
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencep-Phenyleneethynylene unit-conjugated dimeric zinc-phthalocyanines in bulk heterojunction solar cells: a comparative experimental and theoretical study†
Gülenay
Tunç
a,
Gizem Gümüşgöz
Çelik
 a,
Betül
Canımkurbey
b,
Burcu
Dedeoglu
a and
Ayşe Gül
Gürek
*a
a,
Betül
Canımkurbey
b,
Burcu
Dedeoglu
a and
Ayşe Gül
Gürek
*a
aFaculty of Science Department of Chemistry, Gebze Technical University, 41400 Gebze, Kocaeli, Turkey. E-mail: gurek@gtu.edu.tr
bPolatli Faculty of Arts and Sciences, Department of Physic, Ankara Hacı Bayram Veli University, 06900 Ankara, Turkey
First published on 25th April 2025
Abstract
Nowadays, the photosensitivity of certain molecules, particularly phthalocyanines (Pcs), is well-studied. This field has made much progress, and several practical applications exist for these molecules. In this study, p-phenyleneethynylene-bridged two ZnPc dimers containing either bulky tert-butyl (GT57) or tert-butyl thiol (GT60) substituents at the peripheral position were synthesized as a novel donor component for bulk heterojunction (BHJ) solar cell applications. The molecular structure and photophysical properties of dimeric ZnPc derivatives were investigated by combined experimental and theoretical studies. The density functional theory (DFT) method was employed with B3LYP functional and the def2-SVP basis set to examine the designed complexes and calculate geometrical parameters and natural transition orbitals (NTOs). Additionally, the time-dependent density functional theory (TD-DFT) method was employed to investigate the optical properties through the analysis of UV-vis spectra. Dimeric ZnPc derivatives were blended as donor components alongside PCBM as the acceptor material in BHJ solar cells, achieving a maximum power conversion efficiency of 4.05%, for GT60 and compared to GT57, cells based on GT57 exhibited lower photovoltaic performance. These findings are encouraging and highlight the potential for further research on BHJ solar cells employing near-infrared-absorbing, non-aggregated dimeric ZnPc derivatives containing S heteroatoms.
Introduction
Phthalocyanines (Pcs) are commonly used as active components in organic photovoltaic (OPV) and hybrid dye-sensitized solar cells (DSSCs).1–4 This is due to their unique electronic properties, distinctive chemical structure, and broad photo response range between 600 and 800 nm, which coincides with the region of maximum solar flux. As a result, only very thin films are required to absorb a significant portion of solar light. Pc-based OPVs can be fabricated either through vacuum evaporation or solution-processing techniques. However, unsubstituted Pcs are nearly insoluble in standard organic solvents because they tend to aggregate, making solution-based processing difficult. Despite this limitation, unsubstituted Pcs have been successfully used as active layers in vacuum-deposited small molecule solar cells. On the other hand, chemically modified Pcs, which include substituents either at the peripheral or axial positions of the macrocycle, can be processed using solution-based techniques. These modifications not only adjust the electronic properties of the Pcs but also enhance their solubility in organic solvents.In the case of solution-processed bulk heterojunction (BHJ) solar cells, only a limited number of examples involve soluble phthalocyanine (Pc) derivatives blended with soluble acceptor materials to form the BHJ active layer.5–7 One such example, reported by Palomares et al.,8 features a small molecule organic solar cell based on a soluble tetra-tert-butyl zinc phthalocyanine (ZnTBPc)/PCBM blend. This device achieved a short-circuit current density (JSC) of approximately 3.57 mA cm−2, an open-circuit voltage (VOC) of about 0.53 V, and a power conversion efficiency (PCE) of roughly 0.77%. On the other hand, another group reported up to 1.6% efficiencies in solution-processed bulk heterojunction solar cells.9 These cells utilized highly soluble ruthenium phthalocyanines (RuPcs) axially functionalized with pyridyl dendrons up to the third generation and blended with fullerene derivatives, such as PCBM or PC71BM. The findings highlight that further advancements in the design of new Pc macrocycles are necessary to enhance their performance in BHJ solar cells. One key limitation contributing to the low efficiency of Pcs is their VOC. Structural modifications to the Pc molecules, such as introducing different functional groups at specific positions, could help address this issue. These modifications aim to extend the absorption spectrum and fine-tune the electronic configuration of the excited states, offering promising strategies to improve the power conversion efficiency of solution-processed Pc-based BHJ solar cells.
A particularly promising approach that has gained traction in recent years is the synthesis of linker-bound dimeric phthalocyanine molecules. The incorporation of linkers, which are molecular bridges that connect phthalocyanine units, offers several advantages in optimizing the material properties for photovoltaic applications10–12
Linker groups significantly influence the intermolecular and intramolecular interactions of organic compounds in the solid state. As a result, they have garnered considerable attention in the synthesis of dimeric zinc(II) phthalocyanines. These groups play a crucial role in shaping the molecular and structural properties of the dimers, which, in turn, profoundly impact their functional and application-related characteristics.
Although the Pc system has garnered significant scientific and technological interest, the effort invested in developing multi-phthalocyanine arrays has been relatively limited13–16 Nonconjugated Pc dyads have been documented as the two Pc cores being covalently connected via various types of bridges.17 However, there are limited studies on Pc subunits connected through π-conjugated pathways.18–23 Leznoff and colleagues synthesized the first Pc dimeric system featuring a butadiynyl bridge between the chromophores.24 However, their method was limited to the preparating of homodimetallic bisphthalocyanines, resulting in low yield. Later, Torres et al. developed an efficient method for preparing homo- and heterodimetallic bisphthalocyanine complexes with ethynyl and butadiynyl bridges using metal-mediated coupling methodologies.25 Göransson and colleagues then studied long-range electron transfer in zinc–phthalocyanine–oligo(phenylene-ethynylene)-based donor-bridge-acceptor dyads to understand the charge transfer mechanisms for solar energy conversion26
In recent years, zinc Pc dimers with various conjugated systems linking tri-tert-butyl phthalocyanine units have been synthesized as hole transport materials (HTMs) in organic–inorganic hybrid perovskite solar cells. These competitive HTMs linkers, to balance the trade of stability and performance27 include 2,5-thienyl, 2,7-fluorenyl, 3,6-bisthienyl diketopyrrolopyrrole, and 1,4-phenyl.10,28,29 Furthermore, Pc derivatives have also been utilized as a third component in polymer-fullerene-based BHJ solar cells to enhance the light-harvesting capacity of the photoactive layer.30,31
More recently, we have reported an oligo-(p-phenyleneethynylene) π-framework with proper functionalities and substituents could be an essential molecular family for high-efficiency solution-processed OLED devices.32 Our previous study indicated that an oligo-(p-phenyleneethynylene)-framework is a promising linker group for photovoltaic applications, requiring a deeper analysis of the influence of the structure of used molecules on the final performance of OLED devices.
