 34–74, DOI:10.1016/j.phrs.2013.11.002.
- F. Walker, L. Abramowitz, D. Benabderrahmane, X. Duval, V. Descatoire, D. Hénin, T. Lehy and T. Aparicio, Growth factor receptor expression in anal squamous lesions: modifications associated with oncogenic human papillomavirus and human immunodeficiency virus, Hum. Pathol., 2009, 40, 1517–1527, DOI:10.1016/j.humpath.2009.05.010.
- S. Fuse, H. Nakamura, M. Inaba, S. Sato and M. Joshi, Synthesis of Pyrazolofuropyrazine via One-Pot SNAr Reaction and Intramolecular Direct C–H Arylation, Synthesis, 2018, 1493–1498, DOI:10.1055/s-0036-1591885.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions, J. Phys. Chem. A, 2013, 117, 12590–12600, DOI:10.1021/jp408166m.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023, DOI:10.1063/1.456153.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, S. S. Iyengar, J. Tomasi, R. L. Martin, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- W. Pao and J. Chmielecki, Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer, Nat. Rev. Cancer, 2010, 10, 760–774, DOI:10.1038/nrc2947.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Jänne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos,
EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib, N. Engl. J. Med., 2005, 352, 786–792, DOI:10.1056/NEJMoa044238.
- A. Daina and V. Zoete, A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules, ChemMedChem, 2016, 11, 1117–1121, DOI:10.1002/cmdc.201600182.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25. 1, Adv. Drug Delivery Rev., 2001, 46, 3–26, DOI:10.1016/S0169-409X(00)00129-0.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- J. H. Park, Y. Liu, M. A. Lemmon and R. Radhakrishnan, Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain, Biochem. J., 2012, 448, 417–423, DOI:10.1042/BJ20121513.
- M. U. Johansson, V. Zoete, O. Michielin and N. Guex, Defining and searching for structural motifs using DeepView/Swiss-PdbViewer, BMC Bioinf., 2012, 13, 173, DOI:10.1186/1471-2105-13-173.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19, 1639–1662, DOI:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B.
-
P. Bauer, B. Hess and E. Lindahl, GROMACS 2022 Manual, 2022 DOI:10.5281/ZENODO.6103568.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem., 2010, 31, 671–690, DOI:10.1002/jcc.21367.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690, DOI:10.1063/1.448118.
- Y. Lin, D. Pan, J. Li, L. Zhang and X. Shao, Application of Berendsen barostat in dissipative particle dynamics for nonequilibrium dynamic simulation, J. Chem. Phys., 2017, 146, 124108, DOI:10.1063/1.4978807.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472, DOI:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092, DOI:10.1063/1.464397.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101, DOI:10.1063/1.2408420.
- R. Kumari and R. Kumar, Open Source Drug Discovery Consortium, A. Lynn, g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations, J. Chem. Inf. Model., 2014, 54, 1951–1962, DOI:10.1021/ci500020m.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR, LCMS, docking figures and optimised structures of the compounds. See DOI: https://doi.org/10.1039/d5nj01451d |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c










