
Reversing cancer immunosuppression via K+ capture and repolarization of tumor-associated macrophages†
Si-Ye
Tong
a,
Cong-Min
Huo
 a,
Yu-Cheng
Zuo
a,
Shuo
Gao
a,
David Tai
Leong
*b,
Wei
Xue
a and
Jing-Yi
Zhu
*a
a,
Yu-Cheng
Zuo
a,
Shuo
Gao
a,
David Tai
Leong
*b,
Wei
Xue
a and
Jing-Yi
Zhu
*a
aKey Laboratory of Biomaterials of Guangdong Higher Education Institutes, Guangdong Provincial Engineering and Technological Research Center for Drug Carrier Development, Department of Biomedical Engineering, Jinan University, Guangzhou, 510632, China. E-mail: jyzhu@jnu.edu.cn
bDepartment of Chemical and Biomolecular Engineering, Faculty of Engineering, National University of Singapore, Singapore, Singapore. E-mail: cheltwd@nus.edu.sg
First published on 27th March 2025
Abstract
Immunosuppression from the tumor microenvironment plays a key role in the failure of cancer immunotherapy. The presence of potassium ions (K+) from dying tumor cells creates an immunosuppressive environment that encourages tumor-associated macrophages (TAMs) to adopt a pro-tumor M2-like phenotype. Alleviating immune suppression from the high K+ environment might boost innate immunity and fight tumor growth. Herein, disulfide-rich mesoporous silica modified with 18-crown-6 ether was developed as a nanocarrier (D-C) to load ML133, encapsulating with the DiR-embedded macrophage membrane (CM) to create D-C/M@CM/DiR. We first saturated the phagocytosis of the mononuclear phagocyte system (MPS) with blank nanocarriers to enhance the tumor accumulation of D-C/M@CM/DiR, which was coated with the same CM. 18-Crown-6 ether captures K+ to reduce immunosuppression, while ML133 promotes the polarization of TAMs to an anti-tumor M1 phenotype by targeting the K+ channel protein Kir2.1 on their membranes. This strategy activates the anti-tumor immune response and effectively inhibits tumor growth.
New conceptsThe immunosuppressive microenvironment, enriched with potassium ions released from necrotic tumor cells, promotes a pro-tumor M2-like phenotype in tumor-associated macrophages (TAMs). However, few researchers have focused on the influence of potassium ions in the tumor microenvironment when it comes to cancer immunotherapy. This study introduces a novel strategy to overcome the immunosuppressive effects of elevated K+ levels in the tumor microenvironment (TME) by combining plant-derived chloroplast-like vesicles with chondrocyte membranes to modulate the mononuclear phagocyte system (MPS). By using a hybrid membrane vesicle system, the approach effectively saturates the MPS, minimizing premature elimination of nanomedicines by liver and spleen macrophages. Furthermore, the use of 18-crown-6 modified mesoporous silica as a nanocarrier to deliver ML133 selectively repolarizes TAMs to an M1-like phenotype, counteracting the immunosuppressive TME. This strategy differentiates itself from traditional methods by targeting K+-mediated immune modulation and enhancing tumor-targeted therapy without compromising the phagocytic function of the MPS. This work provides new insights into how nanomedicine can be optimized to overcome both immune suppression and inefficient drug retention in cancer therapy. |
Introduction
Research has found that only 20–30% of patients respond to immunotherapy.1–3 The high failure rate of clinical cancer immunotherapy can be attributed to the immunosuppressive microenvironment of solid tumors.4–7 T cells tend to have a limited lifespan, functional exhaustion, and reduced reactivity due to the toxic tumor microenvironment (TME).8–12 Moreover, M2 polarized tumor-associated macrophages (TAMs) are dominant in the tumor stroma and promote tumor growth and metastasis.13–15 Necrotic cell death elevates K+ mediated crosstalk between tumor cells and extracellular components that form the TME.8,16,17 In breast cancer, extracellular potassium levels in the tumor microenvironment are 8–15 times higher than in normal serum.18 The high potassium tumor microenvironment mediates abnormal tumor cell behaviors, including proliferation, migration, invasion, stem cell-like phenotype acquisition, and immune cell dysfunction. Elevated K+ within tumors regulate functional polarization of TAMs by targeting the Kir2.1 inward rectifying K+ channel on TAM surfaces.19 Studies have shown that elevated potassium levels released by dying tumor cells in the TME limit the effector function of tumor-infiltrating lymphocytes (TILs). This ionic imbalance suppresses signaling through AKT and mTOR kinases, thereby inhibiting T-cell effector programs essential for tumor clearance.16 High K+ can impair the ability of the cytotoxic T cells against cancerous cells.20 Thus alleviating immune suppression caused by the high K+ microenvironment in tumors may re-ignite innate immunity against tumor growth.To reduce premature elimination of our nanomedicine by the mononuclear phagocyte system (MPS) in organs like the liver and spleen,20–22 we have used a combination of a plant-derived membrane with chondrocyte membranes.23–27 Therefore, we presume that the use of plant chloroplast-like vesicles may rapidly saturate the phagocytosis of the MPS.28
Currently, reversal of K+ immunosuppression in the TME remains elusive.29,30 Crown ethers are transmembrane artificial ion channels using complementary cavity size and diameter to bind alkali metal ions.31,32 18-Crown-6 selectively binds to K+ ions.33–35 ML133 repolarized the endogenous TAM to the antitumor M1-like phenotype through selectively inhibiting the Kir2.1 channel without affecting other channels.36,37 Here, disulfide-rich mesoporous silica modified with 18-crown-6 ether as a nanocarrier (D-C) to load ML133,38 encapsulating with the DiR-embedded macrophage membrane (CM) to create D-C/M@CM/DiR. DiR is employed for fluorescence tracking and photothermal therapy.39,40 We attempt to saturate MPS phagocytosis using hybrid membrane vesicles made from plant chloroplast-like vesicles and the nanocarrier component CM.41 This allows subsequent nanodrugs with the same nanocarrier CM to effectively target tumors. This study showed the development of a simple, universal strategy to saturate MPS phagocytic function, minimize ineffective retention of nanomedicines in the liver, and alleviate the immunosuppressive effects caused by high K+. In this way, we hope to activate innate immunity and enhance the effectiveness of tumor-targeted therapy (Scheme 1).