Building on these findings, the present paper focuses on synthesizing and characterizing of p-phenyleneethynylene-bridged two ZnPc dimers (GT57 and GT60, Fig. 1) used in BHJ solar cells as new electron donor components. In this molecular structure, 2-ethylhexyloxy (–OCH2CH(C2H5)C4H9) substituents on the bridged phenyl ring enhance solubility, enabling efficient synthesis, purification, and the fabrication of high-quality emissive films. The presence of swallow-tailed bulky substituents at the linker unit between two Pc molecules in a relatively small-sized molecular dimension is expected to impede undesired π–π interactions in the solid state that could otherwise yield excimer formation (redshift inemission) and induce nonradiative decay pathways (low quantum yield). Considering that the low photovoltaic performance of Pc-based BHJ solar cells may be linked to the inhomogeneous morphology of the active layer, characterized by the formation of large domains, the use of non-aggregated Pc can aid in achieving better control over the active layer morphology. Additionally, to gain deeper insight into the impact of sulfur atoms and six bulky tert-butyl substituents at the peripheral positions of dimeric Pcs on their photophysical and electrochemical properties, as well as their photovoltaic performance, theoretical calculations were performed.
 | ||
| Fig. 1 (a) The molecular design of dimeric ZnPcs and (b) the corresponding chemical structures of GT57 and GT60. | ||
1. Experimental
The 4-(tert-buthylthio)phthalonitrile (PN3)33 and 1,4-bis(ethynyl)-2,5-bis(2-ethylhexyloxy)benzene (L1),34 2,9,16,23-tetra-tert-butyl-29H,31H-phthalocyanine (TB4-ZnPc)35 and 2,9,16,23-tetra-tert-butyl-sulfanyl-29H,31H-phthalocyanine (STB4-ZnPc)36 were synthesized according to the literature. The equipment, materials, photophysical, electrochemical measurements, and spectra used for the characterization of the compounds are provided in the ESI.†1.1. Synthesis
Synthetic pathways of GT56, GT58, GT57, GT60 were given in Scheme 1. | ||
| Scheme 1 Synthetic route of molecules (GT56, GT57, GT58, GT60). (i) DMAE(dry), Zn(OAc)2, reflux, 24 h. (ii) Pd(PPh3)2Cl2, CuI, THF, TEA. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dioxane (4:1) mixture as eluent, yielding a blue solid designated as GT56. Yield: 82 mg (24%) 1H NMR (500 MHz, d6-DMSO) δ (ppm): 9.35–8.86 (m, 6H), 8.42–8.21 (m, 5H), 7.91–7.81 (m, 1H), 2.00–1.88 (m, 27H). 13C NMR (125 MHz, d6-DMSO) δ (ppm): 169.99, 158.44, 153.44, 153.14, 152.93, 152.57, 151.79, 150.22, 149.93, 149.28, 149.06, 138.86, 138.66, 138.14, 138.02, 137.02, 136.34, 136.12, 135.88, 133.36, 131.77, 130.59, 127.63, 127.48, 127.41, 127.36, 123.83, 123. 33, 122.66, 122.41, 121.86, 120.18, 119.12, 118.74, 118.40, 40.41 (quaterner C), 36.21 (CH3), 36.17 (CH3), 36.15 (CH3), 36.17 (CH3), 36.12 (CH3), 36.10 (CH3), 36.00 (CH3), 35.91 (CH3). MALDI-TOF (m/z) calcd for C44H39IN8Zn: 872.14; found: 871.735 [M]+. Elemental analysis (%) calcd C, 60.60; H, 4.51; N, 12.85, found C 60.56; H, 4.60; N, 12.35.
:dioxane (3:1) mixture as the mobile phase, yielding GT58 as a blue color solid. Yield: 60 mg (31%). 1H NMR (500 MHz, d6-DMSO) δ (ppm): 9.31–8.72 (m, 6H), 8.59–8.14 (m, 6H), 1.82–1.61 (m, 27H). 13C NMR (125 MHz, d6-DMSO) δ (ppm): 171.94, 171.10, 154.53, 153.98, 145.99, 140.78, 140.47, 140.09, 137.45, 136.37, 134.49, 133.72, 133.33, 128.03, 127.70, 126.06, 125.18, 124.88, 110.97, 105.14, 99.06, 49.93 (quaterner C), 34.33 (CH3). MALDI-TOF (m/z) calcd for C44H39I3N8S3Zn: 966.08; found: 968.320 [M + 2H]+. Elemental analysis (%) calcd C, 54.58; H, 4.06; N, 11.57, found C, 54.36; H, 4.58; N, 11.34.
:dioxane (4:1) mixture as the mobile phase, yielding GT57 as a green solid (12 mg 85%). FT-IR (ATR) νmax/cm−1 3054 (ArC-H), 2958–2860 (aliphatic CH), 2106.5 (C
:dioxane (4:1) mixture as eluent, yielding a blue solid designated as GT56. Yield: 82 mg (24%) 1H NMR (500 MHz, d6-DMSO) δ (ppm): 9.35–8.86 (m, 6H), 8.42–8.21 (m, 5H), 7.91–7.81 (m, 1H), 2.00–1.88 (m, 27H). 13C NMR (125 MHz, d6-DMSO) δ (ppm): 169.99, 158.44, 153.44, 153.14, 152.93, 152.57, 151.79, 150.22, 149.93, 149.28, 149.06, 138.86, 138.66, 138.14, 138.02, 137.02, 136.34, 136.12, 135.88, 133.36, 131.77, 130.59, 127.63, 127.48, 127.41, 127.36, 123.83, 123. 33, 122.66, 122.41, 121.86, 120.18, 119.12, 118.74, 118.40, 40.41 (quaterner C), 36.21 (CH3), 36.17 (CH3), 36.15 (CH3), 36.17 (CH3), 36.12 (CH3), 36.10 (CH3), 36.00 (CH3), 35.91 (CH3). MALDI-TOF (m/z) calcd for C44H39IN8Zn: 872.14; found: 871.735 [M]+. Elemental analysis (%) calcd C, 60.60; H, 4.51; N, 12.85, found C 60.56; H, 4.60; N, 12.35.
:dioxane (3:1) mixture as the mobile phase, yielding GT58 as a blue color solid. Yield: 60 mg (31%). 1H NMR (500 MHz, d6-DMSO) δ (ppm): 9.31–8.72 (m, 6H), 8.59–8.14 (m, 6H), 1.82–1.61 (m, 27H). 13C NMR (125 MHz, d6-DMSO) δ (ppm): 171.94, 171.10, 154.53, 153.98, 145.99, 140.78, 140.47, 140.09, 137.45, 136.37, 134.49, 133.72, 133.33, 128.03, 127.70, 126.06, 125.18, 124.88, 110.97, 105.14, 99.06, 49.93 (quaterner C), 34.33 (CH3). MALDI-TOF (m/z) calcd for C44H39I3N8S3Zn: 966.08; found: 968.320 [M + 2H]+. Elemental analysis (%) calcd C, 54.58; H, 4.06; N, 11.57, found C, 54.36; H, 4.58; N, 11.34.
:dioxane (4:1) mixture as the mobile phase, yielding GT57 as a green solid (12 mg 85%). FT-IR (ATR) νmax/cm−1 3054 (ArC-H), 2958–2860 (aliphatic CH), 2106.5 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1653.2, 1611.7, 1485.8, 1460.2, 1436.7, 1389.6, 1328.4, 1257.3, 1148.3, 1044.8, 1024.4, 919.8, 827.2, 799.9, 747.4, 720.2, 691.3, 674.1. 1H NMR (500 MHz, d8-THF) δ (ppm): 9.50–9.18 (m, 16H, ArH), 8.34–8.21 (m, 8H, ArH), 7.50 (brs, 2H ArH), 4.38, 4.34 (s, 4H, OCH2) 2.01–1.86 (m, 56H, CH3), 1.45 (m, 8H), 1.32 (m, 8H), 1.06–0.97 (m 12H, CH3). 13C NMR (125 MHz, d8-THF) δ (ppm): 154.47, 152.76, 151.35, 145.92, 145.25, 138.99, 138.33, 136.68, 131.35, 125.50, 124.15, 122.37, 119.03, 116.56, 114.48, 112.70, 96.25, 93.26, 72.91, 71.96, 53.13, 40.29, 35.65, 31.67, 31.02, 29.67, 24.83, 23.37, 13.74, 11.38. MALDI-TOF (m/z) calcd for C114H114N16O2Zn2: 1873.04; found: 1871.015 [M-2H]+. Elemental analysis (%) calcd C, 73.18; H, 6.14; N, 11.98, found C, 73.01; H, 6.44; N, 11.55.
