
Framework nucleic acid-programmed aptamer–paclitaxel conjugates as targeted therapeutics for triple-negative breast cancer†
Lin
Li‡
 a,
Pengyao
Wei‡
a,
Tong
Kong
a,
Bo
Yuan
a,
Pan
Fu
a,
Yong
Li
bc,
Yuhui
Wang
*ad,
Jianping
Zheng
*ad and
Kaizhe
Wang
*ad
a,
Pengyao
Wei‡
a,
Tong
Kong
a,
Bo
Yuan
a,
Pan
Fu
a,
Yong
Li
bc,
Yuhui
Wang
*ad,
Jianping
Zheng
*ad and
Kaizhe
Wang
*ad
aNingbo Key Laboratory of Biomedical Imaging Probe Materials and Technology, Ningbo Cixi Institute of Biomedical Engineering, Ningbo Institute of Materials Technology and Engineering of Chinese Academy of Sciences, Ningbo, 315300, P. R. China. E-mail: wangkaizhe@nimte.ac.cn; wangyuhui@nimte.ac.cn; zhengjianping@nimte.ac.cn
bCancer Care Centre, St George Hospital, Kogarah, NSW 2217, Australia
cSt. George and Sutherland Clinical Campuses School of Clinical Medicine UNSW Sydney Kensington, NSW 2052, Australia
dUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 5th March 2025
Abstract
Triple-negative breast cancer (TNBC) is highly invasive with a poor prognosis, and chemotherapy remains the clinical treatment of choice. Paclitaxel is a commonly used first-line chemotherapy drug, but its untargeted distribution poses clinical challenges. Inspired by antibody–drug conjugates, we develop a precisely structured framework nucleic acid-programmed aptamer–paclitaxel conjugate (FAPC) with chemically well-defined paclitaxel loading dosing, enabling the regulation of receptor–aptamer affinity to facilitate tumor-targeted chemotherapy. Utilizing framework nucleic acids as a precise addressing scaffold, we organize the AS1411 aptamer with accurate intermolecular spacing and find that an inter-aptamer spacing of 19.04 nm could enhance the affinity of the FAPC for tumor cells. Then, the multifunctional FAPC can disrupt actin reorganization to achieve cytotoxicity in tumor cells. Furthermore, the AS1411-specifically modified FAPC further enhances the structure-dependent selective accumulation of drugs at tumor sites in a human xenograft model of triple-negative breast cancer, subsequently leading to significantly improved antitumor efficacy and reduced toxicity. The FAPC provides a precisely programmable platform for efficient targeted delivery of chemotherapeutic agents to malignancies.
New conceptsTriple-negative breast cancer (TNBC) is highly invasive with a poor prognosis, and chemotherapy remains the clinical treatment of choice. Paclitaxel is a commonly used first-line chemotherapy drug, but its untargeted distribution poses clinical challenges. In this study, we develop a precisely structured framework nucleic acid-programmed aptamer–paclitaxel conjugate (FAPC) with chemically well-defined paclitaxel loading dosing, enabling the regulation of receptor–aptamer affinity to facilitate tumor-targeted chemotherapy. Notably, the FAPC (i) has a precise nanostructure and programmability, allowing precise loading of chemotherapeutic agents with controlled stoichiometry; (ii) has precise addressability and allows the nanoscale spatial arrangement of aptamers, enabling affinity screening with target cells; (iii) can further enhance the structural-dependent selective accumulation of drugs at tumor sites, significantly improving anti-tumor efficacy and reducing toxicity. Given these advantages, the FAPC provides a precise programmable platform for the development of chemotherapy drugs and efficient target therapy for malignant tumors. |
Introduction
Triple-negative breast cancer (TNBC) is one of the most aggressive malignancies in women, characterized by high malignancy and invasiveness, leading to extremely poor clinical outcomes, with a five-year survival rate of less than 15%.1,2 Conventional treatments, including surgery and radiotherapy, offer limited efficacy, while targeted therapies and immunotherapy are hindered by the complex tumor heterogeneity and immunosuppressive microenvironments.3,4 Hence, chemotherapy remains the primary treatment for TNBC.5,6 Paclitaxel (PTX) is a first-line chemotherapeutic agent for breast cancer,7,8 but its low water solubility and small molecule size lead to low bioavailability and therapeutic efficacy. In clinical settings, PTX is typically bound to albumin to enhance solubility,9–11 but this approach is limited by weak hydrophobic interactions that compromise encapsulation efficiency and stability. More importantly, PTX lacks specificity in targeting cancer cells over normal cells, often leading to serious side effects.12,13 To improve the targeting ability, antibody–drug conjugates (ADCs) have been developed. ADCs combine the targeting precision of antibodies with the therapeutic potential of small-molecule drugs, aiming to improve treatment efficacy.14,15 In the past few decades, various antibodies have been employed for the conjugation with drug molecules.16–18 Although conceptually straightforward, the availability of ADCs for specific drugs remains limited, and PTX-based ADCs have not yet been developed. The development of ADCs has encountered several challenges including unmanageable toxicity, heterogeneity conjugation, restricted drug payload capacity, and poor targeting ability to impede their translation from research to clinical use in this field.Inspired by the ADC strategies, aptamer–drug conjugates (ApDCs) were developed in 2009.19 Aptamers, small single-stranded DNA or RNA sequences with their unique three-dimensional structures, exhibit a high degree of specificity for binding to target proteins or molecules.20,21 Beyond the advantages of ADCs, ApDCs offer additional benefits such as simple synthesis, high chemical stability, low immunogenicity, and ease of molecular engineering, making them highly effective for targeted cancer therapy.22,23 Recently, various aptamer-conjugation strategies have been developed for the targeted delivery of PTX.24 One approach involves the self-assembly of amphiphilic DNA–PTX conjugates into SNA-like micellar nanoparticles through Watson–Crick base pairing, creating a high drug-loading delivery system.25,26 However, this system typically lacks precise control over drug stoichiometry and fails to regulate aptamer binding affinity effectively. Another method involves chemically modifying aptamers with reactive functional groups to conjugate PTX via click reactions, enabling precise control over drug valency.27,28 However, such nucleotide modification significantly impairs its target recognition. Furthermore, the drug loading capacity remains limited because the binding of multiple drug molecules can alter the water solubility of the aptamer, leading to uncontrolled aggregate formation. Thus, there remains a demand to develop a precise and controllable ApDC with both a high payload and strong targeting ability to achieve PTX delivery for the treatment of TNBC.
In contrast to small molecule nucleic acid aptamers, framework nucleic acids (FNAs) serve as precise structural nanomaterials that enable programmable assembly of complex nanostructures.29–31 Their biocompatibility and favorable pharmacokinetic properties have been convincingly demonstrated in vivo, rendering them highly suitable for delivering various drugs.32–35 Notably, as a representative of FNA, DNA origami nanostructures (DONs) with their addressability allow for the spatial organization of multiple functional molecules (including enzymes,36 cytokine,37 peptides,38 nucleic acids,39etc.) into sophisticated geometric patterns with nanometric precision. This capability facilitates detailed investigations into how the structural characteristics of target molecules influence receptor–aptamer binding, aiding in the identification of optimal nano conformations. For example, two-dimensional DNA origami has been utilized as drawing boards to arrange peptide-MHC (pMHC) multimers or CD95 ligands in a specific geometric configuration, thereby enhancing downstream immune signal modulation.40,41 Additionally, DNA origami has been employed to construct aptamer nanoarrays with precise nanometer spacing to modulate their affinity with target proteins, aiming to construct a nanoscale anticoagulant agent.42 These studies provide valuable insights into designing affinity-tunable aptamer conjugates for PTX and other chemotherapeutic drugs.