 | ||
| Scheme 1 (A) Preparation of D-C/M@CM/DiR nanoparticles. (B) Schematic illustration of the mechanism of D-C/M@CM/DiR NPs in cancer immunotherapy. | ||
Results and discussion
Characterization of D-C/M@CM/DiR NPs and in vitro performance
The D-C/M@CM/DiR NPs were prepared through three steps: (a) synthesizing DMON by one-pot method; (b) grafting 18-Crown-6 and loading ML133; and (c) co-extrusion of RAW264.7 macrophage membranes and DiR with D-C/M. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were used to characterize DMON and D-C/M@CM/DiR nanoparticles (Fig. 1A, B and Fig. S1, ESI†). The TEM and SEM images revealed the dendritic structure of DMON, which was obscured following the coating with macrophage membranes. These nanoparticles exhibited a spherical structure, with a distinct surface coating seen in D-C/M@CM/DiR NPs. The hydrodynamic diameter was then evaluated by dynamic light scattering (DLS). The results showed that the particle size of D-C/M was 200 nm and D-C/M@CM was 220 nm (Fig. S2, ESI†). Subsequently, stability investigation was performed and the results indicated that there was no significant alteration in the particle size and PDI values of the NPs upon preservation in water, PBS and DMEM cell culture medium for one week (Fig. 1C and Fig. S3, ESI†), suggesting the potential stability of D-C/M@CM/DiR NPs in vivo under the dilution effect of body fluids. Upon coating with the macrophage cell membrane, the average surface potential changed from −32 mV for D-C/M NPs to −20 mV for D-C/M@CM/DiR NPs (Fig. 1D). The Fourier transform infrared (FT-IR) spectra of D-C/M NPs exhibited absorbed signals at 942, 1084, and 1512 cm−1 (Fig. 1E), corresponding to the characteristic peaks of 18-crown-6, DMON, and ML133, respectively. These results demonstrated the successful modification of 18-Crown-6 and loading of ML133 onto the DMON. Then, the drug loading efficiency of D-C/M@CM/DiR NPs was further studied (Fig. S4, ESI†). The release profiles of ML133 from D-C/M@CM/DiR NPs were investigated at pH = 6.5 and 7.4, respectively. The results depicted in Fig. 1F demonstrated that the release profiles of ML133 were pH-dependent, with a faster release observed at pH = 6.5, indicating that the encapsulation of ML133 by macrophage membranes facilitates its targeted release within the acidic tumor microenvironment. Following 24 hours of dialysis against solutions containing potassium ion (5 mmol L−1 or 45 mmol L−1), the changes in the zeta potential of D-C/M NPs and D-C/M@CM/DiR NPs were analyzed to assess their complexation behavior under varying potassium ion concentrations. As displayed in Fig. 1G, in a neutral environment, it was observed that uncoated D-C/M NPs exhibited an increase in potential under both low and high concentrations of potassium ions, suggesting the robust potassium ion complexation capability. Conversely, minimal fluctuations were noted in the potential of D-C/M@CM/DiR NPs under neutral conditions, highlighting the ability of nanoparticles coated with macrophage cell membranes to retain stability during blood circulation. However, in the acidic environment at pH = 6.5 (Fig. 1H), 18-Crown-6 initiated a discernible impact after the disruption of the macrophage cell membrane coating. D-C/M@CM/DiR NPs displayed an escalation in potential under both low and high concentrations of potassium ions, implying the nanoparticles' inherent potassium ion complexation proficiency. In addition, qualitative analysis of elemental mapping images (Fig. 1I) and energy dispersive spectroscopy (EDS) (Fig. 1J) provided further confirmation of the presence of Si, O, S, C, P, and O in the EDS intensity spectra. These findings support the successful synthesis of the D-C/M@CM/DiR NPs and the expected surface modifications. Nanoparticles with abundant S–S bonds can undergo redox reactions with GSH, resulting in disintegration, as shown by TEM (Fig. 1K). DMON exhibited surface disintegration after 12 h of co-incubation with 10 mM GSH, and its dendrimer framework collapsed after 24 h. After centrifuging the reaction solution and adding DTNB for 10 minutes, a yellow solution with GS-DTNB was produced, confirming that S–S-enriched DMON depleted GSH over time (Fig. 1L and Fig. S5, ESI†). The protein content in the RAW 264.7 membrane plays a key role in the nanoparticles' biological properties. SDS-PAGE results confirmed the successful coating of D-C/M@CM/DiR NPs and synthesis of TK-CM (Fig. S6, ESI†). The photothermal effect of D-C/M@CM/DiR NPs was determined in vitro under 808 nm laser irradiation (Fig. S7, ESI†), showing superior photothermal efficiency compared to free DiR, indicating effective photothermal conversion capacity. | ||
| Fig. 1 In vitro performance of D-C/M@CM/DiR NPs. (A) and (B) TEM images of DMON and D-C/M@CM/DiR NPs. Scale bar = 200 nm. (C) Dynamic light scattering analysis of variation in D-C/M@CM/DiR NP size upon standing in water, PBS, and DMEM for 7 days. (D) Zeta potentials of (a) DMON, (b) D-C/M, and (c) D-C/M@CM/DiR NPs. (E) FTIR spectra of DMON, D/M, D-C, and D-C/M. (F) In vitro release profiles of ML133 loaded in D-C/M@CM/DiR NPs in different pH buffers. Potassium ion complexation capacity of D-C/M NPs and D-C/M@CM/DiR NPs in a neutral environment (G) and in the acidic environment (H). (I) D-C/M@CM/DiR HRTEM image of NPs and mapping of C, O, Si, N, S, and P elements. Scale bar: 100 nm. (J) EDS intensity spectra of D-C/M@CM/DiR NPs. (K) TEM images of DMON dispersed in GSH solution (10 mM) for 0, 12, 24, and 48 h. (L) GSH consumption rate of DMON after incubation with 10 mM GSH for 12, 24, 48, and 72 h (n = 3, mean ± SD). | ||
In vitro TAM repolarization and in vitro antitumor evaluation
As reported, high potassium ion concentration may affect the activity of tumor-associated macrophages (TAMs). Excessive K+ in the tumor microenvironment may restrain TAM anti-tumor polarization. Reducing potassium ion concentration in the tumor microenvironment could reverse TAM polarization from the tumor-supportive M2 to the tumoricidal M1 phenotype, enhancing anti-tumor efficacy. First, mouse RAW264.7 macrophages were pre-polarized to an M2-like phenotype by incubation in a high potassium medium for 24 hours. Subsequently, the repolarization ability of D-C/M@CM/DiR NPs on M2-like phenotypes of macrophages pre-treated with a high potassium medium was evaluated. As depicted in Fig. 2, the results indicated that inhibiting the potassium ion inlet and outlet channel Kir2.1 of RAW264.7 and reducing the concentration of potassium ions in the medium could effectively inhibit macrophage polarization towards the M2 type. Compared to the control, D-C@CM/DiR NPs, and D-C/M@CM/DiR NPs group, D-C/M@CM/DiR NPs significantly increased the expression of CD80, an M1-related marker of RAW264.7 cells, by approximately 4.7-fold (Fig. 2A and B). At the same time, the expression of M2-related marker CD206 was decreased (Fig. 2C and D). These results implied that M2-type macrophages could be effectively repolarized into an M1 phenotype by inhibiting Kir2.1 with ML133, which serves as the potassium ion entry and exit channel in RAW264.7 cells, and concurrently reducing the potassium ion concentration in the medium. Additionally, bone marrow-derived macrophages (BMDMs) were extracted from mouse bone marrow and induced using macrophage colony-stimulating factor (M-CSF) for repolarization experiments. The data presented in Fig. S8 (ESI†) show that D-C/M@CM/DiR nanoparticles effectively induce M2-BMDMs to adopt an M1 phenotype, significantly increasing the M1/M2 ratio, which aligns with the results obtained from RAW264.7 cells. | ||
| Fig. 2 In vitro TAM repolarization and anti-tumor efficacy of D-C/M@CM/DiR NPs. (A) and (C) flow cytometry analysis of F4/80+ cells gated with M1-like (CD80+) and M2-like macrophages (CD206+) after treatment of RAW264.7 macrophages with various NPs. (B) and (D) Quantification of M1 marker (CD80+) and M2 marker (CD206+) expression by flow cytometry from different treatments: (a) Control, (b) D-C@CM/DiR NPs, (c) D/M@CM/DiR NPs, and (d) D-C/M@CM/DiR NPs. (E) Relative viability of 3T3 and 4T1 cells after 24 h incubation with different concentrations of D-C/M@CM/DiR NPs. (F) Sketch depicting the tumor-killing effect of D-C/M@CM/DiR NPs NP-stimulated TAM using the Transwell system after incubation with various NPs for 24 h. Cell viability of 4T1 cells was assessed using the CCK-8 assay. (G) Live/dead staining assay of 4T1 cells using Calcein-AM/PI upon different NP treatment under different irradiation conditions. Scale bar: 50 μm. Data are shown as mean ± SD (n = 3). (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). | ||
Under the premise of good biosafety (Fig. 2E and Fig. S9, S10, ESI†), the survival rate of 3T3 was high even when D-C/M@CM/DiR NPs were used at a concentration of 80 μg mL−1 as determined using the Cell Counting Kit-8 (CCK-8) assay, which indicates non-cytotoxicity to normal cells. Subsequently, a Transwell assay was employed to evaluate the impact of D-C/M@CM/DiR NP-treated M2-M1 on inhibiting the growth of 4T1 cells. 4T1 tumor cells were inoculated in the lower chamber of Transwell plates and co-cultured with the original M2 cells on 0.4 μm Transwell membranes (allowing for the free transport of biologically active molecules) for 24 h, the cytotoxicity of 4T1 cells in the lower chamber was then assessed using standard CCK-8 assays and live/dead staining. As shown in Fig. 2F, D-C@M/D NPs, D-C/M@CM/DiR NPs, and D-C/M@CM/DiR NPs significantly decreased 4T1 cell viability. The CCK-8 assay revealed significant cytotoxicity when 4T1 cells were incubated with repolarized macrophages and D-C/M@CM/DiR NPs + 808. The live/dead assay confirmed that repolarized macrophages effectively eradicated 4T1 cells, consistent with CCK-8 results (Fig. 2G). These findings manifest that the formulation of D-C/M@CM/DiR NPs can regulate the M1-type polarization of differentiated macrophages (M2) and bolster the anti-tumor efficacy of M1 cells. The phenotypic alterations of tumor associated macrophages effectively impede the proliferation of cancer cells.
In vivo targeting and photothermal effects
Tumor targeting plays a crucial role in determining the effectiveness of therapeutic interventions. To investigate whether macrophage camouflage can facilitate the targeted delivery to tumor tissue in vivo and the reticuloendothelial system (RES) blocking effect of TK-CM, a 4T1 tumor model was initially established, and the biological distribution of D-C/M@CM/DiR NPs in mice was studied using a fluorescence imaging system. As depicted in Fig. 3A, the fluorescence signals at the tumor site in all groups of mice exhibited a gradual enhancement over time, culminating in a surge at the 24-hour post-injection. Notably, weak fluorescence was detected at the tumor site at 6 h in the group administrated with only D-C/M@CM/DiR NPs, whereas the other three groups pre-injected with TK-CM displayed obvious fluorescence signals at the tumor site as early as 3 h. Impressively, among these groups, the mice pre-injected with 3 mg TK-CM exhibited the most pronounced fluorescence signal intensity within the initial 3 h, maintaining consistently high levels of fluorescence intensity at the tumor sites throughout the monitoring period. This outcome was further substantiated by the quantitative analysis of fluorescence intensity (Fig. 3B). After observation for 60 h, the tumors of various groups were dissected out and exposed to the fluorescence imaging system. The outcome of ex vivo fluorescence imaging and the quantitative analysis showed a similar tendency to the in vivo findings (Fig. 3C and D). The results indicated that the RES-blocking effect of TK-CM facilitated the rapid delivery of D-C/M@CM/DiR NPs to the tumor site, thereby enhancing their accumulation at the target site. Moreover, the fluorescence intensity of ex vivo organs was also investigated (Fig. 3E and Fig. S11, ESI†), with the liver exhibiting the most intense fluorescence signal, primarily attributed to its crucial role in NP metabolism. Notably, the 3 mg TK-CM injection group consistently displayed significantly higher fluorescence levels both in the liver and the tumor as compared to the other groups throughout the observation period. Encouraged by the above desired outcomes, we further evaluated the in vivo photothermal efficacy of D-C/M@CM/DiR NPs against 4T1 subcutaneous tumors. As illustrated in Fig. 3F, the tumor surface temperature of mice injected with D-C/M@CM/DiR NPs rapidly escalated to approximately 45 °C within 1 minute, reaching above 50 °C within 5 minutes (Fig. S12, ESI†).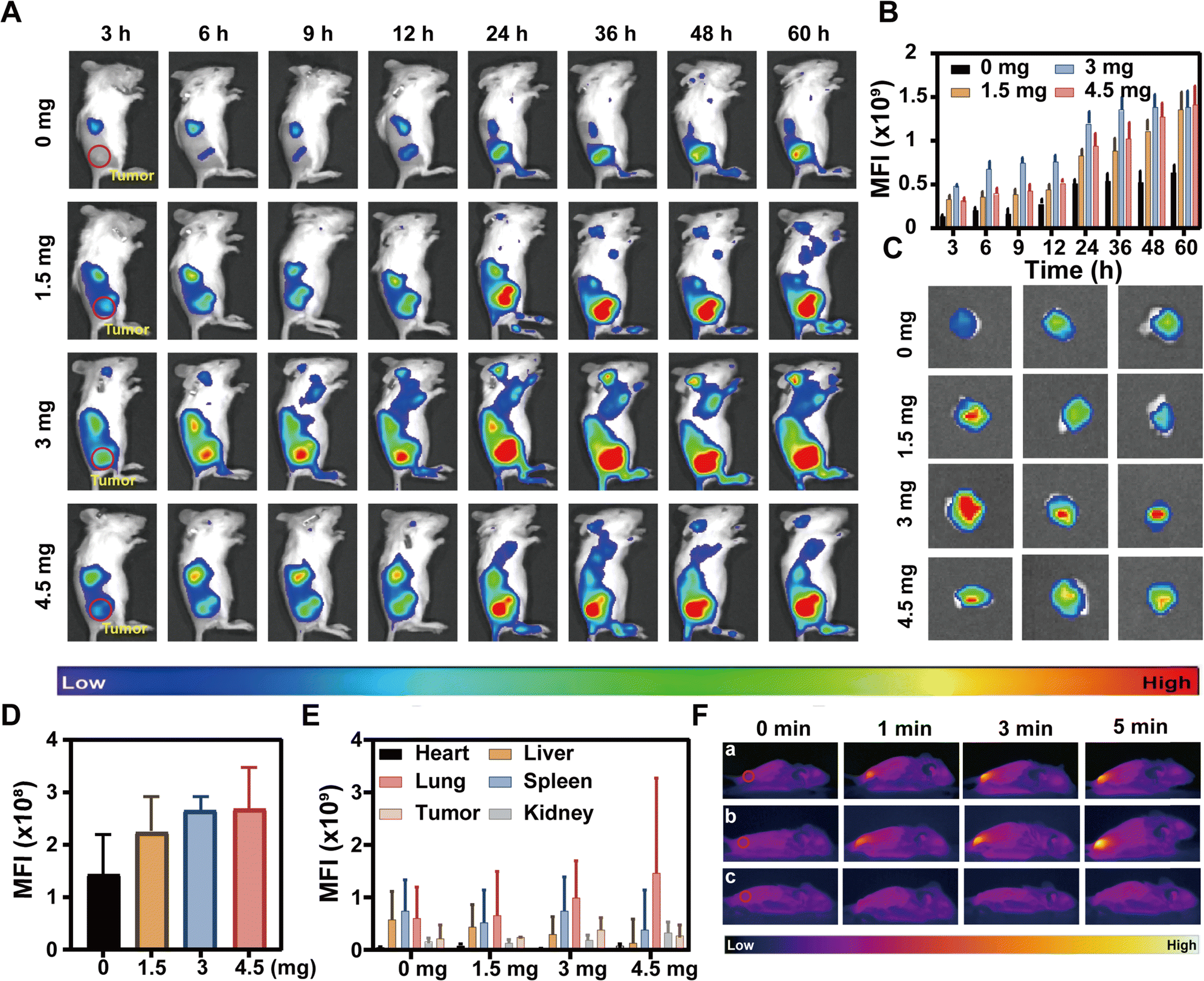 | ||
| Fig. 3 Tumor targeting and in vivo photothermal effects of D-C/M@CM/DiR NPs. Intravenous injection of different concentrations of TK-CM for 1.5 h followed by injection with D-C/M@CM/DiR NPs. (A) In vivo fluorescence images of tumor-bearing mice at different time intervals. (B) Semi-quantitative mean fluorescence intensity (MFI) analysis of tumor sites at different time intervals. (C) Ex vivo fluorescence images of tumor after 60 h observation. (D) Semi-quantitative MFI analysis of tumor after 60 h observation. (E) Semiquantitative mean fluorescence intensity (MFI) analysis at the cancer site at various time intervals after an intravenous injection of D-C/M@CM/DiR NPs. (F) In vivo photothermal images of mice upon intravenous injection with (a) D-C/M@CM/DiR NPs, (b) TK-CM + D-C/M@CM/DiR NPs, and (c) PBS for 24 h with an 808 laser irradiation (1.5 W cm−2). | ||
In vivo anti-tumor efficacy evaluation
Based on the excellent in vitro tumor suppressive effects and in vivo targeting performance, we further established a subcutaneous tumor model using 4T1 cells to investigate their in vivo antitumor efficacy (Fig. 4A). Tumor-bearing mice were randomly divided into six groups: (G1) PBS; (G2) D/M@CM/DiR NPs; (G3) D-C@CM/DiR NPs; (G4) D-C/M@CM/DiR NPs; (G5) D-C/M@CM/DiR NPs with NIR treatment; and (G6) Intravenous injection of TK-CM for 1.5 hours followed by intravenous injection of D-C/M@CM/DiR NPs with NIR treatment. Intravenous injections were administered on the first and third days, and the tumors were exposed to the laser (808 nm, 1.5 W cm−2) for 5 min 24 h after each injection. The treatment effect was assessed by monitoring the tumor size every other day, and the tumor volume and weight were quantified as illustrated in Fig. 4B–F. Rapid tumor growth was observed in the PBS control group. In contrast, the tumor size in the D-C/M@CM/DiR NPs treatment group displayed a significant decline compared to the PBS control group, D-C@CM/DiR NPs group, and D/M@CM/DiR NPs group, mainly attributed to the reversal of K+ immunosuppression in the tumor microenvironment by K+ binder 18-crown-6 and repolarization of the endogenous TAM to an antitumor M1-like phenotype by ML133. Following the injection of TK-CM and irradiation with an NIR laser a further decrease in tumor tissue was observed owing to the MPS saturation effect of TK-CM, leading to increased accumulation of D-C/M@CM/DiR NPs in the tumor. Moreover, the synergistic effects of PTT and repolarization of TAM contributed to this outcome. These findings indicate that the administration of D-C/M@CM/DiR NPs, particularly when used in combination with TK-CM and NIR laser irradiation, significantly suppressed tumor growth in mice. Simultaneously, tumors extracted from mice upon various treatments were subjected to hematoxylin and eosin (H&E) staining, immunohistochemical analysis for proliferation cells (Ki67), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). As displayed in Fig. 4G, compared with the control group, the TK-CM + D-C/M@CM/DiR NPs + NIR treatment group exhibited the most robust tumor regression capability. Our proposed synergistic immunotherapy approach was able to induce effective apoptosis and inhibit tumor proliferation. In addition, none of the groups displayed significant weight fluctuations or notable pathological damage to major organs (Fig. S13, ESI†). The levels of corresponding markers for liver and kidney function remained within the normal ranges (Fig. S10, ESI†). Therefore, these results confirm the biosafety of D-C/M@CM/DiR NPs for tumor therapy. | ||
| Fig. 4 In vivo Anti-tumor effect. (A) Schematic diagram of in vivo experiment design. (B) Photographs of stripped tumors from various treatment groups on day 21. Scale = 1 cm. (C) The tumor weight of various treatment groups on day 21. (D) and (E) Tumor growth curves of 4T1 tumor-bearing BALB/c mice in different treatment groups. (F) Body weight of 4T1 tumor-bearing mice in different groups. Data are displayed as mean ± SD (n = 5). (G) Histological observation of tumor sections in each group, including TUNEL immunofluorescence staining, immunohistochemical Ki67 staining (scale: 200 μm), and H&E staining (scale: 200 μm). Normal nuclei were stained blue, damaged DNA was stained green in TUNEL (scale: 125 μm), and proliferating cells were stained brown in Ki67 staining. (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). | ||
In vivo antitumor immunometabolic mechanisms of NPs
Further study of the immune response in vivo is necessary to clarify the antitumor therapeutic mechanisms of D-C/M@CM/DiR NPs. Additionally, we aim to determine whether treatment with D-C/M@CM/DiR NPs can induce repolarization of M2 macrophages into the M1 phenotype in vivo. Flow cytometry analysis revealed that TAMs within tumor tissues for the PBS control group predominantly exhibited the M2 phenotype, with minimal expression of the M1 phenotype (Fig. 5A and B), resulting in the rapid tumor growth observed in the control group. Both the D-C/M@CM/DiR NPs and D-C@CM/DiR NPs groups demonstrated a remarkable reduction in the M2 phenotype of TAM (Fig. 5C and D), which may be attributed to the inhibition of the potassium channel Kir2.1 on TAM by ML133 as well as the decrease in potassium ion levels in the tumor microenvironment by 18-Crown-6. D-C/M@CM/DiR NP treatment resulted in the higher expression of CD80 (M1-like markers) and lower expression of CD206 (M2-like markers), which was in agreement with findings from in vitro macrophage studies (Fig. 5E), indicating that the synergistic effects of ML133 and 18-Crown-6 can promote macrophage polarization towards the M1 phenotype. Notably, the TK-CM + D-C/M@CM/DiR NPs + NIR group exhibited higher M1-type macrophage counts and lower M2-type macrophage counts compared to the other groups, and especially, there was a significant increase in the M1/M2 ratio from 0.32 (PBS group) to 1.25 (Fig. 5F). Based on these findings, we further evaluated the anti-cancer immune response activated by TAM repolarization via quantitative analysis of cytotoxic T lymphocytes (CD8+ T cells) and Th1 cells (CD4+ T cells) after various treatments. As depicted in Fig. 5G, the infiltration of CD3+ T cells in tumor tissues significantly increased after TK-CM + D-C/M@CM/DiR NPs + NIR treatment compared to other treatments. Notably, the percentage of tumor-infiltrating CD8+ T cells in the TK-CM + D-C/M@CM/DiR NPs + NIR group substantially increased to 22.2%, up to 19-fold higher than the case in the PBS group (1.03%) (Fig. 5H, I and Fig. S14, ESI†). The impact of D-C/M@CM/DiR NPs on the systemic anti-tumor immune response was further investigated. The expression of CD3+ T cells and cytotoxic CD8+ T cells in other treatment groups also surpassed those in the PBS control group, confirming the capability of D-C/M@CM/DiR NPs to activate cytotoxic T lymphocyte infiltration in TME. To identify anti-tumor immune memory responses, effector memory CD4+ and CD8+ T cells in the spleen were measured. As illustrated in Fig. 5J–L, the mice treated with TK-CM + D-C/M@CM/DiR NPs + NIR exhibited the highest T cell levels in splenocytes, further confirming that TK-CM + D-C/M@CM/DiR NPs + NIR treatment has the potential to induce long-lasting immune memory, consequently inhibiting tumor recurrence (Fig. S15, ESI†). As illustrated in Fig. 5M, immunohistochemical results demonstrated that treatment with D-C/M@CM/DiR NPs led to a remarkable augmentation in M1 macrophages, a decrease in M2 macrophages, and an increase in cytotoxic CD8+ T cells in tumor tissues. These results provide additional evidence that D-C/M@CM/DiR NPs can effectively repolarize TAMs and promote the infiltration of cytotoxic T lymphocytes in the TME, which can effectively activate the anti-tumor immune response within the tumor microenvironment. | ||
| Fig. 5 Antitumor immunometabolic mechanisms of D-C/M@CM/DiR NPs in vivo. Representative plots of flow cytometry analysis of intratumoral infiltration of (A) F4/80+CD80+ and (B) F4/80+CD206+ macrophages in TME. Quantification of (C) TAM (F4/80+), (D) M1-like TAM (CD80+ in F4/80+ cells), and (E) M2-like TAM (CD206+ cells in F4/80+ cells) tumor infiltration by flow cytometry. (F) Ratio of M1/M2 TAM in tumor tissues after different treatments. Quantification of (G) T-cell (CD3+), (H) helper T-cell (CD4+ in CD3+ cells), and (I) CTL (CD8+ in CD3+ cells) tumor infiltration detected by flow cytometry. (J) Quantification of splenic infiltration of (potassium ion concentration) T cells (CD3+), (K) helper T cells (CD4+ in CD3+ cells), and (L) CTL (CD8+ in CD3+ cells) by flow cytometry. Data are shown as mean ± SD (n = 3). (M) Representative immunohistochemical images for F4/80, CD206, iNOS and CD8 of tumor sections from different groups: (G1) PBS group, (G2) D/M@CM/DiR NPs group, (G3) D-C@CM/DiR NPs group, (G4) D-C/M@CM/DiR NPs only group, (G5) D-C/M@CM/DiR NPs + 808 group, and (G6) TK-CM + D-C/M@CM/DiR NPs + 808 group. Scale bar: 100 μm. (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). | ||