:ethanol (10:1) mixture as the mobile phase, yielding GT60 as a green solid (11 mg, 86%). FT-IR (ATR) νmax/cm−1 3000 (ArC–H), 2925–2862 (aliphatic CH), 2105.4 (CC), 1605.0, 1485.8, 1388.9, 1363.1, 1334.3, 1256.8, 1138.8, 1041.4, 908.4, 832.1, 745.1, 685.2. 1H NMR (500 MHz, d8-THF-) δ (ppm): 9.16–9.10 (m, 6H, ArH), 8.37–832 (m, 4H, ArH), 7.47–7.42 (m, 16H, ArH), 4.76 (brs, 4H, OCH2), 2.06 (s, 54H, CH3), 1.64–1.62 (m, 10H, CH2 + CH), 1.29 (m 8H, CH2), 0.91–0.88(m, 12H, –CH3) 13C NMR (125 MHz, d8-THF) δ (ppm): 164.92, 153.09, 138.68, 137.97, 135.14, 134.60, 133.59, 131.32, 129.23, 128.01, 127.94, 122.22, 97.20, 89.94, 56.96, 53.74, 46.23, 39.11, 31.90, 30.85, 29.67, 29.39, 28.97, 23.99, 22.59, 22.29, 13.46, 10.44. MALDI-TOF (m/z) calcd for C114H114N16O2S6Zn2: 2063.401; found: 2063.493 [M]+. Elemental analysis (%) calcd C, 66.36; H, 5.57; N, 10.86, found C, 66.14; H, 5.61; N, 10.46.
C), 1653.2, 1611.7, 1485.8, 1460.2, 1436.7, 1389.6, 1328.4, 1257.3, 1148.3, 1044.8, 1024.4, 919.8, 827.2, 799.9, 747.4, 720.2, 691.3, 674.1. 1H NMR (500 MHz, d8-THF) δ (ppm): 9.50–9.18 (m, 16H, ArH), 8.34–8.21 (m, 8H, ArH), 7.50 (brs, 2H ArH), 4.38, 4.34 (s, 4H, OCH2) 2.01–1.86 (m, 56H, CH3), 1.45 (m, 8H), 1.32 (m, 8H), 1.06–0.97 (m 12H, CH3). 13C NMR (125 MHz, d8-THF) δ (ppm): 154.47, 152.76, 151.35, 145.92, 145.25, 138.99, 138.33, 136.68, 131.35, 125.50, 124.15, 122.37, 119.03, 116.56, 114.48, 112.70, 96.25, 93.26, 72.91, 71.96, 53.13, 40.29, 35.65, 31.67, 31.02, 29.67, 24.83, 23.37, 13.74, 11.38. MALDI-TOF (m/z) calcd for C114H114N16O2Zn2: 1873.04; found: 1871.015 [M-2H]+. Elemental analysis (%) calcd C, 73.18; H, 6.14; N, 11.98, found C, 73.01; H, 6.44; N, 11.55.
:ethanol (10:1) mixture as the mobile phase, yielding GT60 as a green solid (11 mg, 86%). FT-IR (ATR) νmax/cm−1 3000 (ArC–H), 2925–2862 (aliphatic CH), 2105.4 (CC), 1605.0, 1485.8, 1388.9, 1363.1, 1334.3, 1256.8, 1138.8, 1041.4, 908.4, 832.1, 745.1, 685.2. 1H NMR (500 MHz, d8-THF-) δ (ppm): 9.16–9.10 (m, 6H, ArH), 8.37–832 (m, 4H, ArH), 7.47–7.42 (m, 16H, ArH), 4.76 (brs, 4H, OCH2), 2.06 (s, 54H, CH3), 1.64–1.62 (m, 10H, CH2 + CH), 1.29 (m 8H, CH2), 0.91–0.88(m, 12H, –CH3) 13C NMR (125 MHz, d8-THF) δ (ppm): 164.92, 153.09, 138.68, 137.97, 135.14, 134.60, 133.59, 131.32, 129.23, 128.01, 127.94, 122.22, 97.20, 89.94, 56.96, 53.74, 46.23, 39.11, 31.90, 30.85, 29.67, 29.39, 28.97, 23.99, 22.59, 22.29, 13.46, 10.44. MALDI-TOF (m/z) calcd for C114H114N16O2S6Zn2: 2063.401; found: 2063.493 [M]+. Elemental analysis (%) calcd C, 66.36; H, 5.57; N, 10.86, found C, 66.14; H, 5.61; N, 10.46.
2. Results and discussion
2.1. Synthesis and structural characterization
The key to the synthesis of ethynyl-bridged dimeric phthalocyaninate complexes via metal-mediated coupling is the availability of an asymmetric iodo phthalocyanine and a linker with appropriately functionalized ethynyl groups such as L1 (Fig. 1). This study reports the synthesizing of dimeric zinc phthalocyanines (GT57, GT60) using 1,4-bis(ethynyl)-2,5-bis(2-ethylhexyloxy)benzene as a linker between two Pc units (Fig. 1). The synthesis of dimeric phthalocyanines GT57 and GT60 was carried out in two steps following the routes depicted in Scheme 1. Firstly, the asymmetric iodo-tert-buthyl or iodo-tert-butylthio phthalocyanine derivatives (GT56, GT58) were obtained utilizing by the cyclotetramerization reaction of iodophthalonitrile (PN-1) with other phthalonitriles (PN-2 and PN-3) in a molar ratio of 1:7, respectively. The phthalonitrile derivatives were used in a 1:7 molar ratio, resulting in the formation of only two products: the desired mono-iodinated asymmetric phthalocyanine derivatives (GT56 or GT58) and symmetric phthalocyanine derivatives containing tert-butyl groups, as outlined in Scheme 1. In the final, Sonogashira cross-coupling step, the dimeric phthalocyanine derivatives (GT57, GT60) were subjected to reactions with mono-iodophthalocyanines GT56 or GT58 and 1,4-bis(ethynyl)-2,5-bis(2-ethylhexyloxy)benzene (L1) as linker group in the presence of a Pd(PPh3)2Cl2 catalyst and a CuI/Et3N co-catalyst/base system in THF, resulting in the formation of the desired products with a yield of 85% and 86% (GT57, GT60) respectively. Highly selective Sonogashira cross-coupling at the iodo-functionalized reaction site was utilized in this process. The selectivity undoubtedly arises from the better reactivity of iodine atoms as compared to others. This facilitates the rate-determining oxidative addition step in the catalytic cycle.
Despite its large molecular weight, Pc derivatives were found to be soluble in common organic solvents such as chloroform and THF, as well as printing-friendly solvents such as toluene. Pcs were purified by thin layer chromatography on silica gel using dioxane:n-hexane (1:3, v/v) solvent mixture. All new compounds were fully characterized using 1H–13C-NMR, MALDI TOF-MS, and FTIR, and optically evaluated by UV/vis and steady-state fluorescence emission spectroscopies Fig. S1–S25 (ESI†).
1H NMR spectra recorded in [d8]THF are very well resolved and all the different protons could be assigned (see the ESI†). However, although the system in which two units of ZnPc are bonded to the L1 moiety, GT57 and GT60, are quite soluble in THF, broad signals were obtained in the 1H NMR spectra even when using a coordinating solvent such as [d8]THF due to the high degree of π–π-stacking and also to the presence of too many regioisomers (see the Fig. S12, S13, S17 and S18, ESI†). Phthalocyanines tend to form aggregates in solutions. Aggregation causes molecules to move closer together, changing the chemical environment of the protons. This can result in broad, indeterminate, and unresolved peaks in the spectrum. However, the integrated signal intensities reflect the number of hydrogen atoms in the compounds.
MALDI-TOF MS spectra are important for verifying the structure of phthalocyanines. The MALDI-TOF MS spectra of the purified monofunctional Pcs were detected molecular ion peaks at m/z 871.735 [M]+, 968.320 [M]+, for GT56, GT58, and 1871.015 [M]+ and 2063.493 [M]+ for GT57 and GT60, respectively (Fig. S2, S6, S11 and S16, ESI†). The absence of signals corresponding to other substituted derivatives in the mass spectra provides significant support for the structural purity of the compounds. Each spectrum offers compelling evidence that reinforces the validity of their proposed structures.
2.2. Photochemical and photophysical measurements
The photophysical and photochemical properties of the prepared dimeric ZnPcs were extensively evaluated using UV-vis absorption, steady-state fluorescence, and time-resolved fluorescence spectroscopies. The UV-vis absorption and fluorescence spectra of the GT57 and GT60 were investigated in the 300–800 nm range using different solvents such as toluene, THF, dioxane, DMSO, and DMF to evaluate their aggregation properties and their photophysical and photochemical characteristics. THF was chosen for the photophysical and photochemical measurements among these solvents due to its superior solubility and the absence of significant changes with solvent variation. All spectral data, including UV-vis spectra obtained at different concentrations in THF, are provided in the ESI† (Fig. S19 and S20).Fig. 2 shows the absorption spectra of the dimers and monomeric ZnPcs (TB4-ZnPc and S-TB4-ZnPc) dissolved in THF. As can be seen, the Soret bands of both dimeric ZnPc derivatives are observed between 300 and 400 nm, while the Q bands of GT57 and GT60 exhibit a bathochromic shift and increased bandwidth compared to their monomeric counterparts, TB4-ZnPc (672 nm)29 and S-TB4-ZnPc (678 nm),36 respectively, due to the more extended conjugation of these systems. In addition, the Q-band maxima of dimer GT57 is split in two peaks in THF solution (Fig. 2a). This is likely caused by the presence of regioisomers and their non-symmetric nature29,37,38 Typically, enhanced molecular stacking results in broader absorption signals and a reduced molar extinction coefficient (ε) due to decreased absorption intensity. However, this trend does not hold for dimer GT60, as the presence of the tert-butyl thio moieties consisting bulky tert-butyl and S heteroatoms on two Pc macrocycles reduces molecular aggregation, resulting in a strong single Q-band at 679 nm.
 | ||
| Fig. 2 (a) UV-vis absorption spectra of dimeric ZnPc derivatives (GT60 and GT57) (b) TB4-ZnPc, S-TB4-ZnPc, GT60, GT57 in THF. | ||
The various solvents were employed to examine the fluorescence emission properties of dimeric ZnPcs (Fig. S21–S25, ESI†). While no significant changes were observed in the fluorescence of the GT60 compound in different solvents, the GT57 compound exhibited strong fluorescence in low-polarity solvents such as THF, dioxane, and toluene. However, a significant decrease in fluorescence intensity was observed in high-polarity solvents like DMF and DMSO. This behavior is attributed to the tendency of high-polarity solvents to promote molecular aggregation or facilitate intermolecular energy transfer, leading to fluorescence quenching. Emission maximums of GT57 and GT60 are observed at 702 nm and 687 nm, in THF respectively.
The fluorescence quantum yield (ΦF) is a key parameter that reflects the efficiency of the fluorescence process and is essential for photophysical applications.39 To calculate the fluorescence quantum yields in THF, the fluorescence area integration of the dimeric ZnPc derivatives (GT60 and GT57) and the reference Unsub-ZnPc ZnPc vs. absorbance were plotted (Fig. 3a). Table 1 shows the obtained fluorescence quantum yields. ΦF values of dimeric ZnPcs GT57 and GT60 were determined to be 0.20 and 0.08 which were lower than that of Unsub-ZnPc. respectively, by the absolute method using an integrating sphere.
Time-resolved fluorescence measurements quantify fluorescence lifetime (τF). The fluorescence lifetimes of GT57 and GT60 were measured in ambient conditions using the time-correlated single photon counting (TCSPC) technique in THF. Both molecules showed single exponential decay, obtaining the values of τF = 2.18 ns for GT57 and τF = 2.96 ns for GT60 (Fig. 3b). These values indicate that there is no significant difference between the fluorescence lifetimes of dimeric phthalocyanine derivatives (GT57 and GT60) and the standard Unsub-ZnPc.
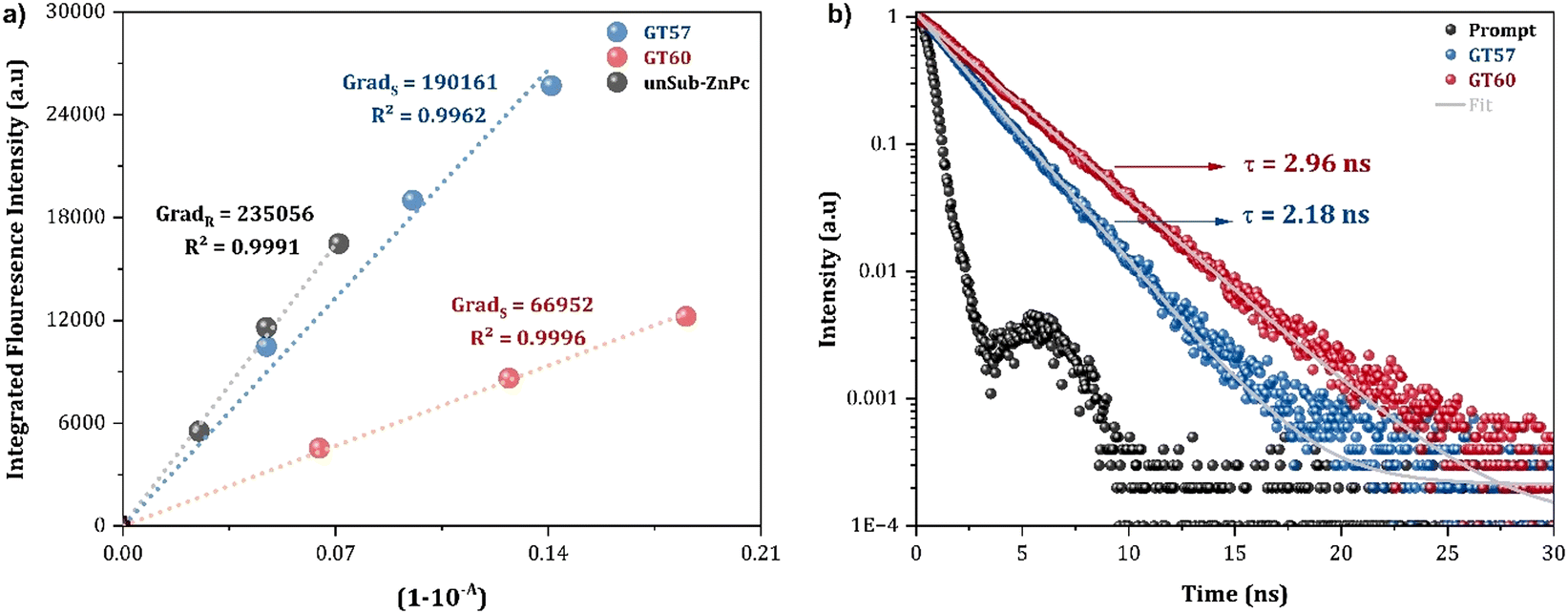 | ||
| Fig. 3 (a) Fluorescence area integrates vs. absorbance of GT57, GT60 and Unsub-ZnPc (b) fluorescence decay curves of GT57 and GT60 in THF (NanoLed λex: 674 nm). | ||
The singlet oxygen generation quantum yield (ΦΔ) represents the number of singlet oxygen molecules produced per photon the photosensitizer absorbs. Optical determination of singlet oxygen was carried out using a direct measurement technique. The direct method involves detecting the intrinsic phosphorescence of singlet oxygen using a near-IR sensitive detector. After excitation with a xenon-arc source at the respective absorption maxima, the phosphorescence spectra of singlet oxygen were recorded for four phthalocyanines in THF at equal absorbance (0.23) to determine ΦΔ directly (Fig. S26, ESI†). The Unsub-ZnPc was used as the reference (Table 1). The highest ΦΔ value, observed for the dimer GT60 complex (0.74), indicates efficient energy transfer to molecular oxygen, which may be linked to its UV behavior. In contrast, GT57 demonstrates singlet oxygen generation comparable to Unsubs-ZnPc, confirming their potential for triplet photosensitized applications.
2.3. Electrochemical measurements
The electrochemical behavior of MPcs was investigated using cyclic voltammetry (CV) to determine the HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) energy level. The measurements were performed using a three-electrode system, consisting of a glassy carbon electrode (GCE) as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl aqueous electrode as the reference electrode. MPcs were prepared at a concentration of 5 × 10−4 M, and all experiments were conducted in THF at 25 °C, with 0.1 M NBu4PF6 (tetrabutylammonium hexafluorophosphate) used as the supporting electrolyte. Before measurements, the solution was purged with N2(g) for 5 minutes. The ferrocene/ferrocenium (Fc/Fc+) redox couple was employed as an internal standard. CVs were recorded at a 100 mV s−1 scan rate and used to calculate the HOMO–LUMO energy level. The voltammograms were referenced against the Fc/Fc+ redox couple and calculated with 4.8 eV below the vacuum level set as the reference level, as shown in Fig. 4. | ||
| Fig. 4 CVs of GT57 and GT60 at 100 mV scan rate on GCE in THF(dry)/0.1 M TBAP at 25 °C. | ||
Zn(II) phthalocyanines essentially showed ligand-centered redox reactions instead of metal-centered ones. Oxidation and reduction processes include electron transfer inside the π-conjugated system. In this case, the initial redox couple identified in the negative scan correlates to the LUMO level, whereas the first redox couple detected in the positive scan corresponds to the HOMO level. The HOMO energy levels of the dimeric phthalocyanine compounds were determined as −4.98 and −5.10 eV while their LUMO levels were found to be −3.28 eV and −3.45 eV for GT57 and GT60, respectively. According to Molina et al., the energy levels of the monomeric ZnPc derivative (TB4-ZnPc) were determined to be −5.05 eV and −3.26 eV.29 No significant change was observed in the energy levels between the dimeric GT57 and the monomeric TB4-ZnPc phthalocyanine derivatives.
2.4. Theoretical calculations
The HOMO–LUMO band gap was calculated for these optimized structures and compared with experimental values (Table S1, ESI†). While CAM-B3LYP and wB97XD overestimated the band gap, B3LYP provided results closest to the experimental data. Additionally, the performance of this methodology in previous studies on macrocyclic systems showed good agreement between theoretical and experimental results.36 Based on both the current benchmark study and our prior experience, B3LYP with the def2-SVP basis set was selected for geometry optimizations in this study. All stationary points have been characterized by a frequency analysis at the same theory level as the geometry optimizations.
The UV-vis electronic absorption spectra in the solution phase were computed using the time-dependent DFT (TD-DFT) method.49 Two different functionals were employed: CAM-B3LYP/def2-SVP, based on ground-state geometries optimized at the same level, and TPSSh/def2-SVP, based on geometries optimized at the B3LYP/def2-SVP level (Table S2, ESI†). The CAM-B3LYP method was chosen due to its established accuracy in excited-state predictions in solar cell applications.47,50,51 However, the results obtained from both functionals were comparable. Since TPSSh had previously been used in similar zinc phthalocyanine systems, we selected this functional to maintain consistency with our earlier study.52 The solvent was modeled using the integral equation formalism model (IEFPCM)53–56 and tetrahydrofuran (THF) was used, as it was as the solvent employed in the experimental part of this study.
In TD-DFT, excited states are depicted as a linear combination of excitations from occupied to virtual Kohn–Sham orbitals. This expression in terms of molecular orbitals can be complicated, as numerous occupied and virtual orbital pairs may contribute to the expansion with comparable significance. An enhancement for gaining a more profound comprehension of the physical characteristics of electronic transitions relies on depicting an excited state through natural transition orbitals (NTOs).57 Thus, NTOs have been computed as implemented in Gaussian.
 | ||
| Fig. 5 Optimized molecular structures of GT57 and GT60 calculated at the B3LYP/def2-SVP level of theory in the gas phase. Hydrogen atoms are omitted for clarity. | ||
The frontier molecular orbitals (HOMO and LUMO) offer essential insights into the chemical behavior, of the systems under investigation. For GT57, both the HOMO and LUMO are delocalized across the dimer, including the bridge, and exhibit a π–π* character (Fig. S28, ESI†). A similar delocalization and electronic nature are also observed for GT60. The calculated HOMO energy values are slightly underestimated, while the LUMO energies are slightly overestimated (Table 3). Although the calculated bandgap values are similar, the bandgap of GT57 is slightly lower than that of GT60, which is consistent with the experimental values (Table 2).
| Compounds | R1a | O1a | ΔE1/2b (V) | HOMOc (eV) | LUMOd (eV) | E opt (V) |
|---|---|---|---|---|---|---|
| a E 1/2 = (Epa + Epc)/2 at 0.100 V s−1. b ΔE1/2 = E1/2 (first oxidation) − E1/2 (first reduction) = HOMO–LUMO gap. c E HOMO = −[(Eox − E1/2(ferrocene)) + 4.8]. d E LUMO = −[(Ered − E1/2(ferrocene)) + 4.8].58 | ||||||
| GT57 | −1.52 | 0.18 | 1.70 | −4.98 | −3.28 | 1.75 |
| GT60 | −1.35 | 0.30 | 1.65 | −5.10 | −3.45 | 1.77 |
| Compounds | E HOMO (eV) | E LUMO (eV) | ΔE (eV) |
|---|---|---|---|
| GT57 | −5.02 | −2.98 | 2.05 |
| GT60 | −5.29 | −3.23 | 2.07 |
We also analyzed the molecular electrostatic potential (MEP) of the molecules, which provides insights into their electron-rich and electron-poor regions. Fig. 6 displays the electrostatic potential maps for GT57 and GT60. An electrophilic region (shown in blue) on the zinc denotes an electron-poor site on the metal atom, which is as expected. In GT60, the zinc atom is more electrophilic due to the presence of tert-butylthio substituents. The NPA charges of the metal atoms in GT57 and GT60 are 1.261 and 1.263, respectively. In contrast, the linker exhibits a strong nucleophilic character, indicated in red.
 | ||
| Fig. 6 Electrostatic potential maps of GT57 and GT60. | ||
 | ||
| Fig. 7 TD-DFT (TPSSh/def2-SVP) predicted absorption spectra for GT57 and GT60 in THF. | ||
The nature of the different excited states can be easily determined by analyzing the corresponding Natural Transition Orbitals (NTOs) (Fig. S29, ESI†). The occupied and virtual NTOs for the transitions in the Q band indicate that these transitions are primarily attributed to a π–π* localization within the phthalocyanine rings, with noticeable delocalization present in both the occupied and virtual NTOs involving the substituents. For the transition responsible for the 400 nm absorption, the occupied and virtual NTOs of GT57 are mainly delocalized over the linker and the benzene substituent connected to the linker of the phthalocyanine. For GT60, we observe two less intense peaks around 415 and 473 nm. For 415 nm transitions, occupied NTOs are delocalized over the linker involving the benzene substituents whereas the virtual NTOs are localized over the phthalocyanine ring. The parameters obtained from TD-DFT calculations are summarized in Table 4.
| Compounds | μ (D) | λ (nm) | E (eV) | f |
|---|---|---|---|---|
| GT57 | 0.48 | 660 | 1.88 | 1.699 |
| 391 | 3.17 | 1.115 | ||
| GT60 | 0.31 | 654 | 1.90 | 1.600 |
| 473 | 2.62 | 0.3514 | ||
| 415 | 2.99 | 0.1515 | ||
2.5. Solar cell device performance
Organic solar cells (OSCs) have garnered significant interest as a cost-effective solution for photovoltaic energy generation. The introduction of the bulk-heterojunction (BHJ) concept has notably improved OSC performance by effectively blending electron donor and acceptor materials into a solution, which is subsequently deposited as a thin film between two electrodes. Over the last decade, substantial advancements have been achieved in enhancing the power conversion efficiency (PCE) of organic BHJ solar cells.In this study, OSCs with the structure ITO-ZnO/P3HT:GT57-GT60:PCBM/MoO3/Al were fabricated, as illustrated in Fig. 8a. Fig. 8 provides an overview of the device architecture, energy band diagram, and the relationship between current density and applied voltage. Current density–voltage (J–V) measurements were conducted using a Keithley 4200 semiconductor characterization system, with a 100 mW cm−2 solar simulator (Thermo Oriel, AM 1.5G) serving as the light source. The photovoltaic parameters of the devices, derived from three distinct blend compositions, are summarized in Table 5, where each parameter represents the average of 24 devices. Representative J–V curves for the fabricated devices are also compared in Fig. 8.
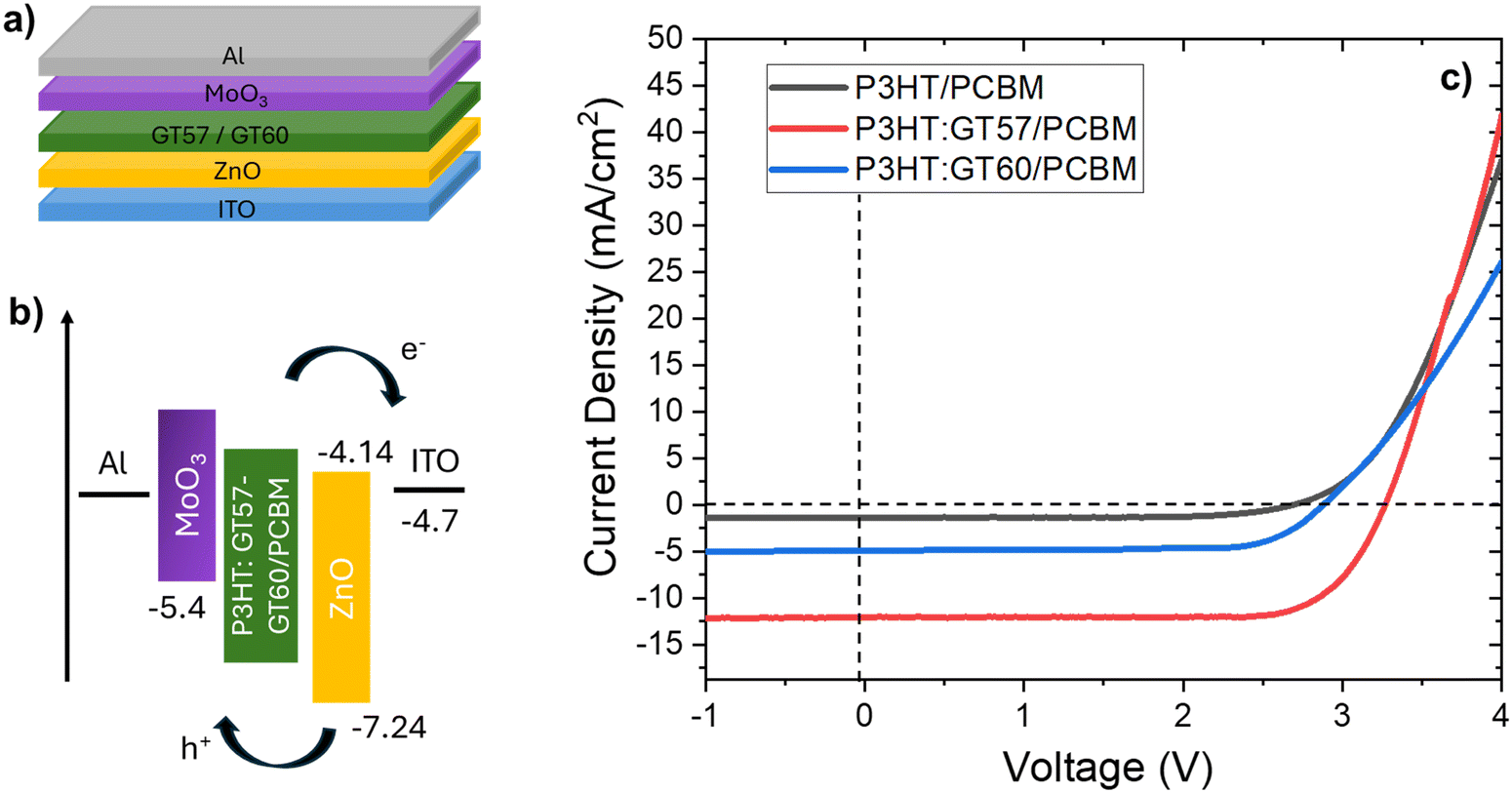 | ||
| Fig. 8 (a) Device structure (b) energy level of materials (c) typical J–V curves of P3HT/PCBM, P3HT:GT57/PCBM, P3HT:GT60/PCBM solar cell devices. | ||
| Vices | FF (%) | V OC (V) | J SC (mA cm−2) | η (%) |
|---|---|---|---|---|
| P3HT/PCBM | 0.56 | 2.67 | 1.43 | 3.68 |
| P3HT:GT57/PCBM | 0.58 | 2.88 | 4.91 | 3.87 |
| P3HT:GT60/PCBM | 0.63 | 3.26 | 12.09 | 4.05 |
The reference P3HT:PCBM device exhibited a VOC of 2.67 V, a JSC of 1.43 mA cm−2, and a fill factor (FF) of 0.56, consistent with reported literature values59,60 Incorporating GT57 and GT60 into the P3HT blend resulted in a significant enhancement in JSC, with only a minor change in FF, leading to PCEs of 3.87% and 4.05% for the P3HT:GT57/PCBM and P3HT:GT60/PCBM devices, respectively (Table 5). Notably, the P3HT:GT60/PCBM device demonstrated an approximately 25% increase in JSC compared to the reference P3HT:PCBM device, underscoring the effectiveness of the material modifications.
The primary performance parameters of organic solar cells include short-circuit current density (JSC), open-circuit voltage (VOC), fill factor (FF), and efficiency (η). The JSC and VOC values are derived from the intersections of the current density–voltage (J–V) curve with the respective axes. The maximum power point (MPP) is identified from the power–voltage (P–V) curve, allowing for the calculation of IMP and VMP. The fill factor and efficiency are computed using the following equations.59

where PS is the power of incident light.

The surface morphology of the active layers was investigated using scanning electron microscopy (SEM) (Fig. 9a and b) and atomic force microscopy (AFM). Fig. 9c and d show the root-mean-square (RMS) roughness values of 1.92 nm for the P3HT:GT57/PCBM blend film and 1.35 nm for the P3HT:GT60/PCBM blend film, respectively. These results indicate a high level of miscibility between the donor materials and the PCBM acceptor. Notably, the P3HT:GT60/PCBM blend film exhibits reduced surface roughness and more pronounced bicontinuous phase separation. This well-defined morphology enhances the interfacial area between the donor and acceptor materials, improving exciton dissociation efficiency and resulting in a higher short-circuit current density (JSC).
 | ||
| Fig. 9 SEM images of (a) GT57 (b) GT60 films on quartz substrate. AFM images of (c) GT57 (d) GT60 films on the quartz substrate. | ||
Both GT57 and GT60 exhibit strong absorption in the visible region and possess similar molecular backbones; however, their photovoltaic performances differ slightly, with GT57 yielding a slightly lower PCE than GT60. The small participation of sulfur atoms in the frontier molecular orbitals may influence the charge separation dynamics, leading to a slightly larger band gap and thus an enhanced PCE conversion. Nevertheless, the difference is so small that both exhibit comparable performance. Furthermore, TD-DFT calculations indicate subtle differences in excited-state behavior.
More broadly, the modest PCEs (∼4%) of both systems reflect several limiting factors beyond intrinsic molecular properties. While bulky tert-butyl and tert-butylthio groups enhance solubility and suppress aggregation, they may also hinder close π–π stacking, which is critical for efficient charge carrier mobility. In addition, potential mismatch in energy level alignment or unfavorable domain sizes within the bulk heterojunction may lead to charge recombination or inefficient exciton dissociation.
To enhance the photovoltaic performance of such ZnPc dimers, further molecular engineering is warranted. Strategies such as modifying the bridge (e.g., incorporating more planar or electron-donating linkers), introducing push–pull architectures to improve charge separation, or tailoring the side chains to modulate packing and morphology could be explored. Moreover, the use of alternative acceptors (e.g., non-fullerene acceptors) and post-processing optimization (e.g., thermal or solvent vapor annealing) could improve the active layer morphology and device efficiency. These directions will be central to our ongoing efforts toward developing next-generation ZnPc-based donor materials for organic photovoltaics.
Conclusion
This study reports the design and synthesis of two novel dimeric zinc phthalocyanines, GT57 and GT60, incorporating bulky tert-butyl and tert-butylthio substituents. The two phthalocyanine units are connected via a 1,4-bis(2-ethylhexyloxy)-2,5-diethylbenzene linker. These molecules were studied to evaluate their effectiveness as donor materials in bulk heterojunction (BHJ) solar cell devices. Solar cells fabricated from a thin evaporated layer of dimeric ZnPcs and a spin-coated layer show short-circuit current densities of ∼1.4 mA cm−2 with open circuit voltages ∼200 mV under 100 mW cm−2 simulated AM 1.5 illumination. The electrochemical analysis and optical properties indicated that all synthesized phthalocyanine derivatives possess appropriate HOMO–LUMO band gap levels for BHJ solar cell applications, which theoretical calculations have further confirmed. The theoretical electronic structures are in good agreement with experimental findings regarding molecular orbital energies. The spectra calculated via TD-DFT enable us to assess the key spectral features of the newly designated dimeric zinc phthalocyanines. This study opens new avenues for utilizing Pc dimers as efficient, stable, and cost-effective hole-transporting materials. Ongoing research is focused on reducing recombination phenomena and optimizing device performance.Previous dimeric phthalocyanines (ZnPcs) faced significant challenges such as weak electronic coupling, poor charge separation, and fast charge recombination, primarily due to the use of flexible or non-conjugated linkers. These limitations hindered their efficiency in solar energy conversion applications. Additionally, the synthesis of dimeric ZnPcs often involves issues like purification difficulties, solubility, and aggregation, which adversely affect the device structure and performance. Addressing these challenges is crucial for optimizing the final product and improving its efficiency in electronic applications.
In this study, 2-ethylhexyloxy (–OCH2CH(C2H5)C4H9) substituents were used as bridges to facilitate ease of solubility and electronic transitions. Additionally, bulky tert-butyl and tert-butylthio groups were employed to prevent aggregation. These structures, as phthalocyanine functional groups, were utilized to enhance donor properties, minimize challenges in synthesis and purification processes, and optimize solubility. Compared to previous studies, this approach demonstrated improved efficiency, making it a more promising strategy for dimeric ZnPcs.
Author contributions
Gülenay Tunç: investigation, formal analysis, visualization, writing – original draft. Gizem Gümüşgöz Çelik: formal analysis, validation, visualization, writing – original draft. Betül Canımkurbey: investigation, formal analysis, visualization, writing – original draft. Burcu Dedeoğlu: methodology, writing – original draft, writing – review & editing. Ayşe Gül Gurek: methodology, writing – original draft, writing – review & editing, supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data are not publicly available due to participant privacy and ethical considerations. Any additional data or materials can be provided upon request to ensure transparency and reproducibility of the research.Conflicts of interest
The authors declare that the are no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The calculations reported in this paper were performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).References
- K. M. Kadish, K. M. Smith, Â. S. Gabriel and R. Guilard, The Porphyrin Handbook, 1st edn, 2002, vol. 7 Search PubMed.
- M. V. Martínez-Díaz, G. De La Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032–4045 RSC.
- F. Silvestri, I. López-Duarte, W. Seitz, L. Beverina, M. V. Martínez-Díaz, T. J. Marks, D. M. Guldi, G. A. Pagani and T. Torres, Chem. Commun., 2009, 4500–4502 RSC.
- A. Varotto, C. Y. Nam, I. Radivojevioc, J. P. C. Tomé, J. A. S. Cavaleiro, C. T. Black and C. M. Drain, J. Am. Chem. Soc., 2010, 132, 2552–2554 CrossRef CAS PubMed.
- Q. Wang, Y. Li, X. Yan, M. Rathi, M. Ropp, D. Galipeau and J. Jiang, Appl. Phys. Lett., 2008, 93(7), 073303 CrossRef.
- A. Sánchez-Díaz, R. Pacios, U. Muñecas, T. Torres and E. Palomares, Org. Electron., 2011, 12, 329–335 CrossRef.
- M. K. R. Fischer, I. López-Duarte, M. M. Wienk, M. V. Martínez-Díaz, R. A. J. Janssen, P. Bäuerle and T. Torres, J. Am. Chem. Soc., 2009, 131, 8669–8676 CrossRef CAS PubMed.
- M. Pegu, D. Molina, M. J. Álvaro-Martins, M. Castillo, L. Ferrer, P. Huang, S. Kazim, Á. Sastre-Santos and S. Ahmad, J. Mater. Chem. C, 2022, 10, 11975–11982 RSC.
- Á. J. Jiménez, F. Spanig, M. S. Rodríguez-Morgade, K. Ohkubo, S. Fukuzuml, D. M. Guldi and T. Torres, Org. Lett., 2007, 9, 2481–2484 CrossRef PubMed.
- H. Ali and J. E. Van Lier, J. Porphyrins Phthalocyanines, 2014, 18, 991–997 CrossRef CAS.
- C. C. Leznoff and A. B. P. Lever, Wiley: Phthalocyanines Prop. Appl., 1996, vol. 1–4, 1568 Search PubMed.
- G. De La Torre, P. Vázquez, F. Agulló-López and T. Torres, J. Mater. Chem., 1998, 8, 1671–1683 RSC.
- M. Hanack and H. Heckmann, Eur. J. Inorg. Chem., 1998, 367–373 CrossRef CAS.
- G. De La Torre, T. Torres and F. Agulló-López, Adv. Mater., 1997, 9, 265–269 CrossRef CAS.
- C. C. Leznoff, H. Lam, S. M. Marcuccio, W. A. Nevin, P. Janda, N. Kobayashi and A. B. P. Lever, J. Chem. Soc., Chem. Commun., 1987, 699–701 RSC.
- N. Kobayashi, M. Numao, R. Kondo, S. ichiro Nakajima and T. Osa, Inorg. Chem., 1991, 30, 2241–2244 CrossRef CAS.
- D. Lelièvre, L. Bosio, J. Simon, J. J. André and F. Bensebaa, J. Am. Chem. Soc., 1992, 114, 4475–4479 CrossRef.
- D. Lelievre, O. Damette and J. Simon, J. Chem. Soc., Chem. Commun., 1993, 939–940 RSC.
- J. Yang and M. R. Van De Mark, Tetrahedron Lett., 1993, 34, 5223–5226 CrossRef CAS.
- N. Kobayashi, P. Janda, H. Lam, W. A. Nevin, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879–890 CrossRef CAS.
- N. Kobayashi, Y. Higashi and T. Osa, J. Chem. Soc., Chem. Commun., 1994, 1785–1786 RSC.
- S. Vigh, H. Lam, P. Janda, A. B. P. Lever, C. C. Leznoff and R. L. Cerny, Can. J. Chem., 1991, 69, 1457–1461 CrossRef CAS.
- E. M. Maya, P. Vázquez and T. Torres, Chem. – Eur. J., 1999, 5, 2004–2013 CrossRef CAS.
- E. Göransson, J. Boixel, J. Jero Me Fortage, D. Jacquemin, H.-C. Becker, E. Blart, L. Hammarströ and F. Odobel, Inorg. Chem., 2012, 51, 11500–11512 CrossRef PubMed.
- Y. H. Chiang, H. H. Chou, W. T. Cheng, Y. R. Li, C. Y. Yeh and P. Chen, ACS Energy Lett., 2018, 3, 1620–1626 CrossRef CAS.
- D. Molina, M. A. Ruiz-Preciado, F. Sadegh, M. J. Álvaro-Martins, M. Grätzel, A. Hagfeldt and Á. Sastre-Santos, J. Porphyrins Phthalocyanines, 2019, 23, 546–553 CrossRef CAS.
- D. Molina, M. A. Ruiz-Preciado, B. Carlsen, F. T. Eickemeyer, B. Yang, N. Flores-Díaz, M. J. Álvaro-Martins, K. Nonomura, A. Hagfeldt and Á. Sastre-Santos, ChemPhotoChem, 2020, 4, 307–314 CrossRef CAS.
- A. C. Yüzer, G. Kurtay, T. Ince, S. Yurtdaş, E. Harputlu, K. Ocakoglu, M. Güllü, C. Tozlu and M. Ince, Mater. Sci. Semicond. Process., 2021, 129, 105777 CrossRef.
- M. M. Stylianakis, D. Konios, G. Viskadouros, D. Vernardou, N. Katsarakis, E. Koudoumas, S. H. Anastasiadis, E. Stratakis and E. Kymakis, Dyes Pigm., 2017, 146, 408–413 CrossRef CAS.
- H. Usta, D. Alimli, R. Ozdemir, E. Tekin, F. Alkan, R. Kacar, A. G. Altas, S. Dabak, A. G. Gürek, E. Mutlugun, A. F. Yazici and A. Can, J. Mater. Chem. C, 2020, 8, 8047–8060 RSC.
- S. Kaipova, H. Dinçer and A. Altindal, J. Coord. Chem., 2015, 68, 717–731 CrossRef CAS.
- H. Usta, D. Alimli, R. Ozdemir, S. Dabak, Y. Zorlu, F. Alkan, E. Tekin and A. Can, ACS Appl. Mater. Interfaces, 2019, 11, 44474–44486 CrossRef CAS PubMed.
- H. Uchida, H. Tanaka, H. Yoshiyama, P. Y. Reddy, S. Nakamura and T. Toru, Synlett, 2002, 1649–1652 CAS.
- G. Tunç, E. Güzel, I. Şişman, V. Ahsen, G. Cárdenas-Jirón and A. G. Gürek, New J. Chem., 2019, 43, 14390–14401 RSC.
- H. Ali and J. E. Van Lier, Tetrahedron Lett., 2014, 55, 4163–4167 CrossRef CAS.
- K. M. Kadish, K. M. Smith, Â. S. Gabriel and R. Guilard, The Porphyrin Handbook, Academic press, Boston, 1st edn, 2002, vol. 7 Search PubMed.
- G. Gümüşgöz Çelik, A. N. Şahin, F. Lafzi, N. Saracoglu, A. Altındal, A. G. Gürek and D. Atilla, Dalton Trans., 2022, 51, 9385–9396 RSC.
- E. T. Saka, M. Durmuş and H. Kantekin, J. Organomet. Chem., 2011, 696, 913–924 CrossRef CAS.
- L. Kaestner, M. Cesson, K. Kassab, T. Christensen, P. D. Edminson, M. J. Cook, I. Chambrier and G. Jori, Photochem. Photobiol., 2003, 2, 660–667 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, D. J. Fox, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford, Wallingford CT, 2013.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- K. Jaffar, Z. M. Elqahtani, Q. Q. Afzal, M. Ans, S. Riaz, M. A. Tahir, J. Iqbal, Z. M. M. Mahmoud, Z. A. Alrowaili and M. S. Al-Buriahi, Polymer, 2022, 245, 124675 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. El Mouhi, A. Slimi, A. Fitri, A. T. Benjelloun, S. ElKhattabi, M. Benzakour, M. Mcharfi and M. Kurban, Phys. B, 2022, 636, 413850 CrossRef CAS.
- R. El Mouhi, O. Daoui, A. Fitri, A. T. Benjelloun, S. El Khattabi, M. Benzakour, M. Mcharfi and M. Kurban, New J. Chem., 2023, 47, 812–827 RSC.
- K. Harmandar, K. Granados-Tavera, M. Gezgin, M. Nebioğlu, İ. Şişman, G. Cárdenas-Jirón, D. Atilla and A. G. Gürek, New J. Chem., 2022, 46, 714–725 RSC.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- J. Tomasi, B. Mennucci and E. Cancès, THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- A. M. Abdelghany, E. M. Abdelrazek, S. I. Badr and M. A. Morsi, Mater. Des., 2016, 97, 532–543 CrossRef CAS.
- W. H. Baek, H. Yang, T. S. Yoon, C. J. Kang, H. H. Lee and Y. S. Kim, Sol. Energy Mater. Sol. Cells, 2009, 93, 1263–1267 CrossRef CAS.
- P. R. Berger and M. Kim, J. Renewable Sustainable Energy, 2018, 10, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nj00636h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |