
Cu–phytic acid nanozyme-induced cuproptosis therapy for the inhibition of tumor growth†
Xiao-Wan Han,
Xu Chen,
Tian-Le Yang,
Ying-Yi Luo,
Rui-Xue Liang*,
San-Qi An* and
Xin-Li Liu *
*
Life Sciences Institute, Guangxi Medical University, Nanning 530021, China
First published on 1st July 2025
Abstract
Cuproptosis has recently received much attention in cancer treatment. However, copper ionophores do not show any obvious clinical efficacy. Although some Cu-based and copper ionophore-loaded nanomaterials have been applied to induce cuproptosis, it is difficult to achieve their clinical translation as they are limited by their complicated composition, harsh synthesis conditions, requirement of external stimuli, and potential biotoxicity. Phytic acid, a naturally occurring organic phosphorus carbohydrate, possesses a distinct antineoplastic effect on multiple types of cancer and high biocompatibility. Based on metal–phosphonate coordination, a novel Cu–phytic acid nanozyme (denoted as CP) with a pH/GSH dual response was fabricated by a “one-pot” method. CP with three enzyme-mimicking activities enhanced cuproptosis therapy through GSH depletion, reactive oxygen species augmentation, hypoxia relief and the attenuation of glycolysis. As a proof of concept, Elesclomol (a copper ionophore)-resistant A549 cells were used to investigate CP-induced cuproptosis for the inhibition of tumor growth in vitro and in vivo.
New conceptsThe study of metal-based nanozymes in tumor therapy predominantly focuses on the construction of nanozymes using transition metals with catalytical properties or noble metals. The combinational approach of a natural carbohydrate with pharmacological effects and metal ions may reform the therapeutic efficacy, modalities, and biosafety of nanozymes in therapy. The advantage of these nanozymes is not limited to the catalytic properties; it would show a multi-pathway effect on disease treatment with the functional ligand introduced. We selected phytic acid, a naturally occurring organic phosphorus carbohydrate with antitumor properties and excellent biocompatibility. The integration of copper and phytic acid in the form of nanozymes can address the limitations of metal ions or phytic acid used alone, such as poor tumor accumulation, rapid blood circulation, and systemic toxicity. More importantly, the nanozyme not only exhibited multienzyme-like activities, but also showed a dual-response to the tumor microenvironment, releasing copper ions and phytic acid to proceed its distinct functions in tumor suppression. Therefore, the Cu–phytic acid nanozyme possesses diversified functions that Cu or phytic acid alone cannot exert. In view of the simple methods of fabrication and inherent biocompatibility and efficacy of the components, this work developed a new perspective on nanozymes in tumor treatment. |
Introduction
Cuproptosis has been identified as a copper-dependent programmed cell death.1,2 However, copper ionophores such as Elesclomol and Disulfiram show unsatisfactory clinical efficacy, probably due to their rapid blood clearance and weak tumor-targeting capacity.3–5 Hence, various Cu-based nanomaterials have been applied to boost intracellular copper accumulation and improve the tumor-targeting capacity of copper ionophores.6–10 Furthermore, the combinational therapies of cuproptosis with other cell deaths (e.g., apoptosis, ferroptosis, and pyroptosis)11–13 or therapeutic strategies (e.g., photothermal/photodynamic/sonodynamic therapy, chemoradiotherapy and immunotherapy) have made breakthrough progress for cancer treatment.14–19 However, the majority of these nanomaterials rely on the complex compositions, harsh preparation conditions, external stimuli, and uncertain biosafety, which hinder their clinical translation. Therefore, developing an effective, simple, and safe cuproptosis inducer remains a significant challenge.Phytic acid (PA) is a naturally polyphosphorylated carbohydrate, which is mostly present in cereals, legumes, and nuts. It has been proven that PA exhibits broad antitumor effects.20–22 PA-doped nanomaterials have been employed in cancer treatment.23–26 Recently, with PA being an attractive building block, the versatility of the metal–PA assembly strategy has been explored.27 In view of the antineoplastic property and high biocompatibility of PA, a Cu–PA nanozyme (CP) based on metal–phosphonate coordination was fabricated by a facile “one-spot” method for cancer treatment via cuproptosis (Scheme 1). The sodium hyaluronate (HA) modification not only regulates the particle size and enhances the enzyme-like property but also augments the tumor-targeting capability of CP via the CD44 receptor pathway.10 As shown in Scheme 1, CP exhibits three enzyme-mimicking activities, including glutathione peroxidase (GPx), peroxidase (POD) and catalase (CAT). First, CP with POD-like activity could generate excess reactive oxygen species (ROS), which would disturb intracellular redox homeostasis, and trigger cell death. Second, CP with CAT-like activity could alleviate tumor hypoxia by catalytic decomposition of the overexpressed H2O2 to O2 at the tumor site.28 Third, CP with GPx-like activity can deplete high levels of glutathione (GSH) in cancer cells, thereby reducing its ability to bind copper ions and releasing more free copper ions to induce cuproptosis.29 In brief, the multiple enzyme-mimicking activities of CP can boost cuproptosis. Moreover, CP exhibited degradation behavior in acidic GSH solution. It is hypothesized that the pH/GSH dual-responsive CP enables the release of Cu2+ ions triggering cuproptosis in cancer cells and reducing side effects on normal tissue.
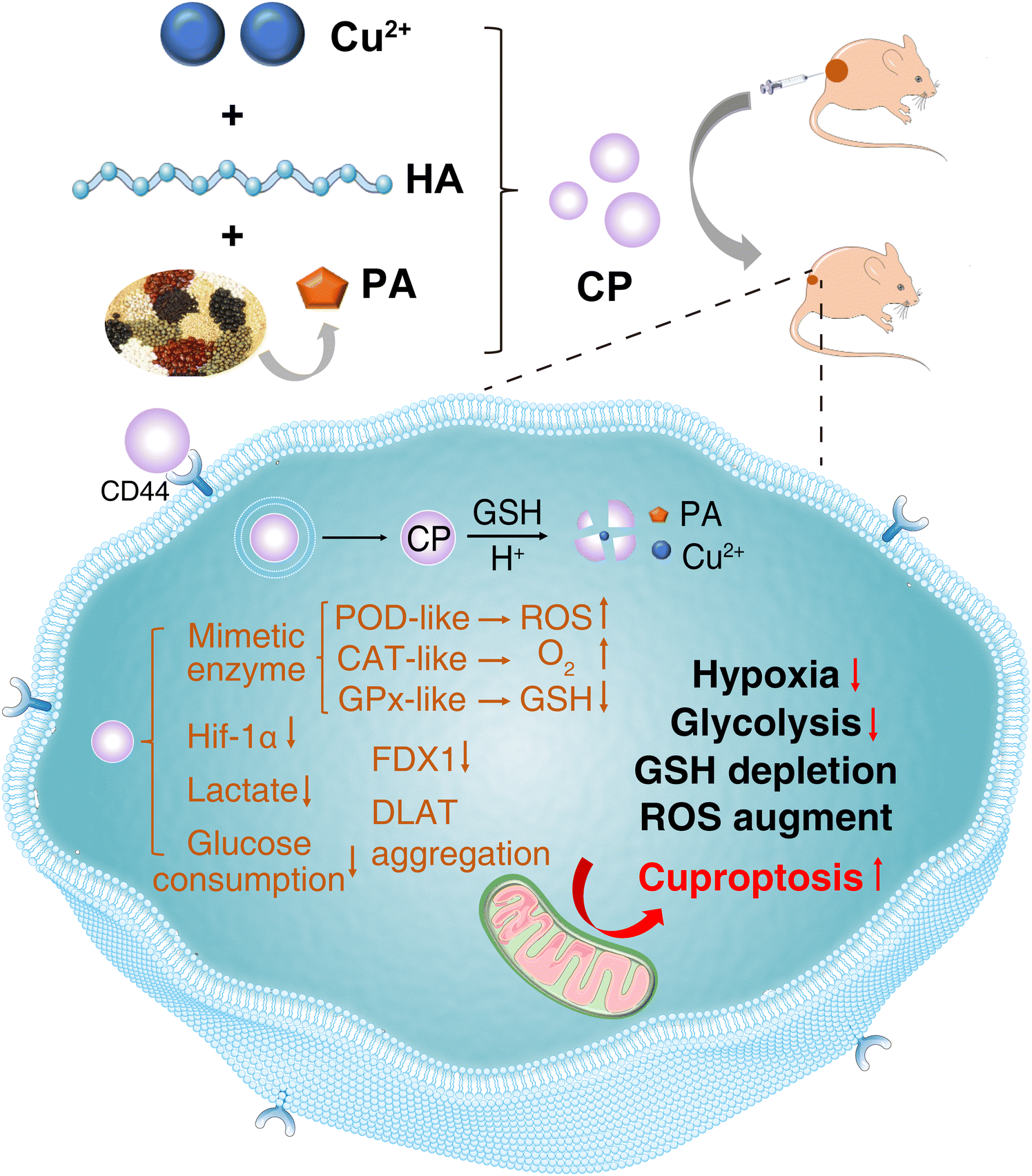 | ||
| Scheme 1 Schematic illustration displaying the synthesis and antitumor mechanism of CP. | ||
Most solid cancers prefer aerobic glycolysis and exhibit local hypoxia, which inhibits cuproptosis in cancer cells.2 Some strategies have been proposed to boost cuproptosis therapy. For instance, some Cu-doped nanomaterials can alleviate tumor hypoxia through catalase immobilization or intrinsic CAT-like activity.30,31 Furthermore, Cu-based nanocomposites can also act as targeted delivery systems of glycolysis inhibitors and genomics tools for the suppression of glycolysis in cancer cells.32–34 However, there is less research on the simultaneous inhibition of hypoxia and glycolysis for cuproptosis-enhancement. Based on current knowledge, it has been reported that hyperbaric oxygen can sensitize cancer stem cells to cuproptosis by regulating hypoxia and glycolysis.35 According to the reports, IP6 (inositol hexakisphosphate, classified as PA) treatment could reduce glycolysis in osteosarcoma and medulloblastoma.36,37 Therefore, we suppose that CP would enhance the sensitivity of tumors to cuproptosis through hypoxia relief and the regulation of glycolysis by its inherent properties.
It has been reported that non-small cell lung cancer (NSCLC) A549 cells are insensitive to Elesclomol-induced cuproptosis.2 As a proof of concept, we assumed that a pH/GSH dual-responsive CP with multienzyme activities would induce cuproptosis in A549 cells and improve the sensitivity of A549 cells to Elesclomol. Furthermore, CP-induced inhibition of A549 cells was investigated in vitro and in vivo.
Results and discussion
Synthesis and characterization
The CP was fabricated by a facile “one-pot” method shown in Experimental Section 4.3. The TEM images revealed that CP exhibited a spherical morphology with an average size of approximately 6.1 ± 1.1 nm (Fig. 1(A) and (B)). However, CP without HA coating (denoted as CPW) agglomerated and presented poor dispersibility (Fig. S1, ESI†). HA modification not only enables CP to acquire a small and narrow size distribution but also improves the dispersibility of CP. The zeta potential of CP was measured to be −42.7 mV, which is more negative than that of CPW (−17.5 mV) (Fig. 1(C)). This confirmed that CP was coated with negatively charged HA. The short-term stability of CP within 72 h was evaluated by using dynamic light scattering (DLS). As shown in Fig. S2 (ESI†), CP was stable in PBS (pH = 7.4) and 10% serum without obvious aggregation. The results exhibited the good stability of CP under physiological conditions. The results of EDS further revealed the elemental composition of CP, indicating the presence of Cu, P, O, C, and N elements (Fig. S3, ESI†). The Cu content in CP was approximately 47.4% (mass ratio), as determined by ICP-OES.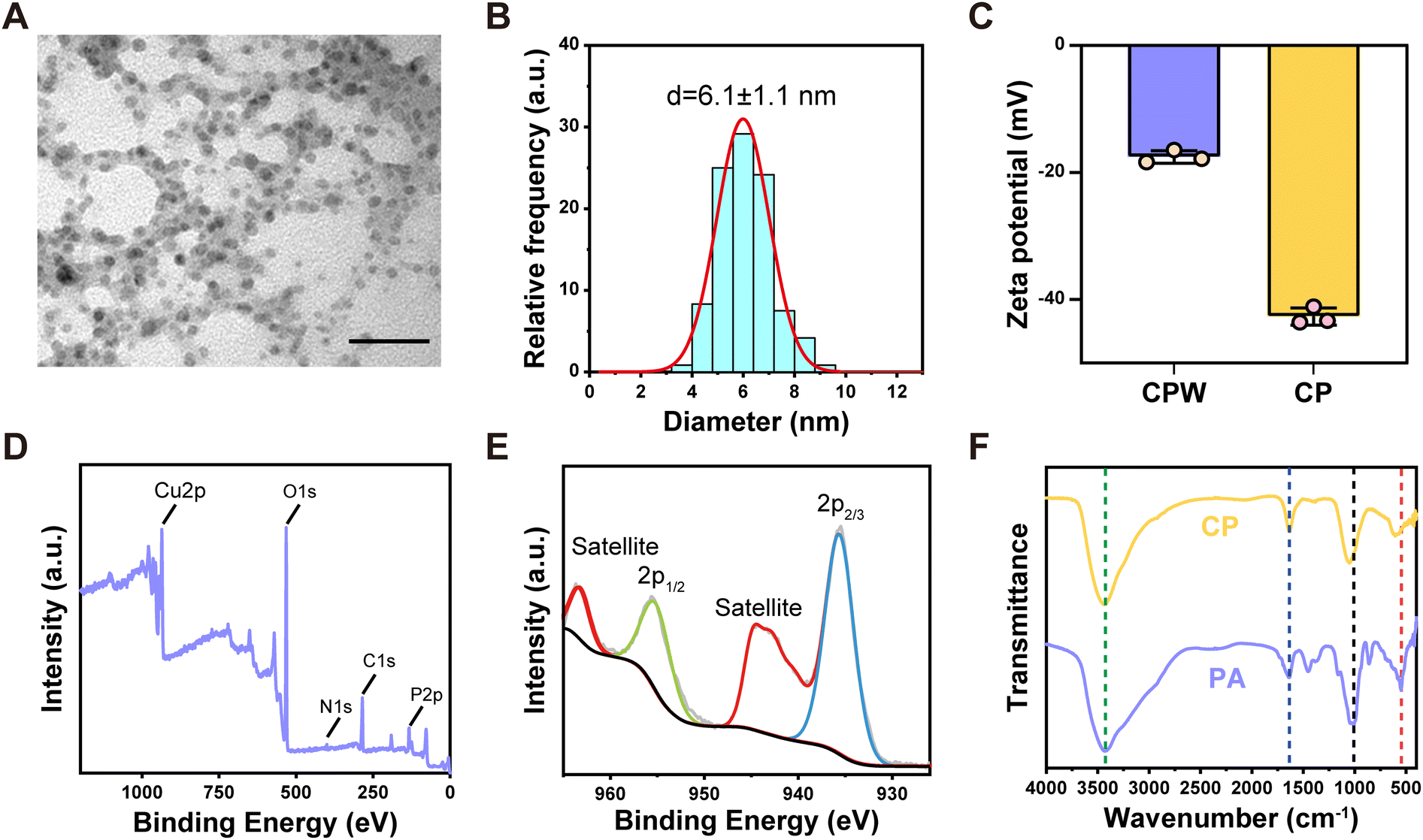 | ||
| Fig. 1 Characterization of CP. (A) The TEM image of CP (scale bar: 50 nm). (B) The particle size analysis of CP. (C) The zeta potential histograms of CPW and CP (mean ± s.d., n = 3). (D) The XPS spectra of CP. (E) High-resolution Cu2p XPS spectra of CP. (F) The FTIR spectra of PA and CP. | ||
There was only one wide peak in the range of 20° to 40° in the XRD pattern of CP, and no other sharp peaks appeared (Fig. S4, ESI†), revealing the amorphous structure of CP. The XPS spectra of CP verified the existence of Cu 2p, P 2p, N 1s, C 1s and O 1s peaks (Fig. 1(D)).24 Furthermore, from the Cu 2p XPS spectrum (Fig. 1(E)), the characteristic peaks at 935.6 eV (Cu 2p3/2) and 955.1 eV (Cu 2p1/2), as well as two satellite peaks are assigned to Cu2+.38 The FTIR spectra of PA (Fig. 1(F)) showed an O–H bond broad absorption peak at ∼3431 cm−1. The characteristic bands at ∼1639 cm−1 and 1455 cm−1 represent P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibration peaks. The most prominent peaks at ∼1010 cm−1, 859 cm−1 and 547 cm−1 are attributed to P–O bond vibration peaks.24,39–42 The addition of Cu2+ lead to some changes in the vibration of COPO3. Compared with PA, some typical peaks (nearby 1455 cm−1 and 859 cm−1) disappeared or shifted to higher frequencies (from 1010 cm−1 to 1050 cm−1, and from 547 cm−1 to 606 cm−1) in the FTIR spectrum of CP, indicating the coordination between PA and Cu2+.27,43 The above results confirmed the successful preparation of CP.
O bond vibration peaks. The most prominent peaks at ∼1010 cm−1, 859 cm−1 and 547 cm−1 are attributed to P–O bond vibration peaks.24,39–42 The addition of Cu2+ lead to some changes in the vibration of COPO3. Compared with PA, some typical peaks (nearby 1455 cm−1 and 859 cm−1) disappeared or shifted to higher frequencies (from 1010 cm−1 to 1050 cm−1, and from 547 cm−1 to 606 cm−1) in the FTIR spectrum of CP, indicating the coordination between PA and Cu2+.27,43 The above results confirmed the successful preparation of CP.
pH/GSH dual response of CP
Fig. 2(A) illustrates that CP obviously agglomerated at pH 5.5. In addition, the entire structure of CP collapsed under acidic pH and GSH conditions (Fig. 2(B)). As shown in Fig. S5A (ESI†), CP degraded at 48 h in acidic GSH solution, while its degradation behaviour could not be observed in PBS solution (pH = 7.4), almost like that in aqueous solution. The particle size distribution of CP under different conditions was measured by DLS (Fig. S5B, ESI†). The hydrodynamic diameters significantly increased in both the acidic GSH group and the acidic group. The particle size distribution of CP in the acidic GSH group became broader than that in the pH 5.5 group, while it did not change significantly in the pH7.4 PBS group. According to the previous TEM and DLS data, these results revealed that CP was stable under physiological conditions and degraded in acidic GSH solution. Then, the Cu release behaviour of CP was studied using ICP-OES. In acidic GSH solution, the release of Cu was approximately 50% at 48 h, while there was no significant release of copper at pH 7.4 (Fig. S5C, ESI†). | ||
| Fig. 2 Verification of CP properties. The TEM images of CP at pH 5.5 (A) and in an acidic GSH solution (10 mM, pH 5.5) (B); scale bar: 100 nm. (C) The UV-vis absorption spectra of oxidized TMB at different concentrations of CP. (D) The UV-vis absorption spectra of the GPx-like capacity of CP under different conditions. (E) The results of GSH depletion by CP at different concentrations. (F) Determination of CAT-like activity of CP at various concentrations. | ||
The results indicated that the pH/GSH dual-responsive CP could offer abundant Cu ion sources to trigger cuproptosis in tumor cells, because malignant tumors have unique features, including a mildly acidic tumor microenvironment and high GSH levels. In contrast, CP has minimal impact on normal cells (pH = 7.4).
Enzyme-like activities of CP
To evaluate the POD-like activity of CP, a TMB assay was conducted to investigate the OH generation (Fig. S6A, ESI†). The TMB + H2O2 and TMB + CP groups had no characteristic absorption peaks, indicating no oxidation reaction. After mixing CP and TMB solution with H2O2 under acidic conditions, OH was produced from the catalytic decomposition of H2O2 by CP. The characteristic peaks at 370 nm and 652 nm, originating from oxidized TMB, gradually increased in a concentration-dependent manner (Fig. 2(C)). Besides, the POD-like activity of the CP NPs was also evaluated using fluorescent spectra. Terephthalic acid (TA) reacts with ˙OH generated from H2O2 decomposition to produce 2-hydroxyterephthalic acid with a fluorescent peak at 425 nm. In the presence of CP, the fluorescence intensity at 425 nm significantly increased in a concentration-dependent manner (Fig. S6B, ESI†), illustrating the POD-like activity of CP. Therefore, the POD-like activity of CP is apparent. It was possible that CP with POD-like activity could utilize intratumoral H2O2 to produce ˙OH, leading to ROS augmentation, and aggravating CP-mediated tumor cell death by cuproptosis and oxidative damage.CP with GPx-like catalytic activity was illustrated by a DNTB chromogenic reaction assay. GSH is oxidized to glutathione disulfide (GSSG) by H2O2 in the presence of CP. The peaks at 326 nm are attributed to GSSG generation. When DNTB reacts with GSH, it yields a yellow production that results in a characteristic peak appearing at 412 nm in the UV-vis spectrum. As shown in Fig. 2(D), a peak appears at 326 nm, and the peak at 412 nm disappears in the CP + GSH + H2O2 group, proving that CP has GPx-like activity. Moreover, a GSH depletion experiment was carried out without the addition of H2O2. When CP reacts with GSH, the peaks at 412 nm gradually decrease, and peaks at 326 nm appear as the concentration of CP increases (Fig. 2(E)). This suggests that CP can efficiently deplete GSH in a dose-dependent manner. Correspondingly, no matter whether in a catalytic experiment or in a depletion experiment, the GSH/GSSG ratios obtained by calculation all decreased (Fig. S7, ESI†). Thereby, CP can cause GSH decline, which could enhance cuproptosis therapy.
When CP was mixed with H2O2 solution, a large number of small bubbles were generated (Fig. S8A, ESI†). This indicated that CP with CAT-like activity can decompose H2O2 to generate O2. Subsequently, the colorimetry results further validated the catalytic property of CP. The principle is that H2O2 can oxidize 4-aminoantipyrine in the presence of DHBS and peroxidase, resulting in a characteristic absorption peak at 505 nm that appears due to oxidized 4-aminoantipyrine formed through the coupled enzymatic reactions. If H2O2 is decomposed by CP, the peak at 505 nm would decrease. As we expected, the peak at 505 nm decreased distinctly after addition of various concentrations of CP, suggesting that CP with CAT-like activity decomposed H2O2 (Fig. 2(F)).
Besides, the oxygen production performance of CP was investigated by an oxygen dissolving assay. A dissolved oxygen meter was employed to determine the generated oxygen content. Obvious oxygen production can be observed when CP was added in H2O2 solution (Fig. S8B, ESI†), while no oxygen was detected in pure H2O2 solution, thus indicating the ability of CP to react with H2O2 for oxygen generation. Therefore, we assumed that the CP may relieve tumor hypoxia by catalytic decomposition of H2O2 to oxygen and boost cuproptosis therapy.
Moreover, to study whether HA modification influences the enzyme-like activities of CP, comparison experiments between CP and CPW were conducted. The results from Fig. S9 (ESI†) suggest that CP exhibited higher enzyme-like activities than CPW. It is speculated that HA modification reduced the size of CP, thereby enhancing its catalytic activity.
Cytotoxicity of CP
The cytotoxicity of CP against A549 NSCLC cells and LO2 human hepatocyte cells was evaluated by a CCK-8 assay. As shown in Fig. 3(A), the cell viability of A549 decreased markedly after treatment with different concentrations of CP. When the concentration of CP was 100 μg ml−1, the cell viability was less than 20%. In contrast, the cell viability of LO2 was still above 80% in the same range. Moreover, CP showed apparent cell mortality in T98G human glioblastoma cells, H1975 NSCLC cells, and MDA-MB231 triple-negative breast cancer cells in a concentration-dependent manner (Fig. S10, ESI†). These results illustrated that CP possessed distinct anticancer capability. | ||
| Fig. 3 In vitro cytotoxicity assessment of CP. (A) Cell viability of A549 cells treated with different concentrations of CP (mean ± s.d., n = 3). (B) Fluorescent microscope images of A549 cells stained with calcein-AM (green) and PI (red) after CP treatment. (C) After various treatments, A549 cells were analyzed by flow cytometry. (D) Representative fluorescence images of DCFH-DA probe-stained A549 cells to detect ROS under different conditions. (E) Relative GSH levels in A549 cells after co-incubation with different concentrations of CP (mean ± s.d., n = 3). (F) HIF-1α immunofluorescence images of tumor tissue from the control group and CP group in animal experimentation (****p < 0.0001). | ||
Calcein-AM/PI double staining was used to evaluate the cytotoxicity of CP. The cells in the CP group presented stronger red fluorescence (dead cells) than in the control group (Fig. 3(B)). As shown in flow cytometry analysis (Fig. 3(C)), the proportion of apoptotic cells significantly increased as the concentration of CP increased. When the concentration of CP reached 100 and 200 μg ml−1, the apoptosis rate was approximately 26% and 39%, respectively, accompanied by cellular necrosis. This indicated that apoptosis also contributed to CP-induced cell death. In brief, the above results proved CP-induced A549 cell death.
Variations in intracellular ROS, GSH Content and HIF-1α expression after CP treatment
To investigate the POD, GPx and CAT-like activities of CP in A549 cells, we evaluated intracellular ROS production, GSH content, and hypoxia-inducible factor-1α (HIF-1α) expression after CP treatment. The ROS levels were estimated by fluorescence microscopy using DCFH-DA probes (Fig. 3(D)). The images demonstrated that the green fluorescence in the CP group was more robust than that in the control group, indicating the elevated ROS production in A549 cells via the POD-like activity of CP.The GSH detection kit was used to assess the intracellular GSH content after different concentrations of CP treatment. Compared to the control group, the relative GSH content decreased to 19% (CP: 200 μg ml−1) and 40% (CP: 100 μg ml−1), respectively (Fig. 3(E)). Therefore, intracellular GSH was apparently consumed by CP.
Hypoxia remarkedly promotes tumor glycolysis, conferring cancer cell resistance to cuproptosis. We assumed that CP with CAT-like activity may relieve hypoxia in A549 cells. HIF-1α protein is upregulated under hypoxic conditions. The immunohistochemistry staining of HIF-1α in A549 tumor tissues that were harvested from animal experiments was used to evaluate the hypoxia relief of CP. It was shown that the red fluorescence intensity was obviously reduced in the CP group in contrast to the control group (Fig. 3(F)), and the corresponding fluorescence intensity ratio of the CP group to the control group is shown in Fig. S11 (ESI†), suggesting that CP could alleviate tumor hypoxia by its CAT-like activity. All the results verified that CP, with three catalytic activities, induced ROS augmentation, GSH depletion, and hypoxia relief in A549 cells, which would enhance cuproptosis therapy.
CP-mediated attenuation of glycolysis
We verified whether CP could abate glycolysis in A549 cells. Based on the Warburg effect, cancer cells take up abundant glucose, produce lactate and release the lactate into the extracellular space. The alterations in glycolysis of A549 cells were investigated through glucose consumption and lactate secretion tests. The culture medium of A549 cells was collected after co-incubation with CP. The glucose and lactate concentrations in the culture medium were measured using a Glucose kit and Lactate assay kit, respectively. The results suggested that CP could abate glycolysis, as evidenced by the lowered glucose consumption and lactate secretion levels after CP treatment compared with the control group (Fig. 4(A) and (B)). According to the relevant literature,30,31,44 PA and copper element have been reported to regulate glycolysis and glucose metabolism. We supposed that the alleviation of glycolysis by CP may be associated with PA and/or copper, which needs further research. Recently, starvation therapy via decreasing the glucose in tumor cells has been proven to boost cuproptosis efficacy.45 Encouraged by this, we hoped that A549 cells would be susceptible to cuproptosis by CP-induced alleviation of glycolysis. | ||
| Fig. 4 CP-induced cuproptosis in vitro. (A) Glucose consumption in A549 cells following the indicated treatment (mean ± s.d., n = 3). (B) Relative lactate levels secreted by A549 cells after the indicated treatments (mean ± s.d., n = 3). (C) Cell viability of CP-treated A549 cells after pretreatment of various cell death inhibitors (mean ± s.d., n = 3). (D) Western blot analysis on the expressions of DLAT and FDX1. (E) Cell viability of A549 cells in various groups, including the control, elesclomol, CP, CP + Elesclomol, and CP + Elesclomol + TTM groups under different concentrations of CP conditions (mean ± s.d., n = 3) (**p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
CP-induced cuproptosis
To examine whether CP induces cuproptosis in A549 cells, first, A549 cells were pretreated with several types of cell death inhibitors, including apoptosis inhibitor (Z-VAD-FMK), ROS inhibitor (NAC), necroptosis inhibitor (Necrostatin-1), autophagy inhibitor (3-MA), ferroptosis inhibitor (Ferrostatin-1) and cuproptosis inhibitor (TTM). Notably, compared with other inhibitors, the cell viability of the CP-treated A549 cells was restored from 28% to 83% after TTM pretreatment (Fig. 4(C)). Therefore, it was proven that CP can induce cuproptosis in A549 cells. Moreover, Z-VAD-FMK and NAC could rescue modest cell killing. It is well-known that ROS induce apoptosis in tumor cells. It further verified that CP with POD-like activity could cause ROS augmentation and lead to apoptosis, which was consistent with the ROS fluorescent test and flow cytometry results.DLAT and FDX1 as the key cuproptosis-related proteins were investigated in A549 cells after CP treatment. Western blot results indicated that the DLAT stripe in 69 kDa, and DLAT oligomers from 130 to 200 kDa appeared obviously in the CP groups, while the oligomers did not appear in the control group (Fig. 4(D)). As shown in Fig. 4(D), the FDX1 expressions in the CP groups were downregulated in a dose-dependent manner. Compared with the control group, the FDX1 expressions were reduced by more than 50% in the CP groups (Fig. S12, ESI†). The above results coincided with the typical features of cuproptosis, including increased DLAT oligomerization and reduced FDX1. Taken together, it is demonstrated that CP effectively induces cuproptosis in A549 cells.
CP Sensitizes A549 cells to elesclomol
Encouraged by the above results, we investigated whether CP boosts Elesclomol-induced cuproptosis in A549 cells. Fig. S13 (ESI†) shows the limited toxicity in A549 cells after Elesclomol coupled with adding Cu2+ treatment, even at a high concentration of 100 nM. These results coincided with literature reports that A549 cells are Elesclomol-resistant. 20 nM of Elesclomol was chosen for the subsequent experiment. Compared to the Elesclomol group, the CP + Elesclomol group exhibited a prominent decline in cell viability as the amounts of CP increased (Fig. 4(E)). When the concentration of CP was 60 μg ml−1, only 19% of the cells survived in the CP + Elesclomol group, whereas the Elesclomol group showed negligible cytotoxicity. Apparently, the combination of CP with Elesclomol exhibited a synergistic effect on the suppression of A549 cells, compared with CP or Elesclomol used alone. Moreover, TTM rescued cell viability with varying degrees in the CP + Elesclomol + TTM group. These results demonstrated that CP could significantly improve the sensitivity of A549 cells to Elesclomol-induced Cuproptosis and enhance the therapeutic efficacy of Elesclomol.In vivo antitumor efficacy of CP
The antitumor activity of CP was evaluated in an A549 tumor-bearing nude mice model. When the tumor volume reached about 100 mm3, the mice were randomly divided into the control group and the CP group. CP (2 mg kg−1) was administered to each mouse by intratumor injection every 2 d, as well as PBS solution as a control. The body weight and tumor volume of the mice were measured before injection. During the treatment period, there was no apparent difference in the body weight of the mice between the CP and the control group. The body weight curve of the mice in the CP group was close to that of the control group (Fig. S14A, ESI†). After 16 days of treatment, the tumor volume and corresponding tumor weight in the CP group were significantly reduced compared with the control group (Fig. 5(A) and (B)), as confirmed by the photos of representative tumor-bearing mice (Fig. S14B, ESI†) and harvested tumors from the two groups (Fig. 5(C)). This suggests that CP exhibited a superior antitumor effect. | ||
| Fig. 5 In vivo antitumor efficacy of CP. (A) Tumor growth curves of A549 tumor-bearing mice in the control group and CP group (mean ± s.d., n = 5). (B) Corresponding tumor weight of the mice after the indicated treatments (mean ± s.d., n = 5). (C) The representative images of the dissected tumors in each group. The H&E (D), TUNEL (E), Ki-67 (F) and DLAT (G) staining images of tumors obtained from the two groups after 16-days of treatment (***p < 0.001, ****p < 0.0001). | ||
Then, tumor tissues were stained by H&E, TUNEL, Ki67 and DLAT to evaluate the therapeutic efficacy. The CP group exhibited massive tumor cell death and serious cellular damage, including loss of nuclei, irregular cell shrinkage, and sparse tissue density, which were more severe than in the control group (Fig. 5(D)). The results were consistent with the tumor growth curve. Moreover, the CP-treated tumors exhibited intensely red fluorescence, indicating CP-mediated apoptosis (Fig. 5(E)). Moreover, the markedly descending Ki67 expression (brown) (Fig. 5(F)) revealed that CP can inhibit tumor proliferation. The consequence was also consistent with the H&E and TUNEL experiment results. Finally, the remarkable augmentation of the red fluorescence verified the CP-induced cuproptosis in the DLAT immunofluorescence staining experiment (Fig. 5(G)). The proportion of positive signals stained by Ki67 and DLAT was quantified. Compared to the control group (50.9%), the Ki67 positive cell rate was 14.5% in the CP group. As for DLAT, the mean fluorescence intensity was 4.8-fold higher relative to that in the control group (Fig. S15, ESI†).
Hypoxia is an inherent characteristic of solid tumors. CP treatment resulted in a decreased expression level of Hif-1a in the tumor section, which was relative to O2 generation from the intratumoral H2O2 catalyzed by CP with CAT-like activity. In addition, CP with POD-like activity could produce excess ROS to induce oxidative stress. As a result, it can prominently induce cell apoptosis, which was proved by TUNEL staining of tumor slices in the CP group. By contrast, the fluorescence intensity of Hif-1a and TUNEL was almost negligible in the control group. The enzyme-mimicking property of CP alleviated tumor hypoxia and exacerbated oxidative stress damage, which effectively reinforced the cuproptosis-induced antitumor effect, as evidenced by the tumor size variation curve of the mice, and H&E, Ki67, and DLAT staining of the tumor tissues after CP treatment. Overall, these data indicated that CP showed superior inhibition of A549 cell growth in vivo.
The results of the major organs (heart, liver, spleen, lung and kidneys) from the H&E staining images showed that there were no obvious histological abnormalities observed between the healthy mice and CP-tread mice (Fig. S16A, ESI†). Afterwards, the results of hematology indices displayed no significant variation detected in the CP group as compared with the control group (Fig. S16B, ESI†). ALT, AST, BUN, and Crea were chosen to evaluate liver function and kidney function (Fig. S16C, ESI†). These indicators showed no significant changes in the CP group, compared to the control group, revealing no severe liver or kidney injury. The results from H&E staining, routine hematological assay and blood biochemical index suggested that CP had good biocompatibility.
Conclusions
In summary, a pH/GSH dual-responsive CP with three enzyme-mimicking activities induced cuproptosis in A549 cells, making A549 cells more vulnerable to Elesclomol. CP enhanced cuproptosis therapy and showed superior antitumor performance due to GSH depletion, increased ROS, hypoxia relief, and glycolysis suppression. Compared with other Cu-based nanomaterials as cuproptosis inducers, our work proposed a promising and therapeutic CP nanodrug with facile synthesis, safety and cost-effective composition. It expands the multiple biological functions of metal–phosphonate nanozymes.Materials and methods
Chemicals and materials
Copper chloride, sodium hyaluronate, glutathione, 3,3,5,5-tetramethylbenzidine (TMB), and 5,5-dithio bis-(2-nitrobenzoic acid) (DTNB) were purchased from Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Phytic acid sodium salt hydrate (C6H18O24P6·xNa + yH2O) was purchased from Macklin Biochemical Co., Ltd (Shanghai, China). 30% H2O2 solution was obtained from Sinopharm Chemical Reagent Co., Ltd. Z-VAD-FMK, ferrostatin-1, necrostatin-1,3-methyladenine, ammonium tetrathiomolybdate (TTM), and Elesclomol were purchased from MedChemExpress Biochemical Technology Co., Ltd (Shanghai, China). Roswell Park Memorial Institute (RPMI) 1640 medium and Dulbecco's modified Eagle's medium (DMEM) were purchased from Thermo Fisher Scientific Inc. (USA). Fetal bovine serum (FBS) was purchased from ExCell Bio. Co., Ltd (Taicang, China). Cell counting kit-8 (CCK-8) was purchased from Biosharp Biotechnology Co., Ltd (Hefei, China). The calcein-AM/PI cell viability/cytotoxicity assay kit, and N-acetyl-L-cysteine (NAC) were purchased from Beyotime Biotech Inc. (Shanghai, China). The annexin V-APC/7-AAD apoptosis kit was purchased from MultiSciences Biotech Co., Ltd (Hangzhou, China). The lactic acid assay kit, and glucose kit (glucose oxidase method) were purchased from Nanjing Jiancheng Bioengineering Institute Co., Ltd. Phosphate buffered saline (PBS, 1×), penicillin–streptomycin solution (100×), trypsin–EDTA solution (0.25% with phenol red), and the glutathione (GSH) content assay kit were purchased from Beijing Solarbio Science & Technology Co., Ltd (Beijing, China). The BCA protein kit, Cell Complete Lysis Buffer, LDS Sample Buffer, QuickBlock blocking buffer, and PVDF membranes were purchased from Beyotime Biotech Inc., (Shanghai, China). Running buffer and precast Tris-Gly 4–12% gels were purchased from Wansheng Haotian Biotechnology Co., Ltd (Shanghai, China). The primary antibodies DLAT and FDX1 were purchased from Abmart Co., Ltd (Shanghai, China). The secondary antibody IgG was purchased from Thermo Fisher Scientific Inc. (Shanghai, China). β-actin antibody was purchased from Zhengneng Biotechnology Co., Ltd (Chengdu, China).Characterization
The morphology of the nanoparticles was detected by transmission electron microscope (H-7650, Hitachi, Japan). The zeta potentials of the samples were obtained by a nano particle size analyzer (Winner601, Jinan Winner Particle, China). An X-ray photoelectron spectrometer (K-Alpha, Thermo Fisher, America) was employed for XPS analysis. Energy dispersive X-ray spectroscopy (VEGA3LMU, Tescan, Czech) was employed to measure the elemental composition in the samples. X-Ray powder diffraction spectroscopy (MiniFlex 600, Rigaku, Japan) was used for XRD analysis. The Cu content was tested by an inductively coupled plasma optical emission spectrometer (720ES, Agilent, USA). Fourier Transform Infrared Spectroscopy (Nicolet iN10, Thermo Fisher Scientific, USA) was applied for the infrared spectra of the samples. The flow cytometry data were acquired on a flow cytometer (FACS Calibur, BD Biosciences, USA). UV-vis spectra of the samples were recorded on a microplate reader (BioTek Synergy H1, Agilent, America). The fluorescence images and immunohistochemical images were observed by fluorescence microscope (Evos FL Auto2, Thermo Fisher Scientific, USA). The WB results were observed by an Odyssey Imaging System (Odyssey CLx, LI-COR Biosciences, USA).Synthesis of CP
CP was synthesized by a “one-pot” method. In brief, 20 μl of CuCl2 solution (150 mM) and 100 μl of sodium hyaluronate solution (1 mg ml−1) were added into 1 ml of deionized water under stirring for 10 min. After the addition of PA solution (60 mM), the mixture solution was agitated for 2 h. Then, the as-prepared CP solution was separated by centrifuging at 6000 rpm for 5 mins, and the resulting precipitates were washed 3 times, followed by drying. Finally, the CP powders obtained were stored at 4 °C.Catalytic activities of CP
The POD-like activity of CP was evaluated using UV-visible absorption and fluorescence spectra.Absorption method
TMB (1 mM) and H2O2 (10 mM) were mixed with various concentrations of CP (10, 20, 50, and 100 μg ml−1) in HAc–NaAc buffer solution (pH = 4.5). The mixture solution was stirred for 30 min. Subsequently, the obtained supernatant was collected and measured using a microplate reader. The absorbance spectra at 370 nm and 652 nm were recorded, respectively.Fluorescent method
After acetate buffer (25 mM, pH 5.5) containing H2O2 (10 mM) and different concentrations of CP were vortexed vigorously and incubated at room temperature for 2 h, TA (0.5 mM) was added. Then, the fluorescence of the mixture was measured.For GPx-like activity detection, the various CP solutions (0, 10, 50 and 100 μg ml−1) were mixed with GSH (1.8 mM) and H2O2 (0.1 mM), respectively, in PBS buffer solution (pH = 7.2) under stirring for 20 min. Following this, the mixture and DTNB (20 mM) were transferred to Tris–HCl buffer solution (pH = 8.3). Finally, the spectra at 326 nm and 412 nm were tested by a microplate reader. For the GSH depletion experiment, the operation was performed using the same procedures as described above without the addition of H2O2.
The CAT-like activity of CP was evaluated using the UV-visible absorption method and dissolved oxygen assay.
Absorption method
CP solutions of various concentrations (0, 10, 50, and 100 μg ml−1) were mixed with H2O2 solution (10 mM) to react for 1 h. After centrifugation, 2.5 μl of the supernatant was added to 250 μl of PBS solution (pH = 7.4) that contained 4-aminoantipyrine (1 mmol l−1), DHBS (2 mmol l−1) and peroxidase (8 kU l−1). The spectra at 505 nm were recorded by a microplate reader.Dissolved oxygen assay
A dissolved oxygen meter was used to monitor the change in oxygen content in the H2O2 solution in real time. The curve of dissolved oxygen content-time was applied to evaluate the oxygen production ability of CP.Cell culture
The A549, H1975, T98G, MDA-MB231, and LO2 cell lines were purchased from the Institute of Chinese Academy of Medical Sciences (Shanghai, China). Based on the cell types, they were cultured in high-glucose Dulbecco's modified Eagle medium (DMEM) or RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin in an incubator at 37 °C with a 5% CO2 humidified atmosphere.Cell viability assay
Cells were seeded and incubated in a 96-well culture plate (6 × 103 cells per well) for 24 h. Then, the cells were treated with various concentrations of CP for 24 h. After that, CCK-8 solution was added to each well. The OD450 value was measured by a microplate reader. Cell viability (%) was calculated according to the manufacturer's protocols.For the live/dead cell double staining assay, according to the manufacturer's protocol, A549 cells were stained with calcein-AM and PI, followed by monitoring with a fluorescence microscope. The Annexin V-APC and 7AAD Apoptosis Kit was applied for flow cytometry analysis.
Determination of ROS production and intracellular GSH
A549 cells were incubated in a 96-well plate (8 × 103 cells per well) and cultured for 24 h. Then, A549 cells were incubated with CP for 24 h. The cells were treated with DCFH-DA (10 μM) for 20 min. After elimination with the unloaded probe, the fluorescence intensities of the cells were observed by fluorescence microscopy.A549 cells (5 × 106) were harvested at 24 h after CP treatment. After the ultrasonication of the cells in an ice-water bath, the cell lysate was collected. The GSH content assay kit was used to measure intracellular GSH, according to the manufacturer's protocols.
Detection of glucose consumption and lactate secretion
CP-mediated inhibition of glycolysis in A549 cells was evaluated by detecting glucose and lactate concentrations in the culture medium. The supernatants of the culture medium were collected after treatment with CP at different concentrations. The glucose and lactate concentrations in the supernatants were measured, respectively, using a glucose kit and lactic acid assay kit, according to the manufacturer's instructions. The glucose consumption was calculated by the reduction of the glucose concentrations in the culture medium.Cell death analysis
A549 cells were preincubated with several types of death inhibitors for 1 h, including apoptosis inhibitor (Z-VAD-FMK), ROS inhibitor N-acetylcysteine (NAC), necroptosis inhibitor (Necrostatin-1), autophagy inhibitor (3-MA), ferroptosis inhibitor (Ferrostatin-1) and cuproptosis inhibitor (Tetrathiomolybdate, TTM). Subsequently, the cells were incubated with CP for 24 h. A CCK-8 assay was then performed using the same procedures as described previously.Western blot assay
A549 cells were seeded in a 6-well plate at a density of 6 × 105 cells per well and incubated for 24 h. After different treatments for 24 h, the cells were harvested and treated with lysis buffer. The protein concentration was quantified using the BCA protein assay. The cell lysates were separated by SDS–polyacrylamide gel electrophoresis, with SDS–PAGE running buffer in precast Tris-Gly 4–20% gels and transferred onto PVDF membranes. Then, the membranes were blocked with blocking buffer for 15 min. Then, the membrane was coated with the primary antibodies: DLAT (1:1000), FDX1 (1:1000) and β-actin (1:5000), followed by overnight incubation at 4 °C. Following this, the membrane was incubated with a secondary antibody (IgG, 1:10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dilution) for 1 h. After the samples were washed three times with TBST, the protein bands were visualized using an LI-COR Odyssey Imaging System.
000 dilution) for 1 h. After the samples were washed three times with TBST, the protein bands were visualized using an LI-COR Odyssey Imaging System.
Animal experimentation
All animal procedures were approved by the Animal Care & Welfare Committee of Guangxi Medical University (no. 202306018).A549 cells were harvested and resuspended in PBS solution, and A549 cells (106 cells per mouse) were subcutaneously inoculated into the right thigh region of BALB/c nude mice. When the tumor volume reached approximately 100 mm3, the tumor-bearing mice were randomly divided into two groups: control group and CP group (n = 5 mice per group). The mice in the CP group received intratumoral administration (dose: 2 mg kg−1), while the control group was treated with PBS solution. The administration was performed every 2 d until the 16th day. During the treatment, the body weight and tumor volume were monitored. The animals were sacrificed by euthanasia on day 16. The tumor volume was calculated as follows: volume = length × width2 × 0.5.
The mice's tumors and major organs (heart, liver, spleen, lung and kidney tissues) were harvested. The tissues were fixed and embedded in paraffin and sectioned into about 4 μm thickness for hematoxylin and eosin (H&E), TUNEL, Ki67, DLAT and HIF-1α staining. The photos were observed under a fluorescence microscope.
For biocompatibility analysis, the whole blood and blood serum samples from healthy mice and experimental mice with CP treatment were collected for blood routine analysis and blood biochemical index. Their major organs (heart, liver, spleen, lungs and kidneys) were collected for H&E analysis.
Statistical analysis
The results were expressed as mean ± standard deviation (SD). Student's t test or one-way analysis of variance (ANOVA) was used to compare differences between two groups or multiple groups. Statistical analyses were performed using the GraphPad Prism software. p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).Author contributions
All authors have read the final manuscript and approved it for publication. Xiao-Wan Han data curation, formal analysis, investigation, software, writing – original draft; Xu Chen formal analysis, software, visualization; Tian-Le Yang formal analysis, visualization, Ying-Yi Luo visualization, software; Rui-Xue Liang conceptualization, supervision; San-Qi An supervision, writing – review and editing; Xin-Li Liu conceptualization, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
All the data that support this study are included in this article and its ESI.†Acknowledgements
This work was financially supported by the Guangxi Natural Science Foundation (no. 2025GXNSFAA069953), Guangxi Science and Technology Base and Talent Project (Guike AD23026189) and Middle/Young aged Teachers’ Research Ability Improvement Project of Guangxi Higher Education (grant no. 2024KY0114).Notes and references
- J. Xie, Y. Yang, Y. Gao and J. He, Mol. Cancer, 2023, 22, 46 CrossRef CAS.
- P. Tsvetkov, S. Coy, B. Petrova, M. Dreishpoon, A. Verma, M. Abdusamad, J. Rossen, L. Joesch-Cohen, R. Humeidi, R. D. Spangler, J. K. Eaton, E. Frenkel, M. Kocak, S. M. Corsello, S. Lutsenko, N. Kanarek, S. Santagata and T. R. Golub, Science, 2022, 375, 1254–1261 CrossRef CAS PubMed.
- P. Zheng, C. Zhou, L. Lu, B. Liu and Y. Ding, J. Exp. Clin. Cancer Res., 2022, 41, 271 CrossRef CAS PubMed.
- Y. Lu, Q. Pan, W. Gao, Y. Pu, K. Luo, B. He and Z. Gu, Biomaterials, 2022, 281, 121335 CrossRef CAS.
- Q. Wang, C.-h Huang, F. S. Wibowo, R. Amin, J. Shen, F. Li and R. J. Babu, ACS Appl. Mater. Interfaces, 2024, 16, 13509–13524 CrossRef CAS.
- S. Y. Lee, J. H. Seo, S. Kim, C. Hwang, D. I. Jeong, J. Park, M. Yang, J. W. Huh and H. J. Cho, Small, 2023, 19, e2301402 CrossRef.
- B. Guo, F. Yang, L. Zhang, Q. Zhao, W. Wang, L. Yin, D. Chen, M. Wang, S. Han, H. Xiao and N. Xing, Adv. Mater., 2023, 35, e2212267 CrossRef.
- F. Hu, J. Huang, T. Bing, W. Mou, D. Li, H. Zhang, Y. Chen, Q. Jin, Y. Yu and Z. Yang, Adv. Sci., 2024, 11, e2309388 CrossRef.
- Y. Lu, Q. Pan, W. Gao, Y. Pu and B. He, J. Mater. Chem. B, 2022, 10, 6296–6306 RSC.
- R. Li, W. Zhao, Z. Han, N. Feng, T. Wu, H. Xiong and W. Jiang, Small, 2024, 20, e2306263 CrossRef.
- G. He, Y. Pan, F. Zeng, S. Qin, X. Luan, Q. Lu, C. Xie, P. Hu, Y. Gao, J. Yang, B. He and Y. Song, ACS Nano, 2024, 18, 9031–9042 CrossRef CAS PubMed.
- Y. Y. Wang, S. L. Li, X. Y. Zhang, F. L. Jiang, Q. L. Guo, P. Jiang and Y. Liu, Small, 2024, 20, e2400254 CrossRef PubMed.
- Y. Zhang, N. Zhang, J. Xing, Y. Sun, X. Jin, C. Shen, L. Cheng, Y. Wang and X. Wang, Biomaterials, 2024, 311, 122675 CrossRef CAS PubMed.