Conclusions
The in vivo results showed that the percentage of tumor-infiltrating CD8+ T cells in the TK-CM + D-C/M@CM/DiR NPs + NIR group was up to 19-fold times higher than that of the case in the PBS group and that led to robust tumor death. Flow cytometry studies elucidated the mechanism by which D-C/M@CM/DiR nanoparticles altered the tumor immune microenvironment. This nanoplatform reversed the immunosuppressive microenvironment caused by the high concentration of potassium ions, providing an alternative strategy to enhance cancer immunotherapy.Materials and methods
Materials and instruments
Triethanolamine (TEA), sodium salicylate (NaSal), ethyl orthosilicate (TEOS), and 3-aminopropyltriethoxysilane (APTSE) were purchased from Aladdin. Ethanol (99.7%) was purchased from Macklin. Cell membrane protein and cytoplasmic protein extraction kits, BCA protein assay kits, SDS-PAGE gel preparation kits, and phenylmethylsulfonyl fluoride (PMSF) were purchased from Beyotime. Fluoride (PMSF) was purchased from Beyotime. Anti-mouse CD3-APC, anti-mouse CD8-FITC, anti-mouse CD4-PE, anti-mouse CD80-FITC, anti-mouse CD206-PE, and anti-mouse F4/80-APC for flow cytometric analysis were purchased from BioLegend. Cell live/dead kits, 1640 medium and DMEM medium were purchased from Keygen BioTECH. All chemicals in this work were commercially available and used directly without further purification.Synthesis of dendritic mesoporous organosilica nanoparticles
DMON was synthesized based on a previous study. Initially, 50 mL of deionized water and 0.25 g of TEA were gently stirred using a magnetic stirrer in an 80 °C oil bath for 30 minutes. Subsequently, 760 mg of CTAB and 168 mg of NaSal were added to the solution, followed by continuous stirring for 30 minutes. Following this, 4 mL of TEOS and 3.2 mL of BTES premix were vigorously stirred into the solution for 12 hours. The resulting products were collected via high-speed centrifugation and washed thrice with ethanol. The collected products were subsequently re-dispersed in 1% NaCl methanol solution at 45 °C, stirred for 6 hours, collected, washed several times with ethanol, and the process was repeated thrice. DMON was finally obtained through high-speed centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes.
000 rpm for 10 minutes.
Study of the GSH depletion ability of DMON
Around 100 mg of DMON was dispersed in 10 mL of a GSH solution (10 mM) and stirred continuously at room temperature. Subsequently, 1 mL of the solution was withdrawn at predetermined time intervals (12, 24, 48, and 72 hours) and centrifuged to collect the supernatant. Next, a 10 mM DTNB solution was added to the supernatant and co-incubated at room temperature for 10 minutes. The absorbance of the characteristic peak at 450 nm was subsequently measured using UV spectroscopy.Synthesis of D-C
1 gram of mesoporous silica DMON was weighed and added to a round-bottomed distillation flask. 100 mL of TL was added and shaken to mix. Then, under N2 protection, 6 mL of Aptes was added using a magnetic stirrer at 110 °C for 24 hours. The resulting mixture was washed several times with anhydrous ethanol, and then filtered to obtain a white powder. The white powder was transferred to a round-bottomed distillation flask. 15 mL of DMF was added. 1 gram of the white powder was weighed and dissolved in DMF. The mixture was dripped slowly into the reaction system (25 drops, with acceleration controlled at about 60 drops per minute). After completion of the addition, stirring of the reaction was continued for 24 hours.The white powder obtained from the previous step was washed several times with anhydrous ethanol, filtered, and then dried under vacuum at 65 °C for 12 hours to obtain carboxyl-functionalized DMON. Next, 10 mg of DMON-COOH was added to 25 mL of DMF. Under stirring, 250 mg each of EDC and NHS were added and activated at room temperature for 4 hours. Subsequently, 60 mg of 186-CPM was added to the activated mixture, and the reaction was conducted at 40 °C for 24 hours. After completion, the products were collected and treated with ethanol. The final product was then washed multiple times with ethanol and dried at 50 °C.
Synthesis of D-C/M
Initially, 1 mg of ML133 was dissolved in 0.5 mL of ethanol and combined with 0.5 mL of an ethanol solution containing D-C NPs (1.0 mg mL−1). ML133 was adsorbed onto the surface of DMON through non-covalent interaction. The mixture was stirred overnight at 25 °C. After stirring, it was centrifuged at 13000 rpm for 10 minutes, resulting in the formation of a precipitate, which was collected as D-C/M NPs. The drug loading capacity (LC%) and encapsulation efficiency (EE%) were calculated using the following formulas:

Macrophage membrane extraction and preparation of D-C/M@CM/DiR NPs
Cells were cultured in 10 cm diameter cell culture dishes following methods described in the previous literature for RAW264.7 cells. After culturing, the cells were separated by centrifugation at 700 × g for 5 minutes using a cell scraper. The collected cells were resuspended in pre-cooled PBS buffer (pH = 7.4) and centrifuged at 600 × g for 5 minutes. The resulting pellet was then suspended in hypotonic lysis buffer containing fluorinated membrane protein extraction reagent and phenylmethanesulfonyl PMSF, and incubated on ice for 10–15 minutes. Subsequently, the cells were subjected to freeze–thaw cycles and further disrupted by centrifugation at 700 × g for 10 minutes at 4 °C. The resulting supernatant was then centrifuged at 14000 × g for 30 minutes to collect cell membrane fragments (RAW-CM). The cell membrane fragments were dispersed in PBS and stored at −80 °C for future use.
Macrophage membrane encapsulation was achieved using the extrusion method. Initially, macrophage membrane fragments and D-C/M NPs (w/w = 1:1) were thoroughly mixed in deionized water. Subsequently, the mixture of macrophage membrane fragments and D-C/M NPs underwent sequential co-extrusion through polycarbonate filter membranes of varying pore sizes (1 μm, 0.8 μm, 0.4 μm) using an Avanti liposome extruder. Finally, the extruded mixture was centrifuged at 12000 rpm for 10 minutes to isolate D-C/M@CM NPs. DiR was then added and incubated for 30 minutes to load it, followed by centrifugation at 12000 rpm for 10 minutes to obtain D-C/M@CM/DiR NPs.
Drug release of D-C/M@CM/DiR NPs
The release profiles of ML133 from D-C/M@CM/DiR NPs under different pH conditions were investigated as follows: initially, 1 mL of the sample solution (1 mg mL−1) was placed inside a dialysis bag with a molecular weight cutoff of 3500 Da. This bag was then immersed in 20 mL of PBS and shaken at 100 rpm at 37 °C. At specified intervals, 1 mL of the sample solution was withdrawn from the dialysis bag. Simultaneously, an equal volume of fresh PBS was added to the external PBS solution to maintain sink conditions. The ML133 concentration in the withdrawn medium was subsequently analyzed using UV-Vis spectrophotometry. This experimental procedure was repeated three times to ensure data reliability and consistency. This methodology allows for the determination of how ML133, encapsulated within D-C/M@CM/DiR NPs, behaves in terms of release under different pH environments, providing insights into its controlled release properties for potential biomedical applications.Preparation of the TK membrane and TK-CM
Taxoid membranes (TK) were isolated from fresh spinach following a previously reported method. Spinach leaves (100 g) were washed and stored overnight at 4 °C. They were homogenized with pre-cooled HEPES buffer (5 mM MgCl2, 50 mM NaCl, and 0.3 M sucrose, pH = 7.6, 300 mL) using a wall breaker (CN-802, China). Leaf fragments were filtered through ten layers of cotton gauze to collect the filtrate. Centrifugation at 8000 rpm for 10 min allowed for the collection of intact chloroplasts. The stromal extract was removed from the supernatant by resuspending in hypotonic lysis buffer (10 mM HEPES, pH = 8.0) for 2 h and centrifuging at 12000 rpm for 30 min. The pellet containing TK was collected, washed with 10 mM HEPES buffer, and suspended in sonicated water for 5 min at 4 °C. After centrifugation at 16000 rpm for 15 min, the supernatant was removed to obtain TK. The TK membrane was washed twice with HEPES buffer and sonicated for 15 min. Subsequently, a mixture of the TK membrane and macrophage membranes (RAW-CM) was extruded through 800 nm, 400 nm, and 200 nm membranes using a micro-extruder to obtain TK-CM.
Characterization of D-C/M@CM/DiR NPs
Morphology and particle size characterization of DMON and D-C/M@CM/DiR NPs was conducted via transmission electron microscopy (TEM) and scanning electron microscope (SEM). The average particle size polydispersity index (PDI) and stability of D-C/M@CM/DiR NPs was measured by dynamic light scattering (DLS), and their zeta potential was analyzed using a zetasizer (Malvern, UK). Fourier transform infrared spectroscopy (FT-IR, Thermo Scientific iN10) and UV-visible spectroscopy (UV-Vis, Shimadzu, Japan) were employed to analyze the UV absorption spectra of D-C/M@CM/DiR NPs and ML133. Temperature changes post-irradiation with a near-infrared laser (808 nm, 1.5 W cm−2) were measured to assess the photothermal effect of D-C/M@CM/DiR NPs (DiR concentration: 40 μg mL−1). Phosphate-buffered saline (PBS) served as a control and was irradiated for 5 minutes, with temperature measurements recorded at 30-second intervals.The membrane proteins of D-C/M@CM/DiR NPs and TK-CM were analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). To prepare the samples, TK, RAW264.7 cell membrane fragments, TK-CM, and D-C/M@CM/DiR NPs were each added to the sample buffer and incubated at 95–100 °C for 10 minutes. Simultaneously, solutions for the electrophoresis gels (12% separation gel, 5% stacking gel) were prepared following the kit's protocol.
After incubation, the treated samples were loaded into separate wells of the SDS-PAGE gel. Constant voltage electrophoresis was conducted at 80 V for the stacking gel and 120 V for the separation gel. Following electrophoresis, the gels were agitated on a shaker for 60 minutes and then stained with Coomassie brilliant blue dye to visualize the protein bands. After staining, the gels were washed multiple times with deionized water to remove excess dye and visualize the protein bands clearly. This method allowed for the characterization and comparison of membrane proteins between TK-CM and D-C/M@CM/DiR NPs, providing insights into their composition and molecular weight distributions.
Cell culture
RAW264.7 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 sodium penicillin G, and 100 mg mL−1 streptomycin sulfate. 4T1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U mL−1 sodium penicillin G, and 100 mg mL−1 streptomycin sulfate. All cells were maintained in a humidified incubator at 37 °C under a 5% CO2 atmosphere.Bone marrow-derived macrophages (BMDMs) were isolated from the tibiae and femora of 6–8-week-old female C57BL/6 mice. Bone marrow cells were flushed out and filtered through a 70-μm mesh. After centrifugation, red blood cells were lysed, and the remaining cells were resuspended in an appropriate growth medium. The cells were then cultured in fresh DMEM supplemented with 10% FBS and 20 ng mL−1 M-CSF for 7 days to induce differentiation into M0-like BMDMs.
In vitro analysis of M2 macrophage repolarization and transwell experiments
Firstly, BMDM and RAW264.7 cells seeded on 24-well plates (1 × 105 cells per well) were both treated with 40 mM high-potassium complete DMEM for 24 hours. Subsequently, the cells were incubated with PBS, D-C/M@CM/DiR NPs (200 μg mL−1), D/M@CM/DiR NPs (200 μg mL−1), and D-C@CM/DiR NPs (200 μg mL−1) in fresh 40 mM high-potassium complete DMEM for another 24 hours. After incubation, the cells were harvested, and stained with APC-anti-F4/80, PerCP-cy5.5-anti-CD206, FITC-anti-CD80 antibodies and PE-anti-CD11b antibodies (Elabscience Biotechnology Co, Ltd, China) for 30 min at 4 °C, washed, resuspended in PBS, and analyzed by flow cytometry.In the 4T1-RAW 264.7 Transwell system, 4T1 cells and RAW 264.7 cells were pre-incubated with 40 mM high-potassium complete DMEM in the lower and upper chambers, respectively. After 24 h of normoxic incubation, the medium in both chambers was replaced with fresh medium containing PBS, D-C/M@CM/DiR NPs (200 μg mL−1), D/M@CM/DiR NPs (200 μg mL−1), and D-C@CM/DiR NPs (200 μg mL−1). In the PTT-treated group, the upper chamber containing RAW 264.7 cells was removed, exposed to a 1.5 W cm−2 808 nm laser for 5 min, and then returned to co-culture with 4T1 cells for an additional 20 h. Subsequently, the CCK-8 assay was performed by adding the kit and incubating at 37 °C for 1 h in the dark, followed by measuring the absorbance at 450 nm using a spectrophotometer.
