
Non-saturated nucleic acid probes with a broad dynamic range†
Xinmiao
Kang‡
,
Yu
Liu‡
,
Dandan
Tian
,
Zuhao
Shen
,
Shihui
Wang
 * and
Xin
Su
*
* and
Xin
Su
*
State Key Laboratory of Organic-Inorganic Composites, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing Key Laboratory of Bioprocess, College of Life Science and Technology Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: xinsu@mail.buct.edu.cn; wangshihui@mail.buct.edu.cn
First published on 14th June 2025
Abstract
Molecular probes are essential for analytical detection but encounter major limitations, such as a limited dynamic range, the need for probe concentrations to exceed target concentrations, and restricted reusability. These issues stem from the irreversible binding of probes to targets, resulting in signal saturation when the target concentration surpasses that of the probe. To overcome these challenges, we developed a non-saturating nucleic acid probe (NSNAP) inspired by dynamic DNA nanotechnology and non-equilibrium chemistry. The NSNAP combines an affinity probe with an enzyme that degrades the target within the probe–target complex, allowing the probe to reset and produce continuous signals before complete target degradation. This design dramatically expands the dynamic range by up to 5000-fold, compared to the typical 81-fold range of conventional probes, and enables the NSNAP to detect targets at concentrations up to 250 times greater than that of the probe. Moreover, the NSNAP is reusable, with experimental validation demonstrating at least seven cycles of reuse, enhancing both cost-effectiveness and sustainability. Exonuclease III or λ Exonucleases can support NSNAP operation, and these enzymes can be pre-loaded with DNA probes. Using the NSNAP, we successfully quantified both viral genes (HIV, HHV, HPV) and bacterial marker genes (oprL, dnaJ, ddl) across concentrations from 1 to 1000 fM in complex biological matrices, achieving strong linear correlations. This innovative approach offers great potential for advancing DNA nanotechnology-based diagnostic tools for scientific research and clinical applications.
New conceptsUsing a non-saturated nucleic acid probe (NSNAP) is a novel strategy in molecular detection by harnessing dynamic DNA nanotechnology and non-equilibrium chemistry to overcome longstanding limitations in quantitative analysis. Traditional molecular probes suffer from signal saturation and narrow dynamic ranges, limiting their ability to accurately quantify targets across varying concentrations. The NSNAP overcomes these limitations by incorporating an enzymatic target depletion mechanism, which continuously removes target molecules and prevents signal saturation—even when target concentrations significantly exceed probe concentration levels. This design enables an ultra-wide dynamic range of up to 5000-fold and allows for the quantification of targets at concentrations up to 250 times higher than that of the probe. Furthermore, the NSNAP exhibits remarkable reusability, maintaining consistent performance over at least seven assay cycles. Collectively, these features position the NSNAP as a powerful, sustainable platform for high-precision nucleic acid quantification, offering broad applicability in clinical diagnostics, infectious disease monitoring, and molecular research. |
1. Introduction
Affinity-based molecular probes play an integral role in biomedical research and clinical diagnostics.1–3 They are capable of specifically recognizing and binding to target molecules or biological structures, thus enabling highly sensitive and specific detection.4,5 Currently, the most commonly employed affinity-based molecular probes include monoclonal antibodies, peptide probes, nucleic acid probes, small molecule probes, nanoparticle probes, and fusion protein probes. Monoclonal antibodies are ideal for various biological assays due to their high specificity and affinity.6 Peptide probes are favored for their ease of synthesis, low cost, and ease of cell penetration.7 Nucleic acid probes achieve specific recognition of target sequences through base pairing, rendering them particularly effective for detecting low-abundance nucleic acid targets.8 Small molecule probes are extensively applied in live cell imaging and drug targeting studies due to their excellent cell penetration ability and versatility.9 Nanoparticle probes are suitable for multi-target detection and imaging due to their multi-functionality and signal enhancement capabilities.10 Fusion protein probes, created by fusing probe molecules with functional proteins, provide functional diversity while maintaining high specificity and ease of production.11 These molecular probes operate through mechanisms such as specific binding, fluorescent labeling, and signal amplification, allowing for efficient detection and analysis of target molecules. As such, they play a pivotal role in advancing biomedical research and improving diagnostic capabilities.12,13Despite their strong performance in many applications, affinity-based molecular probes face significant limitations that constrain their effectiveness and broader applicability in certain fields.14,15 One major challenge lies in the concentration ratio between the probe and target molecule. As shown in Fig. 1A(i), the binding of these probes to the target molecule is typically irreversible at a macroscopic level, allowing detection only when the target molecule's concentration is lower than that of the probe. When the target molecule concentration exceeds the probe's, signal saturation occurs, leading to overlap in signal responses at high target concentrations (Fig. 1A(ii)). This saturation effect hinders quantitative analysis and results in a narrow dynamic range for conventional affinity probes (Fig. 1A(iii)), limiting their utility in analyzing samples with widely varying target concentration spans.16 Furthermore, conventional molecular probes are generally single-use probes, rendering them non-reusable after detection. This not only increases operational costs but also exacerbates environmental burdens, particularly in large-scale or high-frequency applications. Addressing these challenges is critical to expanding the versatility and sustainability of affinity-based molecular probes. To overcome these challenges, researchers have proposed a series of improvement strategies. For instance, probes with broader dynamic response ranges have been developed to enhance target detection performance.17
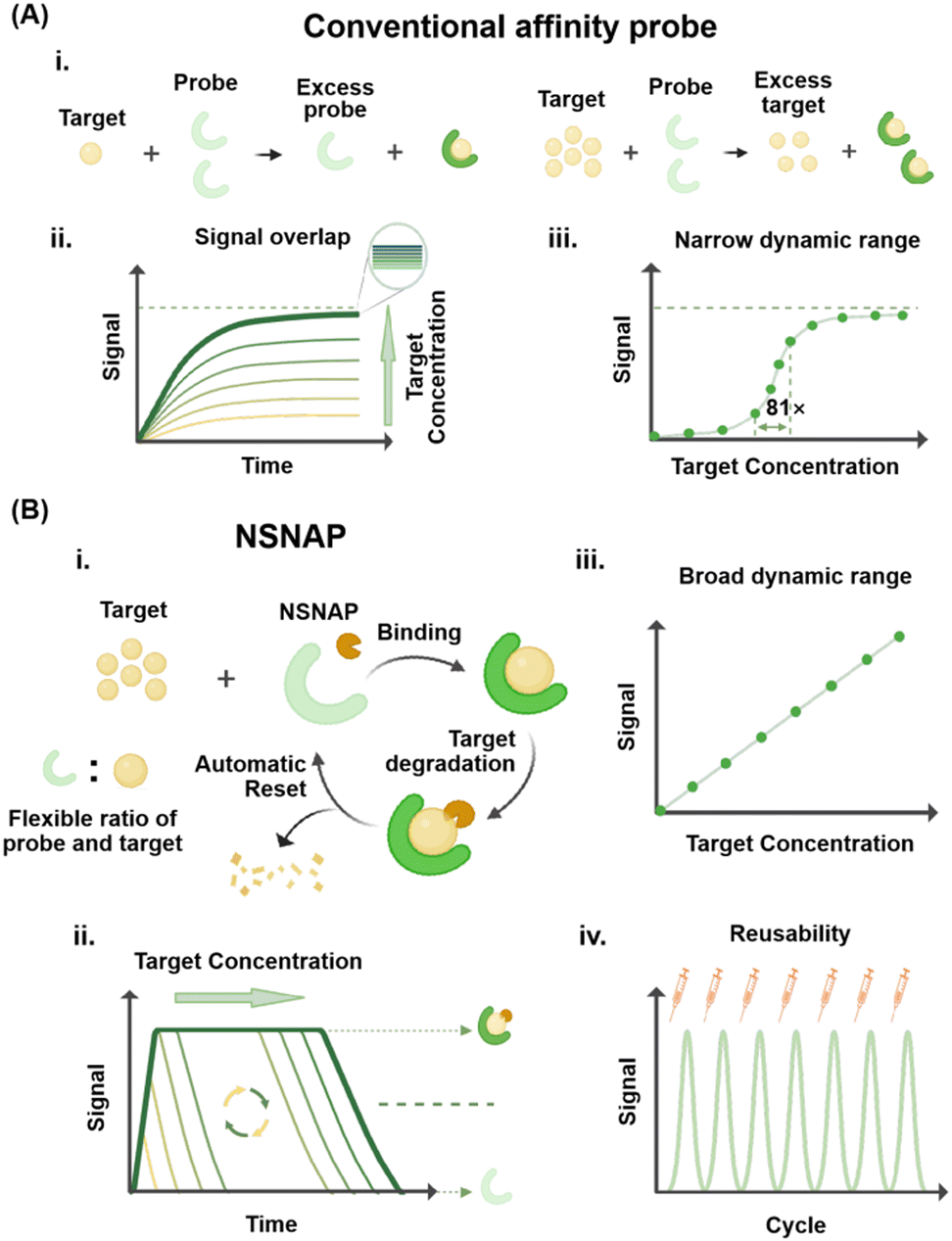 | ||
| Fig. 1 Comparison between the NSNAP and conventional affinity probes. (A) Conventional affinity probe: (i) detection principle of conventional affinity probes. (ii) Signal generated by detecting targets at different concentrations using conventional probes. (iii) Relationship between the signal and the concentration of the target. (B) NSNAP: (i) detection principle of the NSNAP. (ii) Signal generated by detecting targets at different concentrations using the NSNAP. (iii) Relationship between the signal and the concentration of the target. NSNAP exhibits a broad dynamic range. (iv) Signals generated by the repeated use of the NSNAP. | ||
The combination of multiple affinity probes with different equilibrium constants has successfully enabled ultra-wide dynamic range detection, spanning over four orders of magnitude.18,19 Similarly, the introduction of allosteric activators and inhibitors has extended the dynamic response range of probes by approximately three orders of magnitude.20,21 Despite these advances, these approaches may complicate experimental design and lead to the generation of non-specific signals.
In this study, we developed a non-saturated nucleic acid probe (NSNAP) system based on dynamic DNA nanotechnology and non-equilibrium chemistry.22,23 The NSNAP integrates affinity molecular probes with built-in enzymes that degrade target molecules, enabling versatile detection capabilities. As shown in Fig. 1B(i), the NSNAP enables the detection and quantification of targets, regardless of whether the target concentration is below or above that of the probe. This capability arises from the unique mechanism of the NSNAP, which continuously consumes target molecules, maintaining their capacity to bind free targets. This addresses the limitation of conventional affinity-based molecular probes, which require the probe concentration to be higher than that of the target for effective detection.24,25Fig. 1B(ii) demonstrates that the NSNAP produces distinct signal outputs for different target concentrations, with the target concentration exhibiting a linear relationship with the signal (Fig. 1B(iii)). This linearity facilitates quantitative analysis across a broad dynamic range. The minimum detection concentration is consistent with those of conventional affinity-based molecular probes, while the maximum detection concentration significantly exceeds those of conventional affinity-based molecular probes. Moreover, the NSNAP is reusable, maintaining consistent response performance across multiple uses (Fig. 1B(iv)). We showed that both nucleic acid exonucleases, Exo III and λ Exo, can effectively support the NSNAP's operation. The NSNAP overcomes the 81-fold concentration limitation of conventional affinity probes, achieving quantitative detection across a remarkable 5000-fold concentration range. It also enables the detection of target molecules at concentrations up to 250 times higher than that of the probe. Our results further highlight the NSNAP's long-term stability and reusability, with the system demonstrating consistent performance over at least seven uses. To validate its practical applicability, the NSNAP was employed for the quantitative detection of viral genes (HIV, HHV, and HPV) and bacterial marker genes (oprL, dnaJ, and ddl) within a concentration range of 1–1000 fM. The detection results showed a strong linear relationship between the gene concentration and the signal output, with R2 values exceeding 0.90 in all cases. This study underscores the NSNAP's potential to address longstanding challenges in molecular detection, offering a powerful alternative to conventional affinity-based probes. Its wide dynamic range, high reusability, and robust performance open new avenues for advanced molecular detection technologies.
2. Results and discussion
2.1. The non-saturated nucleic acid probe based on exonuclease III (NSNAP-III)
Fluorescent DNA probes are currently extensively employed for the quantitative detection of target molecules. These probes generally consist of two labelled DNA strands: one conjugated with a fluorescent dye and the other with a fluorescence quencher. Their mechanism relies on the hybridization of the target molecule with the probe, which disrupts the interaction between the fluorescent and quencher-labelled strands, thereby releasing a measurable fluorescent signal. Quantification of the target is achieved by correlating the intensity of the emitted fluorescence with the target concentration (Fig. 2A). Despite their utility, conventional fluorescent DNA probes face significant limitations. One major drawback is their susceptibility to saturation, which restricts their effective dynamic range. These probes can only quantify target concentrations within a limited range. Once the target concentration exceeds that of the probe, the fluorescence intensity plateaus and no longer reflects changes in the target concentration (Fig. 2B). | ||
| Fig. 2 Non-saturated nucleic acid probe based on Exo III (NSNAP-III). (A) Schematic of the conventional affinity probe operation. (B) Fluorescence kinetic curves of the conventional affinity probe (500 nM) detecting targets at different concentrations (10 nM–10 μM). (C) Fluorescence intensity of the conventional affinity probe versus target concentration under experimental conditions. (D) Schematic of NSNAP-III operation. The method for calculating fluorescence kinetic curves is detailed in the Note (ESI†). (E) Fluorescence kinetic curves of NSNAP-III (200 nM nucleic acid probe) detecting different target concentrations (10 nM–50 μM). (F) Linear relationship between the area under the fluorescence kinetic curve and target concentration for NSNAP-III detection, with R2 = 0.99. (G) Normalized fluorescence signals generated by 1 μM target detected by NSNAP-III with nucleic acid probes of different concentrations (100–500 nM). (H) Fluorescence signals from NSNAP-III after 7 repetitions, with the corresponding area under the curve at each time point (200 nM nucleic acid probe, 1 μM target). The Exo III concentration used in all experiments was 250 U mL−1, with NEBuffer 1 at 37 °C. Data are means ± s.d. (n = 3 independent experiments). | ||
For instance, as shown in Fig. 2C, a conventional probe at a concentration of 500 nM can accurately detect target concentrations below 500 nM. However, for targets exceeding this threshold, the signal becomes unreliable. Furthermore, a strong linear correlation between the target concentration and fluorescence intensity (R2 = 0.97) is observed only within a narrow range, between 50 nM and 500 nM.
As shown in Fig. 2D, we constructed NSNAP-III based on Exo III. Structurally similar to conventional probes, NSNAP-III consists of two labelled DNA strands, one labelled with a fluorophore and the other with a quencher. These two labelled DNA strands are complementarily paired with an unlabelled DNA strand, positioning the fluorophore and quencher in close proximity to suppress fluorescence.26 The probe system is preloaded with Exo III, a double-stranded DNA-specific exonuclease that catalyses the sequential removal of nucleotides from linear or nicked double-stranded DNA in the 3′ to 5′ direction.27,28 Since Exo III cannot cleave phosphonothioate bonds, the five bases at the 3′ ends of the three DNA strands comprising the probe are modified with phosphonothioates to prevent degradation by Exo III cleavage. Exo III and probes are pre-mixed and directly added to the sample during detection to activate the NSNAP-III system. The molecular probe generates fluorescence via a toehold-mediated strand displacement (TMSD) reaction triggered by the target, while Exo III simultaneously consumes the target to reset the probe.29,30 Theoretically, the strand displacement reaction and enzymatic degradation occur concurrently and synergistically in the NSNAP system. Due to the faster kinetics of strand displacement, the system initially operates in a non-equilibrium state, generating pulse-like fluorescence.31 As the target is progressively consumed, enzymatic degradation becomes dominant, fully degrading the target and resetting the probe. This dynamic transition between the two reactions leads to a characteristic fluorescence curve, where the signal rises during strand displacement and then diminishes as enzymatic degradation takes place. This coordinated mechanism of strand displacement and enzymatic degradation grants NSNAP-III several advantages, including an extended dynamic range, high adaptability to varying nucleic acid probe concentrations, and reusability.
We first verified the feasibility of the system through gel electrophoresis. As shown in Fig. S1 (ESI†), the nucleic acid probe was successfully constructed (Lane 1), and its structure remained stable after the addition of Exo III (Lane 2). The reporter strand band appeared upon the addition of the target (Lane 3) and disappeared in the presence of Exo III (Lane 4). Furthermore, as shown in Fig. S2 (ESI†), the probe retained its structural integrity after multiple dissipation-reset cycles, confirming its high stability and reusability. These results collectively demonstrate that the probe specifically recognizes the target, triggers the release of the reporter strand, and undergoes efficient enzymatic resetting without degradation of the probe structure itself.
We next conducted fluorescence kinetic experiments, starting with the optimization of the enzyme concentration in the NSNAP-III system. The results showed that at low Exo III concentrations, the consumption rate of high-concentration targets was slower. When the Exo III concentration exceeded 250 U mL−1, the target reacted with the probe to produce a distinct signal and was consumed in a shorter time. Therefore, we finally determined the optimal concentration of Exo III to be 250 U mL−1, which can ensure both a distinct signal and a shorter run time of the NSNAP (Fig. S3, ESI†). As shown in Fig. 2E, the area under the fluorescence kinetic curves for NSNAP-III detecting different target concentrations exhibited excellent linearity (Fig. 2F, R2 = 0.99). Notably, NSNAP-III could detect target molecules at concentrations up to 250 times higher than that of its own, achieving a detection range spanning up to 5000-fold, significantly surpassing the performance of conventional affinity probes.
To evaluate the applicability of varying nucleic acid probe concentrations in the NSNAP-III system, we adjusted the probe concentration between 100 and 500 nM while maintaining a fixed target concentration of 1 μM. For consistent comparison across different conditions, fluorescence curves were normalized using min–max scaling, setting the minimum and maximum average fluorescence values to 0% and 100%, respectively. This normalization approach minimized baseline variability and enabled direct comparison of the kinetic curve profiles. Fig. 2G shows that fluorescence kinetic curves for different probe concentrations responded consistently to the same target concentration after normalization. Furthermore, when using nucleic acid probes at various concentrations, NSNAP-III detected targets across a range of 10 nM to 50 μM. The area under the fluorescence curve exhibited a strong linear correlation with target concentration (Fig. S4, ESI†). This indicates that the nucleic acid probes in NSNAP-III have wide concentration applicability.
After that, we assessed the reusability of NSNAP-III. As shown in Fig. 2H, no significant difference was observed in the area under the curve after seven consecutive inputs of the same target quantity. Additionally, NSNAP-III remained available even after one week of storage (Fig. S5, ESI†). To further evaluate enzyme stability, the NSNAP-III detection system was stored at 4 °C and reused on Day 1, Day 3, and Day 7. The AUC remained strongly correlated with the target concentration (R2 = 0.96, 0.95, and 0.93, respectively; Fig. S6, ESI†). These results demonstrate the stability and reusability of the NSNAP-III system, underscoring its potential for practical applications. We also evaluated the specificity of NSNAP-III and its sensitivity to single-base mutations. Experimental results indicated that the NSNAP exhibits good specificity, generating no signal for non-target sequences (Fig. S7A, ESI†). Furthermore, the NSNAP demonstrates high sequence discrimination, effectively distinguishing the target from both single- and multi-base mismatched sequences. As shown in Fig. S7B (ESI†), the fluorescence signal decreases progressively with increasing mismatch number, confirming its robustness in detecting subtle sequence variations.
Finally, we performed kinetic simulations (see Section S1.1 in the ESI† for detailed information on the kinetic simulations), which closely aligned with the experimental results. As shown in Fig. S8A (ESI†), the NSNAP exhibited a broad response range, with NSNAP-III containing 200 nM nucleic acid probes generating signals for targets within the range of 10 nM to 100 μM. Furthermore, a strong linear correlation was observed between the signal intensity and the target concentration, with a correlation coefficient of R2 = 1.000 (Fig. S8B, ESI†). As shown in Fig. S8C (ESI†), the detection of the same concentration of targets using NSNAP-III containing different concentrations of nucleic acid probes all produced normalized signals with consistent trends, confirming the wide applicability of the nucleic acid probe concentrations in the NSNAP. Additionally, as shown in Fig. S8D (ESI†), seven consecutive additions of the target at a concentration of 1 μM to the NSNAP-III system containing 200 nM nucleic acid probe yielded consistent response signals, which effectively demonstrated the reusability of the NSNAP.
2.2. The non-saturated nucleic acid probe based on lambda exonuclease (NSNAP-λ)
To showcase the universality of the NSNAP and the diversity of enzyme options, we constructed NSNAP-λ in this section (as shown in Fig. 3A). The enzyme preloaded in the NSNAP system is λ Exo. λ Exo is a DNA-specific exonuclease that degrades double-stranded DNA in a highly sequential manner, starting from the 5′ end. λ Exo preferentially targets 5′-phosphorylated double-stranded DNA, exhibiting significantly reduced degradation rates for non-phosphorylated substrates.32 | ||
Fig. 3 Non-saturating nucleic acid probe based on λ Exo (NSNAP-λ). (A) Operational schematic of NSNAP-λ. (B) Fluorescence kinetic curves of NSNAP-λ (400 nM nucleic acid probe) detecting different concentrations of targets (10–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM). (C) Linear relationship between the area under the fluorescence kinetic curve and target concentration for NSNAP-λ detection, with R2 = 0.98. (D) Normalized fluorescence signals generated by 1 μM target detected by NSNAP-λ with nucleic acid probes of different concentrations (100–500 nM). (E) Fluorescence signals from NSNAP-λ after 7 repetitions, with corresponding area under the curve at each time point (500 nM nucleic acid probe, 1 μM target). The λ Exo concentration used in all experiments was 500 U mL−1, with lambda exonuclease reaction buffer at 37 °C. Data are means ± s.d. (n = 3 independent experiments). 000 nM). (C) Linear relationship between the area under the fluorescence kinetic curve and target concentration for NSNAP-λ detection, with R2 = 0.98. (D) Normalized fluorescence signals generated by 1 μM target detected by NSNAP-λ with nucleic acid probes of different concentrations (100–500 nM). (E) Fluorescence signals from NSNAP-λ after 7 repetitions, with corresponding area under the curve at each time point (500 nM nucleic acid probe, 1 μM target). The λ Exo concentration used in all experiments was 500 U mL−1, with lambda exonuclease reaction buffer at 37 °C. Data are means ± s.d. (n = 3 independent experiments). | ||
To enhance digestion efficiency, the 5′ end of the input DNA was phosphorylated. Additionally, to ensure probe stability and prevent degradation from λ Exo, the five bases at the 5′ ends of the three DNA strands composing the probe were modified with phosphonothioates.31
Similarly, we first validated the feasibility of the NSNAP-λ system through gel electrophoresis. As shown in Fig. S9 (ESI†), the nucleic acid probe was successfully constructed (Lane 1) and remained stable after the addition of λ Exo (Lane 2). Upon adding the target strand, a strand displacement reaction occurred, forming the target–probe complex and resulting in the disappearance of the target strand band (Lane 3). In the presence of λ Exo, the target strand was consumed, and the probe was reset (Lane 4). These results confirm that NSNAP-λ effectively recognizes the target and resets the probe. Additionally, Fig. S10 (ESI†) shows that the probe retains its structural integrity after one and seven cycles of use with λ Exo, validating that the enzymatic degradation selectively removes the target without affecting the probe itself.
After that, we similarly optimized the enzyme concentration in the NSNAP-λ system. At a λ Exo concentration of 500 U mL−1, target depletion occurred more rapidly and completely. Therefore, the concentration of λ Exo was finalized to be 500 U mL−1 (Fig. S11, ESI†). As shown in Fig. 3B, the area under the fluorescence kinetic curves generated by NSNAP-λ detecting varying target concentrations showed a strong linear correlation with target concentration, with R2 = 0.97 (Fig. 3C). NSNAP-λ demonstrated the ability to detect target molecules at concentrations up to 20 times higher than its own and successfully achieved quantitative detection across a 1000-fold concentration range.
Next, we assessed the system's performance using nucleic acid probes at different concentrations (100–500 nM) to detect a fixed target concentration (1 μM). As shown in Fig. 3D, the normalized fluorescence curves generated under these conditions showed consistent trends. Furthermore, we used NSNAP-λ with nucleic acid probes at different concentrations to detect targets ranging from 10 nM to 10 μM. It was observed that the area under the fluorescence curves exhibited a strong linear relationship with target concentration in all cases (Fig. S12, ESI†). These findings highlight that the nucleic acid probes in NSNAP-λ also have a broad concentration applicability. We then verified the reusability of NSNAP-λ. As shown in Fig. 3E, no significant changes were observed in the area under the curve after seven consecutive inputs of the same target quantity, confirming that NSNAP-λ is reusable.
Finally, kinetic simulations were performed to corroborate the experimental findings (detailed information regarding the kinetic simulation is provided in Section S1.2 of the ESI†). The simulation results were consistent with the experimental data. As shown in Fig. S13A (ESI†), NSNAP-λ containing 500 nM nucleic acid probe generated signals for target concentrations ranging from 10 nM to 10 μM. The signal intensity showed a linear correlation to the target concentration, with R2 = 1.0 (Fig. S13B, ESI†). Additionally, NSNAP-λ systems with varying nucleic acid probe concentrations produced normalized signals with consistent trends when detecting the same target concentration (Fig. S13C, ESI†). As shown in Fig. S13D (ESI†), seven consecutive additions of a 1 μM target to the NSNAP-λ system containing 500 nM nucleic acid probes resulted in consistent signal responses.
2.3. Quantitative analysis of viral and bacterial genes in complex samples using the NSNAP
Quantitative detection enables precise measurement of viral load, a critical parameter for monitoring and managing viral infections. Tracking changes in viral load serves as a key indicator of antiviral therapy effectiveness. In this study, we selected three viral genes – HIV, HPV, and HHV – that exhibit significant variations in the viral load within the human body. Depending on the severity of infection, HIV viral loads in patients can range from as low as 500 copies per mL to over 30000 copies per mL.33 Similarly, HPV and HHV viral loads range from a few copies to several million copies.34,35
As mentioned above, we constructed two non-saturated nucleic acid probes, NSNAP-III and NSNAP-λ, using Exo III and λ Exo enzymes, respectively. These probes were employed for the quantitative detection of HIV, HHV, and HPV genes. As shown in Fig. 4A, the target gene fragments were added in the blood extracted DNA, and amplified by LATE (linear-after-the-exponential)-PCR, an improved asymmetric PCR technique that generates single-stranded products through optimized primer design. Exponential amplification occurs in the initial phase, followed by efficient linear amplification after restriction primer depletion, enhancing PCR efficiency and accuracy.36,37 NSNAPs were then added to the amplification products for quantitative analysis.
 | ||
| Fig. 4 Quantification of viral and bacterial genes using the NSNAP in complex biological matrices. (A) Procedure for detecting viral and bacterial genes using the NSNAP. (B) Quantitative analysis of HIV, HHV, and HPV genes using NSNAP-III, showing linear correlations between the target concentration and the area under the fluorescence curve (AUC), with R2 values of 0.93, 0.90, and 0.91, respectively. (C) Quantitative analysis of HIV, HHV, and HPV genes using NSNAP-λ, with R2 values of 0.93, 0.93, and 0.95. (D) Quantitative analysis of bacterial genes oprL, dnaJ, and ddl (specific to P. aeruginosa, E. cloacae, and E. faecium, respectively) in serum using NSNAP-III, with R2 values of 0.94, 0.97, and 0.93. All reactions used a nucleic acid probe concentration of 50 nM. Exo III and λ Exo were used at concentrations of 250 U mL−1 and 500 U mL−1, respectively. Data are the means ± s.d. (n = 3 independent experiments). | ||
In the NSNAP-III detection system, LATE-PCR amplification was performed on HIV, HHV, and HPV viral genes at concentration gradients of 1000 fM, 100 fM, 10 fM, and 1 fM, respectively. The concentration ratio of the upstream and downstream primers was 40:1. As shown in Fig. S14–S16 (ESI†), the three target genes were successfully amplified, producing single-stranded products (Lane 2). NSNAP-III was then added to the amplified products for fluorescence kinetic detection (Fig. S17A, ESI†). The results demonstrated a good linear relationship between the area under the fluorescence kinetic curve generated by the reaction of NSNAP-III with the amplified genes and the pre-amplification gene concentration, with R2 values of 0.93, 0.90, and 0.91 for HIV, HHV, and HPV, respectively (Fig. 4B).
In the NSNAP-λ detection system, the 3′ ends of the upstream primer strand DNA of the three genes were phosphorylated to ensure successful degradation of the amplified targets by λ Exo. LATE-PCR amplification was carried out for the three viral genes at concentration gradients of 1000 fM, 100 fM, 10 fM, and 1 fM, with an upstream and downstream primer concentration ratio of 40:1. As shown in Fig. S14–S16 (ESI†), the three genes were successfully amplified, and single strands were produced (Lane 5). NSNAP-λ was then added for fluorescence kinetic detection (Fig. S17B, ESI†). The area under the fluorescence kinetic curve, generated by the reaction of NSNAP-λ with the amplified products, showed a good linear relationship with the pre-amplification gene concentrations, with R2 values of 0.93, 0.93, and 0.95 for HIV, HHV, and HPV, respectively (Fig. 4C).
To further validate the applicability of NSNAP-III in real-world complex biological samples, we extended our detection beyond viral targets. We selected three clinically relevant bacterial species—Pseudomonas aeruginosa, Enterobacter cloacae, and Enterococcus faecium—and designed specific nucleic acid probes targeting their marker genes oprL,38dnaJ,39 and ddl,40 respectively. Plasmids containing these target genes were spiked into human serum to simulate realistic clinical detection conditions. Following LATE-PCR amplification of the serum-spiked samples, NSNAP-III was applied for fluorescence kinetic detection (Fig. S17C, ESI†). As shown in Fig. 4D, NSNAP-III successfully quantified these bacterial genes across a wide concentration range of 1–1000 fM, maintaining strong linear correlations between the area under the fluorescence kinetic curves and target concentrations (R2 = 0.94, 0.97, and 0.93). Additionally, as shown in Fig. S18 (ESI†), NSNAP-III maintained consistent performance over three consecutive reuse cycles when detecting the species-specific marker gene dnaJ in serum. These results highlight the high sensitivity, reusability and cross-kingdom applicability of NSNAP-III for detecting both viral and bacterial nucleic acids in complex clinical matrices. A detailed performance comparison with some commercial methods is summarized in Table S6 (ESI†), highlighting the NSNAP's advantage in accessibility and scalability for practical applications.
3. Conclusions
In this study, we developed a non-saturating nucleic acid probe (NSNAP) that leverages dynamic DNA nanotechnology to facilitate wide dynamic range quantitative detection of nucleic acids. The NSNAP effectively addresses the limitations of conventional molecular probes, such as a narrow dynamic range and signal saturation, by incorporating enzymes that deplete targets within the detection system. Unlike conventional affinity probes, the NSNAP achieves a dynamic range expansion of up to 5000-fold, greatly exceeding the typical 81-fold limitation of traditional methods, and allows for the detection of targets at concentrations up to 250 times that of the probe. The NSNAP demonstrated excellent reusability over at least seven detection cycles and showed high sensitivity (1–1000 fM, R2 > 0.90) in quantifying viral (HIV, HPV, HHV) and bacterial (oprL, dnaJ, ddl) genes in human serum. The NSNAP system exhibits robust performance across clinically relevant complex matrices and maintains high stability even under conditions of long-term storage and reuse. Its adaptability, reusability, and cross-kingdom applicability establish the NSNAP as a powerful and cost-efficient platform for molecular diagnostics.Looking forward, further enhancement of the system's sensitivity and operational flexibility is anticipated. Integration with isothermal amplification techniques such as LAMP or RPA could provide amplification-free or simplified workflows, improving applicability for point-of-care diagnostics. Additionally, optimizing the coordination between strand displacement and enzymatic degradation at extreme target concentrations could further extend the dynamic range and detection robustness. These advancements will broaden the utility of the NSNAP in diverse clinical and research settings, supporting rapid, accurate, and scalable nucleic acid detection in real-world applications.
4. Experimental section
4.1. Reagents and materials
All oligonucleotides and primers used in this work were synthesized by Sangon Biotech (Shanghai) Co., Ltd, and their sequences are listed in Table S1 (ESI†). All modified oligonucleotides were purified by HPLC, while unmodified oligonucleotides were purified by PAGE. Enzymes and their corresponding buffers were obtained from New England Biolabs (NEB). Other chemicals used in this work were of analytical grade. DNase/RNase-free deionized water was used in all experiments.Assays for the NSNAP-III system were conducted in 1× NEBuffer 1 (10 mM bis-tris-propane-HCl, 10 mM MgCl2, 1 mM DTT, pH 7). Assays for the NSNAP-λ system were conducted in 1× lambda exonuclease reaction buffer (67 mM glycine–KOH, 2.5 mM MgCl2, 50 μg mL−1 BSA, pH 9.4).
4.2. Kinetics simulations of NSNAP detection systems
The concentration changes of the fluorescence-labeled DNA strand, which remained unquenched, were computationally simulated over time within the NSNAP detection system. The simulations were performed by solving the corresponding differential equations using the Runge–Kutta method. Detailed parameter settings for the simulations are provided in Note S1 (ESI†).4.3. Real-time monitoring of the reactions by the NSNAP
Nucleic acid probes were prepared by mixing the corresponding single strands at equimolar concentrations in buffer within PCR tubes. The strands were annealed in a PCR thermocycler by cooling from 80 °C to 25 °C at a rate of 5 °C per minute. Next, Exo III or λ Exo was added to the annealed strands, and the components were thoroughly mixed to construct NSNAP-III or NSNAP-λ. Targets were then added, and fluorescence signals were recorded immediately in a real-time PCR cycler (Rotor-Gene Q, Qiagen, Germany) at 37 °C with 5-second intervals.4.4. Amplification of virus genes in blood samples
Total DNA was extracted from blood by QIAamp DNA Blood Kits (Qiagen) according to following the manufacturer's instructions. Virus genes were added into the extracted DNA. LATE-PCR was performed in a 50 μL reaction mixture containing 25 μL of Q5 High-Fidelity 2× Master Mix (M0492s, NEB), 1 μM forward primers, 25 nM reverse primers, and target gene DNA. The thermocycling program consisted of an initial denaturation step at 95 °C for 30 seconds, followed by 20 cycles of denaturation at 95 °C for 10 seconds, annealing at 60 °C for 20 seconds, and extension at 72 °C for 20 seconds, with a final extension at 72 °C for 5 minutes. The LATE-PCR products were analyzed by 12% polyacrylamide gel electrophoresis.4.5. Characterization of the fluorescence detection reaction and LATE-PCR products by polyacrylamide gel electrophoresis (PAGE)
The products from the NSNAP system, including those generated from interactions with the target and LATE-PCR amplification products, were analyzed by native 12% polyacrylamide gel electrophoresis (PAGE) (19:1 acrylamide/bisacrylamide). The experiments were performed in TBE buffer (44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA, pH 8.0). A 5 μL sample was mixed with 2 μL of 6× loading buffer, and 6 μL of the resulting mixture was loaded onto the 12% native PAGE gel for electrophoresis. Electrophoresis was carried out at 120 V for 50 minutes at room temperature. After staining with SYBR gold (invitrogen) dissolved in TBE buffer (pH 8.0) for 15 minutes, the gel was photographed using a gel imaging system.
Author contributions
Xinmiao Kang: methodology, formal analysis, data curation, and writing – review and editing. Yu Liu: methodology and formal analysis. Dandan Tian: formal analysis and technique support. Zuhao Shen: formal analysis and technique support. Shihui Wang: methodology, technique support, and writing – review and editing. Xin Su: project supervision, methodology, technique support, data curation, writing – review and editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Access to the data may be subject to restrictions based on ethical considerations or confidentiality agreements.Acknowledgements
This work was supported by the National Natural Science Foundation of China (32271521, 22325801 and 31971361), the State Key Research Development Program of China (2022YFC2603902), the Fundamental Research Funds for the Central Universities (PT2406).Notes and references
- J. Tong, Z. Wang, J. Zhang, R. Gao, X. Liu, Y. Liao, X. Guo and Y. Wei, ACS Appl. Mater. Interfaces, 2024, 16, 58072–58099 CrossRef CAS PubMed.
- S. Jampasa, B. Jikul, C. Kreangkaiwal, W. Khamcharoen, W. Jesadabundit, W. Waiwinya, P. Saelim, T. Phanbunmee, K. Patarakul and O. Chailapakul, Sens. Actuators, B, 2024, 406, 135411 CrossRef CAS.
- J. Pan, W. Xu, W. Li, S. Chen, Y. Dai, S. Yu, Q. Zhou and F. Xia, Anal. Chem., 2023, 95, 420–432 CrossRef CAS PubMed.
- X. Li, B. Zhang, C. Yan, J. Li, S. Wang, X. Wei, X. Jiang, P. Zhou and J. Fang, Nat. Commun., 2019, 10, 2745 CrossRef PubMed.
- R. Khademi, Z. Mohammadi, R. Khademi, A. Saghazadeh and N. Rezaei, Nanoscale Adv., 2023, 5, 571–595 RSC.
- L.-J. Chan, A. Gandhirajan, L. L. Carias, M. H. Dietrich, O. Vadas, R. Visentin, C. T. França, S. Menant, D. Soldati-Favre and I. Mueller, Nat. Commun., 2021, 12, 1538 CrossRef CAS PubMed.
- C. Wu, P. Jiang, W. Su and Y. Yan, Biomacromolecules, 2024, 25, 5609–5629 CrossRef CAS PubMed.
- L. Zhang, H. Zhao, H. Yang and X. Su, Biosens. Bioelectron., 2023, 239, 115622 CrossRef CAS PubMed.
- K. Grover, A. Koblova, A. T. Pezacki, C. J. Chang and E. J. New, Chem. Rev., 2024, 124, 5846–5929 CrossRef CAS PubMed.
- H. H. Chen, Z. Khatun, L. Wei, C. Mekkaoui, D. Patel, S. J. W. Kim, A. Boukhalfa, E. Enoma, L. Meng and Y. I. Chen, Nat. Biomed. Eng., 2022, 6, 1045–1056 CrossRef CAS PubMed.
- R. Taguchi, T. Terai, T. Ueno, T. Komatsu, K. Hanaoka and Y. Urano, Sens. Actuators, B, 2018, 265, 575–581 CrossRef CAS.
- Y. Zhang, G. Zhang, Z. Zeng and K. Pu, Chem. Soc. Rev., 2022, 51, 566–593 RSC.
- Y. Hu, J. Yu, M. Xu and K. Pu, J. Am. Chem. Soc., 2024, 146, 12656–12663 CrossRef CAS PubMed.
- D. Chang, Z. Wang, C. D. Flynn, A. Mahmud, M. Labib, H. Wang, A. Geraili, X. Li, J. Zhang and E. H. Sargent, Nat. Chem., 2023, 15, 773–780 CrossRef CAS PubMed.
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and V. V. Tsukruk, Chem. Rev., 2017, 117, 12942–13038 CrossRef CAS PubMed.
- N. Maganzini, I. Thompson, B. Wilson and H. T. Soh, Nat. Commun., 2022, 13, 7072 CrossRef CAS PubMed.
- S. M. Tabakman, L. Lau, J. T. Robinson, J. Price, S. P. Sherlock, H. Wang, B. Zhang, Z. Chen, S. Tangsombatvisit and J. A. Jarrell, Nat. Commun., 2011, 2, 466 CrossRef PubMed.
- L. Liu, M. Yuan, Y. Jin, G. Zhou, T. Li, L. Li, H. Peng and W. Chen, Anal. Chem., 2021, 93, 8170–8177 CrossRef CAS PubMed.
- M. Vendrell, D. Zhai, J. C. Er and Y.-T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed.
- A. Porchetta, A. Vallée-Bélisle, K. W. Plaxco and F. Ricci, J. Am. Chem. Soc., 2013, 135, 13238–13241 CrossRef CAS PubMed.
- D. Lauzon and A. Vallée-Bélisle, J. Am. Chem. Soc., 2023, 145, 18846–18854 CrossRef CAS PubMed.
- E. Del Grosso, E. Franco, L. J. Prins and F. Ricci, Nat. Chem., 2022, 14, 600–613 CrossRef CAS PubMed.
- J. Yang, T. Zhang, L. Zhang and X. Su, Nanoscale, 2024, 16, 4219–4228 RSC.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- D. Bedeaux, A. Albano and P. Mazur, Phys. A, 1976, 82, 438–462 CrossRef.
- J. Liu, Y. Liu, L. Zhang, S. Fu and X. Su, Biosens. Bioelectron., 2022, 215, 114561 CrossRef CAS PubMed.
- S. Henikoff, Gene, 1984, 28, 351–359 CrossRef CAS PubMed.
- X. Yang, L. Wang, L. Pang, S. Fu, X. Qin, Q. Chen, C. Man and Y. Jiang, Anal. Chim. Acta, 2021, 1181, 338903 CrossRef CAS PubMed.
- P. Irmisch, T. E. Ouldridge and R. Seidel, J. Am. Chem. Soc., 2020, 142, 11451–11463 CrossRef CAS PubMed.
- L. Zhang, J. Chen, M. He and X. Su, Exploration, 2022, 20210265 CrossRef CAS PubMed.
- Z. Qin, Y. Liu, L. Zhang, J. Liu and X. Su, ACS Nano, 2022, 16, 14274–14283 CrossRef CAS PubMed.
- S. Fu, J. Li, J. Chen, L. Zhang, J. Liu, H. Liu and X. Su, Nat. Biotechnol., 2024, 1–12 Search PubMed.
- J. W. Mellors, A. Munoz, J. V. Giorgi, J. B. Margolick, C. J. Tassoni, P. Gupta, L. A. Kingsley, J. A. Todd, A. J. Saah and R. Detels, Ann. Intern. Med., 1997, 126, 946–954 CrossRef CAS PubMed.
- D. Holzinger, M. Schmitt, G. Dyckhoff, A. Benner, M. Pawlita and F. X. Bosch, Cancer Res., 2012, 72, 4993–5003 CrossRef CAS PubMed.
- E. Oksenhendler, G. Carcelain, Y. Aoki, E. Boulanger, A. Maillard, J.-P. Clauvel and F. l Agbalika, Blood, 2000, 96, 2069–2073 CrossRef CAS PubMed.
- J. A. Sanchez, K. E. Pierce, J. E. Rice and L. J. Wangh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1933–1938 CrossRef CAS PubMed.
- L. Zhang, H. Yang, Y. Yan, H. Zhao, D. Han and X. Su, Adv. Mater., 2025, 2413198 CrossRef CAS PubMed.
- D. De Vos, A. Lim Jr, J.-P. Pirnay, M. Struelens, C. Vandenvelde, L. Duinslaeger, A. Vanderkelen and P. Cornelis, J. Clin. Microbiol., 1997, 35, 1295–1299 CrossRef CAS PubMed.
- M. Pavlovic, R. Konrad, A. N. Iwobi, A. Sing, U. Busch and I. Huber, FEMS Microbiol. Lett., 2012, 328, 46–53 CrossRef CAS PubMed.
- T. Nomura, Y. Hashimoto, J. Kurushima, H. Hirakawa, K. Tanimoto, B. Zheng, G. Ruan, F. Xue, J. Liu and J. Hisatsune, J. Microbiol. Methods, 2018, 145, 69–72 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00218d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |