
Real-time, non-destructive monitoring of the aggregation behavior of silver nanoparticles using nano-impact electrochemistry†
Hairong
Hu‡
a,
Yu-An
Li‡
a,
Meijuan
Liu
a,
Wei
Xu
a and
Yi-Ge
Zhou
 *ab
*ab
aState Key Laboratory of Chemo/Biosensing and Chemometrics, Department of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, Hunan Province, China
bGreater Bay Area Institute for Innovation, Hunan University, Guangzhou 511340, China. E-mail: yigezhou@hnu.edu.cn
First published on 20th March 2025
Abstract
Silver nanoparticles (AgNPs) are widely used in daily life, with their aggregation behavior greatly impacting their application potential. Thus, studying the aggregation of AgNPs is crucial for their practical applications. Nano-impact electrochemistry (NIE) has gained significant attention due to its rapid, cost-effective, and in situ analysis capabilities at the single-nanoparticle level. In this study, we propose a method for real-time and non-destructive monitoring of the rapid aggregation behavior of AgNPs within 10 minutes in chlorine-containing acidic media using NIE, a condition particularly relevant to biological systems, such as the antibacterial applications of AgNPs. Under this environment, a thin AgCl layer forms on the surface of AgNPs, interconnecting them and facilitating their aggregation. Therefore, the aggregation behavior of AgNPs can be analyzed by quantifying the electrochemical reduction of the AgCl coating in NIE measurements, allowing insights into aggregation kinetics by tracking the number of aggregated AgNPs over time. This real-time, non-destructive approach to monitoring AgNP aggregation deepens our understanding of their physicochemical properties and dynamic behavior in biological environments, offering valuable insights for optimizing their application in practical settings.
New conceptsThis study employs nano-impact electrochemistry (NIE) to investigate the aggregation behavior of silver nanoparticles (AgNPs) in aqueous solution, leveraging the technique's simplicity, speed, and in situ analysis capabilities. By quantifying the reduction charge of silver chloride (AgCl) coatings on the surface of aggregated nanoparticles in NIE measurements, this method enables efficient and straightforward aggregation monitoring in a real-time and non-destructive manner, which is difficult to achieve with other techniques. This approach provides insights into aggregation kinetics by tracking the number of aggregated AgNPs over time. The real-time, non-destructive monitoring of AgNP aggregation enhances the understanding of their physicochemical properties and dynamic behavior, providing helpful guidance for real-world applications. |
Introduction
In recent years, silver nanoparticles (AgNPs) have emerged as a major focus of research in nanoscience due to their excellent optical properties,1 catalytic activity,2 strong surface plasmon resonance effects,3 and antibacterial capabilities.4 Despite their desirable properties and potential applications across various fields, the broader utilization of AgNPs is hindered by their poor stability, stemming from susceptibility to oxidation and aggregation.5 For instance, environments where AgNPs exhibit antibacterial effects, such as gastric juice,6 bacteria-infected microenvironments,7 and sweat,8 are typically chloride-containing and acidic. Under such circumstances, AgNPs are susceptible to undergo sequential processes, including oxidation, chlorination, and aggregation.9 The resulting increase in particle size and the formation of AgCl, which interconnects AgNPs, can influence the release of soluble silver ions (Ag+),10 thereby impacting their toxicity and antibacterial effectiveness.11,12 Furthermore, aggregation can reduce the effective surface area, bioavailability, and colloidal stability of AgNPs, further compromising their antibacterial efficacy.13 Therefore, studying the aggregation behavior of AgNPs could provide useful insights for enhancing their practical applications.Traditional methods for studying nanoparticle aggregation include light scattering techniques,14–16 ultraviolet-visible (UV-vis) absorption spectroscopy,17,18 and electron microscopy.19 Dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) are two methods commonly used for size characterization in light scattering analysis. While DLS is often used for hydration radius measurement, it can be biased in polydisperse samples.20 NTA provides better size distribution and visualization but is limited when samples change rapidly within the first 2 minutes.9,21 UV-vis spectroscopy detects aggregation-induced absorption shifts but lacks size distribution data. Ex situ techniques like scanning electron microscopy (SEM) and transmission electron microscopy (TEM) offer detailed morphology but disrupt native aggregation during sample preparation. Advanced techniques like in situ TEM22 and single-particle inductively coupled plasma mass spectrometry (sp-ICP-MS)23 provide high-resolution insights but are time-intensive and costly. Consequently, there is an urgent need to develop real-time and efficient methods for monitoring nanoparticle aggregation.
Nano-impact electrochemistry (NIE) is a recently developed electroanalytical technique that leverages the collisions of individual nanoparticles with an ultramicroelectrode by virtue of Brownian motion.24–29 This method offers rapid, low-cost, and in situ analysis at the single-nanoparticle level, making it a promising approach for real-time investigation of nanoparticle aggregation. NIE has been used to monitor the time-dependent aggregation of AgNPs through the exhaustive electro-oxidation of silver within the aggregates.24 However, this approach is not real-time and is destructive, lacking the ability to dynamically observe or maintain the native state of AgNPs, which is crucial for improved process control and further applications. In this work, a novel electrochemical indicator, the reduction charge (Q) of AgCl, is introduced in NIE to monitor the aggregation of AgNPs in real-time and in a non-destructive manner within chlorine-containing acidic environments (Fig. 1). This approach is particularly relevant as AgNPs are widely utilized in biological systems and natural environments, where chlorine and acidic conditions are commonly encountered. The increase in the average integrated charge generated by collisions over time reflects the progressive intensification of AgNP aggregation, enabling the quantification of AgNP number within aggregates. Analyzing the time-dependent evolution of principal aggregates offers insights into the kinetics of AgNP aggregation, revealing a deceleration in the aggregation rate as the aggregate size increases. The in situ, real-time, and non-destructive tracking of AgNP evolution holds great promise for advancing the understanding of their aggregation behavior, thereby broadening their applications, particularly in antibacterial treatments and other biological systems.
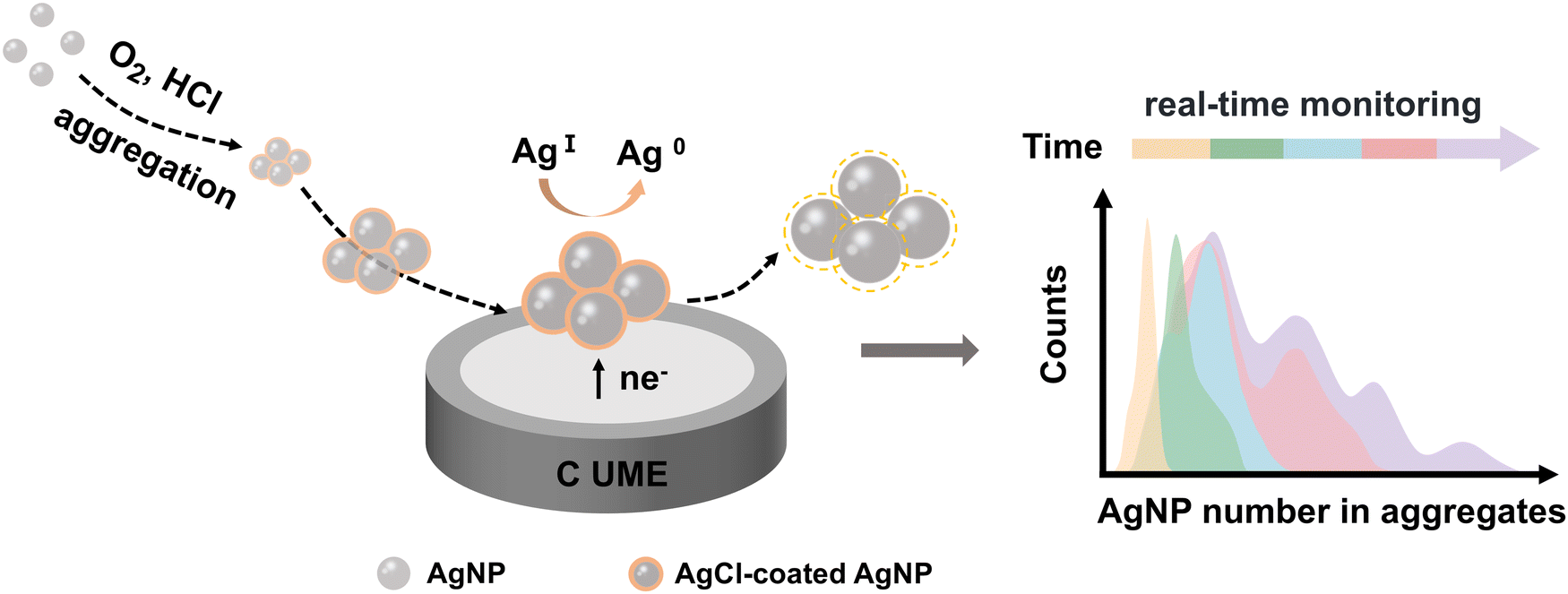 | ||
| Fig. 1 Schematic illustration depicting the real-time monitoring of the aggregation behavior of AgNPs via NIE. | ||
Experimental section
Chemicals and materials
All chemicals and materials were obtained from commercial suppliers and used directly without any prior treatment. Sodium citrate dihydrate (C6H3Na3O7·2H2O, Aladdin Scientific, >99.0%), silver nitrate (AgNO3, Sigma-Aldrich, >99.0%), hydrochloric acid (HCl, Sinopharm Reagent, >99.0%), Sodium hydroborate (NaBH4, Shanghai Titan Scientific, >99.0%), glass carbon electrode (GCE, diameter 3 mm, Gaoss Union), carbon ultramicroelectrode (UME, diameter 7 μm, Shanghai Xianren Instrumentation). All aqueous solutions were prepared by deionized water (resistivity ≥18.2 MΩ cm at 298 K) from a water purification system of Milli-Q for dilution. All the glassware was thoroughly soaked in aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3, 3:1, v/v) overnight before the synthesis experiments, followed by two ultrasonic cleanings with deionized water and subsequent drying for later use.
:HNO3, 3:1, v/v) overnight before the synthesis experiments, followed by two ultrasonic cleanings with deionized water and subsequent drying for later use.
Synthesis of AgNPs
AgNPs were synthesized using the seed-mediated Lee-Meisel method,30 comprising two steps: the synthesis of 4 nm AgNP seeds and subsequent seed growth. First, a solution of 20 mL of 1% (w/v) citrate solution and 75 mL of water was heated to 70 °C for 15 minutes. Subsequently, 1.7 mL of 1% (w/v) AgNO3 solution was added to the mixture, followed by the rapid addition of 2 mL of freshly prepared 0.1% (w/v) NaBH4 solution. The reactants were stirred vigorously at 70 °C for 1 hour. After the reaction was completed, the mixture was cooled to room temperature and then water was added to obtain a transparent light brown-yellow seed solution with a final volume of 100 mL.For the growth step, a mixture of 2 mL of 1% citrate solution and 75 mL of water was heated to boiling for 15 minutes. Next, 10.0 mL of the seed solution was added, followed by the rapid addition of 1.7 mL of 1% AgNO3 solution. The reaction was stirred vigorously under reflux for 1 hour and then cooled to room temperature. Water was added to increase the solution volume to 100 mL. To remove excess reactants, the colloidal solution was centrifuged at 1615 × g for 30 minutes. The supernatant was discarded, and the precipitate was collected and redispersed in deionized water for later use.
Characterization
TEM, high-resolution TEM (HRTEM), and high-angle annular dark-field scanning TEM (HAADF-STEM) images, along with STEM-energy dispersive X-ray spectroscopy (STEM-EDX) element mapping and line scan images, were obtained using a JEM-F200 (JEOL) field emission transmission electron microscope operated at 200 kV. Samples were prepared by depositing them onto a carbon-coated 200-mesh copper TEM grid and allowing them to dry. SEM images were recorded by a JSM-7610FPlus (JEOL) field emission scanning electron microscope at 15 kV. For SEM imaging, samples were drop-cast onto the polished side of a silicon wafer using a drying lamp to accelerate the drying process. UV-vis absorption spectra were recorded using a UV-2600 spectrophotometer (Shimadzu) over a wavelength range of 200–600 nm.NIE experiments
All electrochemical measurements were performed at room temperature using an Autolab PGSTAT 302N (Metrohm-Autolab). The experiments utilized a standard three-electrode setup housed within a custom-built double Faraday cage. A carbon UME with a diameter of 7 μm served as the working electrode, a porous carbon rod was used as the counter electrode, and a saturated calomel electrode (SCE) was used as the reference electrode. The electrolyte consisted of 5 mM HCl (pH 2.3), with 10 pM AgNPs well dispersed in it. Real-time NIE experiments were performed at a potential of −0.04 V vs. SCE at various time intervals, with a sampling interval of 2.3 ms. Each scan lasted 30 seconds without any disruption to the timing.Data processing
The transient current spikes recorded in the chronoamperometric curves from the NIE experiments were analyzed using Origin 10.1 software (OriginLab). The analysis included spike identification and integration, frequency scatter plot generation for statistical evaluation, and Gaussian deconvolution to determine size distributions. Valid signals were identified with a signal-to-noise ratio greater than 3. To perform the statistical analysis on the aggregation data, the peak area integration was first performed using the Polygon Area dialog box, and the integrated charge was plotted as a frequency distribution scatter plot. Next, the deconvolution operation was performed in the Peak Analyzer dialog box. After opening the dialog box, a baseline with a constant value was established. The software's Find Peak function was then used to identify several central peak positions in the raw data, with additional peaks manually detected to compensate for those missed by the automated process. Finally, automatic fitting was performed to obtain deconvolution and sub-distribution curves, continuously improving the fitting accuracy, as indicated by the coefficient of determination (COD, R2).Results and discussions
Characterization of AgNPs
As shown in a typical SEM image (Fig. S1, ESI†), the as-synthesized spherical AgNPs exhibit a uniform size distribution and high monodispersity, with an average diameter of approximately 32 nm. They display a characteristic UV-vis absorption peak at 392 nm.The aggregation mechanism
The standard potential for the reduction of oxygen (O2) to water in acidic media is 1.229 V (O2|H+, H2O) vs. the normal hydrogen electrode (NHE). In chloride-containing acidic solutions (5 mM HCl), the citrate stabilizing the surface of AgNPs is stripped away, making AgNPs readily oxidized by O2 into Ag+ due to the relatively low reduction potential of silver (Ag+|Ag; E0 = +0.7991 V vs. NHE). The oxidation of AgNPs in such a medium can be described as the reaction of metallic silver with protons and O2, as represented by eqn (1): | (1) |
The Ag+ generated by oxidation rapidly reacts with chloride ions to form AgCl on the surfaces of the AgNPs31 as shown in eqn (2), which interconnects AgNPs and facilitates their aggregation.9
| Ag+ + Cl− → AgCl | (2) |
Structural and elemental characterization of AgCl coating on the aggregated AgNPs
The morphologies of aggregates formed after dispersing AgNPs in 5 mM HCl for 5 minutes were observed using TEM, as shown in Fig. 2a. The AgNPs appear interconnected, with some regions showing signs of partial fusion, making it challenging to discern the spherical shape of individual nanoparticles. The HRTEM image in Fig. 2b reveals boundaries between aggregated AgNPs, where distinct Ag (111) lattice fringes are visible. Additionally, a few-nanometer-thick hazy AgCl layer at the boundaries is observed, corresponding to the lattice spacing of AgCl (111). Fig. 2c displays a HAADF-STEM image of a typical AgCl-coated AgNP, along with the corresponding STEM-EDX line scan for Ag and Cl elements across the nanoparticle. The Ag signal intensity varies across the nanoparticle, with a significantly higher signal at the center compared to the edge, attributed to the higher Ag content in the nanoparticle's cross-section as the line scan moved from the edge to the center of the nanoparticle. In contrast, the Cl signal remains nearly constant across the surface, indicating consistent Cl content and suggesting a uniform Cl distribution on the nanoparticle surface. Compositional element mapping of aggregated AgNPs (Fig. 2d–f) further confirms that the Cl element is densely and uniformly distributed on the surface of the aggregated nanoparticles, consistent with the line scan results. These characterizations collectively demonstrate that AgNPs exposed to HCl form aggregates with a thin AgCl coating layer uniformly distributed on each nanoparticle surface, interconnecting the nanoparticles. | ||
| Fig. 2 (a) TEM image showing the morphology of AgNP aggregates. (b) HRTEM image highlighting the boundaries between individual AgNPs within the aggregates. (c) HAADF-STEM image of a single AgCl-coated AgNP (top) and the corresponding STEM-EDX line scan spectra across the nanoparticle (bottom). (d)–(f) STEM-EDX mapping images illustrating the distribution of elements within AgCl-coated AgNP aggregates. | ||
The electrochemical behavior of the precipitated AgCl coating on aggregated AgNPs
To study the electrochemical behavior of the AgCl coating, AgNPs were drop-cast onto a 3 mm glassy carbon electrode (GCE) and exposed to either O2-dissolved or N2-saturated 5 mM HCl for 5 minutes. Cyclic voltammetry (CV) was performed within a potential range of 0.1 V to −0.3 V vs. SCE at a scan rate of 10 mV s−1 (Fig. 3a, red). A distinct reduction peak was observed at approximately −0.04 V, followed by an increase in the catalytic current. In contrast, under identical experimental conditions with N2 saturation, a flat curve was observed, showing neither a reduction peak nor a current increase (Fig. 3a, black). This suggests that the reduction peak at around −0.04 V should be associated with the reduction of AgCl, as reported in previous studies,32 which forms through Ag oxidation and chlorination. Furthermore, the subsequent increase in current is attributed to the oxygen reduction reaction catalyzed by AgNPs exposed after the reduction of AgCl. | ||
| Fig. 3 (a) Cyclic voltammograms of AgNPs-modified GCE in O2-dissolved (red) and in deoxygenated (black) conditions. The electrolyte is 5 mM HCl. Chronoamperometric curves of AgNPs in O2-dissolved HCl (b) and deoxygenated HCl (c) at a potential of −0.04 V based on NIE. | ||