Animal experimentation
SPF-grade BALB/c mice (4–5 weeks old, female) were procured from the Beijing Viton Lihua Laboratory Animal Technology Co., Ltd. The mice were housed at the Animal Management Center of Jinan University. All animal experiments were conducted in accordance with the guidelines and regulations approved by the Laboratory Animal Welfare and Ethics Committee of Jinan University (Approval Number: IACUC-20231106-05).In vivo infrared thermography
Twenty-four hours post-injection of various formulations (PBS, D-C/M@CM/DiR NPs, TK-CM + D-C/M@CM/DiR NPs), the central region of mouse tumors was irradiated using an 808 nm laser (1.5 W cm−2) for 5 minutes. Thermal images corresponding to each formulation were subsequently captured at different time intervals using an infrared camera (FOTRIC 225s# L24).In vivo and in vitro fluorescence imaging
1.5 h after an intravenous injection of TK-CM, an injection of D-C/M@CM/DiR NPs was administered,28in vivo imaging and biological distribution were monitored using an imaging system (IVIS Lumina, USA) at various time points. Subsequently, tumor tissue as well as major organs including the heart, liver, spleen, lungs, kidneys, and others were excised and examined using the same imaging method.In vivo antitumor effects
To assess the antitumor efficacy of D-C/M@CM/DiR NPs, female mice were randomly divided into six groups (n = 5): (a) PBS group, (b) D/M@CM/DiR NPs group, (c) D-C@CM/DiR NPs group, (d) D-C/M@CM/DiR NPs only group, (e) D-C/M@CM/DiR NPs + 808 group, and (f) TK-CM + D-C/M@CM/DiR NPs + 808 group. D-C/M@CM/DiR NPs (100 μL, 186-CPM: 12.5 mg kg−1, ML133: 2 mg kg−1) were administered via tail vein injection on days 0 and 3. Near-infrared (NIR) irradiation (808 nm, 1.5 W cm−2, 5 min) was applied on days 1 and 4 after injection. Tumor volume and body weight were measured every two days for 21 days, with treatments administered every 7 days.Tumor volume was calculated using the formula: volume = 0.5 × length × width2, where V represents tumor volume, L is the longest dimension, and W is the shortest dimension. Mice were euthanized 21 days post-treatment to collect tumor and spleen tissues. Tumor tissues were minced on ice, filtered through a 200 μm cell strainer to obtain single-cell suspensions, centrifuged (1000 rpm, 5 min), washed three times with PBS, and resuspended in PBS. Similarly, spleen tissues were processed to obtain single-cell suspensions, treated with erythrocyte lysis buffer, centrifuged (1000 rpm, 5 min), washed three times with PBS, and resuspended in PBS. Cells were stained with anti-CD3-APC, anti-CD8-FITC, anti-CD4-PE, CD80-FITC, anti-CD206-PE, and anti-F4/80-APC antibodies for 30 min and analyzed for macrophage and T-cell markers. Tumor samples were subjected to hematoxylin and eosin (H&E) staining, immunohistochemical Ki67 staining, and TUNEL immunofluorescence staining.
Safety evaluation of the D-C/M@CM/DiR NP system
To assess safety, the body weight of mice in each group was monitored throughout the treatment period. After 21 days of treatment, mice were euthanized, and major organs (heart, lungs, kidneys, spleen, and liver) were collected and fixed in 4% paraformaldehyde for hematoxylin and eosin (H&E) staining to evaluate in vivo biosafety. Additionally, blood samples were collected for biochemical analysis to assess liver, kidney, and heart function. Serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), blood urea nitrogen (BUN), creatinine (CRE), and creatine kinase (CK) were measured.Statistical analysis
All data were presented as mean ± standard deviation (SD). Differences between experimental groups were analyzed using one-way ANOVA conducted using GraphPad software (Inc., La Jolla, CA, USA). Significance was determined by P-values, where *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.Author contributions
S. Y. Tong synthesized the material and performed all experiments, wrote the manuscript, and analyzed the data. C. M. Huo, Y. C. Zuo, and S. Gao helped conduct experiments related to in vivo imaging and in vivo therapy. J. Y. Zhu supervised the progress of this research work and revised the manuscript. D. T. Leong helped revise the manuscript. W. Xue and J. Y. Zhu provided the financial support. All authors reviewed and discussed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024A1515030001) and the National Natural Science Foundation of China (Grant No. 51903104). The authors gratefully acknowledge the support from the K. C. Wong Education Foundation. The authors thank the Centric Laboratory of Medical College of Jinan University for providing experimental and instrument platforms.References
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18(3), 175–196 Search PubMed.
- W. J. Lesterhuis, J. B. Haanen and C. J. Punt, Nat. Rev. Drug Discovery, 2011, 10(8), 591–600 Search PubMed.
- L. A. Emens, P. A. Ascierto, P. K. Darcy, S. Demaria, A. M. M. Eggermont, W. L. Redmond, B. Seliger and F. M. Marincola, Eur. J. Cancer, 2017, 81, 116–129 CrossRef CAS PubMed.
- S. Taefehshokr, A. Parhizkar, S. Hayati, M. Mousapour, A. Mahmoudpour, L. Eleid, D. Rahmanpour, S. Fattahi, H. Shabani and N. J. P.-R. Taefehshokr, Pathol., Res. Pract., 2022, 229, 153723 Search PubMed.
- Y. N. Chen, Q. Q. Zhou, Z. F. Jia, N. Cheng, S. Zhang, W. D. Chen and L. Wang, Acta Pharm. Sin. B, 2024, 14(9), 3834–3854 CrossRef CAS PubMed.
- Y. Wang, M. Wang, H. X. Wu and R. H. Xu, Cancer Commun., 2021, 41(9), 803–829 Search PubMed.
- Y. Z. Jin, Y. Y. Huang, H. Ren, H. H. Huang, C. Y. Lai, W. J. Wang, Z. Tong, H. Y. Zhang, W. Wu, C. Liu, X. W. Bao, W. J. Fang, H. J. Li, P. Zhao and X. M. Dai, Biomaterials, 2024, 305, 122463 Search PubMed.
- R. Eil, S. K. Vodnala, D. Clever, C. A. Klebanoff, M. Sukumar, J. H. Pan, D. C. Palmer, A. Gros, T. N. Yamamoto and S. J. Patel, Nature, 2016, 537(7621), 539–543 CAS.
- D. Soll, C. F. Chu, S. Sun, V. Lutz, M. Arunkumar, M. Gachechiladze, S. Schäuble, M. Alissa-Alkhalaf, T. Nguyen, M. A. Khalil, I. Garcia-Ribelles, M. Mueller, K. Buder, B. Michalke, G. Panagiotou, K. Ziegler-Martin, P. Benz, P. Schatzlmaier, K. Hiller, H. Stockinger, M. Luu, K. Schober, C. Moosmann, W. W. Schamel, M. Huber and C. E. Zielinski, Nat. Immunol., 2024, 25(10), 1830–1844 Search PubMed.
- M. Wang, M. Chang, C. Li, Q. Chen, Z. Hou, B. Xing and J. J. A. M. Lin, Adv. Mater., 2022, 34(4), 2106010 CAS.
- J. Cai, Q. Shen, Y. Wu, J. Hu, D. Pan, Y. Wu and B. Geng, Adv. Funct. Mater., 2024, 34(52), 2411064 Search PubMed.
- B. Geng, J. Hu, X. He, Z. Zhang, J. Cai, D. Pan and L. Shen, Adv. Mater., 2024, 36(25), 2313670 CAS.
- P. Pathria, T. L. Louis and J. A. Varner, Trends Immunol., 2019, 40(4), 310–327 Search PubMed.
- A. Christofides, L. Strauss, A. Yeo, C. Cao, A. Charest and V. A. Boussiotis, Nat. Immunol., 2022, 23(8), 1148–1156 Search PubMed.
- X. Zhang, S. Li, I. Malik, M. H. Do, L. L. Ji, C. Chou, W. Shi, K. J. Capistrano, J. Zhang, T. W. Hsu, B. G. Nixon, K. Xu, X. X. Wang, A. Ballabio, L. S. Schmidt, W. M. Linehan and M. O. Li, Nature, 2023, 619(7970), 616 CAS.
- F. Baixauli, M. Villa and E. L. Pearce, Science, 2019, 363(6434), 1395–1396 CAS.
- M. Chirra, H. S. Newton, V. S. Gawali, T. M. Wise-Draper, A. A. Chimote and L. Conforti, Cancers, 2022, 14(15), 3564 CAS.
- S. T. Ong, A. S. Ng, X. R. Ng, Z. Zhuang, B. H. S. Wong, P. Prasannan, Y. J. Kok, X. Bi, H. Shim and H. J. B. Wulff, Bioelectricity, 2019, 1(3), 169–179 Search PubMed.
- S. Chen, W. Y. Cui, Z. X. Chi, Q. Xiao, T. Y. Hu, Q. Z. Ye, K. X. Zhu, W. W. Yu, Z. Wang, C. X. Yu, X. Pan, S. Q. Dai, Q. Yang, J. C. Jin, J. Zhang, M. B. Li, D. H. Yang, Q. Z. Yu, Q. Q. Wang, X. F. Yu, W. Yang, X. Zhang, J. B. Qian, K. F. Ding and D. Wang, Cell Metab., 2022, 34(11), 1843 Search PubMed.
- M. A. Erdogan and F. Ines, The role of ion channels in the relationship between the immune system and cancer, Curr. Top. Membr., 2023, 151–198 Search PubMed.
- G. Aizik, N. Waiskopf, M. Agbaria, M. Ben-David-Naim, Y. L. Kalisman, A. Shahar, U. Banin and G. Golomb, Nano Lett., 2019, 19(9), 5844–5852 CAS.
- E. A. Sykes, J. Chen, G. Zheng and W. C. W. Chan, ACS Nano, 2014, 8(6), 5696–5706 CrossRef CAS PubMed.
- A. B. Mirkasymov, I. V. Zelepukin, P. I. Nikitin, M. P. Nikitin and S. M. Deyev, J. Controlled Release, 2021, 330, 111–118 CrossRef CAS PubMed.
- J. McCright, J. Yarmovsky and K. Maisel, Mol. Pharmaceutics, 2023, 21(3), 1160–1169 CrossRef PubMed.
- F. Peng, M. I. Setyawati, J. K. Tee, X. G. Ding, J. P. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nat. Nanotechnol., 2019, 14(3), 279 CrossRef CAS PubMed.
- J. Y. Zhu, C. Sevencan, M. K. Zhang, R. S. A. McCoy, X. G. Ding, J. J. Ye, J. P. Xie, K. Ariga, J. Feng, B. H. Bay and D. T. Leong, ACS Nano, 2020, 14(3), 3259–3271 CrossRef CAS PubMed.
- Y. C. Zuo, C. M. Huo, Y. Chen, P. L. Ding, S. Y. Tong, W. Xue and J. Y. J. A. H. M. Zhu, Adv. Healthcare Mater., 2024, 13(11), 2303779 Search PubMed.
- C. M. Huo, Y. C. Zuo, Y. Chen, L. H. Chen, J. Y. Zhu and W. Xue, Int. J. Biol. Macromol., 2024, 274, 133186 CrossRef CAS PubMed.
- X. F. Pu, L. Li, Z. H. Chen, A. H. Gong, J. Lei, L. R. Zhang and H. Tsai, J. Cancer, 2023, 14(8), 1336–1349 CrossRef CAS PubMed.
- S. T. Ong, A. S. Ng, X. R. Ng, L. Kua, F. Y. X. Lee, S. Q. Tan, H. Shim, P. Prasannan, R. DasGupta, I. B. H. Tan, H. Wulff, K. G. Chandy and N. K. Verma, Biophys. J., 2019, 116(3), 249a Search PubMed.
- X. Zhang, X. Xing, X. Zhao, J. Yang, M. Liu, B. Liu, X. Gao and H. J. C. E. J. Huang, Chem. Eng. J., 2025, 503, 158435 CrossRef CAS.
- J. Li, Y. Shi, C. Qi, B. Zhang, X. Xing, Y. Li, T. Chen, X. Mao, Z. Zuo and X. J. A. C. Zhao, Angew. Chem., 2023, 135(40), e202309918 Search PubMed.
- I. Guberović, M. Marjanović, M. Mioč, K. Ester, I. Martin-Kleiner, T. Šumanovac Ramljak, K. Mlinarić-Majerski and M. J. Sr Kralj, Histochem. Cell Biol., 2018, 8(1), 14467 Search PubMed.
- L. Pang, H. H. Feng, W. Zhong, H. N. Dong, Y. Q. Shen, B. Yu and H. L. Cong, Mater. Des., 2021, 211, 110159 Search PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89(26), 7017–7036 Search PubMed.
- H.-R. Wang, M. Wu, H. Yu, S. Long, A. Stevens, D. W. Engers, H. Sackin, J. S. Daniels, E. S. Dawson, C. R. Hopkins, C. W. Lindsley, M. Li and O. B. McManus, ACS Chem. Biol., 2011, 6(8), 845–856 CrossRef CAS PubMed.
- R. Anton, M. Ghenghea, V. Ristoiu, C. Gattlen, M.-R. Suter, P. A. Cojocaru, A. Popa-Wagner, B. Catalin and A.-F. Deftu, Int. J. Mol. Sci., 2021, 22(4), 2081 Search PubMed.
- T.-Q. Xie, X. Yan, Y.-T. Qin, C. Zhang, X.-K. Jin, Q.-R. Li, Z.-Y. Rao, H. Zhou, W.-H. Chen and X.-Z. Zhang, Nano Lett., 2024,(50), 16132–16142 CrossRef CAS PubMed.
- Y. H. Li, Y. X. Wu, J. T. Chen, J. L. Wan, C. Xiao, J. K. Guan, X. L. Song, S. Y. Li, M. M. Zhang, H. C. Cui, T. T. Li, X. Q. Yang, Z. F. Li and X. L. Yang, Nano Lett., 2019, 19(8), 5806–5817 Search PubMed.
- J. Y. Wang, J. Meng, W. Ran, R. J. Lee, L. S. Teng, P. C. Zhang and Y. P. Li, Nano Lett., 2019, 19(8), 5266–5276 Search PubMed.
- C. Sevencan, R. S. A. McCoy, P. Ravisankar, M. Liu, S. Govindarajan, J. Y. Zhu, B. H. Bay and D. T. Leong, Adv. Ther., 2020, 3(3), 1900201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00050e |
| This journal is © The Royal Society of Chemistry 2025 |