DOI:
10.1039/D5NH00212E
(Communication)
Nanoscale Horiz., 2025,
10, 2094-2103
One-step construction of NH2-UiO-66 based heterojunction photocatalysts for adsorption–photocatalytic synergistic removal of antibiotics†
Received
7th April 2025
, Accepted 20th June 2025
First published on 20th June 2025
Abstract
Metal–organic frameworks (MOFs), as novel crystalline materials, exhibit notable photocatalytic activity and exceptional adsorption capabilities. However, their low charge separation efficiency still limits their photocatalytic performance. Herein, NH2-UiO-66/BiOCl composites were synthesized through a one-step green grinding method, effectively integrating adsorption and photocatalytic degradation mechanisms. The NH2-UiO-66/BiOCl-50% composite demonstrated outstanding degradation efficiency (96.84%) for ciprofloxacin (CIP) within 60 min, with a rate constant (0.0432 min−1) that is 3.6 and 2.1 times those of NH2-UiO-66 and BiOCl, respectively. The removal performance of NH2-UiO-66/BiOCl for CIP and the underlying adsorption–photocatalytic degradation mechanisms were thoroughly investigated in various water environments. The remarkable degradation performance is attributed to the excellent adsorption capacity and the effective formation of a heterojunction between NH2-UiO-66 and BiOCl, which enhances electron–hole separation and transfer efficiency. The stability of the photocatalyst is demonstrated by cyclic testing. Moreover, ˙O2− was identified as the main active species and the photocatalytic mechanisms of the composite were elucidated. This work provides valuable insights into the synergistic integration of adsorption and photocatalytic degradation for the treatment of antibiotic-contaminated wastewater.
New concepts
This work demonstrates the novel concept of synergistically integrating adsorption and photocatalytic degradation mechanisms through heterojunction engineering of MOF-based composites. The NH2-UiO-66/BiOCl composite leverages the dual functionality of MOFs and semiconductor materials. A one-step green grinding synthesis method enables intimate interfacial contact, creating a heterojunction that not only enhances adsorption of pollutants but also promotes spatially separated charge carriers, significantly boosting photocatalytic efficiency. Existing MOF-photocatalyst composites often struggle with inefficient charge transfer and unstable interfaces. This work overcomes these limitations by designing a hierarchical heterostructure where NH2-UiO-66 acts as an electron donor and adsorption reservoir, while BiOCl serves as a hole-rich photocatalyst. The composite achieves 96.84% CIP degradation within 60 minutes, surpassing individual components by 3.6 times (NH2-UiO-66) and 2.1 times (BiOCl), attributed to enhanced electron–hole separation and interfacial charge transfer. It also demonstrates adaptability to diverse antibiotics (tetracycline, sulfamethoxazole) and environmental conditions (inorganic ions, varying concentrations), offering practical versatility absent in most single-component photocatalysts.This work redefines heterojunction engineering by integrating MOFs with semiconductor charge dynamics, emphasizing structural synergy over individual material optimization. It advances the design of multifunctional nanomaterials for complex environmental remediation, bridging the gap between nanoscale interfacial engineering and macroscale pollutant treatment efficiency.
|
1. Introduction
The rapid development of industrialization and human society has led to an increasingly severe problem of environmental pollution. Among them, water contamination is particularly critical.1 The major source of water pollution includes industrial effluents, domestic sewage, agricultural runoff, and medical waste. Previous studies have demonstrated that a large number of drugs are discharged into the water environment annually, such as hormone drugs, anti-inflammatory drugs and antibiotics.2 As a result, scientists have explored various methods for removing antibiotic pollutants from water, including electrochemical methods, Fenton reactions, biological treatment, photocatalysis and adsorption. However, biological treatment, electrochemical methods and Fenton reactions are often hindered by issues such as high costs, long processing times, high energy consumption, and limited reaction conditions.3–7
Adsorption technology is simple and efficient in treating antibiotic sewage. However, it merely transfers the pollutants rather than completely degrading them.8 By contrast, photocatalytic technology has emerged as one of the most promising approaches owing to its green, efficient, and environmentally friendly nature. Adsorption of pollutants plays a critical role in the photocatalytic process by pre-concentrating pollutants on the catalyst surface, thereby enhancing degradation efficiency.9,10 Therefore, designing novel photocatalysts with high adsorption properties is an effective way to improve the photocatalytic activity.
Metal–organic frameworks (MOFs) are innovative crystalline materials composed of metal ions or clusters and organic ligands, and have the advantages of excellent adsorption performance, high surface areas and abundant active sites.11 However, MOFs typically exhibit low photocatalytic efficiency compared to traditional photocatalysts due to their poor charge separation and high carrier recombination rates. The combination of MOFs and semiconductors with appropriate band gaps is beneficial for the carrier separation to improve the photocatalytic efficiency.12,13 Zhang et al.14 prepared Bi/BiO2-X-Bi2O2CO3/BiOCl@Bi-MOF using hydrothermal and photodeposition methods. The Z-scheme heterojunction significantly enhanced electron–hole separation and led to a photocatalytic degradation efficiency of 94.6% for CTC. Luo et al.15 prepared a high-efficiency MIL-88B(Fe)/ZnTi-LDH composite with both adsorption and photocatalytic activities, demonstrating excellent photocatalytic performance due to efficient charge carrier separation. Li et al.16 used a one-step process to prepare Ti–In bimetallic MOFs, combining the advantages of Ti-MOF and In-MOF (large specific surface area) to enhance both adsorption and photocatalytic degradation. Although the combination of MOFs and semiconductors could lead to excellent photocatalytic performance, complex preparation processes involved limit their practical applications. Therefore, it remains a significant challenge to design a green and simple method for preparing photocatalysts with high absorption properties and photocatalytic activity for large-scale applications.
BiOCl is a promising photocatalyst due to its high stability, reactivity and favorable photocatalytic activity. However, its narrow light absorption range, poor adsorption properties, and high carrier recombination rates significantly restrict its practical application in photocatalysis. Interestingly, the light absorption range can be enhanced by compositing materials with a wide light absorption range, and the construction of heterojunctions can promote the separation of carriers, thus improving the photocatalytic activity. Herein, NH2-UiO-66/BiOCl composites were synthesized via a facile one-step green grinding method. NH2-UiO-66 exhibits excellent adsorption performance and its integration with BiOCl results in the formation of a heterojunction that enhances electron–hole separation and transfer efficiency, leading to synergistic effects between adsorption and photocatalytic degradation. Ciprofloxacin (CIP) was chosen as a model molecule to evaluate the adsorption and photocatalytic performance of NH2-UiO-66/BiOCl. This study also explores the factors influencing CIP adsorption and photocatalytic degradation, including the catalyst content, antibiotic concentration, and environmental factors such as inorganic salt ions. And, the adaptation of NH2-UiO-66/BiOCl to different antibiotics, such as tetracycline and sulfamethoxazole, was also discussed. Subsequently, the adsorption mechanism of the composite towards CIP was analyzed, and the generation of active species was examined to deduce the photocatalytic mechanisms. This study provides new insights into the synergism of adsorption and photocatalytic degradation, and the degradation mechanism of CIP.
2. Results and discussion
2.1. Crystal structures, morphologies, and chemical compositions
A three-dimensional flower-like structure is formed by the self-assembly of smooth, impurity-free nanosheets of BiOCl, as shown in Fig. 1b. Fig. 1c and d reveal that NH2-UiO-66 displays a regular octahedral crystal structure with particle size distribution centered around an average diameter of 14.32 ± 3.58 nm. As depicted in Fig. 1e and f, BiOCl acts as a platform and is completely coated with NH2-UiO-66; due to the interaction between charges during the grinding process, the –NH2 group of NH2-UiO-66 is protonated to –NH3+ in acidic environments, which makes the whole NH2-UiO-66 particle positively charged producing a strong electrostatic attraction with the Cl− terminal (negative) of BiOCl. NH2-UiO-66 was successfully coated on the surface of BiOCl due to the minimization of the thermodynamically driven interfacial energy and the promotion of homogeneous compounding of the material during the grinding process (Fig. 1a). The interaction between BiOCl and NH2-UiO-66 leads to the successful formation of a compact heterojunction between the two substances, which promotes charge separation and enhances photocatalytic activity. HR-TEM images of UB-50% are shown in Fig. 1g, in which a lattice fringe with a lattice distance of 0.739 nm corresponds to the (001) facet of BiOCl.17 Furthermore, EDS mapping of UB-50% shows that C, Zr, O, Bi, Cl elements were uniformly distributed (Fig. 1h), confirming the successful synthesis of the sample.
 |
| | Fig. 1 (a) Schematic diagram of the preparation of NH2-UiO-66/BiOCl. SEM images of BiOCl (b), NH2-UiO-66 (d), and UB-50% (e). TEM images of the NH2-UiO-66 (c) and UB-50% (f) and (g). EDS mapping images (h) of UB-50%. | |
In Fig. 2a, the characteristic diffraction peaks at 7.4° and 8.5° correspond to the (111) and (200) planes, respectively, which agree with the calculated characteristic peaks, indicating the successful synthesis of NH2-UiO-66.18 The diffraction peaks of BiOCl at 32.63°, 40.91°, 46.77°, 54.28° and 58.59° correspond to the (110), (112), (200), (211), and (212) crystal planes (JCPDS-06-0249).19 Meanwhile, the peaks of UB-x fit well with the NH2-UiO-66 and BiOCl peaks, confirming the successful combination of both components. The FT-IR spectra of UB-x in Fig. 2b further confirm the successful synthesis of UB-x composites. The two strong vibrational peaks of NH2-BDC located at 1578![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−1 and 1385cm−1 correspond to the carboxyl functional group (–COOH), respectively. The peaks at 1255 cm−1 and 665 cm−1 correspond to the C–N stretching vibrations and the O–H bond in NH2-BDC, respectively.20 For the pristine NH2-UiO-66, the characteristic peak appearing at 765 cm−1 is ascribed to the vibration of the Zr–O bond and the ligands, demonstrating that NH2-UiO-66 was successfully prepared. For BiOCl, the variation band located at 527 cm−1 corresponds to the Bi–O bond stretching vibration.21 All characteristic vibration peaks of NH2-UiO-66 and BiOCl demonstrate the successful preparation of UB-x.
cm−1 and 1385cm−1 correspond to the carboxyl functional group (–COOH), respectively. The peaks at 1255 cm−1 and 665 cm−1 correspond to the C–N stretching vibrations and the O–H bond in NH2-BDC, respectively.20 For the pristine NH2-UiO-66, the characteristic peak appearing at 765 cm−1 is ascribed to the vibration of the Zr–O bond and the ligands, demonstrating that NH2-UiO-66 was successfully prepared. For BiOCl, the variation band located at 527 cm−1 corresponds to the Bi–O bond stretching vibration.21 All characteristic vibration peaks of NH2-UiO-66 and BiOCl demonstrate the successful preparation of UB-x.
 |
| | Fig. 2 The XRD patterns (a) and FT-IR spectra (b) of NH2-UiO-66, BiOCl, and UB-x. The XPS spectra (c) and the high-resolution spectra of (d)–(f): N 1s, Bi 4f and Cl 2p of UB-50%. | |
The X-ray photoelectron spectra (XPS) of NH2-UiO-66, BiOCl and UB-50% are presented in Fig. 2c. Bi, Cl, O, Zr, N and C elements were detected in UB-50% photocatalysts, with no additional impurities, consistent with the EDS results. In Fig. 2d, the N 1s binding energy peaks at 400.6 and 399.45 eV of NH2-UiO-66 correspond to N–H and C–N bonds, respectively.22 Compared to NH2-UiO-66, the characteristic peaks of N–H and C–N for UB-50% were shifted to lower binding energies, which indicates an electron density increase of the N atom of NH2-UiO-66 in UB-50%. In the Bi 4f spectrum of BiOCl (Fig. 2e), the two diffraction peaks located at 164.75 and 159.45eV correspond to the Bi 4f7/2 and Bi 4f5/2 orbits of the trivalent oxidation state of Bi. As shown in Fig. 2f, the two characteristic peaks of Cl 2p at around 199.75 eV and 198.15 eV correspond to Cl 2p3/2 and Cl 2p1/2, respectively.23 When NH2-UiO-66 and BiOCl were combined by wet-grinding, the binding energy peaks of Bi 4f and Cl 2p were shifted to a higher binding energy. The shift of the binding energy peak demonstrates that BiOCl acts as the e− donor and NH2-UiO-66 acts as the e− acceptor in UB-50%.
2.2. Adsorption performance
Fig. 3a–c shows the experimental results of N2 adsorption–desorption. The isotherm of BiOCl belonged to type IV with an H3 shaped hysteresis loop, owing to the slit pores caused by plate slit construction and a crack and wedge structure, which coincided with the SEM results (Fig. 3a).24 NH2-UiO-66 exhibits a type IV isotherm with a H1 shaped hysteresis loop. The pore size of NH2-UiO-66 is mainly concentrated within 5 nm (Fig. 3c), indicating the existence of mesopores.25 In addition, NH2-UiO-66 also exhibits a large specific surface area and high pore volume. NH2-UiO-66 presents an excellent surface area (896.96 m2 g−1). The comparison with other MOFs is shown in Table S1 (ESI†). BiOCl presents a low surface area (4.33 m2 g−1), and the BET surface area of UB-50% is significantly improved compared to that of BiOCl, with a value of 552.11 m2 g−1. In addition, the pore volume of UB-50% is as high as 0.81 cm3 g−1. After the bonding of NH2-UiO-66 and BiOCl by wet-grinding, UB-50% with a large specific surface area and high pore volume inherits the aperture characteristics of NH2-UiO-66, and presents adsorption characteristics with a type-IV isotherm and a H1 shaped hysteresis loop (Fig. 3c). The high specific surface area and porosity of UB-50% are beneficial for the adsorption of CIP molecules.
 |
| | Fig. 3 Nitrogen adsorption–adsorption isotherm and pore size distribution plots of BiOCl (a), NH2-UiO-66 (b) and UB-50% (c). Adsorption kinetics (d) and adsorption isotherms (e). Schematic diagram of the adsorption of NH2-UiO-66 (f). | |
Adsorption of reactants is one of the important processes in photocatalysis. Adsorption kinetics were analyzed using pseudo-first-order and pseudo-second-order models, as shown in Fig. 3d. The adsorption of CIP molecules rapidly increases within the first 10 min, reaching equilibrium after 30 min. The adsorption performances of UB-10%, UB-30%, UB-50% and UB-70% were obviously enhanced compared to that of BiOCl. The relevant parameters calculated using the pseudo-first order and pseudo-second order kinetic models are shown in Table 1. The values of correlation coefficients (R2) of the first-order model of UB-x were smaller than those of the second-order model, which demonstrates that the chemisorption plays a critical role. The Langmuir and Freundlich isotherm models were used to fit the data of adsorption isotherm experiments. As shown in Fig. 3e, R2 of the Langmuir model was higher than that of the Freundlich model (Table 2), which indicates that CIP formed a homogeneous adsorbed monolayer on UB-x.26 According to the Langmuir model, the values of qm of UB-x were all higher than that of BiOCl (6.79 mg g−1). As previously reported, the unsaturated metal Zr sites of NH2-UiO-66 easily interact with carbonyl, hydroxyl and amine groups on CIP molecules, promoting the adsorption of CIP (Fig. 3f).27
Table 1 The best-fit model parameters of CIP adsorption kinetics onto samples
| Sample |
Pseudo-first order |
Pseudo-second order |
|
k
1 (min−1) |
q
e (mg g−1) |
R
2
|
k
2 (g mg−1 min−1) |
q
e (mg g−1) |
R
2
|
| NH2-UiO-66 |
0.2791 |
30.67 |
0.9346 |
0.0236 |
30.64 |
0.9568 |
| BiOCl |
0.0567 |
1.43 |
1 |
0.0094 |
1.81 |
0.9793 |
| UB-10% |
0.2234 |
28.79 |
0.9633 |
0.0190 |
29.63 |
0.8801 |
| UB-30% |
0.1995 |
23.66 |
0.9446 |
0.0171 |
24.44 |
0.9705 |
| UB-50% |
0.1945 |
20.52 |
0.9436 |
0.0164 |
21.26 |
0.9804 |
| UB-70% |
0.1589 |
20.08 |
0.9538 |
0.0154 |
20.75 |
0.9706 |
Table 2 The best-fit model parameters of CIP adsorption isotherms onto samples
| Sample |
Langmuir model |
Freundlich model |
|
q
m (mg g−1) |
K
L (L mg−1) |
R
2
|
K
F (mg g−1)/(mg L−n) |
n
|
R
2
|
| NH2-UiO-66 |
91.20 |
0.0116 |
0.9724 |
4.0429 |
0.6765 |
0.9683 |
| BiOCl |
6.79 |
0.2600 |
0.7855 |
2.0811 |
0.1071 |
0.9047 |
| UB-10% |
79.90 |
0.0162 |
0.9679 |
4.628 |
0.6190 |
0.9541 |
| UB-30% |
68.55 |
0.0175 |
0.9640 |
4.2862 |
0.6016 |
0.9533 |
| UB-50% |
63.09 |
0.0125 |
0.9735 |
3.0673 |
0.6565 |
0.9777 |
| UB-70% |
56.66 |
0.0126 |
0.9921 |
3.2140 |
0.6233 |
0.9695 |
2.3. Optical and photoelectrochemical properties
The optical properties of the materials were studied using UV-visible diffuse reflectance spectroscopy (DRS). As shown in Fig. 4a, UV-vis DRS spectra demonstrated that NH2-UiO-66 and BiOCl materials show characteristic peaks at around 463 nm and 367 nm. However, the absorption edge of UB-50% was redshifted to 468 nm, indicating enhanced visible light absorption due to the formation of the NH2-UiO-66/BiOCl heterojunction. Generally, NH2-UiO-66 and BiOCl are considered as direct bandgap semiconductors, and the corresponding band gaps can be estimated based on the Kubelka–Munk equation with results of 2.82 eV and 3.5 eV for NH2-UiO-66 and BiOCl, respectively (Fig. 4b).28,29 According to Mott–Schottky curves (Fig. 4c and d), NH2-UiO-66 and BiOCl had positive slopes demonstrating that they were n-type semiconductors.30,31 Therefore, the flat band potential was approximately considered to be the fermi level (Ef). Thus, the Ef values of NH2-UiO-66 and BiOCl were −0.34 eV and −0.32 eV (vs. NHE) (Efb-NHE = EAg/AgCl + E0Ag/AgCl), respectively. Furthermore, The VB-XPS of NH2-UiO-66 and BiOCl are shown in Fig. 4e and f. They indicate that the energy gaps between the Ef and the VB were 2.47 eV and 2.34 eV for NH2-UiO-66 and BiOCl, respectively.32 Therefore, the EVB values of NH2-UiO-66 and BiOCl were calculated to be 2.13 eV and 2.02 eV, respectively. According to the equation (ECB = EVB − Eg), the ECB values of NH2-UiO-66 and BiOCl were −0.69 eV and −1.48 eV.33
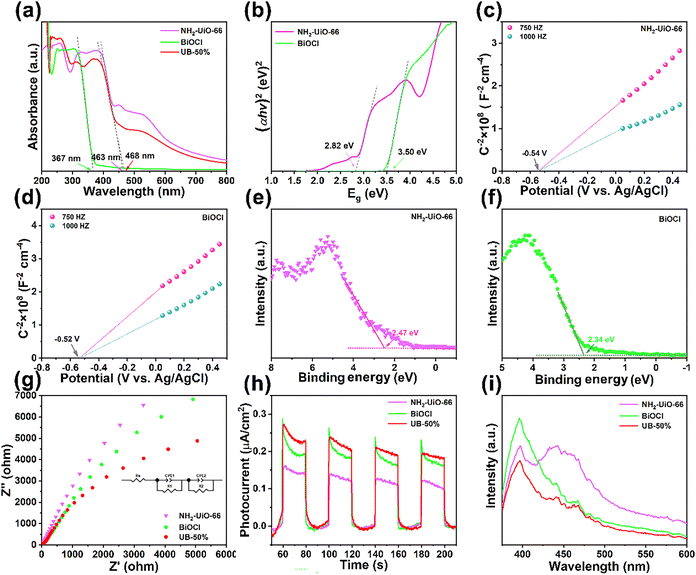 |
| | Fig. 4 UV-vis DRS spectra (a), the (αhν)2versus Eg curves (b), Mott–Schottky plots (c) and (d), VB-XPS spectra (e) and (f), EIS Nyquist spectra (g), transient photocurrent densities (h) and photoluminescence spectra (i). | |
Photoelectrochemical analysis of UB-50% was conducted using transient photocurrent responses, and the electrochemical impedance spectroscopy (EIS) results for equivalent circuits are presented in Table S2 (ESI†). The solution resistance (Rs) was considered as the resistance of the electrolyte/material interface, which was determined by the intercept on the Z′ axis. The charge transfer resistance (Rct) could be ascertained from the semicircle radius.35 As shown in Fig. 4g, UB-50% showed the minimum semicircle radius, demonstrating the best photogenerated charge separation and transfer efficiency. Transient photocurrent responses were used to further study the charge transfer of UB-50%. As shown in Fig. 4h, UB-50% showed the best photocurrent response.34 Furthermore, the photoluminescence (PL) spectroscopy results showed that UB-50% had the weakest PL intensity, suggesting that the heterojunction generated between NH2-UiO-66 and BiOCl could reduce the recombination of photoinduced carriers and accelerate the transition and separation of e− and h+, which is consistent with the results of EIS and transient photocurrent responses (Fig. 4i).36,37
2.4. Photocatalytic performance
The photocatalytic performance of the as-prepared catalysts was evaluated to determine their light-driven CIP degradation efficiency, and optimal catalyst dosage and pollutant concentration were determined to find the optimal reaction parameters (Fig. 5). The removal efficiency of CIP decreased from 96.84% to 59.50% in 60 min with the concentration of CIP increasing from 10 mg L−1 to 60 mg L−1 (Fig. 5a) because of the insufficient active species generated from the limited amount of UB-50%. According to Fig. 5b, the degradation of CIP by UB-50% increased by about 45% as the catalyst dosage increased from 5 mg to 30 mg. In general, increasing the amount of the catalyst brings more reaction sites, which can promote photocatalytic activity. However, when the photocatalyst dosage was increased from 25 mg to 30 mg, the photodegradation efficiency was not significantly improved. According to previous studies, the increase of the photocatalyst concentration in the solution caused a shielding effect and reduced the light penetration, which was not beneficial to the photoreaction process.
 |
| | Fig. 5 Different concentrations (a), catalyst dosages of CIP (b), photocatalytic activity (c), first-order kinetic plots (d), and rate constants of NH2-UiO-66, BiOCl and UB-x (e). Different antibiotics (f), FT-IR spectra (g), and ion co-existence for photocatalytic activity (h). Schematic diagram of the adsorption of UB-50% (i). | |
Based on the optimized conditions of a CIP concentration of 20 mg L−1 and a catalyst dosage of 25 mg and the comparison of catalysts (NH2-UiO-66/BiOCl/UB-x), the universality, reusability and the photocatalytic performance of UB-50% were systematically studied. As seen in Fig. 5c and d, the degradation efficiency of CIP by BiOCl was only 74.30%. The degradation efficiencies of NH2-UiO-66, UB-10%, UB-30%, UB-50%, and UB-70% were 81.50%, 83.81%, 91.45%, 96.84%, and 91.09%, respectively. In addition, the photodegradation removal rate of CIP by UB-50% was higher than those by NH2-UiO-66 and BiOCl. The illumination time and removal efficiency of UB-x could linearly fit well with the first-order kinetics model (−ln(Ct/C0) = kt).38 The removal rate constants (k) are shown in Fig. 5e. The highest k value was 0.0432 min−1 for UB-50%, which was 3.6 times that of NH2-UiO-66 (0.012 min−1) and 2.1 times that of BiOCl (0.0209 min−1). Furthermore, with the increase of the ratio of NH2-UiO-66/BiOCl, the removal efficiency of CIP increased at first and then decreased, and UB-50% displayed the best degradation efficiency. Initially, the heterojunction of UB-x increases with the content of NH2-UiO-66, promoting the separation of photogenerated charges and migration rates. However, excessive NH2-UiO-66 covering the surface of BiOCl could affect the light absorption of BiOCl, causing the decline of degradation efficiency.
In order to investigate the universality of the UB-50% catalyst, CIP, tetracycline (TC) and sulfamethoxazole (SMX) were chosen as the target pollutants (Fig. 5f). The adsorption efficiency of UB-50% was evaluated in an antibiotic solution obtained by stirring for 60 min, and its adsorption reaches equilibrium in the dark. In addition, the adsorption capacity of UB-50% was less than 2% in SMX, about 60% in CIP and more than 90% in TC. In order to further explore the adsorption mechanism of UB-50%, in the aqueous solution of pH = 7 of UB-50%, the surface electric charge was determined by zeta potential. The zeta potential of UB-50% was negative (Fig. S1, ESI†). The two ionizable groups (carboxyl group and secondary amino group) of CIP could display different forms under acidic or alkaline conditions. CIP and TC existed in an amphoteric state in solution of pH = 7, and the positive charges are slightly more than the negative charges. Three mainly dissociating groups (tricarbonyl, phenolic dione and dimethylamino) of TC easily formed hydrogen bonds and electron donor–acceptor complexes, thus showing hydrophilicity and obvious adsorption.39 Therefore, UB-50% exhibits excellent adsorption for TC. As SMX mainly shows negative charge under pH = 7 condition, it is difficult to adsorb SMX via electrostatic interactions by UB-50%.40 Therefore, UB-50% can show 100% degradation efficiency for TC but only 53.53% for SMX under the above optimal reaction conditions after 60 min. The result indicates its excellent removal ability for positive antibiotics. Moreover, Fig. 5g shows the FT-IR spectra obtained before and after the adsorption of CIP on UB-50%. The absorption peaks at 3428 cm−1 and 3355 cm−1 were ascribed to the asymmetric and symmetric N–H stretching vibrations and N–H bending vibration of aromatic amines. The shift of the characteristic peaks of N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C indicates the existence of the hydrogen-bond interaction and π–π interaction between CIP and UB-50%.41
C indicates the existence of the hydrogen-bond interaction and π–π interaction between CIP and UB-50%.41
2.5. Factors affecting degradation activity
The actual water environment is composed of multiple components; therefore, the representative ions (Mg2+, Ca2+, K+, Cl−, and SO42−) were added to the reaction system to explore the effect of ions on degradation of CIP by UB-50%. As shown in Fig. 5h, UB-50% presented satisfactory adsorption performance in CIP solution with Mg2+, K+, Cl− and SO42− except Ca2+. The results showed that anions (Cl− and SO42−) had little influence on CIP adsorption. Meanwhile, cations coordinate with the functional groups of UB-50%, promoting the process of adsorption, which is mainly due to the cation chelation on CIP after addition of cations leading to rapid adsorption by UB-50%. In addition, the electrostatic repulsion effect of SO42− and Cl−, and negatively charged UB-50% weaken adsorption site competition. It can be concluded that Mg2+, K+, Cl− and SO42− had slightly enhanced the adsorption of CIP. However, Ca2+ likely occupied the adsorption sites of UB-50%, obstructing the adsorption of CIP.42,43 It can be seen that cations (Mg2+, K+ and Ca2+) had slight impacts on the CIP photoreaction, except for Cl− and SO42−. This was because SO42− and Cl− ions could act as radical scavengers, as shown in eqn (1) and (2).44,45 As shown in Fig. 5i, chemisorption (hydrogen bonding and π–π interaction) and physisorption (electrostatic interaction) can be considered as the probable mechanisms of adsorption.| | | SO42−/Cl− + ˙OH → SO4˙−/ClOH˙− + OH− | (1) |
| | | SO42−/ClOH˙−+ h+ → SO4˙−/Cl˙ | (2) |
2.6. The stability and photocatalytic mechanism
In practical applications, the stability and repeatability are of vital importance for photocatalysts. Thus, the stability of UB-50% was studied and the results are shown in Fig. 6a. The photocatalytic activity of UB-50% had no obvious loss after 5 cycles. Additionally, the fresh and used UB-50% tested by XRD revealed that the crystal structure and microstructure had no significant changes (Fig. 6b). The results indicate that UB-50% has excellent stability in degradation of antibiotics. The photodegradation mechanism and active species generated by UB-50% were investigated through radical trapping experiments on CIP. The quenching experiment employed t-butanol (t-BuOH), benzoquinone (BQ), and methanol (MeOH) as scavengers for the detection of ˙OH, ˙O2−, and h+, respectively (Fig. 6c).46 Compared to the removal efficiency of CIP without scavengers, the addition of t-BuOH and MeOH scavengers slightly restrained CIP degradation, indicating that ˙OH and h+ are not the key active species. Meanwhile, the degradation ratios of CIP obviously dropped to 18.39% with the addition of BQ. The results suggest that ˙O2− plays the main role in the photocatalytic degradation of CIP, and ˙OH radicals and h+ play an auxiliary role in the degradation process.
 |
| | Fig. 6 Recycle test (a), XRD patterns (b), active species elimination experiments (c), and the possible mechanisms of UB-50% for CIP removal (d). | |
NH2-UiO-66 and BiOCl as single photocatalysts can still excite active species (˙OH, ˙O2−, and h+) for CIP degradation. However, due to the high photogenerated electron–hole complexation efficiency of single-component photocatalysts (NH2-UiO-66 and BiOCl), the active substances could not be generated efficiently. The energy band structure and the above-mentioned results of UB-50% indicate that NH2-UiO-66 and BiOCl easily form a type II heterojunction (Fig. 6d). After NH2-UiO-66 and BiOCl come into contact, e− flows from high Ef (BiOCl) to low Ef (NH2-UiO-66) until the Ef is aligned. Due to electron accumulation, the NH2-UiO-66 side energy band is negatively charged and bent downward. Due to hole accumulation, the BiOCl side energy band is bent upwards and positively charged, causing the formation of a built-in electric field.47,48 The photogeneration of electron–hole pairs over BiOCl and NH2-UiO-66, respectively, occurred upon the irradiation of UB-50% with full-light (eqn (3)). The photogenerated electrons from the CB position of BiOCl transferred to that of NH2-UiO-66 (eqn (4)), and the holes moved from NH2-UiO-66 to BiOCl (eqn (5)). Subsequently, the electrons on the surface of UB-50% reacted with O2 to generate ˙O2− (eqn (6)) and H2O reacted with the holes to generate ˙OH (eqn (7)), which oxidised CIP into CO2, H2O, and some small organic molecules (eqn (8)).
| | | UB-50% + hν → UB-50% (h+ + e−) | (3) |
| | | e− (BiOCl) → e− (NH2-UiO-66) | (4) |
| | | h+ (NH2-UiO-66) → h+ (BiOCl) | (5) |
| | | e− (NH2-UiO-66) + O2 → ˙O2− | (6) |
| | | h+ (BiOCl) + H2O → ˙OH + H+ | (7) |
| | | •OH + h+ + •O2− (UB-50%) + CIP → degradation products | (8) |
3. Conclusions
In summary, UB-50% (NH2-UiO-66/BiOCl) was synthesized using a facile one-step green grinding method at room temperature. CIP was selected as a model molecule to evaluate the adsorption and photocatalytic performance of NH2-UiO-66/BiOCl. Among the tested composites, UB-50% showed excellent adsorption and photocatalytic properties. The composite exhibited excellent stability, as evidenced by repeatability experiments. The enhanced photocatalytic activities of UB-50% can be attributed to its remarkable adsorption capacity and the suitable construction of a heterojunction, which improves electron–hole separation and transfer efficiency. Additionally, ˙O2− was identified as the main active species. Subsequently, the adsorption and photocatalytic mechanisms of the composite for CIP were analyzed. This work proposes a new approach for achieving adsorption and photocatalytic degradation synergistically.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
The data supporting this article have been included as part of the ESI.†
Acknowledgements
The authors acknowledge funding from the National Natural Science Foundation of China (52303376), the Young Science and Technology Star Project of Shaanxi Province (2024), and The Youth Innovation Team of Shaanxi Universities (24JP027).
References
- J. Wu, X. Y. Yang, X. H. Jia, J. Yang, X. Miao, D. Shao, H. J. Song and Y. Li, Chem. Eng. J., 2023, 471, 144684 Search PubMed.
- X. L. Liu, B. B. Huang, J. Li, B. J. Li and Z. Z. Lou, Mater. Horiz., 2024, 11, 5470–5498 RSC.
- H. Xu, J. R. Li and X. X. Chu, Nanoscale Horiz., 2023, 8, 441–452 RSC.
- T. Guo, X. Chen and L. F. Yin, J. Mater. Chem. A, 2024, 12, 4397–4420 RSC.
- G. Shahnazarova, N. A. H. A. Bast, J. C. Ramirez, J. Nogues, J. Esteve, J. Fraxedas, A. Serra, M. J. Esplandiu and B. Sepulved, Mater. Horiz., 2024, 11, 2206–2216 Search PubMed.
- X. Y. Li, S. H. Lin, R. R. Hu, P. Liang, Q. Y. Wu, B. B. Yang, S. Q. Lin, Q. L. Xu, J. N. Mei and J. Zou, J. Opt. Photonics Res., 2025 DOI:10.47852/bonviewJOPR52023222.
- Y. Li, X. Y. Yang, M. Y. Wang, D. Y. Yue, J. Wu and H. J. Song, Sep. Purif. Technol., 2025, 361, 131567 Search PubMed.
- Y. Li, X. Y. Yang, S. Yan, J. Yang, X. H. Jia and H. J. Song, ACS Appl. Mater. Interfaces, 2024, 16, 1794–1804 CrossRef.
- Y. C. Pan, L. J. Yang, G. Wang, H. Li, S. S. Wang, L. Zhang, W. Y. Wei and J. M. Lu, ACS Appl. Mater. Interfaces, 2024, 16, 48836–48845 CrossRef CAS.
- O. Allam, M. Maghsoodi, S. S. Jang and S. D. Snow, ACS Appl. Mater. Interfaces, 2024, 16, 36215–36223 CrossRef CAS.
- W. Wang, B. Ibarlucea, C. H. Huang, R. H. Dong, M. A. Aiti, S. R. Huang and G. Cuniberti, Nanoscale Horiz., 2024, 9, 1432–1474 RSC.
- X. L. Huang, Y. Xing, H. Jiang, Y. Pu, S. Yang, Z. S. Kang and L. Cai, J. Hazard. Mater., 2024, 472, 134502 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, S. Du, J. L. Ren, H. Jiang, L. B. Zhang and J. T. Zhu, ACS Appl. Mater. Interfaces, 2023, 15, 35884–35894 CrossRef CAS PubMed.
- Y. Zhang, F. Y. Ma, M. H. Ling, H. Zheng, Y. T. Wu and L. Li, Chem. Eng. J., 2023, 464, 142762 CrossRef CAS.
- Y. D. Luo, G. Y. Shi, S. H. Yu, Z. T. Liu, J. W. Yin, M. S. Xue, Q. Sun, F. F. Shen, X. B. Li, Z. Z. Yin, C. Xie and B. Gao, Chem. Eng. J., 2023, 473, 145198 CrossRef CAS.
- M. M. Li, J. W. Yuan, G. Wang, L. J. Yang, J. X. Shao, H. Li and J. M. Lu, Sep. Purif. Technol., 2022, 298, 121658 CrossRef CAS.
- C. Dai, R. Z. Hu, C. M. Wang, Z. Liu, S. J. Zhang, L. D. Yu, Y. Chen and B. Zhang, Nanoscale Horiz., 2020, 5, 857–868 RSC.
- S. Z. Zheng, H. Du, L. X. Yang, M. Tan, N. Y. Li, Y. J. Fu, D. Hao and Q. Wang, J. Hazard. Mater., 2023, 447, 130849 CrossRef CAS PubMed.
- Z. Q. Long, H. L. Wang, K. W. Huang, G. M. Zhang and H. J. Xie, J. Hazard. Mater., 2022, 424, 127554 CrossRef CAS PubMed.
- S. Y. Wang, Z. W. Ai, X. W. Niu, W. J. Yang, R. Kang, Z. G. Lin, A. Waseem, L. Jiao and H. L. Jiang, Adv. Mater., 2023, 35, 2302512 CrossRef CAS PubMed.
- Y. R. Chen, D. Y. Zhu, S. K. Xue, H. Y. Wang, Q. J. Lu, G. H. Ruan, C. X. Zhao and F. Y. Du, Appl. Surf. Sci., 2024, 653, 159337 CrossRef CAS.
- I. Akpinar, X. L. Wang, K. Fahy, F. R. Sha, S. L. Yang, T. W. Kwon, P. J. Das, T. Islamoglu, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2024, 146, 5108–5117 CrossRef CAS PubMed.
- Z. H. Zhao, H. Wang, J. J. Li, X. Y. Qiao, Z. P. Liu, Z. P. Ren, M. L. Yuan and J. Zhang, J. Am. Chem. Soc., 2024, 146, 29441–29449 CrossRef CAS PubMed.
- B. H. Wang, J. F. Chen, J. B. Zhong, J. Z. Li and Y. B. Zhu, Appl. Surf. Sci., 2024, 644, 158750 Search PubMed.
- W. J. Qu, Z. L. Wang, M. Y. Qin, X. J. Yang, F. S. Zhang, Z. Wang, D. D. Ji and D. H. Yu, Sep. Purif. Technol., 2023, 325, 124673 CrossRef CAS.
- L. H. Meng, C. Zhao, T. Y. Wang, H. Y. Chu and C. C. Wang, Sep. Purif. Technol., 2023, 313, 123511 CrossRef CAS.
- Y. L. Dong, H. R. Liu, S. M. Wang, G. W. Guan and Q. Y. Yang, ACS Catal., 2023, 13, 2547–2554 Search PubMed.
- M. Pander, R. Gil-San-Millan, P. Delgado, C. Perona-Bermejo, U. Kostrzewa, K. Kaczkowski, D. J. Kubicki, J. A. R. Navarro and W. Bury, Mater. Horiz., 2023, 10, 1301–1308 RSC.
- S. A. Ali, S. Sarkar and A. K. Patra, ACS Appl. Mater. Interfaces, 2024, 16, 38061–38072 CrossRef CAS PubMed.
- A. Kuila, S. Pichiah, D. Bahnemann and C. Y. Wang, Appl. Catal., B, 2021, 293, 120224 CrossRef CAS.
- Q. Wang, S. Z. Zheng, W. G. Ma, J. Y. Qian, L. Y. Huang, H. Deng, Q. Zhou, S. R. Zheng, S. J. Li, H. Du, Q. Li, D. Hao and G. X. Yang, Appl. Catal., B, 2024, 344, 123669 CrossRef CAS.
- F. Zhao, D. Gao, X. M. Zhu, Y. M. Dong, X. Liu and H. X. Li, Appl. Surf. Sci., 2022, 587, 152780 CrossRef CAS.
- H. S. Zheng, B. Y. Zi, T. Zhou, G. Y. Qiu, Z. G. Luo, Q. J. Lu, A. R. P. Santiago, Y. M. Zhang, J. H. Zhao, J. Zhang, T. W. He and Q. J. Liu, Nanoscale Horiz., 2024, 9, 1532–1542 RSC.
- F. H. Xu, Q. Q. Zhang, R. S. An, L. Li and L. M. Zhou, J. Alloys Compd., 2022, 899, 163324 CrossRef CAS.
- Y. Jiang, S. C. Liu, L. P. Zhang, G. W. Guan, Y. T. Li, S. Ni, R. Y. Jiang, S. T. Zheng, H. R. Liu, H. L. Lan and Q. Y. Yang, Chem. Eng. J., 2024, 494, 153100 CrossRef CAS.
- S. M. Obaidulla, A. Supina, S. Kamal, Y. Khan and M. Kralj, Nanoscale Horiz., 2024, 9, 44–92 RSC.
- R. Li, Y. Li, X. H. Jia, J. Yang, X. Miao, D. Shao, J. Wu and H. J. Song, Desalination, 2024, 574, 117295 CrossRef CAS.
- W. Y. Zhen, Y. Liu, X. D. Jia, L. Wu, C. Wang and X. Jiang, Nanoscale Horiz., 2019, 4, 720–726 RSC.
- Q. Q. Ni, X. Ke, W. J. Qian, Z. Yan, J. D. Luan and W. G. Liu, Appl. Catal., B, 2024, 340, 123226 CrossRef CAS.
- B. Zhang, S. Q. Xia, Z. Y. Wang, W. Q. Li, B. D. Li, H. C. Zhang, Y. Xin, J. Wu, K. J. Ma and X. He, Appl. Catal., B, 2024, 349, 123861 Search PubMed.
- X. Z. Jiang, M. Wang, Z. C. Lou, H. Han, N. N. Yan, Q. B. Guan and L. Xu, ACS Nano, 2024, 18(3), 2290–2301 CrossRef CAS.
- J. H. Gu, Z. Liu, A. Y. Jia, Y. Q. Wang, N. N. Li, Z. S. Liu, Y. X. Li and H. X. Zhang, Sep. Purif. Technol., 2023, 312, 123408 CrossRef CAS.
- J. H. Sha, L. L. Li, Z. X. An, M. X. He, H. Y. Yu, Y. Wang, B. Y. Gao and S. P. Xu, Chem. Eng. J., 2022, 428, 131065 CrossRef CAS.
- M. Abinaya, K. Govindan, M. Kalpana, K. Saravanakumar, S. L. Prabavathi, V. Muthuraj and A. Jang, J. Hazard. Mater., 2020, 397, 122885 Search PubMed.
- L. L. Liang, S. W. Gao, J. C. Zhu, L. J. Wang, Y. N. Xiong, X. F. Xia and L. W. Yang, Chem. Eng. J., 2020, 391, 123599 CrossRef CAS.
- Y. Y. Gu, J. X. Wang, Q. Q. Tang, H. T. Wei, J. Ning, X. F. Lan, X. F. Wang, X. H. Li, Y. G. Jia, S. B. Wang and L. Hao, ACS Catal., 2024, 14, 11262–11272 Search PubMed.
- T. Guo, G. J. Hu, C. W. Wang, J. X. Liu, G. M. Ren and Q. J. Guo, J. Alloys Compd., 2025, 1022, 179965 Search PubMed.
- M. Wen, Q. Guo, X. L. Liu, X. Li, H. P. Li, H. J. Zhan and W. Y. Liu, Polyhedron, 2025, 271, 117420 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Xinyue
Yang
a,
Deyun
Yue
a,
Xiao
Miao
b,
Mengyao
Wang
a and
Haojie
Song
*a,
Xinyue
Yang
a,
Deyun
Yue
a,
Xiao
Miao
b,
Mengyao
Wang
a and
Haojie
Song