- J. Bao, J. Wang, S. Chen, S. Liu, Z. Wang, W. Zhang, C. Zhao, Y. Sha, X. Yang, Y. Li, Y. Zhong and F. Bai, ACS Nano, 2024, 18, 9100–9113 CrossRef CAS.
- N. Yu, J. Zhou, M. Ding, M. Li, S. Peng and J. Li, Angew. Chem., Int. Ed., 2024, 63, e202405639 CrossRef CAS PubMed.
- Z. Yang, Z. Zhao, H. Cheng, Y. Shen, A. Xie and M. Zhu, J. Colloid Interface Sci., 2023, 641, 215–228 CrossRef CAS PubMed.
- Y. Liao, D. Wang, C. Gu, X. Wang, S. Zhu, Z. Zheng, F. Zhang, J. Yan and Z. Gu, Nat. Nanotechnol., 2024, 19, 1892–1902 CrossRef CAS PubMed.
- J. Zhong, X.-C. Lu, X. Zheng, R. K. Kankala, S.-B. Wang and A.-Z. Chen, ACS Mater. Lett., 2024, 6, 4282–4290 CrossRef CAS.
- X. Tian, H. Xu, F. Zhou, X. Gong, S. Tan and Y. He, Chem. Mater., 2024, 36, 815–828 CrossRef CAS.
- I. Vucenik and A. M. Shamsuddin, Nutr. Cancer, 2006, 55, 109–125 CrossRef CAS.
- M. A. Brehm and S. Windhorst, Biochem. Pharmacol., 2019, 163, 206–214 CrossRef CAS PubMed.
- L. Dilworth, D. Stennett and F. Omoruyi, Biomolecules, 2023, 13, 972 CrossRef CAS PubMed.
- Q. Ju, R. Huang, R. Hu, J. Fan, D. Zhang, J. Ding and R. Li, Drug Delivery, 2023, 30, 2181743 CrossRef PubMed.
- Z. Zhou, T. Fan, Y. Yan, S. Zhang, Y. Zhou, H. Deng, X. Cai, J. Xiao, D. Song, Q. Zhang and Y. Cheng, Biomaterials, 2019, 194, 130–138 CrossRef CAS PubMed.
- J. Zhang, L. Peng, Y. Hao, H. Yang, W. Zhao and C. Mao, Adv. Healthcare Mater., 2023, 12, e2300167 CrossRef.
- A. Li, Y. Li, Y. Jia, Y. He, M. Yuan, Z. Hao, Y. He, Y. Fu, J. Zhang, D. Gao, X. Zhang, X. Jiang and W. Tu, Adv. Healthcare Mater., 2024, e2400596, DOI:10.1002/adhm.202400596,.
- W. Xu, Z. Lin, C. J. Kim, Z. Wang, T. Wang, C. Cortez-Jugo and F. Caruso, Sci. Adv., 2024, 10, eads9542 CrossRef CAS.
- M. Li, H. Zhang, Y. Hou, X. Wang, C. Xue, W. Li, K. Cai, Y. Zhao and Z. Luo, Nanoscale Horiz., 2020, 5, 202–217 RSC.
- S. Wang, X. Liu, D. Wei, H. Zhou, J. Zhu, Q. Yu, L. Luo, X. Dai, Y. Jiang, L. Yu, Y. Yang and W. Tan, J. Am. Chem. Soc., 2024, 146, 30033–30045 CrossRef CAS.
- P. Pei, Y. Wang, W. Shen, Q. He, X. Han, C. Zhang, Y. Xie, G. Zhou, Y. Zhao, L. Hu and K. Yang, Aggregate, 2023, 5, e484 CrossRef.
- Y. Huang, X. Liu, J. Zhu, Z. Chen, L. Yu, X. Huang, C. Dong, J. Li, H. Zhou, Y. Yang and W. Tan, J. Am. Chem. Soc., 2024, 146, 13805–13816 CrossRef CAS.
- W. Yang, Y. Wang, Y. Huang, J. Yu, T. Wang, C. Li, L. Yang, P. Zhang, L. Shi, Y. Yin, K. Tao and R. Li, Biomed. Pharmacother., 2023, 159, 114301 CrossRef CAS.
- C. Yan, Y. Liu, G. Zhao, H. Yang, H. Lv, G. Li, Y. Li, Y. Fu, F. Sun, Y. Feng, Y. Li and Z. Zhao, Acta Pharm. Sin. B, 2024, 14, 2281–2297 CrossRef CAS PubMed.
- X. Luan, S. Du, Z. Liu, B. He, Y. Pan, J. Yang, Y. Li, D. Kong, X. Han and Y. Song, Adv. Funct. Mater., 2025, 35, 2416285 CrossRef CAS.
- C. Xiao, J. Li, A. Hua, X. Wang, S. Li, Z. Li, C. Xu, Z. Zhang, X. Yang and Z. Li, Research, 2024, 7, 0335 CrossRef CAS PubMed.
- L. Ren, E. S. Hong, A. Mendoza, S. Issaq, C. Tran Hoang, M. Lizardo, A. LeBlanc and C. Khanna, Oncotarget, 2017, 8, 38541–38553 CrossRef PubMed.
- S. Badodi, N. Pomella, X. Zhang, G. Rosser, J. Whittingham, M. V. Niklison-Chirou, Y. M. Lim, S. Brandner, G. Morrison, S. M. Pollard, C. D. Bennett, S. C. Clifford, A. Peet, M. A. Basson and S. Marino, Nat. Commun., 2021, 12, 2148 CrossRef CAS.
- C. Zhang, G. Chu, Z. Ruan, N. Tang, C. Song, Q. Li, W. Zhou, J. Jin, H. Haick, Y. Chen and D. Cui, ACS Nano, 2022, 16, 16584–16597 CrossRef CAS.
- H. Sakai, Y. Ikemoto, T. Kinoshita, T. Moriwaki and K. T. Yoshida, Vib. Spectrosc., 2017, 92, 215–219 CrossRef CAS.
- A. Zając, L. Dymińska, J. Lorenc, S. M. Kaczmarek, G. Leniec, M. Ptak and J. Hanuza, J. Biol. Inorg. Chem., 2019, 24, 11–20 CrossRef.
- Z. Tian, J. Zhao, S. Zhao, H. Li, Z. Guo, Z. Liang, J. Li, Y. Qu, D. Chen and L. Liu, Nano Res., 2022, 15, 4334–4343 CrossRef CAS.
- L. De Carli, E. Schnitzler, M. Ionashiro, B. Szpoganicz and N. D. Rosso, J. Braz. Chem. Soc., 2009, 20, 1515–1522 CrossRef CAS.
- X. Quan, C. Ouyang, Y. Pan, C. Zhang, Z. Wu, Z. Hong and M. Zhi, Nanotechnology, 2020, 31, 415602 CrossRef CAS PubMed.
- K. Zha, M. Tan, Y. Hu, W. Hu, S. Zhang, Y. Zhao, Z. Lin, W. Zhang, H. Xue, B. Mi, W. Zhou, Q. Feng, F. Cao and G. Liu, Bioact. Mater., 2024, 37, 424–438 CAS.
- Y. Xu, Y. Wu, X. Zheng, D. Wang, H. Ni, W. Chen and K. Wang, Adv. Sci., 2024, e2411378, DOI:10.1002/advs.202411378.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00183h |
| This journal is © The Royal Society of Chemistry 2025 |