The reduction of the AgCl coating formed on the aggregated AgNPs was further investigated using NIE measurements. A carbon ultramicroelectrode (UME) with a diameter of 7 μm was used as the working electrode for NIE to minimize the background current. Chronoamperometric scans were conducted at the potential of −0.04 V after dispersing AgNPs in an O2-dissolved 5 mM HCl solution for 5 minutes (Fig. 3b), displaying abundant reductive transient currents.
Control experiments were performed in an N2-saturated solution with AgNPs, where only a flat curve was observed (Fig. 3c). Additional control experiments were conducted in O2-dissolved HCl without AgNPs at −0.04 V and with AgNPs but at an applied potential of 0.1 V, where AgCl reduction does not occur (Fig. S2, ESI†). In both cases, no transient currents were observed, confirming that the reductive spikes originate from faradaic electron transfer during AgCl reduction, triggered by nanoparticles/aggregates collisions with the UME. These results demonstrate that AgCl can serve as an indicator for tracking the dynamic evolution of AgNPs.
Real-time monitoring of AgNP aggregation over time via NIE
To monitor the aggregation of AgNPs in chlorine-containing acidic solutions in situ, NIE experiments were conducted on AgNP aggregates over time under a potential of −0.04 V. Fig. 4a–e show the chronoamperometric curves after 1 minute, 3 minutes, 5 minutes, 7 minutes, and 9 minutes of aggregation, respectively. Each reductive spike on the current curve corresponds to the reduction of the AgCl coating on the aggregated AgNPs resulting from the collisions of individual aggregates with the carbon UME. It is observed that as the aggregation time increases, the area under the reductive spikes (representing charge, Q) also increases. The average integral charge of the obtained spikes (200 events) at different aggregation times-1 min, 3 min, 5 min, 7 min, and 9 min-was determined to be 0.11 pC, 0.27 pC, 0.45 pC, 0.63 pC, and 0.79 pC, respectively. Provided that the AgNP aggregation elevates with time (Fig. S3, ESI†), the reduction charge of AgCl in NIE should be positively correlated with the degree of aggregation. Fig. 4f–j present enlarged views of representative spikes corresponding to Fig. 4a–e, respectively, demonstrating a greater variation in the magnitude of the charge with longer aggregation times. This observation suggests that the distribution of aggregates becomes more complex over time. | ||
| Fig. 4 Chronoamperometric curves (a)–(e) and representative current traces (f)–(j) obtained in NIE at −0.04 V vs. SCE for aggregates formed in O2-dissolved 5 mM HCl solution at different times: 1 minute (a) and (f), 3 minutes (b) and (g), 5 minutes (c) and (h), 7 minutes (d) and (i), and 9 minutes (e) and (j). | ||
Statistical distribution of AgNP number in aggregates over time
To better understand the aggregation behavior, it is essential to quantify the number of AgNPs in aggregates. To achieve this, the morphology of the AgCl coating at different aggregation times should be examined to understand how the amount of AgCl correlates with the aggregation process. TEM characterization was conducted on aggregates with aggregation times of 1, 3, 5, 7, and 9 minutes, respectively (Fig. S4a–e, ESI†), revealing that AgCl was uniformly coated on the surfaces of the AgNPs, with the thickness of the AgCl coating layer remaining largely consistent. This observation can be attributed to the rapid formation of the AgCl layer on the surfaces of the AgNPs, which inhibits further oxidation of the silver beneath the AgCl shell. The formation of larger aggregates over time can therefore be attributed to the collisions of AgCl@AgNPs, which initially form small aggregates through Brownian motion and subsequently continue to collide and grow into larger aggregates.33 In this context, the determination of AgCl can be used to quantify the number of aggregated AgNPs. Thus, the complete reduction of the AgCl coating serves as the basis for this quantification. To verify whether the AgCl coating is fully reduced, a more positive potential of 0.025 V was applied for NIE after 5 minutes of aggregation (Fig. S5, ESI†). At this potential, AgCl reduction can still occur (Fig. 3a). Compared to the spikes obtained at −0.04 V (Fig. 4c and h), the spikes at 0.025 V exhibit longer duration, lower current, and almost equal average charge (0.47 pC vs. 0.45 pC). This suggests that the interaction between the aggregates and the carbon UME follows a collision-sticking type rather than a collision-bounce-off type. The lower current at the more positive potential is attributed to a weaker driving force for the AgCl reduction reaction, and the termination of the spike results from the exhaustion of the AgCl coating. Therefore, this observation confirms that the AgCl in the aggregates can be completely reduced.To establish the relationship between the reduced charge of AgCl from individual aggregates and the number of AgNPs within the aggregates, the thickness of the AgCl coating on the AgNPs must be determined. The statistical distribution of the AgCl coating thickness after 5 minutes of aggregation was used as a representative measure, revealing an average thickness of 2.4 nm (Fig. S6, ESI†). Since each aggregate contains several AgNPs coated with an AgCl shell, the integral reduction charge (Q) during individual aggregate collisions can be correlated to the number of AgNPs within the aggregate (n) using eqn (3), assuming a spherical approximation for the AgNPs and the aggregates:
 | (3) |
 | ||
| Fig. 5 The distribution of the number of aggregated AgNPs in aggregates as determined by NIE, recorded at 1 minute (a), 3 minutes (b), 5 minutes (c), 7 minutes (d), and 9 minutes (e), along with the deconvolution into sub-distributions centered on distinct aggregate forms. | ||
At the 1-minute mark, the size distribution of the aggregates is relatively simple, primarily consisting of 2-mers and 4-mers, with 4-mers comprising the majority (Fig. 5a). As aggregation continues for 3 and 5 minutes (Fig. 5b and c), the size distribution shifts toward larger aggregates, and the proportion of 2-mers and 4-mers decreases rapidly. At 3 minutes, 8-mers become the predominant aggregate type, with larger aggregates, such as 12-mers, also emerging. By 5 minutes, the proportion of 8-mers decreases compared to 3 minutes, and 16-mers becomes the dominant form. After 7 minutes of aggregation (Fig. 5d), the size distribution of the aggregates becomes more complex, and the variation among them increases. The proportion of 16-mers decreases slightly but still accounts for a large share, while the proportion of larger 32-mers increases significantly. At 9 minutes, the proportion of 32-mers further increased, accompanied by the emergence of even larger aggregates, including 48-mers and 64-mers (Fig. 5e). These results suggest that the AgNP exhibits a diversity of aggregation behaviors, with some AgNPs forming larger aggregates while others remain in smaller aggregated structures. The deconvolution statistical analysis reveals that AgNPs undergo rapid and diverse aggregation within just 9 minutes in an acidic medium containing chloride ions at a pH of 2.3.
To verify the reliability of the NIE method in monitoring nanoparticle aggregation, SEM was used to assist in characterizing the aggregation of AgNPs. We obtained 150 SEM images of the aggregates after 5 minutes of aggregation and performed deconvolution on them (Fig. S8, ESI†). The statistical distribution of the aggregation number n obtained from both SEM and NIE experiments is quite similar, confirming the validity of using the AgCl reduction charge of the transient currents in NIE for statistical analysis.
Analysis of AgNP aggregation kinetics
To further analyze the aggregation kinetics, we listed the proportion of aggregates at different aggregation times in Table S1 (ESI†) and conducted a statistical analysis based on the main aggregation forms (Fig. 6). The evolution of aggregates over time revealed that the proportion of 4-mers decreased to zero within 9 minutes. Meanwhile, the proportions of 8-mers and 16-mers initially increased before declining, whereas the proportion of 32-mers continuously increased. This behavior can be explained by the formation of larger aggregates through the successive aggregation of smaller ones. Among them, the 4-mers exhibited the fastest rate of decrease, followed by the 8-mers, while the 16-mers showed the slowest decline. Further aggregation into larger aggregates occurred at a significantly slower rate due to the reduced nanoparticle/aggregate number density and the lower diffusion rates of larger aggregates compared to smaller ones. Consequently, over a longer aggregation period (7 to 9 minutes), the aggregates displayed a distinctly polydisperse distribution. We calculated the rates for 4-mers + 4-mers (4+4), 8+8, and 16+16 based on the proportion of aggregates at different times and found that the 4+4 aggregation has the fastest rate. Therefore, we inferred that 2+2 aggregation occurs at an even higher rate, which agrees well with the prior studies.34 Moreover, the proportion of 2-mers at 1 minute is low, suggesting that there is essentially no aggregation process of 2+2 or 1+1 during the 4+4 aggregation process. Thus, we approximated that 4+4 represents the basic aggregation reaction, and its rate constant can be calculated using the second-order reaction formula as follows: 3 × 10−2 pM−1 s−1. | ||
| Fig. 6 The proportions of the main aggregates at different aggregation times. | ||
Conclusions
In summary, this study demonstrates the application of the NIE technique for real-time, non-destructive monitoring of AgNP aggregation in acidic, chloride-containing media. This electrochemical environment closely resembles biological systems, such as those involved in the antibacterial applications of AgNPs. Under these conditions, a thin AgCl layer forms on the surface of AgNPs, interconnecting them and facilitating aggregation. Through NIE, the aggregation behavior is investigated by quantifying the number of aggregated AgNPs over time, based on the integral charge of AgCl reduction during aggregate collisions with the electrode. The dynamic evolution of different aggregate forms is captured through deconvolution analysis of the number of aggregated AgNPs, shedding light on the aggregation kinetics. It can be concluded that the aggregation process is relatively rapid during the initial stage. However, as the aggregate size increases, the aggregation rate gradually decreases. This study enhances the understanding of the physicochemical properties and dynamic behavior of AgNPs in biological environments, providing valuable insights for optimizing their utilization in real-world applications.Author contributions
Conceptualization: Y.-G. Z., H. R. H., and Y.-A. L.; funding acquisition: Y.-G. Z.; investigation: H. R. H., Y.-A. L.; experiment: H. R. H., Y.-A. L. and M. J. L.; data analysis: H. R. H and W. X.; manuscript draft: H. R. H and Y.-A. L.; manuscript editing: Y.-G. Z.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Natural Science Foundation of China (22222404) and the Science and Technology Project of Hunan Province (2024JJ3005, 2024RC1039, 2021NK1020, 2021SK1020).References
- A. D. Kurdekar, P. Chettri, R. Kurnoothala, C. S. Manohar, S. Srivastava and K. C. Vishnubhatla, J. Mater. Chem. C, 2023, 11, 16234–16246 RSC.
- M. Pagliaro, C. Della Pina, F. Mauriello and R. Ciriminna, Catalysts, 2020, 10 Search PubMed.
- Q. Yue, L. Wang, H. Fan, Y. Zhao, C. Wei, C. Pei, Q. Song, X. Huang and H. Li, Inorg. Chem., 2021, 60, 4226–4235 CrossRef CAS PubMed.
- S. Tang and J. Zheng, Adv. Healthcare Mater., 2018, 7, 1701503 CrossRef PubMed.
- G. Habibullah, J. Viktorova, P. Ulbrich and T. Ruml, RSC Adv., 2022, 12, 30386–30403 RSC.
- M. Qi, X. Wang, J. Chen, Y. Liu, Y. Liu, J. Jia, L. Li, T. Yue, L. Gao, B. Yan, B. Zhao and M. Xu, ACS Nano, 2023, 17, 8851–8865 Search PubMed.
- B. A. Chambers, A. R. M. N. Afrooz, S. Bae, N. Aich, L. Katz, N. B. Saleh and M. J. Kirisits, Environ. Sci. Technol., 2014, 48, 761–769 CrossRef CAS PubMed.
- P. Dhandapani, M. Santhoshkumar, J. Narenkumar, M. S. AlSalhi, P. A. Kumar, S. Devanesan, S. Kokilaramani and A. Rajasekar, Bioprocess Biosyst. Eng., 2022, 45, 1825–1837 CrossRef CAS PubMed.
- J. L. Axson, D. I. Stark, A. L. Bondy, S. S. Capracotta, A. D. Maynard, M. A. Philbert, I. L. Bergin and A. P. Ault, J. Phys. Chem. C, 2015, 119, 20632–20641 CrossRef CAS PubMed.
- X. Li and J. J. Lenhart, Environ. Sci. Technol., 2012, 46, 5378–5386 CrossRef CAS PubMed.
- J. M. Zook, M. D. Halter, D. Cleveland and S. E. Long, J. Nanopart. Res., 2012, 14, 1165 Search PubMed.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 Search PubMed.
- L. Kvítek, A. Panáček, J. Soukupová, M. Kolář, R. Večeřová, R. Prucek, M. Holecová and R. Zbořil, J. Phys. Chem. C, 2008, 112, 5825–5834 CrossRef.
- K. Mehrabi, B. Nowack, Y. Arroyo Rojas Dasilva and D. M. Mitrano, Environ. Sci. Technol., 2017, 51, 5611–5621 CrossRef CAS PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796–810 Search PubMed.
- D. He, M. W. Bligh and T. D. Waite, Environ. Sci. Technol., 2013, 47, 9148–9156 CAS.
- I. Fernando and Y. Zhou, Chemosphere, 2019, 216, 297–305 CrossRef CAS PubMed.
- K. Afshinnia, I. Gibson, R. Merrifield and M. Baalousha, Sci. Total Environ., 2016, 557–558, 395–403 CAS.
- B. Michen, C. Geers, D. Vanhecke, C. Endes, B. Rothen-Rutishauser, S. Balog and A. Petri-Fink, Sci. Rep., 2015, 5, 9793 Search PubMed.
- C. M. Maguire, M. Rösslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732–745 Search PubMed.
- J. H. Shi, J. L. Axson, I. L. Bergin and A. P. Ault, Anal. Chem., 2020, 92, 12257–12264 Search PubMed.
- X. Li, F. Qin, X. Chen, A. Sheng, Z. Wang and J. Liu, Environ. Sci. Technol., 2019, 53, 2416–2425 CAS.
- M. Mansor, S. Drabesch, T. Bayer, A. V. Le, A. Chauhan and A. Kappler, Environ. Sci. Technol., 2021, 8 Search PubMed.
- N. V. Rees, Y. G. Zhou and R. G. Compton, Chemphyschem.: Eur. J. Chem., Phys. Phys. Chem., 2011, 12, 1645–1647 CrossRef CAS PubMed.
- J. Ellison, K. Tschulik, E. J. E. Stuart, K. Jurkschat, D. Omanović, M. Uhlemann, A. Crossley and R. G. Compton, ChemistryOpen, 2013, 2, 69–75 CrossRef CAS PubMed.
- E. J. E. Stuart, N. V. Rees, J. T. Cullen and R. G. Compton, Nanoscale, 2013, 5, 174–177 RSC.
- E. J. E. Stuart, K. Tschulik, C. Batchelor-McAuley and R. G. Compton, ACS Nano, 2014, 8, 7648–7654 CrossRef CAS PubMed.
- X. Li, C. Batchelor-McAuley and R. G. Compton, ACS Sens., 2019, 4, 464–470 CrossRef CAS PubMed.
- Y.-Y. Bai, Y.-J. Yang, Y. Xu, X.-Y. Yang and Z.-L. Zhang, Anal. Chem., 2023, 95, 4429–4434 CrossRef CAS PubMed.
- Y. Wan, Z. Guo, X. Jiang, K. Fang, X. Lu, Y. Zhang and N. Gu, J. Colloid Interface Sci., 2013, 394, 263–268 CrossRef CAS PubMed.
- Y. Yang, N. Zhang, Q. You, X. Chen, Y. Zhang and L. Zhu, Water Res., 2023, 240, 120111 CrossRef CAS PubMed.
- R. Hao, Y. Fan and B. Zhang, J. Electrochem. Soc., 2016, 163, H3145 CrossRef CAS.
- M. Polimeno, C. Kim and F. Blanchette, ACS Omega, 2022, 7, 40826–40835 CrossRef CAS PubMed.
- W. Zhang, J. Crittenden, K. Li and Y. Chen, Environ. Sci. Technol., 2012, 46, 7054–7062 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00019j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |