
A highly selective mercury ion electrochemical detection based on the enhancement of oxidase-like activity by mercury on electrodeposited palladium nanoparticles@reduced graphene oxide
Zhiguang
Liu
 a,
Miaomiao
Li
b,
Xiaofang
Zheng
b,
Xiaolin
Jia
b and
Yujing
Guo
*a
a,
Miaomiao
Li
b,
Xiaofang
Zheng
b,
Xiaolin
Jia
b and
Yujing
Guo
*a
aInstitute of Environmental Science, Shanxi University, Taiyuan, 030006, China. E-mail: guoyj@sxu.edu.cn
bCollege of Chemistry and Chemical Engineering, Shanxi University, Taiyuan, 030006, China
First published on 27th November 2024
Abstract
As the toxic mercury ions (Hg2+) widely present in rivers and soil threaten human health, it is essential to develop various methods to detect and monitor Hg2+. At present, increasing numbers of nanozymes with peroxidase-like or oxidase-like activity have been exploited to develop the colorimetric detection of Hg2+. However, research on the electrochemical detection of Hg2+ by nanozymes is still rarely reported. Herein, on the basis of our previous research on palladium-based nanozymes with oxidase-like activity, graphene and palladium nanoparticles (PdNPs) were electrochemically deposited on the electrode surface. Then, the enhancement effect of palladium–mercury binding on the oxidase-like activity of electrodeposited PdNPs was studied for the first time. Moreover, it was found that only Hg2+ can enhance the catalytic oxidation of TMB compared to other common metal ions. Based on these properties, a highly selective, convenient and eco-friendly electrochemical Hg2+ sensor has been successfully developed, which has a wide linear range of 1.0–40 μM and a low LOD of 0.33 μM. Additionally, the proposed method shows acceptable recovery in real sample tests, indicating promising prospects in the field test and water pollution monitoring of Hg2+.
1. Introduction
Heavy metal ions are common environmental pollutants, which mainly originate from industrial and agricultural production processes, such as mining, industrial chemicals, fertilizers and pesticides.1 Heavy metal ions present in soil and rivers can enter the human body through the food chain and cause harm to human health.2 Mercury ion (Hg2+) is one of the most poisonous heavy metal ions. Even trace amounts of mercury ions can cause severe damage to the human body. Particularly, it may get converted into the extremely toxic methylmercury in the environment, which results in serious harm to the human brain, kidney, nervous system and endocrine system, and may even result in death.3,4 Therefore, the sensitive and selective detection of Hg2+ is quite essential for protecting human health.In the past few decades, a number of common detection techniques for Hg2+ have been continuously applied, including atomic absorption/emission spectrometry (AAS/AES),5,6 inductively coupled plasma-mass spectrometry (ICP-MS),7 UV-visible spectrophotometry (UV-Vis),8 atomic fluorescence spectrometry (AFS)9 and surface enhanced Raman scattering (SERS).10 Although these technologies are accurate and sensitive for the detection of Hg2+, they require bulky instruments, complex operating procedures and sample pretreatment processes that cannot meet the needs of on-site detection and instant recognition of Hg2+ leakage. Therefore, an electrochemical detection method, which is sensitive and simple in operation and has an easy to develop portable equipment for field detection, has attracted enormous attention as an alternative solution.
In recent years, with the rapid progress of nanoscience, various nanomaterials have been explored for electrode surface modification to realize the comprehensive improvement of electrochemical detection performance.11–14 Benefiting from their outstanding electrocatalysis and large specific surface area, hundreds of nanomaterials, such as gold nanoparticles (AuNPs),15 Ag@Au core–shell nanoparticles (Ag@Au CSNPs),16 platinum nanotube arrays (PtNAs),17 carbon nanotubes (CNTs),18 graphene,19 β-cyclodextrin-modified Pd nanoparticles (β-CY-PdNPs),20 GO-ZnO-CdS,21 and ZnS quantum dots (ZnS QDs),22 have been successfully applied on the surface of the electrode to optimize the electrochemical performance for Hg2+ detection. A universal conclusion has been reached that nanomaterials significantly improve the sensitivity of Hg2+ detection. However, the specific recognition and highly selective detection of Hg2+ still remains a substantial challenge in electrochemical sensors. Typically, enzymes and DNA are commonly selected as recognition elements to construct Hg2+ sensors, such as a catalase that is inhibited by Hg2+,23 and more specifically, a thymine-rich single-stranded DNA that forms T–Hg2+–T complexes easily.24 Most natural enzymes and DNA have inherent drawbacks of a complex electrode modification process and instability in an acid–base environment. Hence, there is a growing demand to find stable nanomaterials for the highly selective detection of Hg2+.
Nanozymes are expected to replace natural enzymes in the fields of chemistry,25 medicine26 and biosensors27 due to their good catalytic activity, high stability against denaturation, and benefits of large-scale production. Increasing numbers of nanozymes with peroxidase or oxidase activity, including AuNPs,28 platinum nanoparticles (PtNPs),29 palladium nanosheets (PdNSs),30 metal oxides,31 metal organic frameworks (MOFs),32 covalent organic frameworks (COFs),33 and molybdenum disulfide nanocomposites34 have been extensively used for the highly specific colorimetric sensing of Hg2+. Most of these approaches exploit the enhancement effect of Hg2+ on the catalytic activities of nanozymes. In particular, the peroxidase-like activity of palladium-based nanozymes can be specifically stimulated by Hg2+.30,35 Moreover, our previous research found that the oxidase activity of Pd-MOF can also be specifically improved by Hg2+.32 However, there are few reports so far on the electrochemical detection of Hg2+ using this principle.
In the present work, a highly selective Hg2+ electrochemical sensor based on the enhancement of oxidase-like activity by mercury on electrodeposited palladium nanoparticles and reduced graphene oxide (ePdNPs@rGO) was successfully constructed. As electrodeposition is a simple, fast, and green approach,36 rGO was firstly electrodeposited onto the glassy carbon electrode (GCE) as an exceptional supporting material to afford remarkable electrical conductivity and a large specific surface area.37–39 PdNPs were subsequently electrodeposited onto the surface of graphene to obtain an ePdNPs@rGO-modified electrode (ePdNPs@rGO/GCE) that is able to electrocatalyze the oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB). With the addition of Hg2+, Pd amalgam nanoparticles were formed, resulting in further improvement of the oxidase-like activity and enhancement of the oxidation peak current of TMB (Scheme 1). The results show that the as-constructed electrochemical sensor exhibited outstanding selectivity and good sensitivity for Hg2+, which could be attributed to the specific interaction between palladium and mercury, as well as the uniform and dense electrodeposition of PdNPs on the graphene surface.
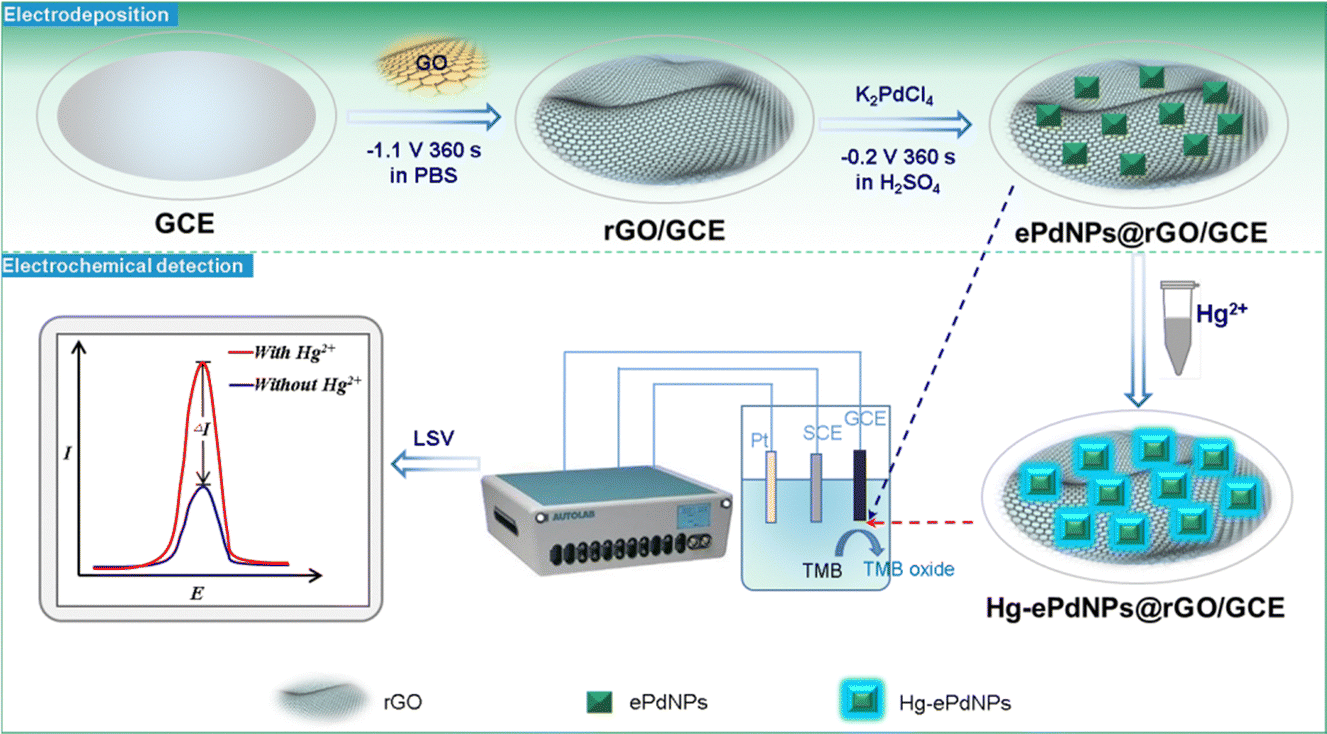 | ||
| Scheme 1 Schematic for the electrodeposition of ePdNPs@rGO and electrochemical detection for Hg2+ based on the Pd–Hg binding improved oxidase-like activity of PdNPs. | ||
2. Methods
2.1. Materials
Monolayer graphene oxide (GO, O wt% > 48%) powder was purchased from Suiheng Graphene Technology Co., LTD (Shenzhen, China). Potassium palladium(II) chloride (K2PdCl4) was obtained from Aladdin-reagent (Shanghai, China). TMB was obtained from Sigma-Aldrich (St. Louis, MO, USA). Mercuric chloride was obtained from Beijing Chemical Works. The electrolyte solutions used in the experiment include 0.5 M H2SO4 solution, 5 mM [Fe(CN)6]−3/−4 solution containing 0.10 M KCl, 0.10 M phosphate buffered saline (PBS, pH = 7.4) and 0.20 M acetic acid–sodium acetate (HAc–NaAc) buffer solution with different pH values. All chemicals and reagents were of analytical grade. Millipore Milli-Q ultrapure water (18.2 MΩ cm) was used to prepare all of the solutions.2.2. Apparatus
X-ray photoelectron spectroscopy (XPS) testing was carried out on an ESCALABM-KII 250 photoelectron spectrometer (VG Co.). Scanning electron microscopy (SEM) images were attained with a TESCAN MIRA. The ePdNPs@rGO was deposited on a fluorine-doped tin oxide (FTO) substrate, and X-ray diffractometer (XRD) patterns were recorded with Rigaku (SmartLab SE, Japan) at a scan rate of 5° per min with a scanning range of 20°–80° using Cu-Kα radiation (λ = 0.154 nm, 40 kV, 40 mA). Fourier transform infrared (FT-IR) spectra were recorded on a FT-IR-Tensor 27 spectrometer (Bruker, Germany). All the electrochemical experiments were performed on an electrochemical workstation (Autolab PGSTAT302N, Metrohm Co.) with a traditional three-electrode system with the GCE (3.0 mm in diameter) as the working electrode, Pt foil as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode.2.3. Electrodeposition of graphene
GO powder was dispersed in water with ultrasonication for 40 minutes to obtain 2 mg mL−1 of a brown GO aqueous suspension, which was diluted with PBS (0.10 M, pH = 7.4) to form 1 mg mL−1 of the GO suspension in PBS for electrodeposition. Prior to any deposition, the GCE was carefully polished with 0.3 μm and 0.05 μm thin alumina slurry in turn and placed into the [Fe(CN)6]−3/−4 solution to carry out cyclic voltammetry (CV) scans at 20 mV s−1 to determine the state of the electrode surface. Then, 6 mL of the above GO suspension in PBS was added into an electrolytic cell with a volume of 10 mL, and the polished GCE was immersed. Subsequently, a CV scan was carried out with a single cycle (100 mV s−1) from −0.8 V to 0.1 V, and then a constant negative potential was applied for a certain time with magnetic stirring. In this way, the electrochemically reduced graphene oxide-modified GCE (rGO/GCE) was obtained.2.4. Electrodeposition of palladium nanoparticles
First, 4.95 mL of 0.5 M H2SO4 solution and 50 μL of 10 mg mL−1 of K2PdCl4 solution were mixed in an electrolytic cell with a volume of 10 mL to obtain 0.1 mg mL−1 K2PdCl4 electrolyte for the electrodeposition of PdNPs. Then, the prepared rGO/GCE was immersed in the K2PdCl4 electrolyte, and a constant negative potential was applied with magnetic stirring for a certain time. Palladium ions were reduced and thus deposited on the surface of rGO/GCE to obtain the ePdNPs@rGO/GCE.2.5. Electrochemical determination of Hg2+
After the above deposition, the ePdNPs@rGO/GCE was transferred into 4.8 mL of HAc–NaAc buffer solution (0.20 M, pH = 4.0) and characterized by CV from −0.5 V to 0.7 V at 20 mV s−1. Then, 12 μL of fresh TMB solution (40 mM) was added into the HAc–NaAc buffer solution (the final concentration of TMB is 0.1 mM), and incubated for a few min with stirring. Afterwards, linear sweep voltammetry (LSV) from 0.35 V to 0.75 V (100 mV s−1) was performed, and the value of the peak current was recorded as I0. Then, a certain concentration of Hg2+ solution was added, and continued to stir for enrichment of Hg2+ for a certain time. The same LSV was then swept again, recording the peak current value as I.3. Results and discussion
3.1. Morphological characterization of the ePdNPs@rGO/GCE