Here, we develop a framework nucleic acid-programmed aptamer–paclitaxel conjugate (FAPC) for targeted therapy of TNBC. The six-helical-bundle DNA origami nanostructure (6-HB DON)43 is pursued as a precise addressing scaffold for ApDCs, offering exceptional control over the spatial arrangement of the targeting AS1411 aptamer and chemically well-defined PTX chemotherapeutic drugs. Specifically, while keeping the number of aptamers constant, we find that the FAPC with an aptamer spacing of 19.04 nm exhibits the highest target efficiency in TNBC cells. Due to the optimized spacing between high-affinity aptamers, the multifunctional FAPC induces efficient PTX cellular internalization and disrupts actin reorganization to achieve cytotoxicity in tumor cells. Furthermore, the AS1411-specifically modified FAPC further enhances the structure-dependent selective accumulation of drugs at tumor sites in a human xenograft model of triple-negative breast cancer, subsequently leading to significantly improved antitumor efficacy and reduced toxicity.
Results and discussion
Herein, we design a programming FAPC with chemically well-defined PTX chemotherapeutic drugs and AS1411 aptamer with precise control of spacing, all framed by a long linear topology of 6-HB DONs. 6HB-DONs with passive targeting to tumor sites can provide higher cellular uptake efficiency44 and longer circulation half-life in vivo.45 6-HB DONs provide two orders of magnitude higher drug payload capacity and allow for the loading of both the nano-drugs and targeting aptamer via chemical modification (Scheme 1). AS1411 is a nucleic acid aptamer that specifically targets nucleolin, a protein overexpressed on the surface of TNBC cells. Briefly, 6-HB DONs are prepared by assembling an M13 bacteriophage genome DNA strand with multiple staple strands. Extended DNA strands (L1) on the 6-HB DONs are designed to hybridize with the DNA–PTX conjugates, anchoring PTX molecules to the 6-HB DONs surface. The number of L1 strands determines the stoichiometry of PTX loading. Similarly, extended DNA strands (L2) were designed to hybridize into DNA–AS1411 conjugates, allowing precise control over the spacing of AS1411 aptamers on the 6-HB DONs by strategically positioning L2 sites.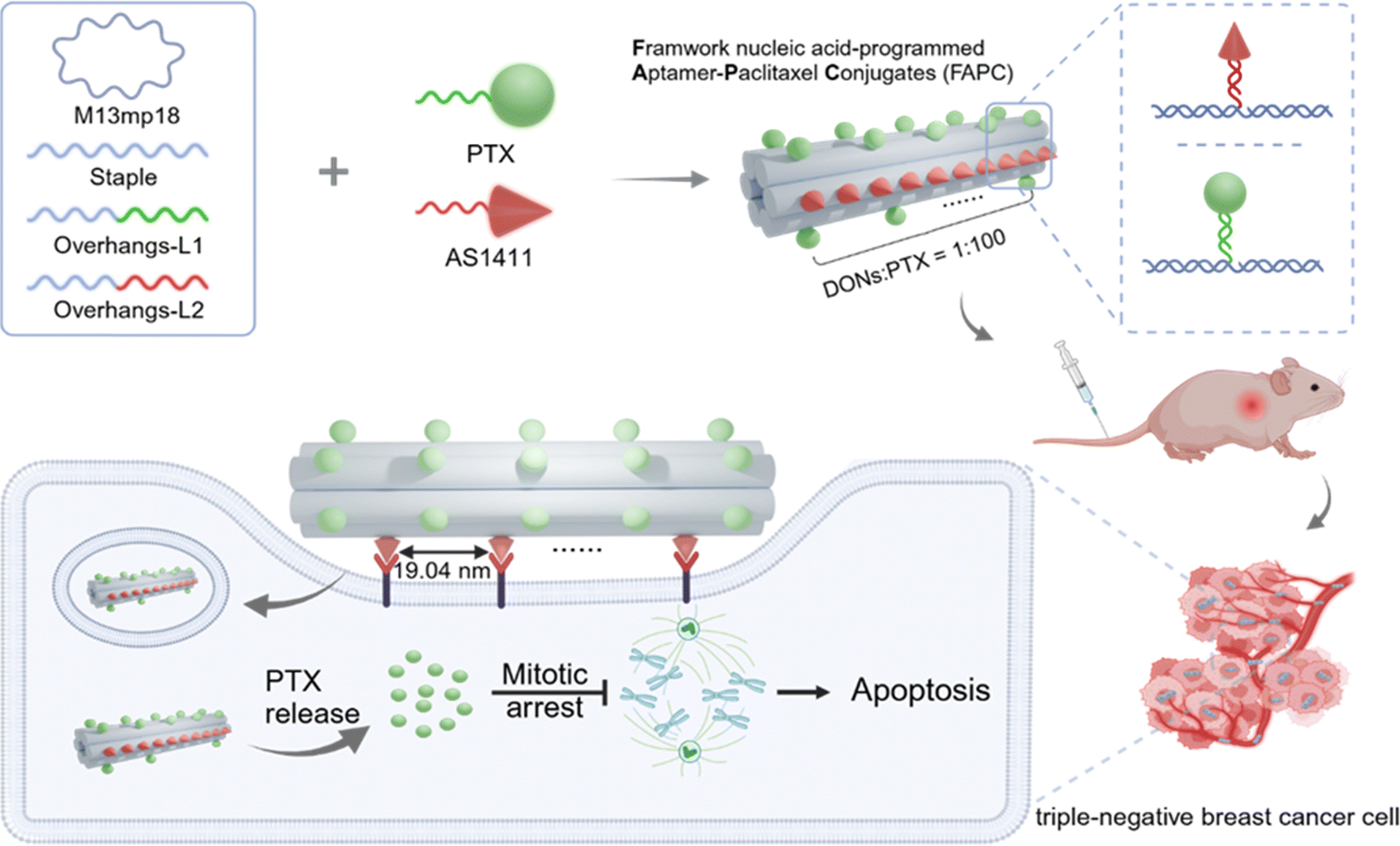 | ||
| Scheme 1 Design of FAPC nanostructure. Illustration of the construction of the FAPC nanostructure based on a DNA origami technique. Schematic representation of utilizing the FAPC nanostructure for TNBC therapy. | ||
Synthesis and characterization of FAPC nanostructures
We constructed the 6-HB DON-based FNA using a previously reported procedure46,47 (the sequences are shown in Table S1, ESI†). To prepare a paclitaxel (PTX) prodrug, we esterified PTX with 10-undecynoic acid according to a previous report,48 as confirmed by nuclear magnetic resonance (NMR) spectroscopy (Fig. S1, ESI†). Subsequently, we conjugated the PTX prodrug to an azide-modified oligonucleotide using a copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction,49 resulting in the formation of a DNA–PTX strand (Fig. S2, ESI†). PAGE analysis revealed that the DNA–PTX conjugate migrated more slowly than unmodified DNA, indicating successful conjugation (Fig. 1A). MALDI-TOF mass spectrometry validated the exact molecular weight of DNA–PTX with 6245.8, closely matching the theoretical value of 6245 (Fig. S3, ESI†). We systematically investigated the optimal reaction stoichiometry and found that the DNA–PTX production yield reached 95% at a CuBr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ssDNA molar ratio of 50:1, demonstrating superior efficiency compared to other stoichiometric ratios (Fig. S4, ESI†). We quantified the concentration of DNA–PTX using standard calculations27 (Fig. S5, ESI†). The DNA–PTX was then hybridized to capture strands on the DON surface, forming a chemically defined DON–PTX complex. Similarly, the AS1411 aptamer was attached to designated sites on the DON via complementary capture strands, allowing precise spatial positioning. Agarose gel electrophoresis showed the clear bands of three 6-HB DON-based conjugates migrated relatively slower than 6-HB DONs, indicating successful PTX and AS1411 aptamer grafting. FAPC nanostructures (DONs–AS1411–PTX) migrated more slowly than DONs–AS1411 and DONs–PTX, confirming the assembly of PTX and AS1411 aptamers onto DONs (Fig. 1B).
:ssDNA molar ratio of 50:1, demonstrating superior efficiency compared to other stoichiometric ratios (Fig. S4, ESI†). We quantified the concentration of DNA–PTX using standard calculations27 (Fig. S5, ESI†). The DNA–PTX was then hybridized to capture strands on the DON surface, forming a chemically defined DON–PTX complex. Similarly, the AS1411 aptamer was attached to designated sites on the DON via complementary capture strands, allowing precise spatial positioning. Agarose gel electrophoresis showed the clear bands of three 6-HB DON-based conjugates migrated relatively slower than 6-HB DONs, indicating successful PTX and AS1411 aptamer grafting. FAPC nanostructures (DONs–AS1411–PTX) migrated more slowly than DONs–AS1411 and DONs–PTX, confirming the assembly of PTX and AS1411 aptamers onto DONs (Fig. 1B).
 | ||
| Fig. 1 The characterization of FAPC nanostructures. (A) 17% non-denaturing polyacrylamide gel electrophoresis (PAGE) of the single DNA strand and PTX anchored DNA strand. (B) 1.5% agarose gel electrophoresis analysis of different molecule anchored DON nanostructures. Lanes 1–6: 5 kb DNA marker, M13, 6-HB DONs, DONs–AS1411, DONs–PTX, FAPC (DONs–AS1411–PTX). (C) Representative AFM images of FAPC nanostructures (left). The length value (down, right) is obtained from the dashed black line in C (up, right). (D) Frequency of the length obtained from the experimental data in (C). | ||
Atomic force microscopy (AFM) imaging revealed that the FAPC nanostructure maintained a long tubular morphology, with a length of 381.40 ± 9.76 nm, consistent with the design of DONs (Fig. 1C and D and Fig. S6, ESI†). This indicated that grafting of AS1411 and PTX did not impact the self-assembly process of DONs. Taken together, these results indicate that FAPC with precise and controllable structures were synthesized.
Modulation of cellular internalization of FAPC via controlling AS1411 aptamer spacing
To assess the stability of FAPC nanostructures, we incubated FAPC with 10% fetal bovine serum (FBS) for 6–12 h at 37 °C. Agarose gel electrophoresis results showed that FAPC still displayed an intact band even for 12 h incubation (to ≈80% of the starting amount), while DNA–PTX exhibited partial degradation (to ≈40% of the starting amount) after 6 h (Fig. S7, ESI†). Next, we investigated the impact of inter-spacing of AS1411 aptamer target molecules on the internalization efficiency of FAPC nanostructures. Maintaining a constant number of 10 aptamers, we fabricated FAPC nanostructures with inter-aptamer distances of 9.52 nm, 19.04 nm, 28.56 nm, and 38.08 nm, respectively. After 8 h incubation, we evaluated the cellular internalization efficiency of Cy5-labeled FAPC nanostructures using confocal imaging and flow cytometry. Compared to other groups, the FAPC with inter-spacing of AS1411 molecules at 19.04 nm spacing exhibited the strongest fluorescence signal (Fig. 2A and B). These results were further confirmed by flow-cytometric analysis, indicating 19.04 nm was the optimum spacing of FAPC cellular internalization (Fig. 2C and D). Therefore, we chose FAPC with inter-spacing of AS1411 molecules at 19.04 nm spacing for subsequent studies. | ||
| Fig. 2 The cellular uptake of FAPC with different inter-spacing AS1411 aptamers. (A) Confocal microscopy images of MDA-MB-231 cells incubated with Cy5 labeled FAPC (50 nM) with different aptamer spacing for 8 h. Scale bar, 20 μm. (B) Corresponding average fluorescence intensity of cells in confocal microscopy images. (C) Flow cytometry-based cellular uptake analysis of tumor cells incubated with FAPC for 8 h. (D) Quantification of the flow cytometry data in (C). Data are shown as mean ± SD of three independent experiments. (E) Confocal microscopy images of MDA-MB-231 cells and NIH-3T3 cells incubated with Cy5 labeled ss-DNA, DONs–PTX, and DONs–PTX–AS1411 for 8 h. The concentration of DNA nanostructures was 50 nM. Scale bar: 20 μm. (F) Flow cytometry-based cellular uptake analysis of MDA-MB-231 cells and NIH-3T3 cells incubated with the ss-DNA, DONs–PTX, and DONs–PTX–AS1411 nanostructure for 8 h. | ||
Next, we evaluated the targeting capability of FAPC toward TNBC cells in vitro. A human triple-negative breast cancer cell line (MDA-MB-231) that overexpress nucleolin on their membrane and a normal fibroblast cell line (NIH-3T3) were incubated with Cy5 labeled DNA–PTX, DONs–PTX, and FAPC for 8 h. Confocal imaging revealed that MDA-MB-231 cells exhibiting a higher fluorescence signal was observed with DONs–PTX compared with DNA–PTX (Fig. 2E and Fig. S8, ESI†). Cells treated with FAPC showed even stronger fluorescence signals than those treated with DONs–PTX, indicating that AS1411 aptamers endow DONs–PTX with an active targeting ability and enhance the capacity of cellular internalization. In contrast, NIH-3T3 cells displayed no significant differences in fluorescence among the three treatments, suggesting that FAPC selectively targets MDA-MB-231 cells. Then, we incubated M13, DONs–PTX, and FAPC with both cell lines. Flow analysis results showed that MDA-MB-231 cells treated with DONs–PTX exhibited higher fluorescence signals than those treated with M13, which could be ascribed to the advantage of the Dons’ architecture (Fig. 2F). Cells treated with FAPC showed even stronger fluorescence signals than those treated with DONs–PTX, consistent with the confocal imaging results.
The cytotoxicity by FAPC nanostructures in triple-negative breast cancer cells
PTX disrupts the microtubule dynamics, inhibiting the mitosis of cancer cells and triggering apoptosis in cancer cells.50,51 To assess the impact of the FAPC on cellular microtubule dynamics, we analyzed the orientation and anisotropy of actin fibers. The actin fibers of cells were stained with FITC-labeled phalloidin and imaged by confocal laser scanning microscopy (Fig. 3A). We analyzed the orientation plots of the cytoskeleton structure generated by ImageJ, in which the same color represents the same orientation (Fig. 3A). Compared to the monotonous color presented in the control group, cells treated with DONs–PTX and FAPC showed colored orientation plots, while cells treated with PTX and DNA–PTX showed a monotonous color with dispersed orientation distribution. The results demonstrate that DONs–PTX and FAPC disrupt the cellular cytoskeleton. Furthermore, FAPC-treated cells had significantly lower anisotropy values than those treated with DON–PTX, indicating that the FAPC displays the greatest ability to disrupt the cellular cytoskeleton (Fig. 3C). Taken together, FAPC targeted delivery of PTX to cancer cells enhances the destruction of microtubule dynamics. | ||
| Fig. 3 Regulation of cellular cytoskeleton reorganization and cytotoxicity in triple-negative breast cancer cells by FAPC. (A) CLSM images of actin fibers (phalloidin staining) in MDA-MB-231 cells and the corresponding orientation of actin fibers highlighted with Orientation J (ImageJ plugin) in which the different colors indicate different orientations of actin filaments, as per the given color map (B) and the microdomain distributions for actin filament orientations. Scale bar, 5 μm. (C) Quantitative analysis of the anisotropy of actin from confocal images (n = 50 cells per condition). The violin plots represent single cells. (D) The cytotoxicity induced free PTX, ssDNA–PTX, DONs–PTX, and FAPC loaded with 1 μM PTX toward tumor cells. | ||
Next, we further examined the cell-killing ability of the FAPC using a CCK-8 assay. Cells were incubated with free PTX, DNA–PTX, DONs–PTX, and FAPC for 24 h, each containing equivalent PTX concentrations (Fig. 3D). The result showed that cells treated with DONs–PTX exhibited lower viability compared to those treated with free PTX or DNA–PTX, indicating that the 6-HB DONs nanocarrier enhances PTX's cell-killing effect. More importantly, the FAPC-treated group presented significantly reduced cell viability compared to the DONs–PTX group, indicating that the AS1411 aptamer enhances the cell-killing ability of DONs–PTX by promoting cell internalization. These results demonstrated that FAPC serves as an effective nanoplatform for delivering PTX, enhancing its cell-killing ability against triple-negative breast cancer.
Targeted antitumor activity of FAPC in vivo
Inspired by the remarkable targeted performance in vitro, we further evaluated the tumor-targeting capability of FAPC nanostructures in vivo. Cy5.5-labeled M13, 6-HB DONs, and FAPC nanostructures were injected via the tail vein in xenograft breast cancer models, and fluorescence signals were monitored using an IVIS Imaging Spectrum System at various time points. The dynamic biodistribution of Cy5.5-labeled M13, 6-HBDONs, and FAPC nanostructures in vivo was tracked over 24 h (Fig. 4A and B). Mice treated with 6-HBDONs exhibited significantly stronger fluorescence at tumor sites compared to those treated with M13, indicating enhanced accumulation and retention of DNA origami nanostructures in tumors. In particular, the FAPC-treated group showed even higher fluorescence intensity at tumor sites than the 6-HB DONs-treated group. To further investigate the biodistribution, mice were sacrificed at 24 h post-injection, and major tissues (heart, liver, spleen, lung, kidneys) and tumor were dissected for fluorescence imaging. Tumors from 6-HB DON-treated mice exhibited stronger fluorescence than those from M13-treated mice. Ex vivo fluorescence imaging revealed that FAPC nanostructures were mainly accumulated in the tumor with minimal distribution in the liver and kidneys, while 6-HB DONs were accumulated in tumors and the liver and kidneys (Fig. 4C and D). These results suggest that the FAPC exhibits active targeting after modification with the AS1411 aptamer at specific distances. | ||
| Fig. 4 The biodistribution of the FAPC nanostructures. (A) In vivo images of MDA-MB-231 tumor-bearing mice before and after intravenous injection of Cy5.5 labeled M13 (40 nM, 100 μL), 6HB-DONs (40 nM, 100 μL), and FAPC nanostructures (40 nM, 100 μL). (B) Quantitative analysis of the relative fluorescence light intensity in tumors. (C) After 24 h injection, the animals were sacrificed and tumor tissues, as well as major organs (heart, liver, spleen, lung, and kidney), were collected for ex vivo imaging. (D) Fluorescence intensities of individual organs from mice treated with different groups. | ||
To systematically evaluate the antitumor activity of FAPC nanostructures, MDA-MB-231 cell-xenografted mice were established and administered saline, free PTX, DONs–PTX, and FAPC nanostructures via the tail vein every 3 days with equivalent amounts of PTX (Fig. 5A). The tumor volumes and body weight of mice were recorded every 3 days during the treatment to evaluate the therapeutic efficacy. PTX-treated groups elicited moderate tumor-inhibiting efficacy, while the volume of tumors in the saline-treated group sharply increased (Fig. 5B–D). In particular, the DON–PTX-treated groups exhibited a great ability to suppress tumor growth compared with the free PTX-treated groups, indicating 6-HB DON delivery improves the PTX accumulation in the tumor site. Notably, the FAPC-treated groups exhibited a great ability to suppress tumor growth compared with the DON–PTX-treated groups, indicating aptamer loading indeed enhanced the therapeutic efficacy. Additionally, changes in body weight were monitored during the treatment period. Compared to the control group, mice in the FAPC-treated group showed no significant change in body weight, while the PTX-treated group exhibited a significant decrease, indicating that 6-HB DONs reduced the toxicity of PTX (Fig. 5E). Next, the tumor tissues were further analyzed by histological studies. Hematoxylin and eosin (H&E) staining revealed nuclear fragmentation and a decrease in the number of tumor cells in the FAPC-treated group. TUNEL staining verified more apoptotic cells in the FAPC-treated group by stronger green fluorescence signals (Fig. 5F). These results indicate that FAPC exhibits significant therapeutic effects in a triple-negative breast cancer xenograft mouse model.
 | ||
| Fig. 5 The targeted antitumor activity of FAPC in vivo. (A) Illustration of the treatment process. (B) Tumor weight of excised tumors after the treatments. (C) Tumor volume changes of MDA-MB 231 xenograft bearing mice after intravenous injection of saline, free PTX (0.04 mg kg−1), DONs–PTX (2.39 mg kg−1), and DONs–AS1411–PTX (2.39 mg kg−1) every 3 days for 6 times (n = 3 for each group). (D) Representative photographs of excised tumors after the last drug administration. (E) Body weight changes of mice during the treatments. (F) H&E staining and TUNEL staining of the tumor slices excised from MDA-MB 231 xenograft-bearing mice after the last drug administration. Scale bar, 50 μm. | ||
Conclusions
In summary, we developed a precise programmable framework nucleic acid-based aptamer–paclitaxel conjugate for the treatment of triple-negative breast cancer. The developed multifunctional FNA-based FAPC shows several distinct advantages. First, the FAPC has a precise nanostructure and programmability, allowing precise loading of chemotherapeutic agents with controlled stoichiometry. Second, the FAPC has precise addressability and allows the nanoscale spatial arrangement of aptamers, enabling affinity screening with target cells. Third, the FAPC can further enhance the structural-dependent selective accumulation of drugs at tumor sites, significantly improving anti-tumor efficacy and reducing toxicity. Given these advantages, the FAPC provides a precise programmable platform for the development of chemotherapy drugs and efficient target therapy for malignant tumors.Materials and methods
Materials
PTX was purchased from Shanghai Jinhe Bio-Pharmaceutical Co., Ltd (Shanghai, China). Undec-10-yonic acid, 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC), 4-dimethylaminopyridine (DMAP), tert-butanol (t-BuOH) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Cuprous bromide was purchased from Sigma Aldrich Co. Ltd (China). Dichloromethane (99.9%, SuperDry, with molecular sieves, water ≤ 30 ppm), tris[(1-benzyl1H-1,2,3-triazol-4-yl)methyl]amine (TBAB), and dimethyl sulfoxide (DMSO) were purchased from J&K Scientific Ltd (Shanghai, China). The single-stranded DNA scaffold (M13 ssDNA) was purchased from Cnbioruler (Suzhou, China). All oligonucleotides were purchased from Sangon Biotech (Shanghai, China). The single strands were stored in 96-well plates with a concentration of 100 μM and used without further purification. Dulbecco's modified Eagle's medium (DMEM), phosphate-buffered saline (PBS), antibiotics (penicillin/streptomycin), and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, USA). Hoechst 33342 was purchased from Dojindo Laboratories (Kumamoto, Japan). Cell Counting Kit-8 was purchased from Glpbio. Actin-Tracker Green-488 was purchased from the Beyotime Institute of Biotechnology. All other chemicals or materials were purchased from Sigma-Aldrich and used as received unless stated otherwise. The ultrapure water was used in all experiments.Synthesis of the paclitaxel anchored DNA strand (DNA–PTX)
Synthesis of pro-PTX. PTX (204.38 mg, 239.35 μM, 1.0 equiv.), under-10-yonic acid (65.44 mg, 359.03 μM, 1.5 equiv.), a catalytic quantity of N,N-dimethylaminopyridine (DMAP, 43.86 mg, 359.03 μM, 1.5 equiv.) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 68.83 mg, 359.03 μM, 1.5 equiv.) in dry dichloromethane (1 L) was added. The resulting solution was left to stir under nitrogen at 25 °C for 20 h. The reaction mixture was then concentrated in vacuo to give a yellow solid. The pro-PTX solution (dissolved in 50% ethanol, 1 mM, 3 μL), cuprous bromide, and TBAB were dissolved in H2O/DMSO/t-BuOH, 4:3:1 (5 mM, 10 μL) and mixed with azide modified DNA single strand (5′-N3-AGATCATTGATCGTGG-3′) solution (0.1 μM, 10 μL) under 500 ramps for 6 h at 30 °C. Then, the mixture was sonicated for 20 min and centrifuged to remove paclitaxel and other reactants.
Quantification of DNA–PTX
17% non-denaturing PAGE gel was prepared by using the 40% acrylamide (29:1, acrylamide/bisacrylamide, 3.75 mL) solution, 1 × TAE/Mg2+ buffer (10 mM), ultrapure water (5.25 mL), ammonium persulfate (75 μL) and tetramethyl ethylenediamine (TEMED) (7.5 μL). DNA–PTX and different concentrations of ss-DNA were mixed with loading dye (2 μL) and then analyzed at 4 °C (100 V) for about 45 min. The bands were stained with gel red, visualized by UV exposure (254 nm), and photographed by UVP Bio-Imaging Systems.
Synthesis of 6-HB DONs
6-HB DONs were designed and assembled according to Rothemund's and Yan's methods.46,47 Briefly, M13mp18 and 168 staple strands were assembled in 1 × TAE/Mg2+ buffer (40 mM tris, 20 mM acetic acid, 2 mM EDTA, and 12.5 mM magnesium acetate, pH 8.3), with a molar ratio of M13mp18 to each staple strand of 1:5, and these strands were assembled in TAE/Mg2+ buffer by using a thermocycler (Veriti 96-Well Thermal Cycler, Thermofisher), slowly annealing from 95 °C to 25 °C for 8 h. The annealed solution was centrifuged using a 100 kDa MWCO ultrafiltration tube at a speed of 3000 g for 15 min, and this process was repeated 3 times to remove excess un-assembled staple strands. The concentration of 6-HB DONs was measured by the absorbance peak at 260 nm using a UV spectrophotometer (NanoPhotomer, Implen) with a molar extinction coefficient of 1.091 × 108 M−1 cm−1.
Synthesis of FAPC nanostructure
The DNA nanostructures with protruding handles (capture strands for AS1411aptamer and PTX) were designed based on the free 6-HB DONs nanostructures. Briefly, the molar ratio of M13 DNA (20 nM) to helper strands (staple stands and capture stands) is 1:5, and these strands were assembled in TAE/Mg2+ buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate, pH 8.3) by using the thermocycler (Veriti 96-Well Thermal Cycler, Thermofisher), slowly annealing from 95 °C to 25 °C for 8 h. For purification, the MWCO 100 kDa ultrafiltration filter was used under the centrifuging speed of 3000 g 3 times to remove the free staple stands and capture strands. The purified FAPC nanostructures were characterized by agarose gel electrophoresis and AFM imaging.
Agarose gel electrophoresis characterization of the FAPC nanostructure
Each sample was mixed with loading dye (1 μL) and analyzed using 1.2% agarose gel at 4 °C (100 V) for 60 min in TAE/Mg2+ buffer. The bands were stained with GelRed, visualized by UV exposure (254 nm), and photographed by a UV spectrophotometer.AFM characterization of the FAPC nanostructure
5 μL solution of sample absorbed for 10 min onto a freshly cleaved mica surface substrate. Subsequently, the sample drop was washed off by ultrapure water and then dried with compressed air. AFM imaging was performed using ScanAsyst mode in Dimension FastScan AFM (Bruker).Stability of the FAPC nanostructure
Each sample (50 nM, 10 μL) was incubated with DMEM culture medium containing 10% fetal bovine serum (FBS) at 37 °C for 0 h, 6 h, and 12 h, respectively. After incubation, each sample was mixed with loading dye (1 μL) and analyzed using 1.2% agarose gel at 4 °C (100 V) for 45 min in TAE/Mg2+ buffer. The bands were stained with GelRed, visualized by UV exposure (254 nm), and photographed by a UV spectrophotometer. The images were analyzed by ImageJ.Cell culture
MDA-MB-231 cells (breast cancer cell line) and NIH-3T3 cells (nucleolin-negative normal cells) were purchased from the Chinese Academy of Sciences Institute of Cell Resource Center (Shanghai, China). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) medium (containing 10% FBS, 100 U ml−1 penicillin, and 100 U ml−1 streptomycin) in a humidified atmosphere containing 5% CO2 at 37 °C.Cell uptake of the FAPC nanostructure
Confocal laser scanning microscopy (CLSM) and flow cytometry were used to investigate the cellular internalization behaviors of FAPC. For CLSM imaging, MDA-MB-231 cells and NIH-3T3 cells (3 × 104 in 200 μL of medium) were seeded in 35 mm confocal dishes and cultured overnight. Then the cells were incubated with Cy5 labeled ss-DNA (50 nM, 200 μL), 6-HB DONs (DONs origami 5 nM, 200 μL), DONs–AS1411 (DONs origami 5 nM, 200 μL) for 8 h at 37 °C, respectively. Subsequently, the cells were incubated with 4% paraformaldehyde for 15 min, then washed with PBS three times and imaged by laser confocal fluorescent microscopy (DFC 450C, Leica, Germany). For flow cytometry, MDA-MB-231 cells and NIH-3T3 cells (3 × 105 in 1 mL of medium) were seeded in 6 well culture plates and cultured overnight. Then the cells were incubated with Cy5 labeled ss-DNA (50 nM, 400 μL), DONs (DONs origami 5 nM, 400 μL), and DONs–AS1411 (DONs origami 5 nM, 400 μL) for 8 h, respectively. Thereafter, the culture medium was removed and the cells were washed three times with PBS. The amounts of intracellular fluorescent signals were quantified using a flow cytometry system (FACSCalibur, BD, USA). Finally, the cell populations were analyzed with FlowJo software (Tree Star, OR, USA).Cellular internalization studies of AS1411 aptamer spacing on the FAPC nanostructure
CLSM and flow cytometry were used to investigate the impact of the AS1411 aptamer spacer on cellular internalization. For CLSM imaging, MDA-MB-231 cells (3 × 104 in 200 μL of medium) were seeded in 35 mm confocal dishes and cultured overnight. Then the cells were incubated with an equivalent dosage of Cy5 labeled DONs–AS1411 (aptamer spacing, 9.52 nm, 19.04 nm, 28.56 nm, 38.08 nm, respectively) for 8 h at 37 °C, respectively. Subsequently, cells were stained with Hoechst 33342 fluorescence dye for 15 min. After that, the cells were incubated with 4% paraformaldehyde for 15 min and were washed by PBS three times and imaged by laser confocal fluorescent microscopy (DFC 450C, Leica, Germany). For flow cytometry, MDA-MB-231 cells (3 × 105 in 1 mL of medium) were seeded in 6 well culture plates and cultured overnight. Then the cells were incubated with an equivalent dosage of Cy5 labeled DONs–AS1411 (aptamer spacing, 9.52 nm, 19.04 nm, 28.56 nm, 38.08 nm, respectively) for 8 h at 37 °C, respectively. Thereafter, the culture medium was removed and the cells were washed three times with PBS. The amounts of intracellular fluorescent signals were quantified using a flow cytometry system (FACSCalibur, BD, USA). Finally, the cell populations were analyzed with FlowJo software (Tree Star, OR, USA).Cell viability assay
The cytotoxicity of FAPC was tested by cell counting kit-8 (CCK-8) assay. MDA-MB-231 cells (1 × 104 in 100 μL of medium) were seeded in 96-well plates and cultured overnight. Then the cells were incubated with free PTX, DNA–PTX, DONs–PTX, and FAPC for 24 h. The different formulations of the drug were adjusted to equal PTX concentration before they were added. The untreated cells were used as a control. The cells were washed with PBS and incubated with 100 μL DMEM medium with 10% CCK-8 reagent for 2 h. The absorbance values at 450 nm were measured by an automated plate reader (iMark (168-1130), Biorad, USA).Cytoskeleton analysis and measurement of actin filament orientation
MAD-MB-231 cells were seeded in 35 mm confocal dishes and cultured overnight. Subsequently, the cells were incubated with PBS, DNA–PTX (500 nM, 200 μL), DONs–PTX (DONs origami 5 nM, 200 μL), and FAPC (DONs origami 5 nM, 200 μL) in DMEM medium containing 10% FBS for 24 h at 37 °C. After being washed twice with PBS, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed three times with precooling PBS, permeabilized with 0.3% Triton X-100 for 5 min, washed three times with PBS again, and then incubated with FITC labeled phalloidin for 1 h. After the samples were washed twice with PBS, fluorescence images (λex = 488 nm, λem = 505 nm) of cells were captured by laser confocal fluorescent microscopy (DFC 450C, Leica, Germany). The counting of cell ruffles was performed on 50 cells in each experiment. The images of cells with FITC-Phalloidin stained actin filaments were analyzed using ImageJ with the Orientation J plugin and the actin anisotropy per cell was quantified using Fibril Tool.Establishment of the MDA-MB-231 tumor-bearing mice model
Female 5-week BALB/c nude mice were purchased from Hangzhou Ziyuan Experimental Animal Technology Co., Ltd. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Wenzhou Medical University and approved by the Animal Ethics Committee of Wenzhou Medical University (Ningbo, China). The subcutaneous tumor models of the breast tumor were established according to a previously described method.52 The tumors were allowed to grow for a volume of 50 mm3 to 200 mm3 for in vivo biodistribution imaging experiments and antitumor studies, respectively. Tumor volumes were calculated by the following formula: tumor volume = 0.5 × ab2, where a and b are the longest and the shortest diameters of the tumor, respectively.The biodistribution of the FAPC nanostructure in vivo
Wen the tumor size reached 200 mm3 or so, the mice were randomly divided into 3 groups (n = 3 per group). Each group was administered with free Cy5.5-M13 (40 nM, 100 μL), Cy5.5-DONs (40 nM, 100 μL), and Cy5.5-FAPC (40 nM, 100 μL) via tail vein injection, respectively. At 1 h, 3 h, 6 h, 8 h, 12 h, and 24 h post-injection, the mice were imaged by an IVIS Imaging Spectrum System and analyzed by IVIS Living Imaging 3.0 software (IVIS Lumina XRMS III, PerkinElmer, USA). After the last time point, the mice were sacrificed with major organs and tumors harvested for ex vivo imaging.In vivo antitumor evaluation
When the tumor volume of BALB/c mice reached 50 mm3 or so, the tumor-bearing mice were randomly divided into four groups (n = 3 for each group). The mice were intravenously injected with saline as the control, free PTX, DONs–PTX, and FAPC with a PTX dosage of 0.04 mg kg−1 which was administered every 3 days during the 18-day treatment. The observation lasted for 21 days, during which the tumor size of each mouse was measured using a digital vernier caliper across its longest (a) and shortest (b) diameters. Meanwhile, the body weight of each tumor-bearing mouse was also recorded. On day 21, the mice were sacrificed. Tumors and major organs were excised for snapshotting and weight measurement and then stored at −80 °C. Tumors were fixed in 4% paraformaldehyde, and cryosections were stained with TUNEL and eosin (H&E) for histological evaluation.Statistical Analysis
All data were collected in triplicate and reported as means ± SD by GraphPad Prism 8 (San Diego, CA). Comparison between the groups was performed by two-way ANOVA with Tukey's multiple comparison test or Student's t-test. *p values < 0.05, **p values < 0.01, and ***p values < 0.001.Author contributions
L. Li: methodology, data curation, writing – original draft; P. Wei: methodology, data curation; T. Kong: formal analysis; B. Yuan: visualization and conceptualization; P. Fu: investigation, writing – review and editing; Y. Ling: investigation; Y. Wang and J. Zheng: supervision, funding acquisition, writing – review and editing; K. Wang: conceptualization, project administration, writing – review & editing, and supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22105124), the Ningbo 3315 Innovation Teams Program (No. 2019A-14-C), the Joint Funds of the Zhejiang Provincial Natural Science Foundation of China under Grant (No. LBZ24H80001), the Natural Science Foundation of Ningbo City (No. 2022J046, 2023J350), the “Science and Innovation Yongjiang 2035” Key R&D Programme of Ningbo (2024Z183), and the Ningbo Yongjiang Talent Introduction Programme (No. 2023A-114-G).References
- A. C. Garrido-Castro, N. U. Lin and K. Polyak, Cancer Discovery, 2019, 9(2), 176–198 Search PubMed.
- A. Goldhirsch, W. C. Wood, A. S. Coates, R. D. Gelber, B. Thurlimann, H. J. Senn and M. Panel, Ann. Oncol., 2011, 22(8), 1736–1747 CrossRef CAS PubMed.
- A. Lee and M. B. A. Djamgoz, Cancer Treat. Rev., 2018, 62, 110–122 Search PubMed.
- A. Bosch, P. Eroles, R. Zaragoza, J. R. Viña and A. Lluch, Cancer Treat. Rev., 2010, 36(3), 206–215 CrossRef CAS PubMed.
- Y. Li, H. Zhang, Y. Merkher, L. Chen, N. Liu, S. Leonov and Y. Chen, J. Hematol. Oncol., 2022, 15(1), 121 Search PubMed.
- F. Poggio, M. Bruzzone, M. Ceppi, N. F. Ponde, G. L. Valle, L. D. Mastro, E. D. Azambuja and M. Lambertini, Ann. Oncol., 2018, 29(7), 1497–1508 CrossRef CAS PubMed.
- P. Schmid, H. S. Rugo, S. Adams, A. Schneeweiss, C. H. Barrios, H. Iwata, V. Dieras, V. Henschel, L. Molinero, S. Y. Chui, A. Husain, E. Winer, S. Lio and L. A. Emens, Lancet Oncol., 2020, 21(1), 44–59 CrossRef CAS PubMed.
- X. Qian, H. Yang, Z. Ye, B. Gao, Z. Qian, Y. Ding, Z. Mao, Y. Du and W. Wang, ACS Nano, 2024, 18(24), 15864–15877 CrossRef CAS PubMed.
- Q. Chen, C. Liang, C. Wang and Z. Liu, Adv. Mater., 2015, 27(5), 903–910 CrossRef CAS PubMed.
- C. Wang, X. Xu, P. Zhang, S. Xiong, J. Yuan, X. Gao, W. Guan, F. Wang, X. Li, H. Dou and G. X. Xu, J. Nanobiotechnol., 2022, 20(1), 319 CrossRef CAS PubMed.
- T. Wang, J. Ding, Z. Chen, Z. Zhang, Y. Rong, G. Li, C. He and X. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 9868–9879 CrossRef CAS PubMed.
- M. Untch, C. Jackisch, A. Schneeweiss, B. Conrad, B. Aktas, C. Denkert, H. Eidtmann, H. Wiebringhaus, S. Kummel, J. Hilfrich, M. Warm, S. Paepke, M. Just, C. Hanusch, J. Hackmann, J. Blohmer, M. Clemens, S. arb-Esfahani, W. D. Schmitt, B. Gerber, K. Engels, V. Nekljudova, S. Loibl and G. V. Minckwitz, Lancet Oncol., 2016, 17(3), 345–356 Search PubMed.
- C. Zielinski, I. Lang, M. Inbar, Z. Kahan, R. Greil, S. Beslija, S. M. Stemmer, Z. Zvirbule, G. G. Steger, B. Melichar, T. Pienkwski, D. Sirbu, L. Petruzelka, A. Eniu, B. Nisenbaum, M. Dank, R. Anghel, D. Messinger and T. Brodowicz, Lancet Oncol., 2016, 17(9), 1230–1239 CrossRef CAS PubMed.
- Z. Fu, S. Li, S. Han, C. Shi and Y. Zhang, Target. Ther., 2022, 7(1), 93 CAS.
- C. H. Chau, P. S. Steeg and W. D. Figg, Lancet, 2019, 394(10200), 793–804 CrossRef CAS PubMed.
- J. Y. Li, S. R. Perry, V. Muniz-Medina, X. Wang, L. K. Wetzel, M. C. Rebelatto, M. J. M. Hinrichs, B. Z. Bezabeh, R. L. Fleming, N. Dimasi, H. Feng, D. Toader, A. Q. Yuan, L. Xu, J. Lin, C. Gao, H. Wu, R. Dixit, J. K. Osbourn and S. R. Coats, Cancer Cell, 2019, 35(6), 948–949 Search PubMed.
- H. Lu, D. Wang, S. Kazane, T. Javahishvili, F. Tian, F. Song, A. Sellers, B. Barnett and P. G. Schultz, J. Am. Chem. Soc., 2013, 135(37), 13885–13891 CrossRef CAS PubMed.
- S. F. Yu, B. Zheng, M. Go, J. Lau, S. Spencer, H. Raab, R. Soriano, S. Jhunjhunwala, R. Cohen, M. Caruso, P. Polakis, J. Flygare and A. G. Polson, Clin. Cancer Res., 2015, 21(14), 3298–3306 CrossRef CAS PubMed.
- Y. F. Huang, D. Shangguan, H. Liu, J. A. Phillips, X. Zhang, Y. Chen and W. Tan, ChemBioChem, 2009, 10(5), 862–868 CrossRef CAS PubMed.
- C. Liang, B. Guo, H. Wu, N. Shao, D. Li, J. Liu, L. Dang, C. Wang, H. Li, S. Li, W. K. Lau, Y. Cao, Z. Yang, C. Lu, X. He, D. W. T. Au, X. Pan, B. Zhang, C. Lu, H. Zhang, K. Yue, A. Qian, P. Shang, J. Xu, L. Xiao, Z. Bian, W. Tan, Z. Liang, F. He, L. Zhang, A. Lu and G. Zhang, Nat. Med., 2015, 21(3), 288–294 CrossRef CAS PubMed.
- C. Ji, J. Wei, L. Zhang, X. Hou, J. Tan, Q. Yuan and W. Tan, Chem. Rev., 2023, 123(22), 12471–12506 Search PubMed.
- X. Wu, J. Chen, M. Wu and J. X. Zhao, Theranostics, 2015, 5(4), 322–344 Search PubMed.
- B. Huang, W. Lai, C. Yeh, Y. Tseng, K. Pech, P. Yang and E. Lin, ACS Nano, 2024, 18(41), 27905–27916 CrossRef CAS PubMed.
- J. He, Q. Duan, C. Ran, T. Fu, Y. Liu and W. Tan, Acta Pharm. Sin. B, 2023, 13(4), 1358–1370 Search PubMed.
- Y. Guo, J. Zhang, F. Ding, G. Pan, J. Li, J. Feng, X. Zhu and C. Zhang, Adv. Mater., 2019, 31(16), e1807533 CrossRef PubMed.
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138(34), 10834–10837 Search PubMed.
- J. He, T. Peng, Y. Peng, L. Ai, Z. Deng, X. Q. Wang and W. Tan, J. Am. Chem. Soc., 2020, 142(6), 2699–2703 Search PubMed.
- L. Zhu, J. Yang, Y. Ma, X. Zhu and C. Zhang, J. Am. Chem. Soc., 2022, 144(4), 1493–1497 CrossRef CAS PubMed.
- J. Zeng, W. Fu, Z. Qi, Q. Zhu, H. He, C. Huang, H. Zuo and C. Mao, Small, 2019, 15(26), e1805552 CrossRef PubMed.
- E. Benson, R. C. Marzo, J. Bath and A. Turberfield, J. Sci. Robot., 2022, 7(65), eabn5459 CrossRef PubMed.
- Y. Liu, Z. Dai, X. Xie, B. Li, S. Jia, Q. Li, M. Li, C. Fan and X. Liu, J. Am. Chem. Soc., 2024, 146(8), 5461–5469 CrossRef CAS PubMed.
- Q. Jiang, Y. Shang, Y. Xie and B. Ding, Adv. Mater., 2024, 36(22), e2301035 Search PubMed.
- D. Jiang, Z. Ge, H. J. Im, C. G. England, D. Ni, J. Hou, L. Zhang, C. J. Kutyreff, Y. Yan, Y. Liu, S. Y. Cho, J. W. Engle, J. Shi, P. Huang, C. Fan, H. Yan and W. Cai, Nat. Biomed. Eng., 2018, 2(11), 865–877 Search PubMed.
- M. Safarkhani, S. Ahmadi, H. Ipakchi, M. R. Saeb, P. Makvandi, M. E. Warkiani, N. Rabiee and Y. Huh, Adv. Sci., 2024, e2401617 CrossRef PubMed.
- Z. Wang, L. Song, Q. Liu, R. Tian, Y. Shang, F. Liu, S. Liu, S. Zhao, Z. Han, J. Sun, Q. Jiang and B. Ding, Angew. Chem., Int. Ed., 2020, 60(5), 2594–2598 CrossRef PubMed.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. Zhou, G. Nie, H. Yan, B. Ding and Y. Zhao, Nat. Biotechnol., 2018, 36(3), 258–264 CrossRef CAS PubMed.
- Y. Fan, C. Wang, W. Dai, Y. Zhou, G. Lu, W. Li, L. Li and T. Lin, ACS Appl. Mater. Interfaces, 2024, 16, 38979–38988 CrossRef CAS PubMed.
- W. Tang, T. Tong, H. Wang, X. Lu, C. Yang, Y. Wu, Y. Wang, J. Liu and B. Ding, Angew. Chem., Int. Ed., 2023, 62(51), e202315093 CrossRef CAS PubMed.
- J. Xiong, Z. He, L. Wang, C. Fan and J. Chao, J. Am. Chem. Soc., 2024, 146(9), 6317–6325 CrossRef CAS PubMed.
- Y. Sun, L. Yan, J. Sun, M. Xiao, W. Lai, G. Song, L. Li, C. Fan and H. Pei, Nat. Commun., 2022, 13(1), 3916 CrossRef CAS PubMed.
- L. Li, J. Yin, W. Ma, L. Tang, J. Zou, L. Yang, T. Du, Y. Zhao, L. Wang, Z. Yang, C. Fan, J. Chao and X. Chen, Nat. Mater., 2024, 23(7), 993–1001 Search PubMed.
- S. Zhao, R. Tian, J. Wu, S. Liu, Y. Wang, M. Wen, Y. Shang, Q. Liu, Y. Li, Y. Guo, Z. Wang, T. Wang, Y. Zhao, H. Zhao, H. Cao, Y. Su, J. Sun, Q. Jiang and B. Ding, Nat. Commun., 2021, 12(1), 358 CrossRef CAS PubMed.
- S. M. Douglas, J. J. Chou and W. M. Shih, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(16), 6644 CrossRef CAS PubMed.
- P. Wang, M. A. Rahman, Z. Zhao, K. Weiss, C. Zhang, Z. Chen, S. J. Hurwitz, Z. G. Chen, D. M. Shin and Y. Ke, J. Am. Chem. Soc., 2018, 140(7), 2478–2484 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2(4), 249–255 CrossRef CAS PubMed.
- L. A. Stearns, R. Chhabra, J. Sharma, Y. Liu, W. T. Petuskey, H. Yan and J. C. Chaput, Angew. Chem., Int. Ed., 2009, 48(45), 8494–8496 Search PubMed.
- P. W. Rothemund, Nature, 2006, 440(7082), 297–302 Search PubMed.
- R. A. Petrov, S. Y. Maklakova, Y. A. Ivanenkov, S. A. Petrov, O. V. Sergeeva, E. Y. Yamansarov, I. V. Saltykova, I. I. Kireev, I. B. Alieva, E. V. Deyneka, A. A. Sofronova, A. V. Aladinskaia, A. V. Trofimenko, R. S. Yamidanov, S. V. Kovalev, V. E. Kotelianski, T. S. Zatsepin, E. K. Beloglazkina and A. G. Majouga, Bioorg. Med. Chem. Lett., 2018, 28(3), 382–387 CrossRef CAS PubMed.
- M. Pilkington-Miksa, D. Arosio, L. Battistini, L. Belvisi, M. D. Matteo, F. Vasile, P. Burreddu, P. Carta, G. Rassu, P. Perego, N. Carenini, F. Zunino, M. D. Cesare, V. Castiglioni, E. Scanziani, C. Scolastico, G. Casiraghi, F. Zanardi and L. Manzoni, Bioconjugate Chem., 2012, 23(8), 1610–1622 Search PubMed.
- M. A. Jordan, R. J. Toso and L. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90(20), 9552–9556 Search PubMed.
- A. C. Von, P. Wadsworth and M. A. Jordan, Mol. Biol. Cell, 1999, 10(4), 947–959 Search PubMed.
- J. Du, Y. Zhang, Z. Jin, H. Wu, J. Cang, Y. Shen, F. Miao, A. Zhang, Y. Zhang, J. Zhang and G. Teng, Mater. Sci. Eng., C, 2020, 116, 111188 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00652f |
| ‡ Lin Li and Pengyao Wei contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |