
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal–organic framework with chiral nanochannels for enantioselective fluorescence switching of amino alcohols†
Ritu
Ladhi
,
Arshminder Kaur
Dhillon
and
Monika
Singh
 *
*
Institute of Nano Science and Technology, Knowledge City, Sector-81, Mohali-140306, Punjab, India. E-mail: monika@inst.ac.in
First published on 8th July 2025
Abstract
The origin of chirality in metal–organic frameworks (MOFs) from achiral linkers remains largely unpredictable, as it mainly stems from the intrinsic arrangement of structural components within the framework. To date, only a handful of reports have explored the achiral ligands for chiral MOF synthesis; their potential for chiral molecular recognition remains a fascinating area of research. Here, we have synthesised a 3-D Co-bpy MOF with chiral nanopores using an achiral linker, which undergoes spontaneous chirality induction through asymmetric crystallization. The MOF crystallizes in a P41212 chiral space group, featuring distinctive 4-fold helical chains generating chiral nanochannels along the c-axis. Remarkably, this structural arrangement enables Co-bpy to exhibit enantioselectivity among chiral amino alcohols by “turn-off” and “turn-off–on” type fluorescence responses for each, with a high enantioselectivity factor.
Introduction
Chirality, a subtle force of nature, where objects differ from their mirror images, plays a fundamental role in chemistry, physics, and biology.1–4 Chiral molecules typically exist as enantiomeric pairs, and in many cases, one enantiomer can have therapeutic effects while the other may be inactive or lethal. Therefore, the resolution of racemic mixtures is both important and challenging.5,6 Recently, Chiral Metal–Organic Frameworks (CMOFs) have been receiving a lot of attention due to their outstanding properties and a wide range of applications across various fields such as asymmetric catalysis,7,8 enantiomeric sensing and separation,9 magnetism and second-order nonlinear optics (NLO).10–12 In general, chirality is incorporated into MOFs by: (1) direct synthesis with an enantiopure chiral ligand, (2) post-synthetic modification with chiral linkers, and (3) spontaneous resolution or chiral induction using an achiral ligand.13 Among these, synthesizing CMOFs with enantiopure ligands is often more expensive due to the cost of chiral precursors and the complexity of their resolution. Moreover, research on the self-assembly of CMOFs from achiral ligands remains limited.14 To date, the understanding of how CMOFs can be constructed from achiral ligands remains both limited and unpredictable.15,16 In some cases, achiral building blocks can undergo a unique spatial arrangement, such as the formation of helical chains, that gives rise to chiral structural motifs. This phenomenon is termed as “spontaneous resolution” or “symmetry breaking”.1,17,18 Additionally, supramolecular interactions such as π–π stacking and hydrogen bonding are also believed to play a role in inducing chirality.19,20Over the past two decades, significant progress has been made in developing CMOF-based enantioselective sensors for detecting various chiral compounds, including amino alcohols, amino acids, and α-hydroxy acids.2,21,22 However, these CMOF sensors are mainly synthesized using enantiopure ligands, which limits scalability and increases cost. Hence, developing CMOF sensors using achiral organic ligands is highly demanding and needs more exploration.23
The widespread use of chiral amino alcohols in pharmaceuticals, the synthesis of chiral catalysts for asymmetric synthesis, and the production of fine chemicals in the agrochemical industry has driven interest in developing analytical approaches that enable the quantitative enantioselective assessment of these compounds in aqueous environments.22,24–26 Various techniques, including circular dichroism (CD),2,27 high-performance liquid chromatography (HPLC),28 nuclear magnetic resonance (NMR),29 optical sensors,30,31 and electrochemical sensing,32 have been employed for this purpose. Among these, optical sensors offer ease of operation, rapid screening capability, and the ability to provide direct visual feedback.33,34 For instance, Xiao and co-workers synthesized a homochiral UiO-66 MOF by integrating chiral amino propanol (namely, S-1 (L-AP@UiO-66-(COOH)2) and R-1 (D-AP@UiO-66-(COOH)2)), which showed a “turn-on” fluorescence response in the presence of L/D-phenylalaninol with enantioselectivity factors of 1.89 and 0.67 towards S-1 and R-1, respectively.22 Han et al. introduced N-benzylquininium chloride and Tb+3 into a Zn-MOF to create chirality and luminescence enhancement. This bifunctional MOF enabled quantitative enantioselective recognition of a mixture of stereoisomers cinchonine and cinchonidine, (Ksv value ratio of 1.4), and also of amino alcohols.5 Despite significant advances in the chiral detection of amino alcohols, the development of chiral MOF sensors constructed from achiral linkers with well-defined chiral nanochannels or nanopores is still scarcely investigated.
Herein, we report a chiral nanoporous 3D [Co(4,4′-bpy) (HCOO)2] (Co-bpy) MOF {4,4′-bpy = 4,4′-bipyridine} that displays a symmetry-breaking crystallization phenomenon during hydrothermal synthesis. Interestingly, it undergoes the spontaneous resolution process in the absence of any external chiral source. In addition, this MOF is highly water stable and shows excellent chiral sensing of amino alcohols by fluorescence, with remarkable selectivity. To the best of our knowledge, this is the first report that employs an achiral ligand to demonstrate chiral recognition of amino alcohol enantiomers.
Results and discussion
Structural description of Co-bpy
Co-bpy was synthesized using the same method described in our previous work.35 It crystallizes in the tetragonal crystal system with the P41212 chiral space group. A Flack parameter of −0.037(16) for Co-bpy suggests that the crystal is enantiomerically pure despite being synthesized from an achiral reagent (Table S1†).36,37 The asymmetric unit of the framework consists of one Co(II) center atom, one HCOO− formate linker, and half of a 4,4′-bipyridyl ligand (Fig. S1a†). The presence of crystallographic 41 screw axes, as indicated by its space group, contributes to the helicity of the MOF. As shown in Fig. (1a and b), the formate group bridging two adjacent Co atoms results in an infinite left-handed helical chain extending along the c-axis with a pitch length of 16.19 Å. Each helical chain functions as a secondary building block and connects to its adjacent neighbors with the same handedness (diamond network, as shown in Fig. S1b and d†) via a bipyridyl bridge, leading to an overall 3-D framework. Additionally, the twisted conformation of the 4,4′-bipyridine organic linker also contributes to helix generation. Typically, 4,4′-bpy is considered achiral due to the unrestricted rotation of its two pyridine rings around the σ-bond. However, within the MOF, these axial chiral conformations become “locked” effectively inducing and stabilizing chirality. Also, the fixed dihedral angle of 47.1° between the pyridine rings (Fig. 1c) plays a crucial role in shaping the chiral behavior of the framework. Therefore, the 41 screw axis and the stereochemical configuration of the bipyridyl ligand are factors contributing to the presence of chirality in the framework.38 As shown in Fig. S1c,† there are nanosize tubules with an opening of 9.1 × 9.1 Å, along the c-axis. Topologically (shown in Fig. 1d), the entire network is regarded as a uninodal 6-c net with a point symbol of {510 64 7}, featuring the rare jsm topology.39 | ||
| Fig. 1 (a) 3-D framework of Co-bpy viewed down along the c-axis with helical channels (color code: C, gray; O, red; N, royal blue; Co, light blue; H, white). (b) The infinite Co-formate 41 helical chain along the c-axis, with a pitch length of 16.19 Å. (c) The axially chiral conformations of 4,4′-bpy and the angle between the two pyridine rings. (d) The 3-dimensional framework of Co-bpy possessing the jsm topological net. | ||
As shown in Fig. S2,† the simulated pattern from SCXRD (Single Crystal X-ray Diffraction) matches well with the experimental PXRD (Powder X-ray Diffraction), indicating the bulk phase purity of the crystals. Also, to check the water stability of the MOF, it was immersed in water for 24 h and then its PXRD pattern was recorded (Fig. S3†). Since all peaks are retained, Co-bpy exhibits excellent water stability and can be explored under real-world experimental conditions. Further to check the optical activity of the MOF, CD spectra were recorded for bulk samples from ten different batches of solvothermal synthesis (Fig. S4†). All showed a positive Cotton effect around 200 nm and between 230 and 240 nm, suggesting homochirality in the MOF. However, the differences in intensity likely reflect inherent differences in crystal quality between batches. The CD signals are attributed to the four-fold helical chains, intra-ligand (π–π*) transitions of the 4,4′-bpy ligand and ligand-to-metal charge transfer (LMCT, n–π*).2
The UV-vis absorption spectrum of Co-bpy (Fig. S5a†) displays bands at 203 nm and 240 nm, which correspond to the intraligand (π–π*) transitions of the 4,4′-bipyridine ligand. Additionally, a shoulder peak around 300 nm is attributed to a ligand-to-metal charge transfer (n–π*) transition. An absorption peak at 500 nm features 4T1g(F) to 4T1g(P) electronic transitions within the cobalt ion.40 In infrared (IR) spectra (Fig. S5b†), a weak absorption band detected at 3045 cm−1 corresponds to the aromatic C–H stretching vibrations. The peaks observed around 2860 cm−1 are attributed to the aliphatic C–H stretching modes. Furthermore, the characteristic vibrational bands appearing at 1560, 1337, and 1219 cm−1 are associated with the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, C–C, and C–N bonds within the pyridine rings. In addition, the bands at 1613 cm−1 and 1390 cm−1 are assigned to the asymmetric and symmetric stretching vibrations of the carboxylate (COO−) groups, respectively.40 The BET (Brunauer–Emmett–Teller) surface area of Co-bpy was found to be 19.161 m2 g−1 (Fig. S5d†). Thermogravimetric analysis (TGA) was performed under a nitrogen atmosphere over a temperature range of 25 °C to 800 °C. The results indicate that the Co-bpy framework maintains its thermal stability up to approximately 250 °C (Fig. S5c†)
C, CN, C–C, and C–N bonds within the pyridine rings. In addition, the bands at 1613 cm−1 and 1390 cm−1 are assigned to the asymmetric and symmetric stretching vibrations of the carboxylate (COO−) groups, respectively.40 The BET (Brunauer–Emmett–Teller) surface area of Co-bpy was found to be 19.161 m2 g−1 (Fig. S5d†). Thermogravimetric analysis (TGA) was performed under a nitrogen atmosphere over a temperature range of 25 °C to 800 °C. The results indicate that the Co-bpy framework maintains its thermal stability up to approximately 250 °C (Fig. S5c†)
Photoluminescence studies
Due to its inherent chirality and exceptional stability in water, Co-bpy stands out as a promising candidate for enantioselective fluorescence sensing in aqueous environments. To investigate its enantioselective capabilities, we initially examined its response to the enantiomers R- and S-valinol, both of which are chiral amino alcohols. The fluorescence spectrum of Co-bpy was recorded at an excitation wavelength of 325 nm, giving an emission at 431 nm (Fig. S6†).For the fluorescence titration studies, 5 mM aqueous solutions of each enantiomer were prepared and gradually added to 500 μL Co-bpy aqueous suspensions (2 mg in 2 mL). In the case of S-valinol, a progressive quenching of fluorescence was observed upon incremental addition, indicating a distinct “turn-off” fluorescence response (Fig. 2a). Conversely, when R-valinol was added, the fluorescence intensity initially decreased for concentrations between 0 and 1.2 mM. Beyond this concentration, however, the emission intensity began to recover, followed by a blue shift, revealing a notable “turn-off–on” behaviour (Fig. 2b). These contrasting responses highlight Co-bpy's remarkable ability to distinguish between enantiomers through fluorescence modulation.
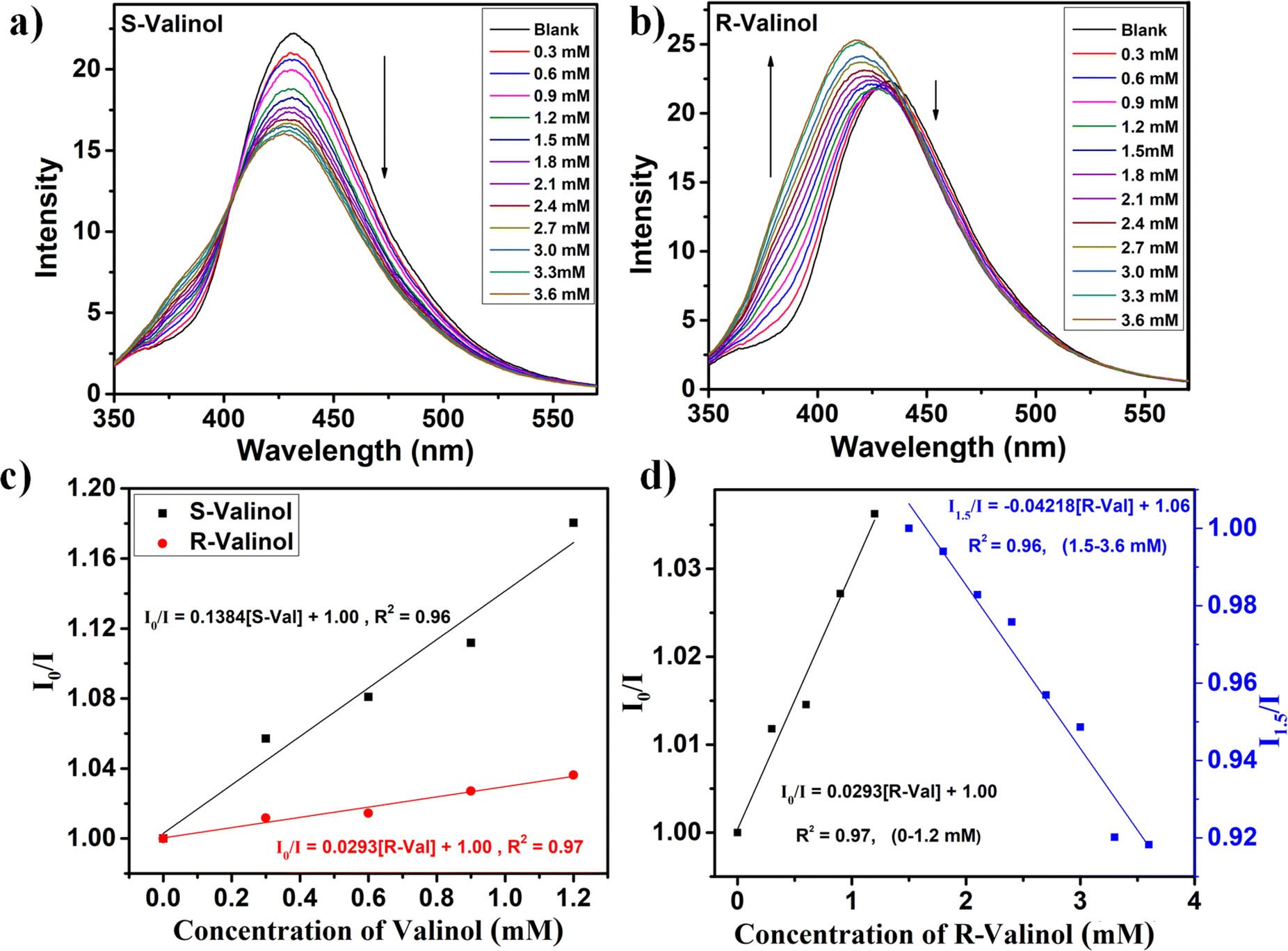 | ||
| Fig. 2 Emission spectrum of Co-bpy dispersed in water upon gradual addition of (a) S-valinol and (b) R-valinol. (c) The Stern–Volmer quenching plot for R- and S-valinol towards Co-bpy (0–1.2 mM). (d) Linear relation between the fluorescence quenching (0–1.2 mM) and enhancing (1.5–3.6 mM) efficiency of R-valinol towards the Co-bpy probe at λemi = 431 nm (upon λexc = 325 nm). | ||
To quantify the quenching and enhancement efficiencies, fluorescence data were analyzed using the Stern–Volmer (SV) equation: I0/I = KSV [Q] + 1, where I0 and I represent fluorescence intensities before and after analyte addition, KSV is the quenching constant, and [Q] is the molar concentration of the analyte. For S-valinol, which induces a “turn-off” effect, the calculated KSV value was 138.4 M−1, indicating strong quenching efficiency (Fig. 2c). In the case of R-valinol, where a biphasic “turn-off–on” response was observed, both stages were analyzed separately.41 During the quenching phase (0 to 1.2 mM, with I0 defined as the fluorescence intensity of neat Co-bpy), a KSV value of 29.3 M−1 was obtained. In the enhancement phase (1.5 to 3.6 mM, with I0 defined as the intensity of Co-bpy at 1.5 mM) an enhancement efficiency of 42.1 M−1 was calculated (Fig. 2d). By comparing the quenching constants of both enantiomers (Fig. 2c), the enantio discrimination value ΔK (ΔK = KSV (S-valinol) − KSV (R-valinol)) was calculated to be 109.1 M−1, and the enantioselectivity factor α (α = KSV (S-valinol)/KSV (R-valinol)) was found to be 4.7.23 This α value is significantly higher than that reported for a chiral Cd-MOF sensor (α = 3.1) used for the enantioselective detection of R-/S-valinol (Table S4†).5
These analytical results demonstrate that Co-bpy exhibits outstanding enantioselectivity toward R- and S-valinol. In addition to its high α value, the substantial ΔK value further confirms its strong ability to discriminate between the two enantiomers.
The limit of detection (LoD) was determined using the equation LoD = 3σ/m, where σ denotes the standard deviation based on five blank measurements of Co-bpy's emission intensity, and m refers to the slope obtained from the linear fit of the plot of fluorescence intensity versus analyte concentration. Based on this calculation, the LoD was found to be 142.8 × 10−5 M for R-valinol and 346 × 10−6 M for S-valinol with Co-bpy (Fig. S7†), demonstrating the probe's good sensitivity.
Furthermore, a linear correlation was observed between the fluorescence quenching intensity (I0/I) and the enantiomeric excess (ee) of R-valinol when Co-bpy was exposed to 2 mM R-valinol solutions with varying R-enantiomer content from 100% to −100% (Fig. 3a). This finding indicates that Co-bpy can be effectively employed for quantifying the enantiomeric excess (ee) of valinol samples through a simple fluorescence quenching measurement. The combination of a low detection limit, high enantioselectivity, and rapid optical response highlights Co-bpy as a promising candidate for enantioselective detection of R- and S-valinol in aqueous environments. Additionally, to test the recyclability, the framework was subjected to multiple sensing cycles. The regenerated Co-bpy consistently exhibited comparable fluorescence quenching and enhancement for S-valinol and R-valinol, respectively, across five consecutive sensing regeneration cycles (Fig. S10†), highlighting the framework's robustness and reliable performance as recyclable sensors. To assess the reproducibility of the sensing behavior, titration experiments were conducted using Co-bpy MOF samples synthesized from four independent batches. In all cases, the material exhibited a “turn-off” fluorescence response toward S-valinol and a characteristic “turn-off–on” response toward R-valinol (Fig. S11–S14†). However, subtle variations were observed in the intensities and the shoulder region around 360–370 nm. These differences suggest that, despite identical synthetic procedures, minor batch-to-batch variations such as differences in crystal size or residual solvent molecules can subtly influence the local photophysical environment within the MOF, as also observed in the CD spectra.
 | ||
| Fig. 3 (a) Fluorescence intensity ratio of Co-bpy versus ee values of R-valinol (2 mM). (b) PXRD patterns of Co-bpy before and after immersing in R- and S-valinol solution for 24 h. | ||
Mechanistic insight into the sensing
To elucidate the underlying sensing mechanism, PXRD patterns were analysed after immersing the MOF in R- and S-valinol solution for 24 h (Fig. 3b). The results confirmed that the structural integrity of Co-bpy remained intact after exposure to both R- and S-valinol, indicating that the observed fluorescence changes are not due to framework degradation or collapse. Furthermore, to investigate the possibility of Förster resonance energy transfer (FRET), the UV-vis absorption spectra of the analytes and the emission spectrum of Co-bpy were analyzed. As shown in Fig. S8,† the absorption spectra of the amino alcohols exhibit no significant overlap with the emission spectrum of Co-bpy, indicating the absence of FRET. Notably, the UV-vis absorption spectra of all amino alcohols overlap with the excitation spectrum of Co-bpy (Fig. S8†), suggesting that the analytes may absorb photons at the excitation wavelength. This competitive absorption results in fluorescence quenching.42 To understand the photoelectron transfer process (PET), the HOMO and LUMO energy levels for the analytes and bipyridine ligand43 were calculated. As depicted in Fig. S9† and summarized in Table S2,† the HOMO energy level of the bipyridine ligand is lower than those of the amino alcohol analytes. This energy alignment favors electron transfer from the HOMO of the analytes (acting as electron donors) to the HOMO of the photoexcited MOF ligand (serving as the electron acceptor), resulting in fluorescence quenching.44To further investigate the molecular-level interactions, DFT (density functional theory) calculations were carried out to assess the interactions of R- and S-valinol with the MOF fragment (Table S3†). Possible host–guest conformations were modelled, and their corresponding interaction energies were calculated. The difference in interaction energies is calculated by using the formula: ΔE = EMOF+analyte − EMOF − Eanalyte, where EMOF+analyte, EMOF, and Eanalyte are the energies of MOF + analyte, MOF, and pure analyte, respectively.
The results revealed that Co-bpy exhibits a stronger interaction with S-valinol (408.2 kcal mol−1) than with R-valinol (778.5 kcal mol−1). We believe that the high enantioselectivity toward S-valinol primarily arises from its interaction with the chiral inner pore space of the MOF, which offers optimal hydrogen bonding with the formate and bipyridyl linkers of Co-bpy.21 Moreover, these host–guest interactions between the MOF and S-valinol also promote the photoinduced electron transfer, causing fluorescence quenching (“turn-off”).42,45 In contrast, the weaker interaction of R-valinol with the MOF suppresses this PET process, enabling fluorescence recovery (“turn-off–on”). Thus, the difference in chiral fluorescence response is governed by the extent to which each enantiomer interacts with the MOF.
To highlight the broad applicability of the MOF, its enantioselective sensing properties were evaluated for two other chiral amino alcohols, including R/S-phenyl glycinol and R/S-alaninol, as shown in Fig. 4. Here also, Co-bpy exhibited distinct and selective fluorescence responses: a “turn-off–on” behavior for R-phenyl glycinol and “turn-off” for S-phenyl glycinol. In the case of alaninol, a “turn-off–on” behavior was observed for S-alaninol and “turn-off” for R-alaninol, which is the opposite trend compared to R/S-valinol. This reversal highlights that the sensing ability of the MOF is highly dependent on the specific host–guest interactions dictated by the structural and stereoelectronic features of each analyte. The calculated Stern–Volmer quenching constant values are provided in Table S4.† For R/S-phenyl glycinol, the enantiodiscrimination value ΔK was found to be 99.6 M−1, with an enantioselectivity factor (α) of 6.2. In the case of R/S-alaninol, ΔK was 35.6 M−1, and α was 1.5. The enantioselectivity factor (α) follows the order: alaninol (1.5) < valinol (4.7) < phenyl glycinol (6.2), which correlates with the increasing size and rigidity of these amino alcohols, illustrating the combined influence of the molecular structure and stereochemical conformation on enantioselectivity. These results underscore the versatility of Co-bpy in recognizing diverse chiral amino alcohols.
 | ||
| Fig. 4 Emission spectrum of Co-bpy dispersed in water upon gradual addition of (a) S-phenyl glycinol, (b) R-phenyl glycinol, (d) R-alaninol, and (e) S-alaninol. The Stern–Volmer quenching plot for (c) R- and S-phenyl glycinol and (f) R- and S-alaninol at λemi = 431 nm (upon λexc = 325 nm). | ||
Conclusions
To summarise, a highly water-stable chiral nanoporous Co-bpy MOF has been synthesised using an achiral precursor and without adding any chiral template or inducer. The origin of chirality in the MOF is attributed to the presence of a 41 screw axis and the stereochemical configuration of the bipyridyl linker. Interestingly, Co-bpy exhibits outstanding chiral recognition of chiral amino alcohols in water, by demonstrating distinguished “turn-on” and “turn-off–on” type fluorescence behavior, along with a high enantioselectivity factor (α), which is rare on its own. Thus, this work paves the way for the rational design of chiral MOFs from achiral building blocks, which offers a rapid, straightforward, and cost-effective platform for detecting chiral molecules in water.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MS, RL, and AKD acknowledge INST for PhD fellowships. MS thanks INST for financial and infrastructural support. MS also acknowledges SERB-DST for the project EEQ/2022/000149.Notes and references
- Y. Tao, C. Yang, H. Fang, H.-D. Bian, X. Xu and F.-P. Huang, Cryst. Growth Des., 2019, 19, 3358–3364 CrossRef CAS.
- R.-X. Yao, H.-H. Fu, B. Yu and X.-M. Zhang, Inorg. Chem. Front., 2018, 5, 153–159 RSC.
- P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2019, 55, 91–94 RSC.
- K. K. Bisht, B. Parmar, Y. Rachuri, A. C. Kathalikattil and E. Suresh, CrystEngComm, 2015, 17, 5341–5356 RSC.
- Z. Han, K. Wang, Y. Guo, W. Chen, J. Zhang, X. Zhang, G. Siligardi, S. Yang, Z. Zhou, P. Sun, W. Shi and P. Cheng, Nat. Commun., 2019, 10, 5117 CrossRef PubMed.
- Y. Wang, J. Xu, Y. Wang and H. Chen, Chem. Soc. Rev., 2013, 42, 2930–2962 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- D. N. Dybtsev and K. P. Bryliakov, Coord. Chem. Rev., 2021, 437, 213845 CrossRef CAS.
- J. Liu, S. Mukherjee, F. Wang, R. A. Fischer and J. Zhang, Chem. Soc. Rev., 2021, 50, 5706–5745 RSC.
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
- X. Huang, Q. Li, X. Xiao, S. Jia, Y. Li, Z. Duan, L. Bai, Z. Yuan, L. Li, Z. Lin and Y. Zhao, Inorg. Chem., 2018, 57, 6210–6213 CrossRef CAS PubMed.
- R. Medishetty, J. K. Zaręba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- W. Gong, Z. Chen, J. Dong, Y. Liu and Y. Cui, Chem. Rev., 2022, 122, 9078–9144 CrossRef CAS PubMed.
- P. C. Rao, S. P. Chaudhary, D. Kuznetsov and S. Mandal, Inorg. Chem., 2016, 55, 12669–12674 CrossRef CAS PubMed.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CrossRef CAS PubMed.
- C. J. Kepert, T. J. Prior and M. J. Rosseinsky, J. Am. Chem. Soc., 2000, 122, 5158–5168 CrossRef CAS.
- E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, J. Am. Chem. Soc., 2004, 126, 1419–1429 CrossRef CAS PubMed.
- J. Gal, Nat. Chem., 2017, 9, 604–605 CrossRef CAS PubMed.
- S. Wu, Y. Wu, Q. Kang, H. Zhang, L. Long, Z. Zheng, R. Huang and L. Zheng, Angew. Chem., Int. Ed., 2007, 46, 8475–8479 CrossRef CAS PubMed.
- J. Zhang and X. Bu, Chem. Commun., 2009, 206–208 RSC.
- M. M. Wanderley, C. Wang, C.-D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050–9053 CrossRef CAS PubMed.
- J. Xiao, X. Wang, X. Xu, F. Tian and Z. Liu, Analyst, 2021, 146, 937–942 RSC.
- Q. Zhang, M. Lei, F. Kong and Y. Yang, Chem. Commun., 2018, 54, 10901–10904 RSC.
- T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, John Wiley & Sons, 2010 Search PubMed.
- C. Liu, L. Zhang, L. Cao, Y. Xiong, Y. Ma, R. Cheng and J. Ye, Commun. Chem., 2022, 5, 1–10 CrossRef PubMed.
- S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353–356 CrossRef CAS PubMed.
- L.-L. Xu, H.-F. Zhang, M. Li, S. W. Ng, J.-H. Feng, J.-G. Mao and D. Li, J. Am. Chem. Soc., 2018, 140, 11569–11572 CrossRef CAS PubMed.
- A. Cavazzini, L. Pasti, A. Massi, N. Marchetti and F. Dondi, Anal. Chim. Acta, 2011, 706, 205–222 CrossRef CAS PubMed.
- L. Tohala, F. Oukacine, C. Ravelet and E. Peyrin, Anal. Chem., 2015, 87, 5491–5495 CrossRef CAS PubMed.
- S. Thoonen, P. Siripanich, L. Hua, H. M. Tay, P. Ramkissoon, T. A. Smith, M. Lessio and C. Hua, Inorg. Chem. Front., 2024, 11, 3877–3888 RSC.
- J.-Y. Yue, L.-P. Song, Y.-H. Shi, L. Zhang, Z.-X. Pan, P. Yang, Y. Ma and B. Tang, Anal. Chem., 2023, 95, 11078–11084 CrossRef CAS PubMed.
- X. Niu, R. Zhao, M. Yuan, Y. Liu, X. Yang, H. Li, H. Xu and K. Wang, ACS Sens., 2024, 9, 4069–4078 CrossRef CAS PubMed.
- Y.-W. Zhao, L.-E. Guo, F.-Q. Zhang, J. Yao and X.-M. Zhang, ACS Appl. Mater. Interfaces, 2021, 13(17), 20821–20829 CrossRef CAS PubMed.
- X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918–4959 CrossRef CAS PubMed.
- R. Ladhi, D. Rani and M. Singh, New J. Chem., 2023, 47, 9714–9720 RSC.
- X. Zhang, B. Li and J. Zhang, Inorg. Chem., 2016, 55, 3378–3383 CrossRef CAS PubMed.
- H. D. Flack, Acta Crystallogr. A, 1983, 39, 876–881 CrossRef.
- J.-L. Liu, X. Bao, J.-D. Leng, Z.-J. Lin and M.-L. Tong, Cryst. Growth Des., 2011, 11, 2398–2403 CrossRef CAS.
- Q. Yang, Q. Zhao, K. Tang, X. Zhang, Y. Li and Z. Chen, Polyhedron, 2015, 90, 23–27 CrossRef CAS.
- X. Gao, C. Wang, F.-Y. Bai, Q. Guan, J.-X. Wang, S.-Y. Wei, X.-X. Zhang, X.-T. Xu and Y. Xing, J. Inorg. Organomet. Polym., 2015, 25, 1088–1102 CrossRef CAS.
- Y.-P. Li, X.-H. Zhu, S.-N. Li, Y.-C. Jiang, M.-C. Hu and Q.-G. Zhai, ACS Appl. Mater. Interfaces, 2019, 11, 11338–11348 CrossRef CAS PubMed.
- Z. Han, T. Sun, R.-R. Liang, Y. Guo, Y. Yang, M. Wang, Y. Mao, P. R. Taylor, W. Shi, K.-Y. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 15446–15452 CrossRef CAS PubMed.
- S. Bhattacharjee, S. Bera, R. Das, D. Chakraborty, A. Basu, P. Banerjee, S. Ghosh and A. Bhaumik, ACS Appl. Mater. Interfaces, 2022, 14, 20907–20918 CrossRef CAS PubMed.
- A. Mallick, B. Garai, M. A. Addicoat, P. St. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC.
- Z. Han, K.-Y. Wang, M. Wang and W. Shi, Acc. Chem. Res., 2025, 58, 625–634 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00504c |
| This journal is © The Royal Society of Chemistry 2025 |