The morphology of the ePdNPs@rGO/GCE was investigated via in situ SEM. Fig. 1a shows a large number of graphene lamellar structures with typical fold and wrinkled characteristics, which indicates the successful electrodeposition of graphene on the surface of GCE. As shown in Fig. 1b, the graphene surface is completely covered with PdNPs, especially at the edges and wrinkled areas. Fig. 1c shows the magnified image of the morphology, which demonstrates that the PdNPs are very evenly and densely distributed in a flat area of the graphene surface with an average size of about 20 nm. At the edges and ridges, PdNPs are more numerous and larger in size, which is evaluated to be around 40 to 50 nm. This could be ascribed to the electrodeposited graphene that has outstanding electric conductivity, in addition to the strong anchoring effect between PdNPs and the graphene surface, which prevents agglomeration of the nanoparticles.36,40 This structure is conducive to the enrichment of Hg2+ and the enhancement of the catalytic performance. Therefore, a successful electrodeposition of graphene and PdNPs on the surface GCE was verified by the SEM images.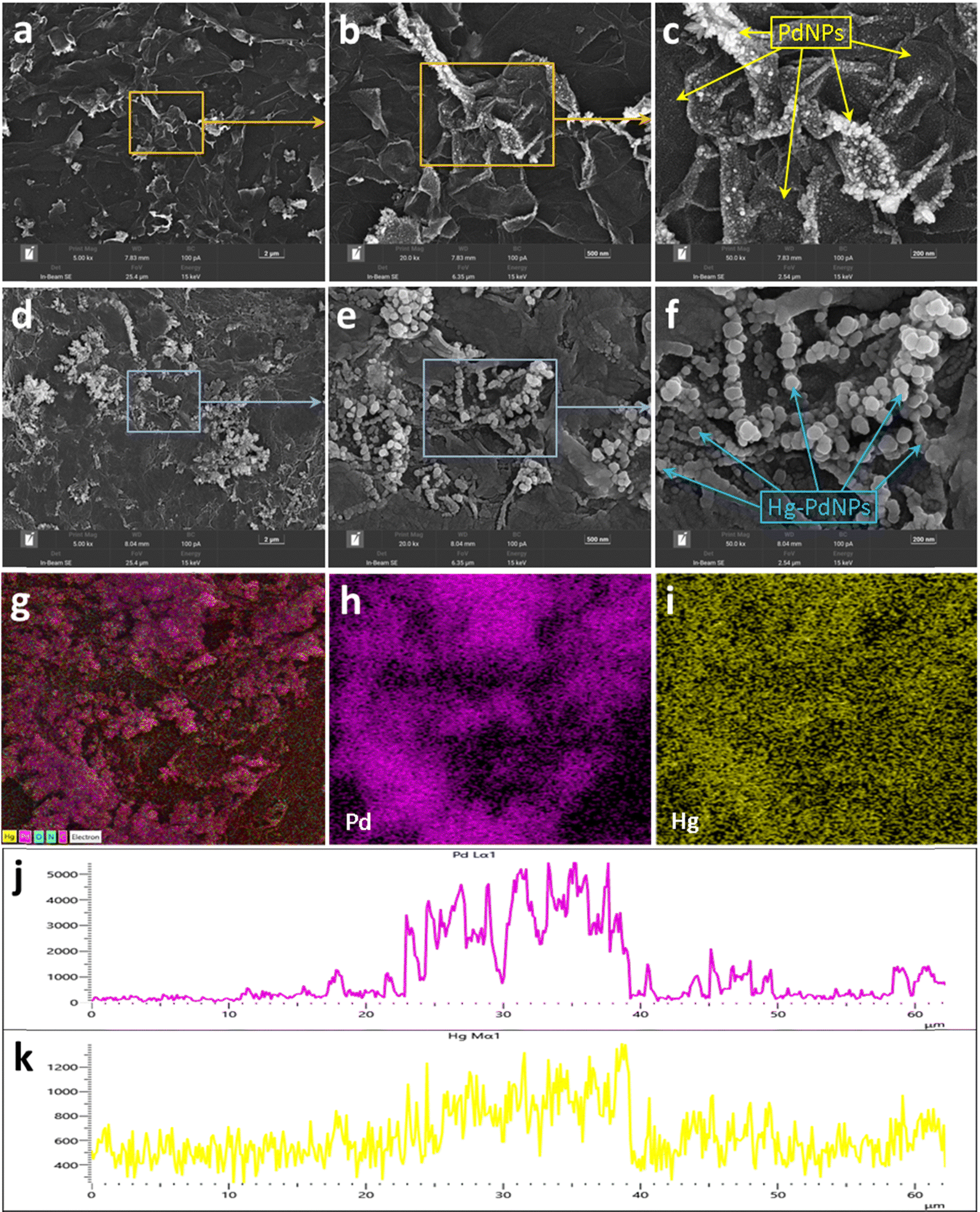 | ||
| Fig. 1 SEM images of ePdNPs@rGO/GCE before (a)–(c) and after (d)–(f) the addition of Hg2+ with different resolutions. EDS images of ePdNPs@rGO/GCE with mercury, including map (g)–(i) and line (j) and (k) scanning. | ||
Subsequently, SEM analysis of the ePdNPs@rGO/GCE with mercury was performed to further reveal the morphologic changes after the addition of Hg2+. As shown in Fig. 1d–f, the surface of the ePdNPs@rGO/GCE appears to be covered by a film. The densely grown PdNPs on the flat areas of graphene seem to be wholly covered by a “quilt”, while the PdNPs on the edges and ridges are also coated with a layer of film, resulting in an apparent shape change from the original angular form into a sphere, and a size increase from 40–50 nm to about 70–80 nm in diameter. This morphology may be due to the mercury film being reduced from Hg2+, covered or encased on the surface of PdNPs, and finally forming a palladium amalgam nanostructure, which possesses higher catalytic activity to TMB than that of the ePdNPs@rGO. Additionally, energy dispersive spectroscopy (EDS) was carried out. The elemental mapping (Fig. 1g–i) study shows that a large amount of palladium and mercury are fully dispersed on the graphene surface, and the position of mercury and palladium is relatively coincident (as shown in Fig. 1j and k), which also suggests the formation of a palladium amalgam.
XPS was carried out to investigate the elemental composition, and confirm the valence states of palladium and mercury on the surface of ePdNPs@rGO/GCE after the enrichment of Hg2+. Fig. 2a shows the survey XPS spectra containing C, N, O, Pd and Hg, which further verified the successful electrodeposition of the PdNPs and enrichment of Hg on the electrode surface. Fig. 2b demonstrates the peaks of Pd 3d. The peaks at 334.9 eV and 340.2 eV are attributed to Pd0 3d5/2 and Pd0 3d3/2, and another two peaks at 337.4 eV and 342.8 eV are assigned to Pd2+ 3d5/2 and Pd2+ 3d3/2, respectively. These features strongly support that most palladium exists in the electrodeposited zero-valent form, while a small amount may be oxidized in the air to exist in the divalent form.
 | ||
| Fig. 2 (a) XPS survey spectra of the ePdNPs@rGO composite. XPS spectra of Pd 3d (b) and Hg 4f (c). (d) XRD patterns of the ePdNPs@rGO composite along with the standard peaks of C (PDF#41-1487), Pd (PDF#46-1043) and SnO2 (PDF#41-1445). (e) FT-IR spectra of rGO and the ePdNPs@rGO nanocomposite. | ||
Fig. 2c exhibits the peaks of Hg 4f. The two peaks at 99.8 eV and 103.1 eV are assigned to Hg2+ 4f7/2 and Hg2+ 4f5/2, respectively, which are attributed to the adsorption of Hg2+ on the surface of the ePdNPs@rGO/GCE. Moreover, the other two peaks located at 99.1 eV and 101.7 eV are ascribed to Hg0 4f7/2 and Hg0 4f5/2, respectively, illustrating that most of the Hg2+ was reduced to zero valency by rGO and combined with palladium to form a palladium amalgam on the surface of the ePdNPs@rGO/GCE. The newly formed Pd amalgam lamella could change the structure and chemical properties of the ePdNPs@rGO/GCE surface, which improved the catalytic oxidation capacity of the system.
Fig. 2d shows the XRD patterns of the ePdNPs@rGO and the FTO substrate. Graphene presented a sharp characteristic diffraction peak at 26.5°, which corresponds to the (002) plane of multilayer graphene (PDF#41-1487). This diffraction peak was generated by the stacking of graphene sheets. The peaks at about 40.1°, 46.3° and 68.5° are assigned to the (111), (200) and (220) lattice planes of the fcc crystalline structure of PdNPs, respectively (PDF#46-1043).41 This indicates that graphene and palladium have been reduced and deposited on the electrode surface. Other characteristic diffraction peaks should be assigned to the lattice planes of the substrate SnO2, respectively (PDF#41-1445).
Fig. 2e shows the FT-IR spectra of rGO (red curve) and the ePdNPs@rGO (blue curve) nanocomposite. The absorption bands generated at about 1073 cm−1, 1631 cm−1 and 3435 cm−1 correspond to the –C–O-stretching, –C–O–H-deformation of the carboxyl groups, and –O–H stretching vibrations of the hydroxyl groups, respectively, which exist on the rGO sheets. The typical absorption band at 1387 cm−1 arises due to the vibration of the graphene sheet skeletal structure. In the spectrum of ePdNPs@rGO, the band arising at about 577 cm−1 corresponds to the vibration of –Pd–O–,42 indicating the formation of palladium oxide. Other absorption bands appearing at 1179 cm−1 and 884 cm−1 should be assigned to the formation of bonds between palladium oxides and the carboxylic group, as well as epoxy groups present on the rGO sheets. The results suggest that palladium nanoparticles have been deposited on the graphene surface, and some of them might be oxidized when they are exposed to the air.
3.2. Electrodeposition of GO
In order to ascertain the optimal potential for GO deposition on GCE, a GCE was placed into a GO suspension in PBS and a single cycle of CV was initially scanned within a potential window of +0.1 V to −1.8 V. As shown in Fig. 3a (inset), a typical irreversible reductive peak occurs at −1.1 V, indicating that GO was reduced to rGO at the GCE surface. When the potential is more negative than −1.2 V, the reduction current increases dramatically, representing the intensification of hydrogen evolution. Therefore, a series of potentials from −0.9 V to −1.2 V were used as constant potential for graphene deposition, and the deposited electrodes were each characterized with CV in [Fe(CN)6]−3/−4 solution. As shown in Fig. 3a, when the applied deposition potential was changed from −0.9 V to −1.1 V, the redox peak current of [Fe(CN)6]−3/−4 gradually increased. This suggested that a more negative potential was beneficial to the reduction and deposition of GO, resulting in more rGO with higher conductivity being deposited on the GCE surface. However, when the deposition potential reached −1.2 V, the peak current of [Fe(CN)6]−3/−4 decreased sharply, indicating that the amount of deposited graphene suddenly decreased significantly. This can be associated with bubbles formed by large amounts of hydrogen evolution that even strip away the rGO that has already been deposited. At this time, the broken rGO film on the electrode surface can be observed with the naked eye, which further confirms the damage of a large amount of hydrogen evolution to the deposited rGO film. Consequently, the optimal potential for GO electrodeposition was found to be −1.1 V.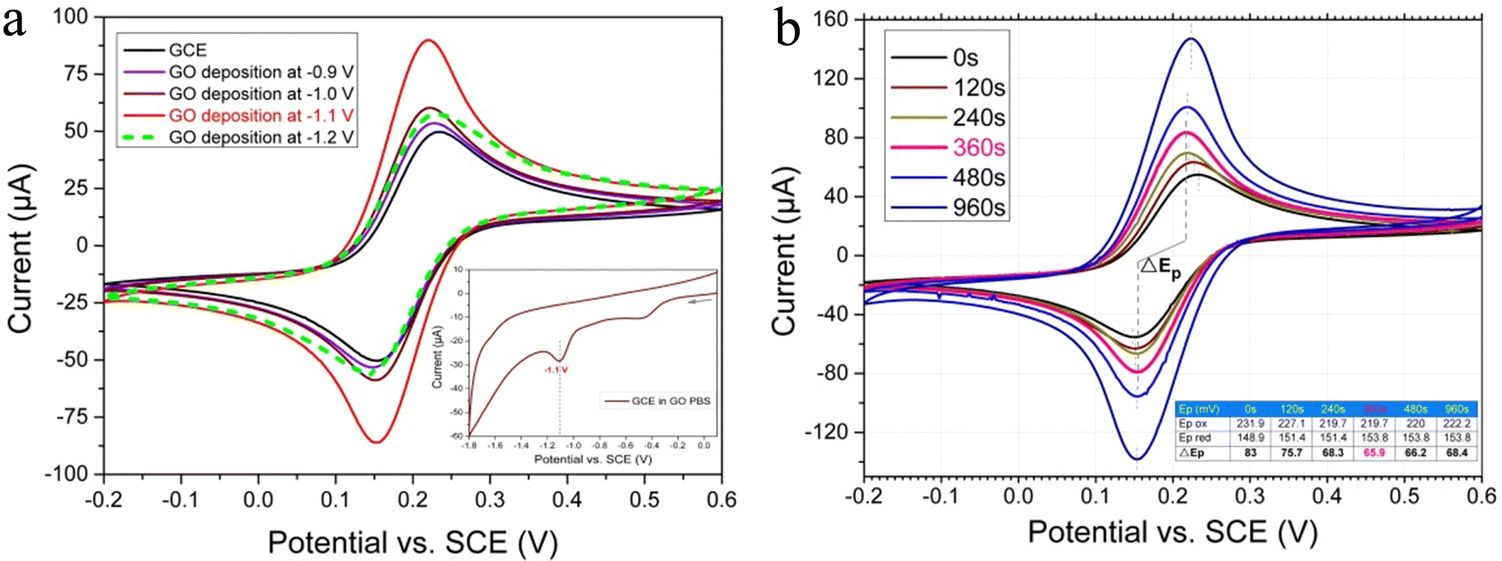 | ||
| Fig. 3 CV characterization of rGO/GCE in [Fe(CN)6]−3/−4 solution after deposition of GO at different potentials (a) and different times (b). | ||
Deposition time, another important factor affecting GO deposition, was examined in detail. A series of time periods from 0 s to 960 s were tested for GO deposition under potential of −1.1 V. Likewise, the deposited electrodes were characterized with CV in [Fe(CN)6]−3/−4 solution, respectively. As shown in Fig. 3b, with the gradual extension of the deposition time, the redox peak current of [Fe(CN)6]−3/−4 continues to increase, suggesting that increasingly more rGO was deposited. Nevertheless, by comparing the redox peak potential difference (ΔEp) of [Fe(CN)6]−3/−4 (usually, a smaller ΔEp means a higher electric conductivity), it can be found that the ΔEp decreased from 83 mV of the bare GCE to a minimum of 65.9 mV after 360 s deposition. Meanwhile, it increased to 68.4 mV after 960 s deposition (as shown in Fig. 3b (inset table)). The results indicate that after 360 seconds of deposition, the electrode conductivity reaches the best value due to the outstanding electrical conductivity and suitable thickness of the deposited rGO. Meanwhile, a deposition for an extremely long time leads to a significantly thick graphene film, resulting in a decrease in conductivity. Hence, 360 s was selected as an optimal time for GO electrodeposition.
3.3. Enhancement effect of graphene on the deposition of palladium
Deposition of PdNPs was initially studied using CV in 0.5 M of H2SO4 solution with 0.1 mg mL−1 of K2PdCl4. The cyclic voltammogram shown in Fig. 4a compares the electrochemical behavior of palladium on the rGO/GCE (red solid line) and bare GCE (blue dash line). For the rGO/GCE, during the forward scan toward negative potentials, the cathodic current starts to increase gradually at about 0.38 V, suggesting the beginning of Pd nucleation on the surface of rGO. A distinct cathodic peak subsequently forms at around 0.2 V, owing to the rapid deposition of palladium on rGO surface and the resulting dramatic drop in the palladium concentration near the rGO surface, i.e., the diffusion process prevails in the system. Once the potential is scanned beyond −0.3 V, the cathodic current increased sharply, which is associated with the predictable strong hydrogen evolution catalyzed by a large amount of PdNPs deposited on the rGO surface. In contrast, palladium deposition on a clean GCE surface has a substantial decrease in the amount of PdNPs compared to that on the rGO surface, indicating the enhancement effect of rGO through its considerable specific surface area and outstanding conductivity. As a control, when the rGO/GCE was placed in H2SO4 solution without K2PdCl4 for the CV scan (green solid line in Fig. 4a), there was no indication of any deposition or hydrogen evolution current. This proved again that the cathodic peak at 0.2 V in H2SO4 solution with K2PdCl4 should be assigned to palladium deposition. Therefore, the applied potential for PdNPs deposition was limited at −0.2 V, so as to avoid the interference of hydrogen evolution on palladium deposition.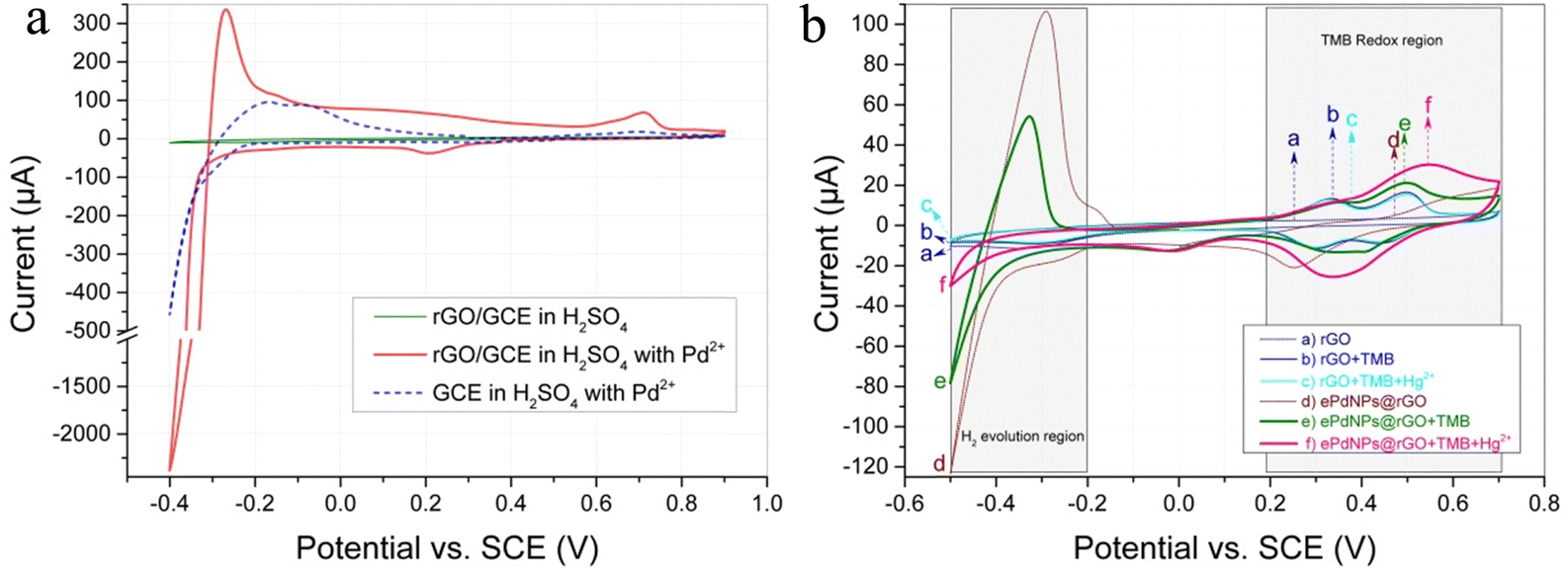 | ||
| Fig. 4 (a) CV characterization of palladium in H2SO4 solution with or without 0.1 mg mL−1 of K2PdCl4 on the rGO/GCE (red solid line) and bare GCE (blue dash line). (b) CV comparison diagram of rGO/GCE and ePdNPs@rGO/GCE before (a) and (d) and after (b) and (e) the addition of TMB with (c) and (f) or without Hg2+. | ||
3.4. Enhancement effect of mercury on the electrocatalysis for TMB
CV was performed to investigate the effect of mercury on the electrocatalysis of TMB by rGO/GCE and ePdNPs@rGO/GCE, respectively. As can be seen in Fig. 4b, the CV scan (line d) of the ePdNPs@rGO/GCE in HAc–NaAc buffer solution shows a significant current of hydrogen evolution in the H2 evolution region, compared to that of the rGO/GCE (line a). When TMB (0.1 mM) was added to the HAc–NaAc buffer solution, the CV scan (line e) of the ePdNPs@rGO/GCE shows less current of hydrogen evolution due to the TMB adsorbed on the surface PdNPs, blocking the catalytic reduction of H+. Meanwhile, the oxide peak current of TMB on the ePdNPs@rGO/GCE (line e, in the TMB redox region) is higher than that on the rGO/GCE (line b, in the TMB redox region), which obviously benefited from the higher catalytic oxidation capacity of PdNPs for TMB compared with that of rGO. Subsequently, Hg2+ was further added and enriched onto the surface of the ePdNPs@rGO/GCE or rGO/GCE by stirring. The oxide peak current of TMB on the Hg-ePdNPs@rGO/GCE (line f, in the TMB redox region) was further improved compared to that on the ePdNPs@rGO/GCE (line e). However, no change in the oxide peak current of TMB on Hg-rGO/GCE (line c) was observed compared to rGO/GCE (line b). This phenomenon is consistent with our previous research,32 and fully proves that Pd–Hg binding can efficiently enhance the oxidase-like activity of PdNPs, while the mimicked oxidase activity of rGO cannot be improved by Hg2+. Therefore, it is possible to detect the Hg2+ concentration by the enhancement of the TMB oxidation peak current after the addition of Hg2+ on the PdNPs surface.3.5. Optimization of conditions for Hg2+ detection
The amount of palladium deposition, which is mainly determined by the deposition time, is crucial to the binding of Hg2+. Thus, the deposition time of palladium was optimized. As shown in Fig. 5a, the oxidation peak current of TMB increases gradually with increasing palladium deposition time, which reached the maximum after 360 seconds of deposition. This indicates that the PdNPs on the electrode surface has the highest catalytic activity for TMB after 360 seconds of deposition. Therefore, 360 seconds was selected for palladium deposition. Furthermore, the oxidation peak current of TMB is influenced by the pH value of the HAc–NaAc buffer solution and accumulation time of TMB. Fig. 5b illustrates that TMB has the maximum oxidation peak current at pH 4.0, so pH 4.0 was selected as the best pH value for detection. Fig. 5c demonstrates that the oxidation peak current of TMB reaches equilibrium after the addition of TMB and stirring for 3 minutes. For stability, 5 minutes was taken as the enrichment time of TMB. More importantly, when Hg2+ are added to HAc–NaAc buffer solution, they need time to reach the electrode surface and bind to PdNPs. Fig. 5d shows that after 5 minutes of stirring enrichment, the peak current of TMB does not increase, suggesting that the surface of PdNPs is saturated with Hg2+. As a result, 5 minutes was chosen as the optimal enrichment time of Hg2+.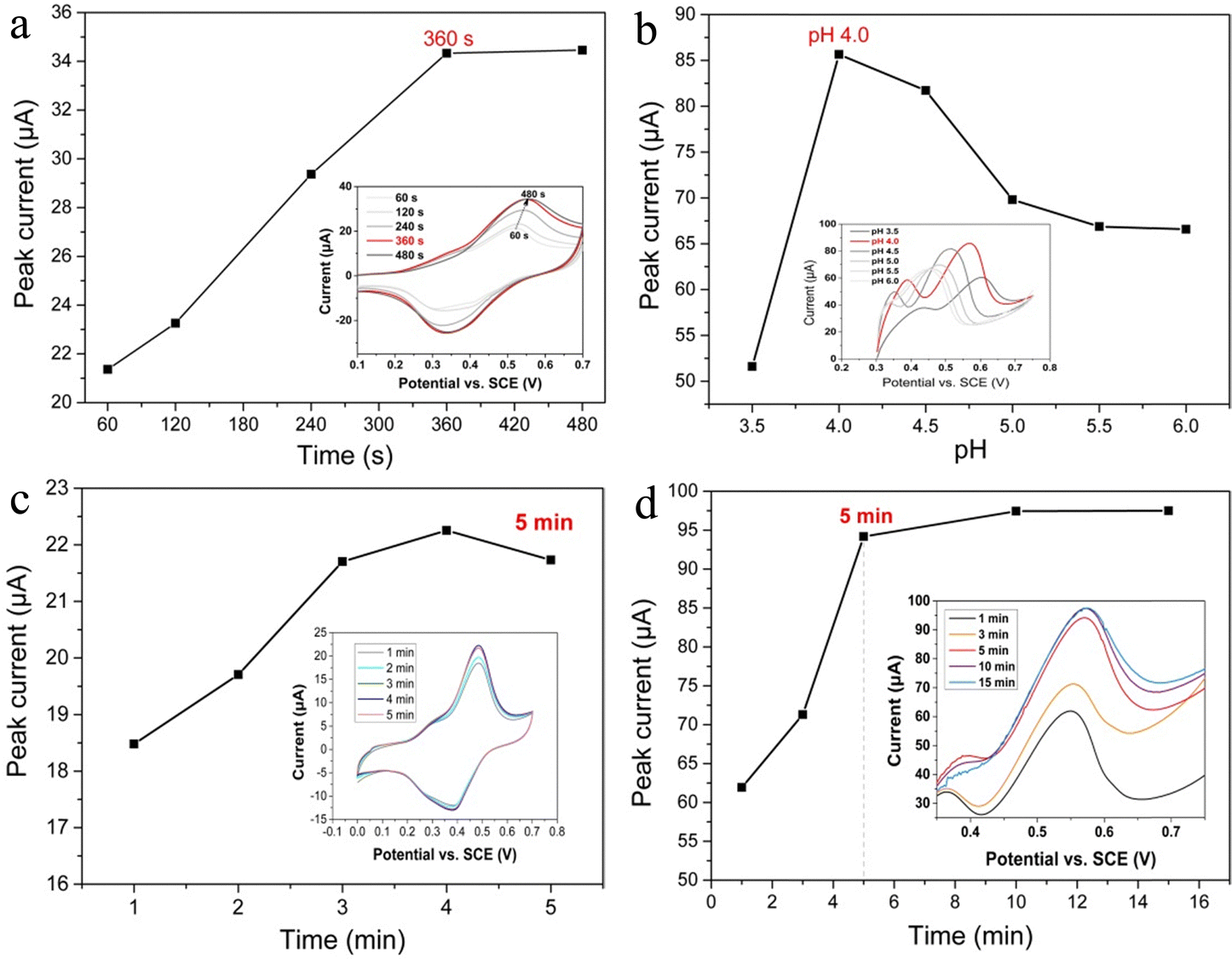 | ||
| Fig. 5 Oxidation peak current plots of TMB at different (a) Pd deposition times (inset: CV curves of TMB on ePdNPs@rGO/GCE with different deposition times of Pd), (b) pH (inset: LSV curves of TMB on ePdNPs@rGO/GCE with different pH), (c) TMB accumulation times (inset: CV curves of TMB on ePdNPs@rGO/GCE at different stirring times after the addition of TMB) and (d) Hg2+ accumulation times (inset: LSV curves of TMB on ePdNPs@rGO/GCE at different stirring times after the addition of Hg2+). | ||
3.6. Electrochemical detection of Hg2+
Based on the improved oxidase-like activity of PdNPs by Hg–Pd binding, the enhancement effect of different Hg2+concentrations on the TMB oxidation peak current on ePdNPs@rGO/GCE was studied by LSV under the optimal experimental conditions (Fig. 6a). The peak oxidation current of TMB on the ePdNPs@rGO/GCE before and after adding Hg2+ was recorded as I0 and I, respectively. The difference between I0 and I was obtained as ΔI. It was found that there is a good linear relationship between the value of ΔI and the concentration of Hg2+ in the range of 1.0–40 μM with a corresponding regression equation of ΔI = −0.0444 + 0.4769 CHg2+ (R2 = 0.9924) (Fig. 6b). The limit of detection (LOD) of Hg2+ was as low as 0.33 μM (S/N = 3). Therefore, the proposed electrochemical detection method for Hg2+ is comparable to some electrochemical sensors reported previously without using DNA and natural enzymes, as described in Table 1. The sensitivity of the method is not as good as the reported methods in the literature. However, the selectivity of the method is very high due to the enhanced catalytic activity originating from the Pd–Hg specific binding. Moreover, this method could be used for the detection of Hg2+ in water samples where the Hg2+ concentration is greater than 1 μM. | ||
| Fig. 6 (a) LSV of TMB oxidation on ePdNPs@rGO/GCE in HAc–NaAc buffer solution containing different concentrations of Hg2+ and (b) linear correlation between the difference of the peak current (ΔI) and the concentration of Hg2+. | ||
3.7. Selectivity, reproducibility and stability of Hg2+ detection
In order to investigate the selectivity of the method to Hg2+, the ePdNPs@rGO/GCE was immersed in HAc–NaAc buffer solution containing 0.1 mM of TMB, and the oxidation peak current of TMB was recorded as I0 by LSV. Then, different kinds of metal ions (chlorine saline), including Mg2+, Ca2+, Co2+, Mn2+, Ni2+, Cd2+, Pb2+, Cu2+, Fe3+ and Hg2+ (20 μM) were added, respectively. After 5 minutes of stirring, LSV was performed again to obtain their oxidation peak currents of TMB (I). The difference (ΔI) between I0 and I was attained (as shown in Fig. 7a). It is worth mentioning that all of the ΔI from the various interfering metal ions are negative, i.e., the peak currents slightly drop, which could be attributed to their inhibition effect on the oxidase-like activity of PdNPs. Conversely, the ΔI from Hg2+ increased substantially, which is obviously due to the improvement of the oxidase-like activity of PdNPs caused by the Pd–Hg specific binding. Hence, the proposed electrochemical detection has excellent selectivity for Hg2+. | ||
| Fig. 7 (a) Difference of the TMB (0.1 mM) peak current (ΔI) comparison from the various metal ions at the concentration of 20 μM. (b) Reproducibility of the sensor by detecting 10 μM of Hg2+ solution using five different ePdNPs@rGO/GCEs. (c) Stability of the ePdNPs@rGO/GCE by testing 20 μM of Hg2+ solution before and after two weeks. | ||
The reproducibility of the electrochemical sensor is an important index to evaluate its practical application value. In this work, the electrodes were modified five times respectively with ePdNPs@rGO to detect the same concentration (10 μM) of Hg2+ solution. As shown in Fig. 7b, the relative standard deviation (RSD) of the testing results after five times was 10%, suggesting that the prepared electrochemical sensor had good reproducibility.
Additionally, the stability of the ePdNPs@rGO/GCE was tested. A freshly modified GCE with ePdNPs@rGO was used to detect the Hg2+ solution (20 μM), and the current signal was recorded. An electrode modified in the same way was then encapsulated in a centrifuge tube to prevent air oxidation and stored at 4 °C. After two weeks, the Hg2+ solution (20 μM) was tested again to obtain the current signal. As shown in Fig. 7c, the electrochemical response decreased within an acceptable range, indicating that the long-term storage stability of the sensor was acceptable.
3.8. Analysis of Hg2+ in real samples
To estimate the practicability of the detecting system for actual environmental samples, river and tap water, as well as soil extract samples were used to measure the Hg2+ concentration. The river sample was collected from the Taiyuan section of Fen River, and the tap water and soil samples came from Taiyuan City. The river water sample was filtered by filter paper before measurement. The soil sample was soaked in 1% dilute nitric acid, and then filtered to obtain the sample to be tested. No Hg2+ was found in these samples after preliminary tests by this proposed electrochemical method, so the recovery rate was examined by standard addition method. Different concentrations of Hg2+ (10 and 20 μM) were added to the water samples, respectively. The measured results are presented in Table 2. The recovery ratio of Hg2+ varied from 84.9% in the soil extract to 114.0% in the tap water, indicating an acceptable response for the electrochemical sensing of Hg2+ in practical sample analysis.| Sample | Added (μM) | Found (μM) | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| Fen river | 10.0 | 9.8 | 11.2 | 97.6 |
| 20.0 | 19.4 | 7.8 | 97.1 | |
| Tap water | 10.0 | 11.4 | 8.0 | 114.0 |
| 20.0 | 22.2 | 9.5 | 111.0 | |
| Soil extract | 10.0 | 8.5 | 6.1 | 84.9 |
| 20.0 | 18.1 | 15.8 | 90.5 |
4. Conclusions
In summary, a highly selective, economical and eco-friendly electrochemical Hg2+ sensor was successfully developed based on the enhancement of the oxidase-like activity of ePdNPs@rGO by Pd–Hg binding. The outstanding advantage of the Hg2+ sensor is that it achieves excellent selectivity for Hg2+ electrochemical detection without the use of DNA and natural enzymes. Another merit of the method is its simplicity and environmental friendliness. Only common and mild reagents are used for the preparation of the ePdNPs@rGO/GCE by electrodeposition. Furthermore, the method can realize the rapid detection of Hg2+, as the entire process from the electrodeposition of the nanocomposites to the detection of the Hg2+ takes less than half an hour. In addition, the proposed Hg2+ sensor has a wide linear range of 1.0–40 μM and a low LOD of 0.33 μM. Notably, the developed electrochemical Hg2+ sensor is easier to commercialize because the strategy is cheaper and faster, and the electrode modification is more convenient. It has a widespread application prospect in the field detection of water and soil pollution. In order to further improve the sensitivity of the detection method, palladium nanomaterials with various morphologies and doped with other metals will be electrochemically synthesized on the electrode surface to modulate the activity of nanozymes, and much work toward this direction is underway in our laboratory.Author contributions
All authors contributed to the study conception and design. Zhiguang Liu: the author contributed to the construction of the electrochemical sensor, and wrote the main manuscript text and prepared Fig. 3–7 and Tables 1 and 2. Miaomiao Li: the author performed data analysis and prepared Fig. 1. Xiaofang Zheng: the author performed data analysis and prepared Fig. 2. Xiaolin Jia: the author prepared Scheme 1. Yujing Guo: the author provided conceptualization and supervision of the project. All authors have read and reviewed the final manuscript.Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research work was supported by the National Natural Science Foundation of China (No. 21775095), the Fundamental Research Program of Shanxi Province (No. 202203021211312), Shanxi Hundred Talent Program (2019) and Shanxi Scholarship Council of China (No. 2021-013).References
- L. Shi, Y. Li, Z. P. Liu, T. D. James and Y. T. Long, Talanta, 2012, 100, 401–404 CrossRef CAS PubMed.
- E. Eksin, A. Erdem, T. Fafal and B. Kivçak, Electroanal, 2019, 31, 1075–1082 CrossRef CAS.
- N. Salandari-Jolge, A. A. Ensafi and B. Rezaei, Sens. Actuators, B, 2021, 331, 129426 CrossRef CAS.
- S. Huang, W. J. Wang, F. F. Cheng, H. Q. Yao and J. J. Zhu, Sens. Actuators, B, 2017, 242, 347–354 CrossRef CAS.
- S. Jamshidi, M. K. Rofouei, S. Seidi and Å. Emmer, Sep. Sci. Technol., 2020, 55, 1505–1514 CrossRef CAS.
- Q. L. Zhou, P. R. Guo, J. C. Pan, Y. Q. Lei and N. Liu, Chin. J. Anal. Chem., 2016, 44, 1270–1276 CAS.
- C. H. Lin, C. K. Su and Y. C. Sun, Microchem. J., 2020, 154, 104569 CrossRef CAS.
- C. H. Yin, J. Iqbal, H. L. Hu, B. X. Liu, L. Zhang, B. L. Zhu and Y. P. Du, J. Hazard. Mater., 2012, 233, 207–212 CrossRef PubMed.
- D. Y. Chen, L. Lu, H. Zhang, B. Lu, J. L. Feng and D. Zeng, Anal. Sci., 2021, 37, 1235–1240 CrossRef CAS.
- Y. Shi, N. Chen, Y. Y. Su, H. Y. Wang and Y. He, Nanoscale, 2018, 10, 4010–4018 RSC.
- T. Liu, Z. Y. Chu and W. Q. Jin, J. Mater. Chem. B, 2019, 7, 3620–3632 RSC.
- N. K. Ravikumar and P. Perumal, J. Mater. Res., 2024, 39, 2744–2756 CrossRef CAS.
- N. G. Balasubramaniyan and P. Perumal, Anal. Methods, 2024, 16, 624–638 RSC.
- M. Narayanan, N. P. S. Chauhan and P. Perumal, RSC Adv., 2023, 13, 5565–5575 RSC.
- Y. Y. Zhang, G. L. Chu, Y. M. Guo, W. P. Zhao, Q. Q. Yang and X. Sun, J. Electroanal. Chem., 2018, 824, 201–206 CrossRef CAS.
- A. T. E. Vilian, A. Shahzad, J. Chung, S. R. Choe, W. S. Kim, Y. S. Huh, T. Yu and Y. K. Han, Microchim. Acta, 2017, 184, 3547–3556 CrossRef.
- J. Y. Luo, D. F. Jiang, T. Liu, J. M. Peng, Z. Y. Chu and W. Q. Jin, Biosens. Bioelectron., 2018, 104, 1–7 CrossRef CAS.
- L. Shi, Y. Wang, Z. Y. Chu, Y. Yin, D. Y. Jiang, J. Y. Luo, S. Ding and W. Q. Jin, J. Mater. Chem. B, 2017, 5, 1073–1080 RSC.
- L. Shi, Y. Wang, S. M. Ding, Z. Y. Chu, Y. Yin, D. F. Jiang, J. Y. Luo and W. Q. Jin, Biosens. Bioelectron., 2017, 89, 871–879 CrossRef CAS.
- Y. J. Hu, Z. M. Liu, H. J. Zhan and Z. Q. Shen, Anal. Methods, 2018, 10, 767–774 RSC.
- B. Zhang, H. Y. Meng, X. Wang, H. H. Chang and W. L. Wei, Sens. Actuators, B, 2017, 253, 495–501 CrossRef CAS.
- Y. Wang, P. P. Wang, Y. Wu and J. W. Di, Sens. Actuators, B, 2018, 254, 910–915 CrossRef CAS.
- B. Elsebai, M. E. Ghica, M. N. Abbas and C. M. A. Brett, J. Hazard. Mater., 2017, 340, 344–350 CrossRef CAS PubMed.
- D. M. Wang, Q. Q. Gai, R. F. Huang and X. W. Zheng, Biosens. Bioelectron., 2017, 98, 134–139 CrossRef CAS PubMed.
- Q. M. Chen, X. D. Zhang, S. Q. Li, J. K. Tan, C. J. Xu and Y. M. Huang, Chem. Eng. J., 2020, 395, 125130 CrossRef CAS.
- M. Wang, M. Y. Chang, Q. Chen, D. M. Wang, C. X. Li, Z. Y. Hou, J. Lin, D. Y. Jin and B. G. Xing, Biomaterials, 2020, 252, 120093 CrossRef CAS.
- Y. Tao, Y. H. Lin, Z. Z. Huang, J. S. Ren and X. G. Qu, Adv. Mater., 2013, 25, 2594–2599 CrossRef CAS.
- Y. W. Wang, Q. Liu, L. X. Wang, S. R. Tang, H. H. Yang and H. B. Song, Microchim. Acta, 2019, 186, 7 CrossRef PubMed.
- Y. Zhao, H. Qiang and Z. B. Chen, Microchim. Acta, 2017, 184, 107–115 CrossRef CAS.
- L. L. Kong, C. C. Wang, W. J. Yang, L. Zhou and S. H. Wei, J. Hazard. Mater., 2022, 427, 128135 CrossRef CAS.
- A. A. B. Christus, A. Ravikumar, P. Panneerselvam and K. Radhakrishnan, Appl. Surf. Sci., 2018, 449, 669–676 CrossRef CAS.
- Z. G. Liu, R. X. Niu, M. M. Li, Z. P. Li and Y. J. Guo, Microchim. Acta, 2024, 191, 352 CrossRef CAS PubMed.
- W. Li, Y. Li, H. L. Qian, X. Zhao, C. X. Yang and X. P. Yan, Talanta, 2019, 204, 224–228 CrossRef CAS PubMed.
- C. M. Ma, Y. Ma, Y. F. Sun, Y. Lu, E. L. Tian, J. F. Lan, J. L. Li, W. C. Ye and H. X. Zhang, J. Colloid Interface Sci., 2019, 537, 554–561 CrossRef CAS.
- S. T. Zhang, D. X. Zhang, X. H. Zhang, D. H. Shang, Z. H. Xue, D. L. Shan and X. Q. Lu, Anal. Chem., 2017, 89, 3538–3544 CrossRef CAS.
- C. B. Liu, K. Wang, S. L. Luo, Y. H. Tang and L. Y. Chen, Small, 2011, 7, 1203–1206 CrossRef CAS PubMed.
- J. X. Wei, Z. H. Chen and Z. F. Tong, J. Colloid Interface Sci., 2021, 596, 22–33 CrossRef CAS.
- Y. G. Wang, G. H. Zhao, G. Y. Zhang, Y. Zhang, H. Wang, W. Cao, T. D. Li and Q. Wei, Sens. Actuators, B, 2020, 319, 128313 CrossRef CAS.
- J. Jiao, M. X. Pan, X. R. Liu, J. Liu, B. S. Li and Q. Chen, Nanomaterials, 2020, 10, 499 CrossRef CAS PubMed.
- Z. Q. Yao, R. R. Yue, C. Y. Zhai, F. X. Jiang, H. W. Wang, Y. K. Du, C. Y. Wang and P. Yang, Int. J. Hydrogen Energy, 2013, 38, 6368–6376 CrossRef CAS.
- Q. Zhao, D. Bu, Z. Li, X. Zhang and L. Di, Nanomaterials, 2021, 11, 1341 CrossRef CAS PubMed.
- K. Arora, S. Srivastava, P. R. Solanki and N. K. Puri, IEEE Sens. J., 2019, 19, 8262–8271 CAS.
- K. B. R. Teodoro, F. L. Migliorini, M. H. M. Facure and D. S. Correa, Carbohydr. Polym., 2019, 207, 747–754 CrossRef CAS PubMed.
- M.-P. N. Bui, J. Brockgreitens, S. Ahmed and A. Abbas, Biosens. Bioelectron., 2016, 85, 280–286 CrossRef CAS PubMed.
- J. Cui, S. Xu and L. Wang, Sci. China Mater., 2017, 60, 352–360 CrossRef CAS.
- A. Sanchez-Calvo, M. T. Fernandez-Abedul, M. C. Blanco-Lopez and A. Costa-Garcia, Sens. Actuators, B, 2019, 290, 87–92 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |