
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Di- and tri-valent metal complexes with tris-amide-functionalised 1,4,7-triazacyclononane chelators†
Charley
O’Callaghan
 ,
Victoria K.
Greenacre
and
Gillian
Reid
*
,
Victoria K.
Greenacre
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 8th February 2025
Abstract
The reactions of a series of divalent 3d metal ions (Co, Ni, Cu, Zn) with two tris-amide functionalised tacn ligands, {PhNHC(O)CH2}3-tacn (1) and {iPrNHC(O)CH2CH2}3-tacn (2), in alcohol solution are described. The resulting complexes, [M(1)](NO3)2 and [M(2)](NO3)2 are characterised by elemental analysis, mass spectrometry, IR, UV-vis, 1H and 13C{1H} NMR spectroscopy, as appropriate, and by single crystal X-ray analysis for four representative examples. In all cases the ligands behave as hexadentate chelators to the divalent metal ion, with N3O3 donor sets through the tacn N-donor atoms and the O-bound carboxamide pendant arms. However, the reaction of 1 with Co(NO3)2·6H2O produces the Co(III) complex, [Co(1-H)](NO3)2, via air oxidation. The X-ray crystal structure of this complex confirms a distorted octahedral N4O2 coordination environment at cobalt(III) through the three facial tacn amine groups, the anionic N atom from one deprotonated amide group, and two O-bound carboxamides. In comparison, the coordination of 1 towards the trivalent group 13 nitrates, M(NO3)·9H2O (M = Ga and In) at room temperature in MeOH yields the distorted octahedral [M(1)](NO3)3 salts initially (from NMR and IR spectroscopy and elemental analysis data) as colourless solids. However, they are less stable than the divalent complexes, undergoing slow amide hydrolysis in MeOH at room temperature over several hours, or more rapidly with heating. This process occurs more readily with Ga(III) than with the less Lewis acidic In(III) analogue. The crystal structure of one hydrolysis product, [Ga(3)](NO3), is also reported (3 = {PhNHC(O)CH2-tacn-(CH2CO2)2}2−), in which two amide arms from 1 are converted to carboxylates.
Introduction
The tacn (1,4,7-triazacyclononane) macrocycle has been used widely in coordination chemistry over the past decades due to the suitability of the nine-membered ring for facial coordination to a wide range of metal ions in different oxidation states and from across the periodic table.1 A further attraction is its ability to accommodate various pendant functional groups via reaction at the secondary amines, leading to ligands that can serve as potent metal chelators. For example, N-functionalisation has been used to introduce neutral donor groups, as well as groups such as carboxylic acids and phosphinic acids,2 that readily deprotonate, providing anionic donor groups. Overall, this provides an exceptional degree of control of the ligand denticity and donor environment, leading to highly tuneable metal binding characteristics. Consequently, complexes incorporating N-functionalised tacn ligands have been the focus of considerable attention for binding various metal radionuclides for imaging and therapy,3–11 to create metal complexes as ‘scaffolds’ for the incorporation of radiofluorine towards positron emission tomography (PET) imaging strategies.12 Recent work has reported combined tacn-based tracers incorporating both 177Lu and 18F towards theragnostic applications,13 as well as for novel luminescent14 and magnetic resonance imaging15 probes.We have reported the coordination of the tris-amide-functionalised tacn derivatives, 1 and 2 (L), with metal trifluoride fragments, using the molecular FeF3·3H2O, [MF3(OH2)2(dmso)], M = Al, Ga, In, precursors in alcohol to form fac-[MF3(1)] and fac-[MF3(2)] in good yield. These complexes involve distorted octahedral F3N3 coordination through the tacn N3 donor atoms only, with retention of the mutually fac fluorides and the amide pendant arms involved in H-bonding interactions with the coordinated fluorides. This was supported by their spectroscopic analysis and subsequently confirmed for [GaF3(1)] and [InF3(2)] by X-ray crystallography.16 We also demonstrated the radiofluorination of [GaF3(1)] in MeOH solution via18F/19F isotopic exchange, by the addition of [18F]F− in water. Upon heating briefly (10 min/80 °C), the target radio-product, [Ga18FF2(1)], was obtained in ∼20% radiochemical yield and shown to have promising radiochemical stability over several hours when formulated in EtOH/H2O or EtOH/phosphate buffered saline solution (Scheme 1).16
 | ||
| Scheme 1 the tris-amide-functionalised tacn-derivatives, 1 and 2, used in this work, together with the carboxylate ligand derivative, 3, formed by hydrolysis of 1 in the presence of the trivalent Ga(III) ions (see text below). | ||
Consideration of the wider literature associated with amide-substituted tacn ligands has revealed a range of possible coordination modes and behaviours, with work mainly focused on 3d metal ions.17–22 For example, Chaudhuri and co-workers have described a series of di-, tri- and tetra-valent complexes with the tacn ring bearing three –CH2C(O)NH2 or –CH2C(O)NHMe pendant arms.17 All of these show hexadentate coordination, most commonly via an N3O3 donor set, i.e. through the tacn N atoms and the O atoms from the pendant carboxamides, with geometries intermediate between octahedral and trigonal prismatic. Under pH control, deprotonation of an amide N–H was observed with Cr(III), leading to a switch from O- to N-coordination of this amide to the Cr(III) ion. They also demonstrated that with Cr(III) or upon oxidation of Fe(II) to Fe(III), hydrolysis of between one and three of the amide functions to carboxylate occurs.17
We were therefore interested to investigate how the tris-amide ligands 1 and 2 behave towards both divalent transition metal ions, as well as to compare their coordination towards M(III) (M = Ga, In) using aquo cations (via M(NO3)3·9H2O) rather than the metal trifluoride precursors used in our earlier work.16 We report here the preparation and characterisation of a series of new divalent transition metal and trivalent main group complexes incorporating the potentially hexadentate tris-amide tacn ligands, 1 and 2. The reactions of 1 with the trivalent M(NO3)3·9H2O salts, M = In, Ga, are also discussed.
Experimental
Infrared spectra were recorded as Nujol mulls between CsI plates using a PerkinElmer Spectrum 100 spectrometer over the range 4000–200 cm−1. Positive ion electrospray mass spectra (ESI+ MS) were recorded in MeOH using a Waters (Manchester, UK) Acquity TQD tandem quadrupole mass spectrometer. Samples were introduced to the mass spectrometer via an Acquity H-Class quaternary solvent manager (with TUV detector at 254 nm, sample and column manager). For diamagnetic complexes, 1H and 13C{1H} NMR spectra were recorded from CD3OD solutions (unless otherwise stated) using a Bruker AV400 spectrometer and referenced to SiMe4via the residual protio-solvent resonance (1H and 13C). Duplicate microanalyses were out-sourced to Medac Ltd. While majority of measurements are within ±0.4% of the theoretical value, in a few cases the values are slightly outside this range, reflecting the inherent variability of microanalytical measurements across different facilities.23 The complexes of ligand 2 are extremely hygroscopic, rapidly changing from free-flowing powders to sticky solids; since our microanalytical measurements are out-sourced, we were unable to obtain reliable data for a few of the complexes, although the spectroscopic and structural data are consistent with the formulations quoted. Solution UV-vis spectra were obtained in 1 cm quartz cells from a MeOH solution using a PerkinElmer Lambda 750S spectrophotometer.Ligands 1 and 2 were prepared and purified using the reported methods.16 Metal salts were obtained from Sigma and used as received.
Preparations
[Co(1-H)](NO3)2
1 (0.030 mg, 0.057 mmol) and Co(NO3)2·6H2O (0.016 g, 0.057 mmol) were dissolved in anhydrous MeOH. The pink solution was stirred at room temperature overnight. The solvent volume was then decreased in vacuo to ca. 1 mL, and Et2O was added, causing precipitation of a dark pink solid, which was filtered and dried in vacuo. Yield: 0.029 g, 72%. Analysis required for C30H35CoN8O9·3H2O·½Et2O·3H2O: C, 47.94; H, 5.78; N, 13.98%. Found: C, 47.99, H, 5.24, N, 13.50%. ESI+ MS (CH3OH): found: 293.7 (expected for [Co(1-H)]2+: m/z = 293.6). IR (Nujol, ν/cm−1): 3293w, 3272w (NH), 1626m, 1595m (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). UV-vis (MeOH):
O). UV-vis (MeOH): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 (ε/mol−1 dm3 cm−1) = 19
/cm−1 (ε/mol−1 dm3 cm−1) = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 (46). 1H NMR spectrum (d4-MeOH): shows a spectrum in the typical chemical shift range as expected, consistent with l.s. Co(III), however, the spectrum appears to contain more than one species, most likely due to partial hydrolysis of the amide groups in solution. Crystals suitable for X-ray analysis were obtained by slow evaporation from a solution in MeOH over a few days.
200 (46). 1H NMR spectrum (d4-MeOH): shows a spectrum in the typical chemical shift range as expected, consistent with l.s. Co(III), however, the spectrum appears to contain more than one species, most likely due to partial hydrolysis of the amide groups in solution. Crystals suitable for X-ray analysis were obtained by slow evaporation from a solution in MeOH over a few days.
[Ni(1)](NO3)2
A solution of Ni(NO3)2·6H2O (0.027 g, 0.095 mmol) in MeOH (5 mL) was added to a solution of 1 (0.050 g, 0.095 mmol) in MeOH (5 mL). The purple coloured solution was stirred overnight. The solvent volume was reduced in vacuo by ca. 50% and Et2O was added, causing precipitation of a purple solid, which was isolated via filtration and dried in vacuo. Yield: 0.054 g, 87%. Analysis required for C30H36N8NiO9·¼Et2O·½H2O: C, 50.74; H, 5.59; N, 14.79%. Found: C, 51.09, H, 5.34, N, 14.46%. ESI+ MS (CH3OH): found: 293.3 (calculated for [Ni(L1)]2+: m/z = 294.4). IR (Nujol, ν/cm−1): 3400 br, 3250 br (OH), 3192 w, 3130 w (NH), 1682 m (HOH), 1621 s, 1596 s (CO). UV-vis (MeOH): /cm−1 (ε/mol−1 dm3 cm−1) = 28500 (46), 17800 (30), 12500 (34), 10700 (54). Crystals suitable for X-ray diffraction were grown via the vapour diffusion of Et2O into a methanol solution containing the product.
[Cu(1)](NO3)2
A solution of Cu(NO3)2·3H2O (0.046 g, 0.190 mmol) in MeOH (5 mL) was added to a solution of 1 (0.100 g, 0.190 mmol) in MeOH (5 mL) and stirred for 3 h at room temperature. The solvent volume was reduced in vacuo and excess Et2O was added, causing precipitation of a blue solid, which was isolated via filtration and dried in vacuo. Yield: 0.101 g, 74%. Analysis required for C30H36CuN8O9·1½H2O: C, 48.48; H, 5.29; N, 15.08. Found: C, 48.88; H, 5.49; N, 14.71%. ESI+ MS (CH3OH): found: 295.9 (expected for [Cu(L1)]2+: m/z = 296.1). IR (Nujol, ν/cm−1): 3400 br, 3200 br (OH), 3143 w (NH), 1652 sh (HOH), 1621 s, 1594 s (CO). UV-vis (MeOH): /cm−1 (ε/mol−1 dm3 cm−1) = 13600 (95). Crystals suitable for X-ray diffraction were grown via the vapour diffusion of Et2O into a methanol solution containing the product.
[Zn(1)](NO3)2
A solution of Zn(NO3)2·6H2O (0.057 mg, 0.191 mmol) in MeOH (5 mL) was added to a solution of 1 (0.100 g, 0.191 mmol) in MeOH (5 mL). This colourless was stirred overnight. The solvent volume was then reduced in vacuo to approx. 2 mL, and excess Et2O was added, causing precipitation of a white solid, which was isolated via filtration and dried in vacuo. Yield: 0.096 g, 70%. Analysis required for C30H36N8O9Zn·1½H2O: C, 48.36; H, 5.28; N, 15.04. Found: C, 48.19; H, 5.54; N, 14.50%. 1H NMR (295 K, CD3OD): δ (ppm) = 7.66–7.63 (m, [6H], Ar![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) ), 7.41–7.37 (m, [6H], Ar), 7.26–7.22 (m, [3H], Ar), 4.85 (H2O), 4.04 (s, [6H], C2), 3.21–3.13 (m, [6H], tacn-C2), 3.01–2.93 (m, [6H], tacn-C2). 13C{1H} NMR (295 K, CD3OD): δ (ppm) = 173.6 (CO), 137.8 (ArC), 130.4 (ArC), 127.5 (ArC), 122.5 (ArC), 59.9 (CH2), 52.2 (tacn-CH2). ESI+ MS (CH3OH): 296.4 (expected for [Zn(L1)]2+: m/z = 297.0). IR (Nujol, ν/cm−1): 3400 br, 3200 br (OH), 3214 w, 3154 w (NH), 1695 sh (HOH), 1626 s, 1596 s (CO).
), 7.41–7.37 (m, [6H], Ar), 7.26–7.22 (m, [3H], Ar), 4.85 (H2O), 4.04 (s, [6H], C2), 3.21–3.13 (m, [6H], tacn-C2), 3.01–2.93 (m, [6H], tacn-C2). 13C{1H} NMR (295 K, CD3OD): δ (ppm) = 173.6 (CO), 137.8 (ArC), 130.4 (ArC), 127.5 (ArC), 122.5 (ArC), 59.9 (CH2), 52.2 (tacn-CH2). ESI+ MS (CH3OH): 296.4 (expected for [Zn(L1)]2+: m/z = 297.0). IR (Nujol, ν/cm−1): 3400 br, 3200 br (OH), 3214 w, 3154 w (NH), 1695 sh (HOH), 1626 s, 1596 s (CO).
[Ni(2)](NO3)2
A solution of Ni(NO3)2·6H2O [0.016 g, 0.053 mmol] in MeOH (5 mL) was added to a solution of 2 (0.025 g, 0.053 mmol) in MeOH (5 mL). This was stirred for 2 h, changing from pale green to pale purple. The volume was reduced to ∼1–2 mL in vacuo and excess Et2O was added, causing precipitation of a hygroscopic purple solid, which was isolated via filtration and dried in vacuo. Yield: 0.027 g, 81%. Analysis required for C24H48N8NiO9·2½H2O: C, 41.39; H, 7.67; N, 16.09. Found: C, 41.51; H, 7.58; N, 16.42%. ESI+ MS (CH3OH): 263.3 [expected for Ni(L2)]2+: (m/z = 263.2). UV-vis (MeOH):/cm−1 (ε/mol−1 dm3 cm−1) = 28300 (43), 17600 (22), 12700 (30), 10800 (62). IR (Nujol, ν/cm−1): 3300 v br (OH) 3100 v br (NH), 1640 s (HOH), 1610 s, 1564 s (CO). Crystals suitable for X-ray diffraction were grown via the vapour diffusion of Et2O into a methanol solution containing the product.
[Cu(2)](NO3)2
A solution of Cu(NO3)2·3H2O (0.029 g, 0.119 mmol) in MeOH (5 mL) was added to a solution of 2 (0.056 g, 0.119 mmol) in MeOH (5 mL). The blue-coloured solution was stirred overnight. The solvent volume was reduced in vacuo and excess Et2O was added, causing precipitation of a dark blue solid. The solid was isolated via filtration and dried in vacuo. Yield: 0.059 g, 75%. ESI+ MS (CH3OH): 266.0 [expected for Cu(L2)]2+: (m/z = 266.1). IR (Nujol, ν/cm−1): 3400 br, 3200 br (OH), 3102 br (NH), 1639 sh (HOH), 1609 m, 1594 m (CO). UV-vis (MeOH): /cm−1 (ε/mol−1 dm3 cm−1) = 13000 (101). Crystals suitable for X-ray diffraction were grown via the slow evaporation of a Et2O/MeOH solution.
[Zn(2)](NO3)2
A solution of Zn(NO3)2·6H2O (0.022 g, 0.075 mmol) in MeOH (5 mL) was added to a solution of L2 (0.035 g, 0.075 mmol) in MeOH (5 mL). The colourless solution was left to stir overnight. The solvent volume was reduced in vacuo and excess Et2O was added, causing precipitation of a white solid, which was isolated via filtration and dried in vacuo. Yield: 0.037 g, 76%. Analysis required for C24H48N9O9Zn.2H2O: C, 41.53, H,7.55, N, 16.14%. Found: C, 41.17, H, 7.12, N, 16.55%. 1H NMR (295 K, CD3OD): δ (ppm) = 4.85 (H2O), 4.04–3.94 (septet, 3JH–H = 6.5 Hz, [3H], iPr-C), 2.93–2.90 (br m, [6H], C2), 2.88–2.80 (br m, [8H], tacn-C2), 2.79–2.64 (br m, [4H], tacn-C2), 2.64–2.52 (br m, [6H], C2), 1.10–1.08 (d, 3JH–H = 6.6 Hz, [18H], iPr2-C3). 13C{1H} NMR (295 K, CD3OD): δ (ppm) = 176.4 (CO), 56.2 (tacn-CH2), 55.0 (CH2), 43.5 (iPr-![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) H), 31.8 (H2), 22.3 (iPr-H3). ESI+ MS (CH3OH): 266.2 (expected for [Zn(L2)]2+: m/z = 266.2). IR (Nujol, ν/cm−1): 3500 br, 3280 w (OH), 3114 br (NH), 1631 m (HOH), 1601 s, 1575 m (CO).
H), 31.8 (H2), 22.3 (iPr-H3). ESI+ MS (CH3OH): 266.2 (expected for [Zn(L2)]2+: m/z = 266.2). IR (Nujol, ν/cm−1): 3500 br, 3280 w (OH), 3114 br (NH), 1631 m (HOH), 1601 s, 1575 m (CO).
[Ga(1)](NO3)3
A solution of Ga(NO3)3·9H2O (0.024 g, 0.095 mmol) in MeOH (3 mL) was added to a solution of 1 (0.050 g, 0.095 mmol) in MeOH (3 mL). This was stirred at room temperature for ca. 12 h. The solvent volume was then reduced in vacuo, and Et2O was added causing precipitation of a white solid, which was isolated via filtration and dried in vacuo. Yield: 0.035 g, 47%. Analysis required for C30H36GaN9O12·Et2O: C, 47.57, H, 5.40, N, 14.68%. Found: C, 47.61, H, 5.41, N, 14.41%. 1H NMR (295 K, CD3OD): δ (ppm) = 7.40–7.47 (m, [6H], Ar), 7.03–6.94 (m, [9H], Ar), 4.85 (s, H2O), 3.87 (s, [6H], C2), 3.21–3.09 (br m, [12H], tacn-C2). IR (Nujol, ν/cm−1): 3400 br (OH), 3059 w (NH), 1686 sh, 1625 m, 1596 m (CO).
[In(1)](NO3)3
A solution of In(NO3)3·9H2O (0.057 g, 0.160 mmol) in MeOH (5 mL) was added to a solution of 1 (0.085 g, 0.160 mmol) in MeOH (5 mL) and the reaction was stirred at room temperature overnight, leaving a clear, colourless solution. The solvent volume was then reduced in vacuo, and Et2O was added, causing precipitation of an off-white solid. This was isolated via filtration and dried in vacuo. Yield: 0.112 g, 84%. Analysis required for C30H36InN9O12·1/3Et2O: C, 44.00, H, 4.62, N, 14.80%. Found: C, 43.69, H, 4.53, N, 14.35%. 1H NMR (295 K, CD3OD): δ (ppm) = 7.67–7.46 (m, [7H], Ar), 7.36–7.25 (m, [6H], Ar), 7.16–7.06 (m, [1H], Ar), 4.85 (H2O), 4.22 (br s, [6H], C2), 3.44–3.35 (br m, [6H], tacn-C2), 3.25–3.06 (br m, [6H], tacn-C2). IR (Nujol, ν/cm−1): 3450 br (OH), 3206 w, 3151 w (NH), (HOH), 1622 s, 1594 s, 1574 s (CO).
X-ray crystallography
For several of the complexes, crystals suitable for single crystal X-ray analysis were obtained as described in the Experimental section. Data collections used a Rigaku UG2 goniometer equipped with a Rigaku HyPix-6000HE hybrid pixel detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with HF Varimax optics (100 μm focus) with the crystal held at 100 K, or a Rigaku UG2 goniometer equipped with a Rigaku Hypix 6000 HE detector mounted at the window of an FR-E+SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with (Arc)Sec VHF Varimax confocal mirrors (70 μm focus), with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(T/S/L)97, SHELX2013, SHELX-2014/7 or olex2.refine via Olex224 or NoSpherA225 (for [Ni(1)](NO3)2·1½CH3OH, [Cu(2))](NO3)2 and [Ga(3)]((NO3)·1½H2O). Structure solution and refinement was mostly routine except for [Ni(1)](NO3)2·1½CH3OH where some disorder was evident in one pendant arm and was modelled using split site occupancies, and [Co(1-H)](NO3)2·2MeOH, [Cu(1)](NO3)2·1¼Et2O·MeOH and [Ni(2)](NO3)2·0.249H2O where there was some disorder in a solvent or a nitrate anion, which were modelled accordingly. For [Ni(2)](NO3)2, a solvent mask was applied to account for residual electron density, corresponding to 0.249 H2O per formula unit. Similarly, a solvent mask was used to account for the solvent in [Ga(3)](NO3)·1½H2O, [Ni(1)](NO3)2·1½MeOH and [Cu(1)](NO3)2·1¼Et2O·MeOH (for one MeOH and 0.25 Et2O; the fully occupied Et2O was identified in the difference map and refined normally). Further details are provided in the relevant cif files. The crystallographic parameters are given in Table S1 (ESI†).Results and discussion
Reactions of 1 and 2 with divalent 3d metal ions
The divalent transition metal complexes of 1 and 2, [M(L)](NO3)2, were prepared by the direct addition of the relevant metal nitrate precursor, M(NO3)2·xH2O, M = Ni, Cu, Zn, to the ligand in methanol (Scheme 2). The solutions were stirred at room temperature overnight, and the complexes were subsequently isolated as powdered solids in good yield. For M = Co, the reaction with ligand 1 formed the Co(III) complex, [Co(1-H)](NO3)2, shown in Scheme 2via air-oxidation – as discussed below. | ||
| Scheme 2 Synthesis of the complexes formed from reaction of 1 or 2 with the M(NO3)2·xH2O precursors (M = Co, Ni, Cu, Zn). | ||
Characterisation of the new complexes used elemental analysis, IR, UV-vis spectroscopy, ESI+ mass spectrometry, 1H and 13C{1H} NMR spectroscopy (for the Zn(II) species), as appropriate, and single crystal X-ray structure determinations for representative examples. The complexes show a strong tendency to incorporate H-bonded solvent (mainly MeOH and/or H2O), which was also confirmed from both the crystallographic analyses and the IR spectra; this hampered efforts to obtain satisfactory elemental analyses for some of the complexes, especially those with ligand 2. The spectroscopic and structural data for these complexes are consistent with the expected hexadentate coordination of 1 and 2 giving distorted octahedral Ni(II), Cu(II) and Zn(II) complexes, and the metrics and UV-visible spectroscopic parameters are in very good agreement with that for related amide-functionalised tacn complexes in the literature which contain the same donor set.17 In the case of [Co(1-H)](NO3)2, the presence of a low spin d6 Co(III) is also supported by the observation of a 1H NMR spectrum in the typical chemical shift range, although, in addition to [Co(1-H)]2+, further hydrolysis of the amide groups occurs readily in solution, hence it is likely that the spectrum contains a mixture of products.
To confirm the coordination environments and investigate both the influence of the metal dn configurations and the effect of the different amide linking groups from the tacn N-donor atoms, X-ray crystallographic studies were undertaken on five examples, [Co(1-H)](NO3)2 (Fig. 1) and [M(L)](NO3)2 (M = Ni, Cu; L = 1 and 2), which are shown in Fig. 2 and 3. Key bond distances and angles involving the tacn ring and the five- or six-membered chelate rings involving the pendant amides formed by 1 and 2, respectively, are presented in Table 1.
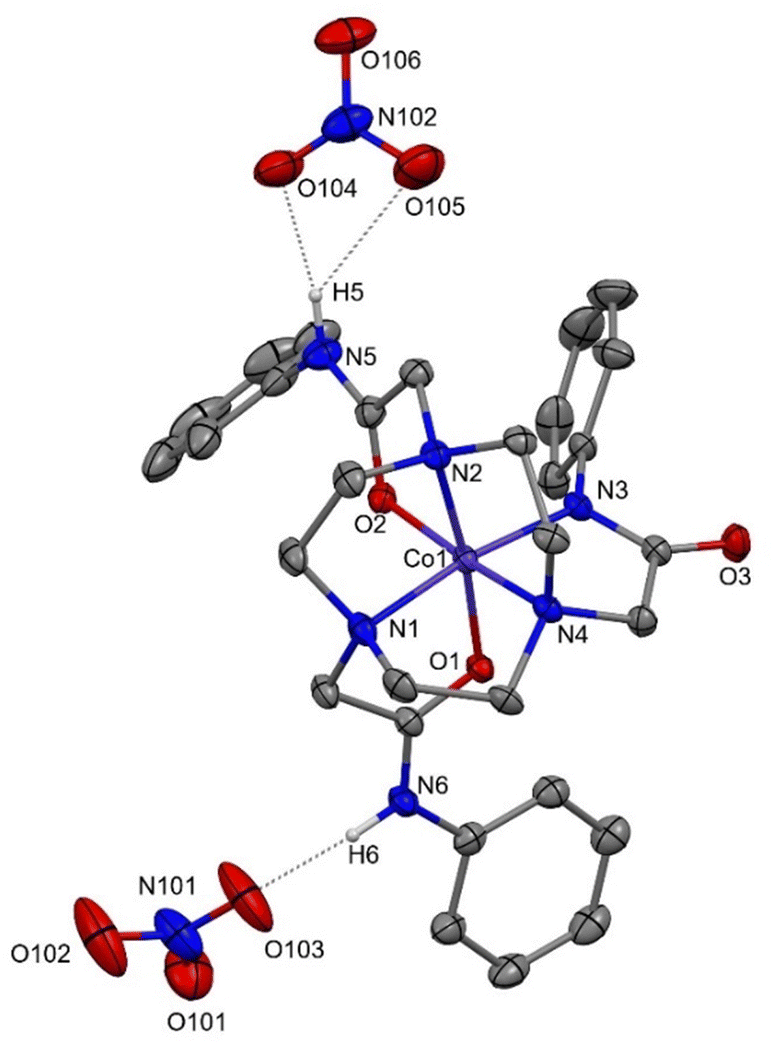 | ||
| Fig. 1 View of the structure of [Co(1-H)](NO3)2·2MeOH showing the atom numbering scheme and the H-bonding interactions between the nitrate anions and the amide N5 and N6 atoms of the carboxamide bound amide arms. Ellipsoids are shown at 50% probability, lattice MeOH and H atoms, except those on the amide N atoms, are omitted for clarity. H-bond distances to the nitrate anions: N5⋯O104 = 2.74, N6⋯O103′ = 2.736, O107⋯O105 = 2.764 Å. | ||
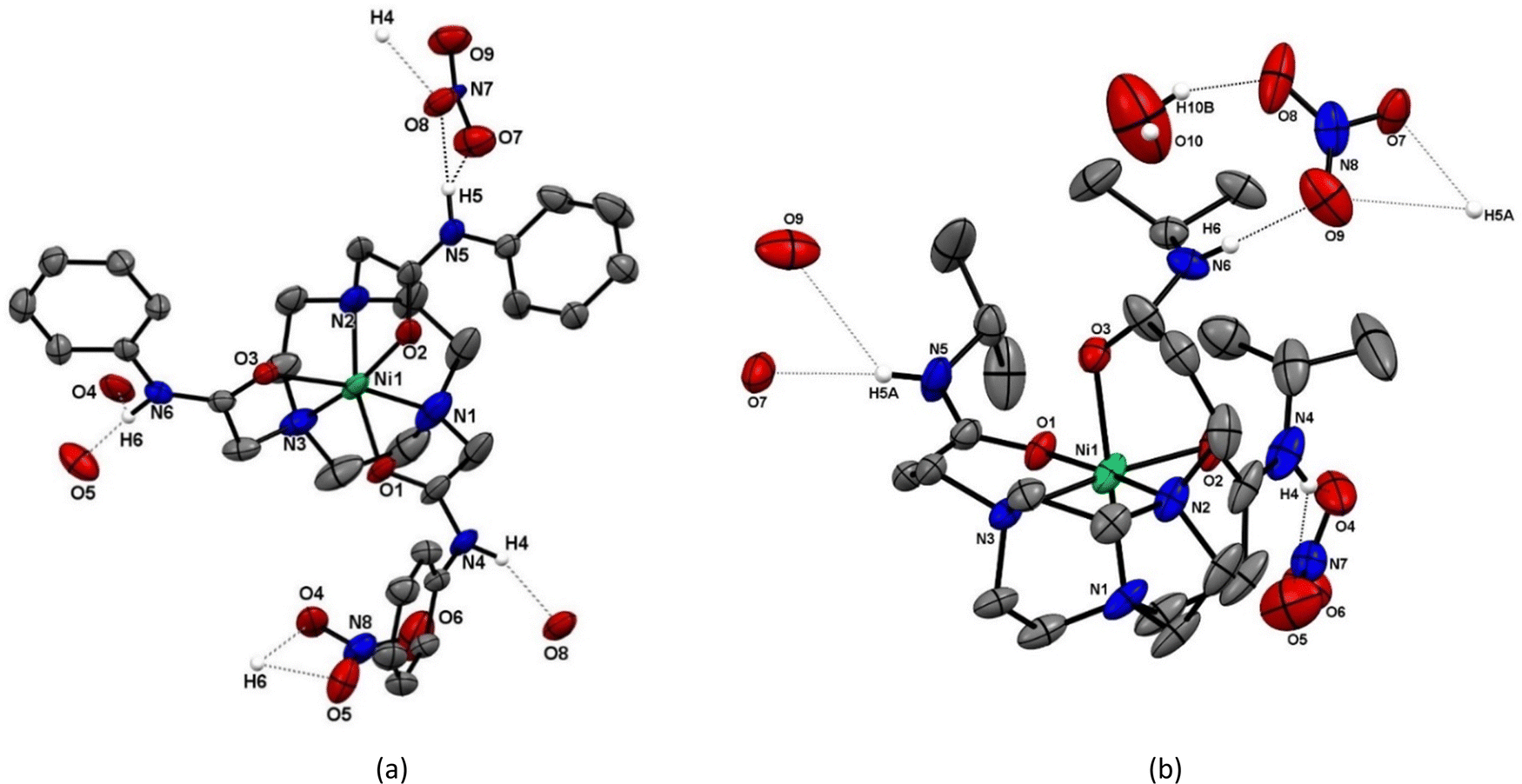 | ||
| Fig. 2 Views of the structures present in (a) [Ni(1)](NO3)2·1½CH3OH and (b) [Ni(2)](NO3)2·0.249H2O showing the atom numbering schemes and the H-bonding interactions between nitrate anions and each of the amide N–H groups (N4⋯O8′ = 2.80, N5⋯O8 = 2.79 Å in [Ni(1)](NO3)2; N6⋯O9 = 2.861(4), N6A⋯O10 = 2.836(16), O10⋯O8 = 2.879(14) Å in [Ni(2)](NO3)2). Ellipsoids are shown at 50% probability and H atoms, except those on the amide N atoms, are omitted for clarity. | ||
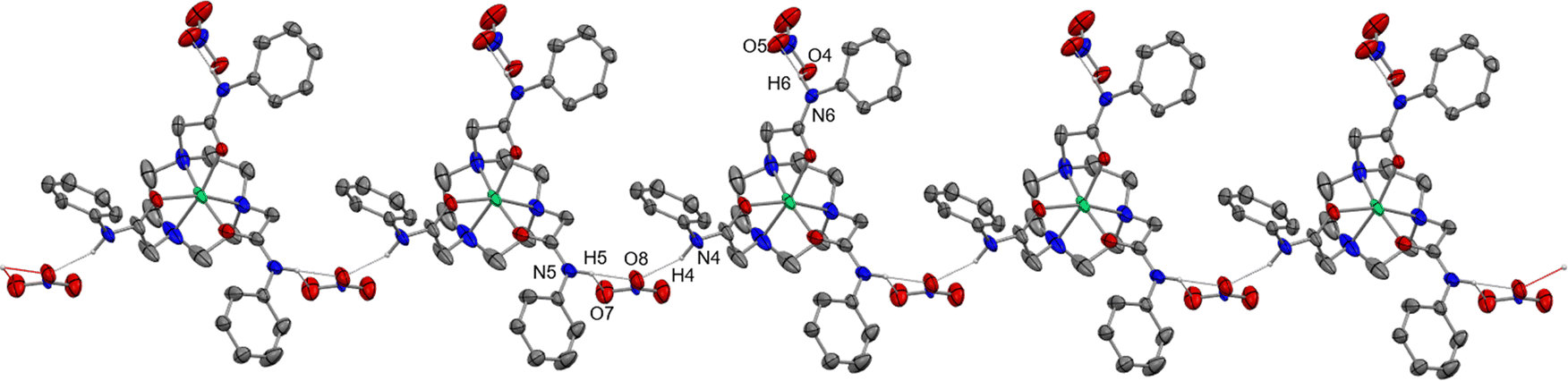 | ||
| Fig. 3 View of a portion of the 1D chain formed through the H-bonding in [Ni(1)](NO3)2. | ||
| Complex | d(M–N)/Å | d(M–O)/Å | ∠N–M–N/° | ∠N–M–O/° | Twist angle, θa/° |
|---|---|---|---|---|---|
| a The twist angle, θ, between the triangular N3 face from the coordinated tacn and the opposite triangular face containing the amide/carboxamide/carboxylate pendant donor groups; trigonal prismatic: θ = 0°; octahedral: θ = 60°.26 | |||||
| [Co(1-H)](NO3)2·2MeOH | Co–N1 = 1.948(2) | Co–O1 = 1.9222(16) | N1–Co–N2 = 88.68(8) | N2–Co–O2 = 85.12(8) | 52.26(6) |
| Co–N2 = 1.9221(19) | Co–O2 = 1.9349(17) | N1–Co–N4 = 88.46(8) | N1–Co–O1 = 85.31(8) | ||
| Co–N3 = 1.9288(19) | N2–Co–N4 = 89.45(8) | ||||
| Co–N4 = 1.928(2) | N3–Co–N4 = 84.96(8) | ||||
| [Ni(1)](NO3)2·1½MeOH | Ni–N1 = 2.079(2) | Ni–O1 = 2.0558(16) | N1–Ni–N2 = 85.50(9) | N1–Ni–O1 = 82.38(8) | 46.84(6) |
| Ni–N2 = 2.072(2) | Ni–O2 = 2.032(2) | N1–Ni–N3 = 85.79(12) | N2–Ni–O2 = 82.63(9) | ||
| Ni–N3 = 2.049(3) | Ni–O3 = 2.0805(15) | N2–Ni–N3 = 85.93(9) | N3–Ni–O3 = 81.62(8) | ||
| [Ni(2)](NO3)2·0.249H2O | Ni–N1 = 2.137(2) | Ni–O1 = 2.0653(16) | N1–Ni–N2 = 83.93(9) | N1–Ni–O2 = 91.37(9) | 56.79(6) |
| Ni–N2 = 2.142(2) | Ni–O2 = 2.078(2) | N1–Ni–N3 = 83.55(9) | N2–Ni–O3 = 90.15(8) | ||
| Ni–N3 = 2.137(2) | Ni–O3 = 2.0643(18) | N2–Ni–N3 = 83.36(8) | N3–Ni–O1 = 91.71(7) | ||
| [Cu(1)](NO3)2·1¼Et2O·MeOH | Cu1–N1 = 2.116(2) | Cu1–O1 = 2.1937(19) | N2–Cu1 N1 = 84.14(8) | N1–Cu1–O1 = 77.64(8) | |
| Cu1–N2 = 2.043(2) | Cu1–O2 = 2.0194(19) | N3–Cu1 N1 = 81.31(8) | N2–Cu1–O2 = 81.70(8) | 26.15(6) | |
| Cu1–N3 = 2.197(2) | Cu1–O3 = 2.0615(19) | N3–Cu1 N2 = 83.26(8) | N3–Cu1–O3 = 78.13(8) | ||
| Cu2–N7 = 2.090(3) | Cu2–O4 = 1.9892(18) | N7–Cu2–N8 = 82.38(9) | N8–Cu2–O4 = 81.43(8) | ||
| Cu2–N8 = 2.204(2) | Cu2–O5 = 2.0454(19) | N7–Cu2–N9 = 85.52(9) | N9–Cu2–O5 = 82.82(8) | 46.28(6) | |
| Cu2–N9 = 2.020(2) | Cu2–O6 = 2.213(2) | N8–Cu2–N9 = 85.59(8) | N7–Cu2–O6 = 79.76(8) | ||
| Cu3–N13 = 2.125(2) | Cu3–O7 = 2.0622(18) | N14–Cu3–N13 = 80.94(8) | N15–Cu3–O7 = 80.93(8) | ||
| Cu3–N14 = 2.178(2) | Cu3–O8 = 2.1632(19) | N15–Cu3–N13 = 83.02(8) | N13–Cu3–O8 = 78.45(8) | 27.15(6) | |
| Cu3–N15 = 2.076(2) | Cu3–O9 = 2.1196(19) | N15–Cu3–N14 = 82.69(8) | N14–Cu3–O9 = 77.64(8) | ||
| [Cu(2)](NO3)2 | Cu–N1 = 2.3231(8) | Cu–O1 = 1.9678(6) | N1–Cu–N2 = 81.02(3) | N1–Cu–O1 = 89.62(3) | 56.12(2) |
| Cu–N2 = 2.0845(8) | Cu–O2 = 2.3599(7) | N1–Cu–N3 = 84.59(3) | N2–Cu–O2 = 89.48(3) | ||
| Cu–N3 = 2.0184(7) | Cu–O3 = 1.9780(6) | N2–Cu–N3 = 87.07(3) | N3–Cu–O3 = 93.69(3) | ||
| [Ga(3)](NO3)·1.5H2O | Ga–N1 = 2.081(3) | Ga–O1 = 1.911(2) | N1–Ga–N2 = 85.10(11) | N1–Ga–O1 = 83.71(11) | 46.80(8) |
| Ga–N2 = 2.071(3) | Ga–O2 = 1.976(2) | N1–Ga–N3 = 84.28(11) | N2–Ga–O3 = 84.23(11) | ||
| Ga–N3 = 2.099(3) | Ga–O3 = 1.911(2) | N2–Ga–N3 = 84.59(12) | N3–Ga–O2 = 81.41(11) | ||
For [Co(1-H)](NO3)2, the donor set at the Co atom is N4O2, corresponding to three fac tacn N-donor atoms, two carboxamide O-donor atoms and one N atom from a deprotonated amide N donor atom; each of the two nitrate counter-anions are also engaged in H-bonding interactions with one of the ‘intact’, O-bonded (amide)NH functions. Comparison of the M–N(tacn) bond distances (ca. 1.93 Å for M = Co) with those in the other transition metal complexes with reported here (Table 1), and to related Co(II) and Co(III) complexes with tacn ligands, strongly supports the assignment as Co(III). For example, the Co(II) cation, [Co(tacn)2]2+, has d(Co–N ca. 2.2) Å,27 some 0.2 Å longer than d(Co–N) in Co(III) complexes with various tacn derivatives.28
The four Ni(II) and Cu(II) complexes (Fig. 2 and 3) adopt similar structures, each involving hexadentate N3O3 coordination via the tacn N-donor atoms and three pendant carboxamide O-atoms. In these cases, one nitrate anion is engaged in H-bonding interactions with each amide N–H group.
In the [Ni(1)]2+ and [Ni(2)]2+ cations (Fig. 2) the ligands are hexadentate, with twist angles of 46.84(6) and 56.79(6)°, respectively (Table 1), consistent with geometries closer to octahedral than trigonal prismatic in both cases, although as expected the shorter linker in 1 leads to a smaller twist angle. The metrics for [Ni(2)]2+ are comparable to those reported for [Ni(tcet)](ClO4)2 (tcet = 3,3′,3′′-(1,4,7-triazacyclononane-1,4,7-triyl)tripropanamide), which also forms six-membered chelate rings to the O-bound carboxamides.29 The different pendant arms in 1 and 2 give rise to differences in the typical N–Ni–N and N–Ni–O(amide) angles (Table 1). As expected, the five-membered chelate rings in [Ni(1)]2+ involving the amide O atoms result in much more acute N–Ni–O angles (ca. 82°) compared to the analogue with ligand 2 (six-membered chelate rings), with <N–Ni–O ca. 91°. The Ni–N bonds are also slightly shorter and the N–Ni–N angles slightly larger in [Ni(1)]2+ compared to [Ni(2)]2+. In both of the Ni(II) complexes each amide N–H group is H-bonded to a nitrate anion, with N(amide)⋯O(nitrate) distances ca. 2.8 Å, giving rise to infinite 1D chains (Fig. 3 and Fig. S10, ESI†).
As for Ni(II), both Cu(II) complexes (Fig. 4) involve coordination via an N3O3 donor set, although the angles subtended at the metal are quite different. The twist angles, θ, measured for the three crystallographically independent [Cu(1)]2+ cations are, Cu1 = 26.15(6), Cu2 = 46.28(6), Cu3 = 27.15(6)°. Thus, the Cu1 and Cu3 centred cations have geometries closer to trigonal prismatic, while the Cu2 centred cation the twist angle is much larger, indicating it is closer to octahedral. This can be seen in Fig. S11 (ESI†). The twist angle, θ = 56.12(2)° for the [Cu(2)]2+ cation, consistent with a distorted octahedron and in line with the larger chelate bite angle involving the amide pendant arms of 2 due to the extra CH2 more readily accommodating the octahedral arrangement cf. that in ligand 1.
 | ||
| Fig. 4 Views of the structures of (a) [Cu(1)](NO3)2·1¼Et2O·MeOH (note that there are three crystallographically independent cations and six nitrate anions in the asymmetric unit) and (b) [Cu(2)](NO3)2 showing the atom numbering schemes and the H-bonding interactions between nitrate anions and each of the amide N–H groups (for the major component: N4⋯O103 = 2.871(4), N5⋯O107 = 2.811(4), N6⋯O105 = 2.900(6), N10⋯O115 = 2.837(3), N11⋯O111′ = 2.849(3), N12⋯O117′ = 2.846(3) Å in [Cu(1)](NO3)2; N4⋯O4 = 2.8 Å in [Cu(2)](NO3)2). Ellipsoids are shown at 50% probability and H atoms, except those on the amide N atoms, are omitted for clarity. | ||
A Jahn–Teller distortion is normally expected for octahedral d9 Cu(II) complexes. The bond distances for the [Cu(2)]2+ cation indeed show that along the N1–Cu–O2 axis the Cu–N1 bond is longer than the other two Cu–N bonds by ∼0.25 Å, while the Cu–O2 distance shows an even greater lengthening, by ∼0.4 Å, compared to the other Cu–O distances, consistent with a tetragonal elongation, and the increased flexibility provided by the six-membered chelate ring involving O2. For [Cu(1)]2+ evidence for a Jahn–Teller distortion from the bond distances around the metal in the distorted trigonal prismatic Cu1 and Cu3 centred cations is, as expected, much less obvious, with very small differences between the Cu–N and Cu–O bond distances (∼0.05–0.08 Å). However, for the Cu2 centred cation, which adopts a geometry closer to octahedral, there is a more obvious tetragonal elongation along the N8–Cu2–O6 axis. The large difference in the degree of twist in the three crystallographically independent [Cu(1)]2+ cations may be a result of crystal packing and/or weak secondary bonding interactions in the lattice.
As in the Ni(II) complexes, in the Cu(II) complexes each amide N–H group is H-bonded to a nitrate anion, with N(amide)⋯O(nitrate) distances of ca. 2.8 Å. While this gives rise to a 1D chain for [Cu(2)](NO3)2 (Fig. S12, ESI†), for [Cu(1)](NO3)2 with three crystallographically independent Cu species, the Cu1-based cation there is H-bonding to nitrate, but no polymeric array, while the Cu2 and Cu3 based cations link via the nitrates to give 2D sheets, as shown in Fig. 5.
 | ||
| Fig. 5 View of a portion of the 2D sheet observed for [Cu(1)](NO3)2 with bridging NO3− anions H-bonded to the amide N–H groups and linking the crystallographically independent Cu2 (purple) and Cu3 (blue) centred cations; each of the amide N–H groups in the Cu1-centred cations (green) H-bond to one nitrate forming a discrete moiety. | ||
Reaction of 1 with M(NO3)3·9H2O (M = Ga, In)
As discussed above, the fac-MF3 units (M = Ga, Fe and, under some conditions, In) readily coordinate to 1 and 2via the tacn N3-donor set, in all cases leaving the amide arms to engage in H-bonding interactions with the fluoride ligands (and sometimes solvent).16 This contrasts with the literature data on complexes of tris-amide tacn ligands with other trivalent ions, for example, the work of Chaudhuri et al. with Cr(III)17 showing deprotonation of one amide N–H group (and observed in our work, forming the Co(III) cation, [Co(1-H)]2+, as discussed above). In some cases hydrolysis of the amide functions has also been observed with trivalent metal ions.17,30,31We therefore investigated the reactions of 1 to the more Lewis acidic M3+ ions, by reaction of M(NO3)3·9H2O (M = In, Ga) and 1 in MeOH, with gentle heating to promote coordination. For M = In, following work-up of the colourless solution, a white powdered solid was isolated in good yield. Both the elemental analysis and 1H NMR spectrum (CD3OD) were consistent with the formulation [In(1)](NO3)3, with hexadentate coordination of 1. 1H NMR studies also showed that prolonged heating in MeOH (overnight) leads to some decomposition of the complex.
The corresponding reaction with a 1:1 Ga(NO3)3·9H2O:1 ratio, and heating in MeOH led to some white solid forming after ca. 1 h, and a pink solution, suggesting significant decomposition. The solid was very poorly soluble in common solvents, however, the 1H NMR spectrum of the solid isolated from the mother liquor showed multiple resonances, suggesting solvolysis of the amide groups. The reaction was therefore repeated at room temperature and the progress monitored by 1H NMR spectroscopy over a 24 h period. After ca. 2–3 h, the NMR spectrum of the colourless solution indicates one major species, consistent with the target [Ga(1)](NO3)3. Addition of Et2O to a solution produced in this way gave a white solid, and microanalytical data support this formulation. However, extending the reaction time beyond ca. 5 h leads to slow emergence of additional resonances (Fig. S5c, ESI†) resembling those formed when the reaction was heated. Crystals suitable for single crystal X-ray analysis were grown via slow evaporation from a methanol solution of the product over several weeks and was found to be [Ga(3)](NO3) with the hydrolysis of two amide arms. The structure (Fig. 6) reveals a distorted octahedral coordination environment at Ga(III) via the three tacn N-donor atoms, one pendant O-bonded carboxamide pendant arm and two carboxylate groups. The twist angle (θ) in [Ga(3)](NO3)·MeOH is 46.80(8)° and the nitrate anion is H-bonded to the amide N–H group.
 | ||
| Fig. 6 View of the structure of complex present in [Ga(3)](NO3)·1½H2O with the atom numbering scheme (together with a diagram of the cation showing the coordination environment) and showing the H-bonding between the amide NH group and the nitrate counter-anion (HN⋯ONO2 = 3.073(8), 2.918(7) Å). Ellipsoids are drawn at the 50% probability level and H atoms (except the amide NH) and the lattice water are omitted for clarity. | ||
Hydrolysis at the amide functions of 1 on reaction with Ga(NO3)3·9H2O contrasts with the behaviour discussed above for the [MF3(1)] complexes.16 However, as discussed earlier, amide pendant groups can undergo hydrolysis in solution depending upon the reaction conditions, particularly in the presence of Lewis acidic metal ions, as in the case of [Ga(3)](NO3) here. The coordination environment is similar to that used by Shetty et al. for 68Ga radiolabelling, with the mono-amide NOTA ligands containing either a methylacetamide or benzylacetamide function.32 For the complex with the latter, coordination by the amide O-donor atom was observed at low pH, while (deprotonated) N-coordination occurs at higher pH. Very recently, Boros and co-workers31 have also exploited the Lewis acid promoted hydrolysis of the amide pendant function in gallium(III) complexes with ligands closely related to 3. This can be used to activate the release of metal pro-drugs for (radio)pharmaceutical applications.
Conclusions
A series of complexes of the tris-amide tacn ligands 1 and 2 with di- and tri-valent metal ions have been prepared. All of the complexes adopt distorted trigonal prismatic or octahedral coordination via an N3O3 donor set, which is confirmed by spectroscopic analysis and X-ray crystal structure determinations for four examples. Reaction of Co(NO3)2·6H2O with 1 in MeOH led to air oxidation, producing the Co(III) complex, [Co(1-H)](NO3)2, containing an N4O2 donor set, with N-coordination via-one deprotonated amide function. Secondary H-bonding from the amide N–H groups to nitrate anions is evident in all of the structures, leading to extended 1- and 2D networks. The complexes of ligand 1 have slightly smaller twist angles, indicative of a larger trigonal prismatic distortion, compared to those with the more flexible ligand 2, which approximate to distorted octahedral geometries. It is also notable that the twist angles for the complexes of 1 reported here (with terminal Ph substituents) are much larger than for the more sterically compact tacn-{CH2C(O)NH2}3 (Ph vs. H terminal substituents), e.g., [M(tacn-{CH2C(O)NH2}3)](NO3)2, M = Fe: θ = 18.9°; M = Co: θ = 18.6°.32The trivalent complexes, [M(1)](NO3)3 (M = Ga, In) undergo slow hydrolysis either with heating or dissolution in MeOH over an extended period, and confirmed by a structure determination of the six-coordinate [Ga(3)](NO3) where two amide arms have been converted to carboxylates. This process is slower for the In(III) analogue, consistent with its lower Lewis acidity. These results contrast with the trivalent [MF3(1)] (M = Al, Ga, Fe) complexes,16 which involve only κ3-coordination from 1 (through the tacn ring), for which no evidence for hydrolysis was observed.
Author contributions
Project conceptualisation and funding (GR), complex synthesis and characterisation (CO’C, VKG), data analysis, manuscript preparation and reviewing (all authors).Data availability
The spectroscopic data for all of the new ligands and complexes are presented in the ESI† for this manuscript, along with the radiolabelling data. The cif files and checkcifs are available via the CCDC with reference numbers 2405331–2405336.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank the EPSRC for support through the Mithras Programme grant (EP/SO32789/1) and through DTP grant number EP/T517859/1 (CO’C).References
- (a) P. Chaudhuri and K. Wieghardt, Prog. Inorg. Chem., 1986, 35, 329 CrossRef; (b) E. Macedi, A. Bencini, C. Caltagirone and V. Lippolis, Coord. Chem. Rev., 2020, 407, 213151 CrossRef CAS.
- (a) K. Wieghardt, U. Bossek, P. Chaudhuri, W. Herrmann, B. C. Menke and J. Weiss, Inorg. Chem., 1982, 21, 4308 CrossRef CAS; (b) J. Notni, K. Pohle and H.-J. Wester, EJNMMI Res., 2012, 2, 28 CrossRef PubMed; (c) E. Cole, D. Parker, G. Ferguson, J. F. Gallacher and B. Kaitner, J. Chem. Soc., Chem. Commun., 1991, 1473 RSC; (d) E. Cole, R. C. B. Copley, J. A. K. Howard, D. Parker, G. Ferguson, J. F. Gallagher, B. Kaitner, A. Harrison and L. Royle, J. Chem. Soc., Dalton Trans., 1994, 1619 RSC; (e) C. J. Broan, E. Cole, K. J. Jankowski, D. Parker, K. Pulukkody, B. A. Boyce, N. R. A. Beeley, K. Millar and A. T. Millican, Synthesis, 1992, 63 CrossRef CAS.
- (a) J. N. Whetter, D. Śmiłowicz and E. Borosa, Acc. Chem. Res., 2024, 57, 933 CrossRef CAS PubMed; (b) T. Joshi, M. Kubeil, A. Nsubuga, G. Singh, G. Gasser and H. Stephan, ChemPlusChem, 2018, 83, 554 CrossRef CAS PubMed; (c) J. Notni, J. Šimeček, P. Hermann and H.-J. Wester, Chem. – Eur. J., 2011, 17, 14718 CrossRef CAS PubMed; (d) J. Notni, K. Pohle and H.-J. Wester, EJNMMI Res., 2012, 2, 28 CrossRef PubMed.
- (a) P. Laverman, W. McBride, R. Sharkey, A. Eek, L. Joosten, W. Oyen, D. Goldenberg and O. Boerman, J. Nucl. Med., 2010, 51, 454 CrossRef CAS PubMed; (b) W. McBride, C. D’Souza, R. Sharkey, H. Karacay, E. Rossi, C. Chang and D. Goldenberg, Bioconjugate Chem., 2010, 21, 1331 CrossRef CAS PubMed; (c) W. McBride, R. Sharkey, H. Karacay, C. D’Souza, E. Rossi, P. Laverman, C. Chang, O. Boerman and D. Goldenberg, J. Nucl. Med., 2009, 50, 991 CrossRef CAS PubMed; (d) W. McBride, C. D’Souza, H. Karacay, R. Sharkey and D. Goldenberg, Bioconjugate Chem., 2012, 23, 538 CrossRef CAS PubMed; (e) D. Shetty, S. Y. Choi, J. M. Jeong, J. Y. Lee, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J.-K. Chung, M. C. Lee and Y. K. Chung, Chem. Commun., 2011, 47, 9732 RSC.
- W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu and G. Reid, Dalton Trans., 2017, 46, 14519 Search PubMed.
- (a) R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Sci., 2014, 5, 381 RSC; (b) F. M. Monzittu, I. Khan, W. Levason, S. K. Luthra, G. McRobbie and G. Reid, Angew. Chem., Int. Ed., 2018, 57, 6658 CrossRef CAS PubMed.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson and G. Reid, Chem. – Eur. J., 2015, 21, 4688 CrossRef CAS PubMed.
- D. E. Runacres, V. K. Greenacre, J. M. Dyke, J. Grigg, G. Herbert, W. Levason, G. McRobbie and G. Reid, Inorg. Chem., 2024, 62, 20844 CrossRef PubMed.
- P. J. Blower, W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu, T. Mules, G. Reid and M. N. Subhan, Dalton Trans., 2019, 48, 6767 RSC.
- M. S. Woodward, D. E. Runacres, J. Grigg, I. Khan, W. Levason, G. McRobbie and G. Reid, Pure Appl. Chem., 2024, 96, 57 CrossRef CAS.
- J. N. Whetter, B. A. Vaughn, A. J. Koller and E. Boros, Angew. Chem., Int. Ed., 2022, 61, e202114203 CrossRef CAS PubMed.
- (a) S. J. Archibald and L. Allott, EJNMMI Radiopharm. Chem., 2021, 6, 30 CrossRef PubMed; (b) S. Schmitt and E. Moreau, Coord. Chem. Rev., 2023, 480, 215028 CrossRef CAS; (c) W. Levason, F. M. Monzittu and G. Reid, Coord. Chem. Rev., 2019, 391, 90 CrossRef CAS.
- C. A. A. Kelderman, O. M. Glaser, J. N. Whetter, E. Aluicio-Sarduy, J. C. Mixdorf, K. M. Sanders, I. A. Guzei, T. E. Barnhart, J. W. Engle and E. Boros, Chem. Sci., 2024, 15, 17927 RSC.
- (a) A. Garau, A. Bencini, A. J. Blake, C. Caltagirone, L. Conti, F. Isaia, V. Lippolis, R. Montis, P. Mariani and M. A. Scorciapino, Dalton Trans., 2019, 48, 4949 RSC; (b) A. Garau, G. Picci, A. Bencini, C. Caltagirone, L. Conti, V. Lippolis, P. Paoli, G. M. Romano, P. Rossi and M. A. Scorciapino, Dalton Trans., 2022, 51, 8733 RSC; (c) M. Mameli, M. C. Aragoni, M. Arca, M. Atzori, A. Bencini, C. Bazzicalupi, A. J. Blake, C. Caltagirone, F. A. Devillanova, A. Garau, M. B. Hursthouse, F. Isaia, V. Lippolis and B. Valtancoli, Inorg. Chem., 2009, 48, 9236 CrossRef CAS PubMed.
- (a) F. Koucký, T. Dobrovolná, J. Kotek, I. Císařová, J. Havlíčková, A. Liška, V. Kubíček and P. Hermann, Dalton Trans., 2024, 53, 9267 RSC; (b) S. J. Dorazio, A. O. Olatunde, P. B. Tsitovich and J. R. Morrow, J. Biol. Inorg. Chem., 2014, 19, 191 CrossRef CAS PubMed; (c) S. M. Abozeid, E. M. Snyder, T. Y. Tittiris, C. M. Steuerwald, A. Y. Nazarenko and J. R. Morrow, Inorg. Chem., 2018, 57, 2085 Search PubMed; (d) S. M. Abozeid, E. M. Snyder, A. P. Lopez, C. M. Steuerwald, E. Sylvester, K. M. Ibrahim, R. R. Zaky, H. M. Abou-El-Nadar and J. R. Morrow, Eur. J. Inorg. Chem., 2018, 1902 CrossRef CAS; (e) A. O. Olatunde, C. J. Bond, S. J. Dorazio, J. M. Cox, J. B. Benedict, M. D. Daddario, J. A. Spernyak and J. R. Morrow, Chem. – Eur. J., 2015, 21, 18290 CrossRef CAS PubMed.
- C. O’Callaghan, V. K. Greenacre, R. P. King, J. Grigg, J. M. Herniman, G. McRobbie and G. Reid, Dalton Trans., 2024, 53, 14897 RSC.
- T. Weyhermuller, K. Wieghardt and P. Chaudhuri, J. Chem. Soc., Dalton Trans., 1998, 3805 RSC.
- J. Huskens and A. D. Sherry, J. Chem. Soc., Dalton Trans., 1998, 177 RSC.
- A. J. Blake, I. A. Fallis, A. Heppeler, S. Parsons, S. A. Ross and M. Schröder, J. Chem. Soc., Dalton Trans., 1996, 31 RSC.
- Z. Zhang, Y. He, Q. Zhao, W. Xu, Y.-Z. Li and Z.-L. Wang, Inorg. Chem. Commun., 2006, 9, 269 CrossRef CAS.
- S. Amin, C. Marks, L. M. Toomey, M. R. Churchill and J. R. Morrow, Inorg. Chim. Acta, 1996, 246, 99 CrossRef CAS.
- P. B. Tsitovich and J. R. Morrow, Inorg. Chim. Acta, 2012, 393, 3 CrossRef CAS.
- R. E. H. Kuveke, L. Barwise, Y. van Ingen, K. Vashisth, N. Roberts, S. S. Chitnis, J. L. Dutton, C. D. Martin and R. L. Melen, ACS Cent. Sci., 2022, 8, 855–863 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. L. How, J. A. L. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- (a) F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkotter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675 RSC; (b) L. Midgely, L. J. Bourhis, O. V. Dolomanov, S. Grabowsky, F. Kleemiss, H. Pushmann and N. Peyerimhoff, Acta Crystallogr., 2021, A77, 519 Search PubMed.
- A. A. Pinkerton in The Rare Earths in Modern Science and Technology, ed. G. J. McCarthy, J. J. Rhyne, and H. B. Silber, Springer, Boston, MA DOI:10.1007/978-1-4613-3054-7_26.
- H.-J. Küppers, A. Neves, C. Pomp, D. Ventur, K. Wieghardt, B. Nuber and J. Weiss, Inorg. Chem., 1986, 25, 2400 CrossRef.
- (a) H. Koyama and T. Yoshino, Bull. Chem. Soc. Jpn., 1972, 45, 481 CrossRef CAS; (b) G. H. Searle, D.-N. Wang, S. Larsen and E. Larsen, Acta Chem. Scand., 1992, 46, 38 CrossRef CAS.
- R. Liu, Y. Z. Li, Z. Zhang and Z. L. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1064 CrossRef CAS.
- D. Śmiłowicz, S. Eisenberg, R. LaForest, J. Whetter, A. Hariharan, J. Bordenca, C. J. Johnson and E. Boros, J. Am. Chem. Soc., 2023, 145, 16261 CrossRef PubMed.
- D. Shetty, S. Y. Choi, J. M. Jeong, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J.-K. Chung, M. C. Lee and Y. K. Chung, Eur. J. Inorg. Chem., 2010, 5432 CrossRef CAS.
- P. B. Tsitovich, J. M. Cox, J. B. Benedict and J. R. Morrow, Inorg. Chem., 2016, 55, 700 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectroscopic data associated with the complexes described in this work (Fig. S1–S12) and the table of X-ray parameters (Table S1). CCDC 2405331–2405336. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj05327c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |