
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMucolytic and antibiotic combination therapy using silica-based nanocarriers to eradicate Escherichia coli biofilms†
Anna
Aguilar-Colomer‡
ab,
Carla
Jiménez-Jiménez‡
ab,
Blanca
González
 ab,
Jaime
Esteban
cd,
María
Vallet-Regí
ab,
Montserrat
Colilla
*ab and
Isabel
Izquierdo-Barba
*ab
ab,
Jaime
Esteban
cd,
María
Vallet-Regí
ab,
Montserrat
Colilla
*ab and
Isabel
Izquierdo-Barba
*ab
aDepartamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Universidad Complutense de Madrid, Instituto de Investigación Sanitaria, Hospital 12 de Octubre i+12, Plaza Ramón y Cajal s/n, 28040 Madrid, Spain. E-mail: mcolilla@ucm.es; ibarba@ucm.es
bCentro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Spain
cUnidad de Microbiología Clínica, IIS-Fundación Jiménez Díaz, Avenida de los Reyes Católicos 2, 28040 Madrid, Spain
dCentro de Investigación Biomédica en Red de Enfermedades Infecciosas (CIBERINFEC), Madrid, Spain
First published on 8th April 2025
Abstract
This research provides new insights into the treatment of E. coli biofilm-related infections through the design of new antimicrobial nanoformulations based on mesoporous silica nanoparticles (MSNs) for mucolytic and antibiotic combination therapy against E. coli biofilms. The development of nanosystems with well-defined compartments to house and sequentially deliver different antimicrobial agents was carried out. A relatively simple and direct straightforward approach was carried out, consisting of loading MSNs with levofloxacin (LVX) by an impregnation method followed by external coating with a gelatin shell embedding a mixture of N-acetylcysteine (AC) plus LVX. Thus, the release of the mucolytic agent, AC, at the earliest stage causes disaggregation of the outer mucopolysaccharide layer of the mature E. coli biofilm, as confirmed by confocal laser scanning microscopy studies. This biofilm disruption effect facilitates the antimicrobial action of LVX, which is released in a more sustained manner over longer periods of time than AC, achieving a remarkable reduction (ca. 99.8%) of mature E. coli biofilms. These results are supported by the combined effect of AC and LVX strategically combined in the same nanocarrier. Preliminary in vitro studies with preosteoblastic cells point to the good biocompatibility of these nanosystems.
1 Introduction
Escherichia coli (E. coli) is one of the species most frequently involved in biofilm-associated diseases, particularly relevant in urinary tract and intestinal infections. In addition to increasing the ability to cause recurrent and chronic infections, the E. coli biofilm is also responsible for infections associated with indwelling medical devices, such as urethral and intravascular catheters, as well as prosthetic shunts and grafts.1E. coli biofilms are well-organized three-dimensional communities of microorganisms embedded in a variety of self-produced protective matrices that are often found attached to solid surfaces in moist environments.2,3 This matrix, which can represent more than 90% of the total biomass of the biofilm, is mainly composed of a conglomerate of different types of biopolymers, known as extracellular polymeric substances (EPS), such as exopolysaccharides, proteins, nucleic acids, lipids, nutrients and other metabolites.4–6 The EPS causes biofilm communities to exhibit distinctive characteristics that are not present in planktonic bacteria. The EPS exerts several functions, such as adhesion and aggregation of bacterial cells, acts as a protective barrier against extreme environments (e.g. pH and temperature variations, ultraviolet irradiation, antimicrobial agents, etc.), promotes water retention, facilitates biofilm cohesion, intervenes in the absorption of organic and inorganic compounds as nutrients, favors genetic exchange between biofilm bacteria and is an important source of carbon.4 Besides, the bacterial cells within the biofilm communicate with each other via quorum sensing that regulates the key biochemical factors that allow bacteria to proliferate and reinforce the ensuing infection.7
Biofilms hinder the penetration of conventional antibiotics, making them highly resistant to conventional antimicrobial treatments. In fact, compared to their planktonic analogs, biofilm bacteria show 10 to 1000 times greater resistance to antibiotics8 and are able to evade the action of the host immune system,9 making treatment and eradication of biofilms extremely difficult.10,11
In this challenging scenario, it is urgent to explore and develop alternative therapeutic agents to combat these infectious diseases, especially those caused by the formation of E. coli biofilms. Therefore, several treatments acting at different stages of biofilm development have been investigated, based on anti-adhesion agents,12–14 quorum sensing inhibitors15–17 and biofilm eradication agents, such as phages18–20 and antimicrobial peptides.21,22 In addition, combination therapies based on the coadministration of antibiotics and matrix-disrupting agents,23 including certain enzymes24–26 and mucolytic agents,27–30 have been proposed. Among mucolytic agents, N-acetylcysteine (AC) has experienced a burgeoning boom in recent years due to its ability to decrease biofilm formation in a wide variety of bacteria, markedly decreasing the production of the EPS, prompting the disruption of mature biofilms and facilitating antibiotic penetration. In medical practice, AC is extensively used by oral, intravenous and inhalation routes, showing well-documented safety.31 Besides, different in vitro studies have proposed the coadministration of AC and antibiotics to eradicate different bacterial biofilms, such as methicillin-resistant Staphylococcus aureus, methicillin-sensitive Staphylococcus aureus, methicillin-resistant Staphylococcus epidermidis, vancomycin-resistant Enterococcus, Klebsiella pneumoniae, Acinetobacter baumannii,32,33Enterococcus faecalis and E. coli,34,35 among others.
However, AC displays low bioavailability (below 5%) due to its high affinity to plasma proteins and high clearance rate, which significantly reduces its antibiofilm efficacy.31,36 In this regard, nanotechnology has emerged as a powerful alternative to overcome these limitations, providing the opportunity to use nanocarriers capable of protecting the therapeutic load and improving drug release at the biofilm site.37 In this context, the great versatility of mesoporous silica nanoparticles (MSNs) makes them ideal nanocarriers for various antimicrobial agents.38,39 In fact, MSNs have recently been proposed to design sophisticated combination therapy nanosystems.40–46 Herein, we designed a novel, easy-to-synthesize and versatile biocompatible nanosystem to eradicate mature E. coli biofilms by using a unique nanoplatform based on mesoporous silica nanocarriers to co-deliver AC and an antibiotic, levofloxacin (LVX), as combination therapy. We demonstrate that through a relatively simple synthetic approach, enhanced antibacterial efficacy is achieved by improving the penetration of the antibiotic LVX into the biofilm through the mucopolysaccharide matrix disruptive action of the mucolytic AC. The strengths of this basic research are based on the precise design and exhaustive characterization of the developed nanomaterials, together with the in-depth evaluation of the in vitro disrupting effect on mature E. coli biofilms. The great versatility of MSNs allows loading the different antimicrobial agents into well-defined compartments of these nanocarriers. For this purpose, LVX was loaded into the mesopores of MSNs and then coated with a gelatin layer containing AC alone or a mixture of AC and LVX. The aim was to achieve a sequential delivery of the different antimicrobial agents placed in the different compartments. Thus, the mucolytic agent, AC, would be released in the first stage to disrupt the EPS matrix, and then LVX would be released in a more sustained manner to eradicate the biofilm. A thorough characterization study of the developed nanosystems was carried out, and the kinetics of molecular release from the different compartments were determined. Moreover, the antimicrobial effect of these nanosystems against mature E. coli biofilms was evaluated in vitro by confocal laser scanning microscopy and biofilm viability assays. Furthermore, preliminary biocompatibility studies were performed with preosteoblastic cells.
2 Experimental section
2.1. Reagents and equipment
Tetraethylorthosilicate (TEOS), cetyltrimethylammonium bromide (CTAB), gelatin Ph Eur, levofloxacin (LVX), N-acetylcysteine (AC) and fluorescein sodium salt were purchased from Sigma-Aldrich. Other chemicals (ammonium nitrate, phosphate buffered saline solution (PBS 10×), absolute EtOH, NaOH, etc.) were of the highest quality available on the market and were used as received. All these compounds were used without further purification. Deionized water was further purified by passage through a Milli-Q Advantage A-10 purification system (Millipore Corporation), achieving a final resistivity of 18.2 MΩ cm.The analytical methods used to characterize the synthesized compounds were as follows: N2 adsorption porosimetry, thermogravimetric and differential thermal analysis (TGA-DTA), chemical microanalysis, Fourier transform infrared spectroscopy (FTIR), transmission electron microscopy (TEM), zeta (ζ)-potential, dynamic light scattering (DLS) and fluorescence spectroscopy. The equipment and conditions used are described in the ESI.†
2.2. Materials synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min and the isolated nanoparticles were washed several times with water, EtOH and finally dried. Ion exchange was chosen as the method for surfactant removal. Thus, 1 g of surfactant-containing MSNs was well-dispersed in 350 mL of an extracting solution consisting of 10 g L−1 NH4NO3 in EtOH/H2O (95:5, v/v). The suspension was stirred at 80 °C overnight, and then the solid was thoroughly washed with water, H2O/EtOH (50:50 (v/v)) and absolute EtOH, respectively. The extraction procedure was repeated for 2 h, and the solid was washed the same way, yielding surfactant-free MSNs, referred to as MSN. TEM characterization of pristine MSNs showed nearly spherical nanoparticles with a fairly homogeneous diameter distribution, with 168 nm being the maximum of the statistically calculated size distribution (see Fig. S1 in the ESI†).
000 rpm for 20 min and the isolated nanoparticles were washed several times with water, EtOH and finally dried. Ion exchange was chosen as the method for surfactant removal. Thus, 1 g of surfactant-containing MSNs was well-dispersed in 350 mL of an extracting solution consisting of 10 g L−1 NH4NO3 in EtOH/H2O (95:5, v/v). The suspension was stirred at 80 °C overnight, and then the solid was thoroughly washed with water, H2O/EtOH (50:50 (v/v)) and absolute EtOH, respectively. The extraction procedure was repeated for 2 h, and the solid was washed the same way, yielding surfactant-free MSNs, referred to as MSN. TEM characterization of pristine MSNs showed nearly spherical nanoparticles with a fairly homogeneous diameter distribution, with 168 nm being the maximum of the statistically calculated size distribution (see Fig. S1 in the ESI†).
2.3. Drug loading and in vial release assays
The synthetic strategies used to prepare the different materials are shown in Scheme 1.
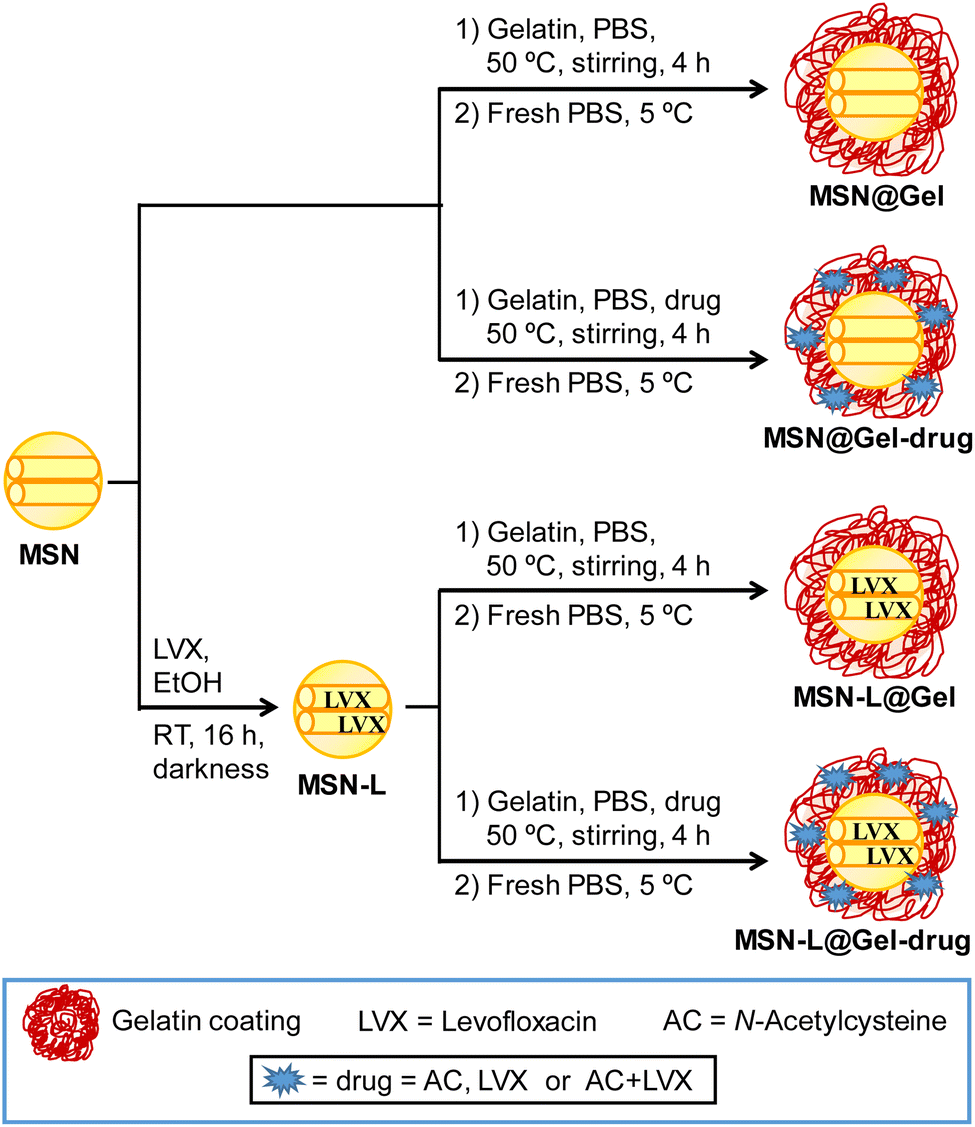 | ||
| Scheme 1 Synthetic strategies used to obtain MSN@Gel nanosystems. Different materials were prepared, taking into account the presence or absence of levofloxacin (LVX) inside the MSN mesopores and the incorporation of LVX, N-acetylcysteine (AC) or the combination of LVX and AC in the gelatin shell. | ||
2.4. Microbiological assays
2.4.3.1 Confocal laser scanning microscopy assay. After 24 h of incubation with the nanosystems, the glass coverslips were washed once with sterile PBS 1× and then 0.5 mL of LB medium was added. Then, 1.5 μL (1
:1 propidium iodide/SYTO) of the Live/Dead® Bacterial Viability Kit (Backlight™) was added and, after 5 min, 5 μL of Calcofluor White Stain (Sigma Aldrich) solution was added to stain the protective mucopolysaccharide matrix of the biofilm (extracellular matrix) in blue. Both reactants were incubated for 10 min at room temperature. Controls containing untreated bacterial biofilms were also stained. Biofilms were examined using an Olympus FV1200 confocal microscope, and eight photographs (60× magnification) were taken for each sample. Confocal images were evaluated and quantified using the ImageJ Fiji software (National Institute of Health, Bethesda, MD). All images are representative of three independent experiments.
2.4.3.2 Biofilm viability assay. After 24 h of incubation with the nanosystems, the wells were washed once with sterile PBS 1× and sonication was carried out for 10 min in a low-power bath sonicator (Selecta, Spain) to break and disperse the biofilm in a total volume of 1 mL of PBS 1×.38,48 Serial dilutions of the disaggregated biofilms were made in PBS 1× for the quantification of bacteria using the drop plate method.49 Five drops of each solution were inoculated onto Tryptic Soy Agar (TSA, Sigma Aldrich) plates, which were incubated for 24 h at 37 °C. The mean count of the 5 drops of each dilution was calculated, and then the average counting for all dilutions was calculated following the procedure described elsewhere.49 The percentage of biofilm growth relative to untreated controls was determined after 24 h. Data are presented as mean ± SD from three independent experiments.
2.5. In vitro biocompatibility tests
2.6. Statistical analysis
In vitro data are expressed as mean ± SD from three or four independent experiments. One-way ANOVA followed by Dunnett's post hoc tests was used to determine statistical significance. In all the statistical evaluations, P < 0.05 was considered as statistically significant. Statistical analyses of drug molecule release, microbiological and cellular results were performed using the Graphpad Prism program (Graphpad software, USA).3 Results and discussion
3.1. Synthesis of the MSN@Gel nanosystems containing levofloxacin or N-acetylcysteine plus levofloxacin
The MSN@Gel nanosystems were prepared using the different synthetic approaches, as displayed in Scheme 1. The samples were fully characterized by using different techniques such as TEM, ζ- potential, DLS, FTIR, chemical microanalysis and TGA-DTA, as described above. Fig. 1 shows the TEM study of these nanosystems in the absence and presence of the two drugs (AC and LVX). To observe the external gelatin coating of the nanoparticles, staining with 1% phosphotungstic acid (PTA) was carried out, which allowed the organic part of these nanosystems to be detected with greater contrast. TEM images corresponding to the unstained material (Fig. 1A and C) showed quasi-spherical nanoparticles of approximately 150 nm in size with a mesoporous arrangement typical of a MCM-41 structure.50 These results evidenced that MSN@Gel nanosystems retained their morphology and mesoporous structure after external coating with gelatin. The presence of this outer coating became evident after staining with PTA (Fig. 1B and D), where the gelatin coating was observed as a darker area around the nanoparticle. In this regard, the sample without AC (MSN@Gel) presented a thinner coating, allowing the visualization of the mesoporous arrangement of MSNs even after staining with PTA (Fig. 1B). However, in the presence of AC (MSN-L@GelL-AC), a thicker organic coating was observed, which even occupied the interparticle gaps, and did not allow the mesoporous arrangement to be clearly seen after staining with PTA (Fig. 1D). | ||
| Fig. 1 TEM images of MSN@Gel and MSN-L@GelL-AC. Micrographs of the unstained (A and C) and PTA-stained (B and D) nanosystems were recorded to detect the organic coating. The gelatin coating is observed as a darker area around the nanoparticle (pointed with red arrows). Interparticle voids (empty or gelatin-filled) are marked with white arrows. | ||
For the purpose of obtaining information about the surface charge and hydrodynamic size of the different MSN@Gel nanosystems, ζ-potential and DLS measurements were performed on suspensions of these materials in water (Table 1 and Fig. S2 in the ESI†).
| Nanosystem | ζ-potential (mV) | D H (nm) | Organic matter (%) |
|---|---|---|---|
| MSN | −22.8 ± 0.3 | 190 ± 15 | 4.4 |
| MSN-LVX | −21.6 ± 0.2 | 220 ± 18 | 6.3 |
| MSN-L@Gel | −15.2 ± 0.4 | 250 ± 19 | 32.8 |
| MSN-L@GelL | −15.7 ± 0.3 | 258 ± 23 | 33.9 |
| MSN-L@GelAC | −16.2 ± 0.3 | 255 ± 20 | 29.2 |
| MSN-L@GelL-AC | −16.1 ± 0.6 | 259 ± 17 | 30.1 |
The MSN sample showed a potential value of −22.8 mV. However, this value slightly shifted towards less negative values in the MSN@Gel nanosystems, which was due to the ionizable groups present in the gelatin (eqn (1) and (2)), corroborating the efficiency of the coating process.
| R–NH2 + H2O ⇌ R–NH3+ + OH− pKa ≈ 6.5 | (1) |
| R–COOH + H2O ⇌ R–COO− + H3O+ pKa ≈ 4.7 | (2) |
The DLS measurements were carried out to determine hydrodynamic diameter (DH) distributions with a maximum of ca. 200 nm for the samples without gelatin and ca. 260 nm for the samples with gelatin (Table 1). The increase in the DH of MSN@Gel nanosystems compared to the pristine MSN sample confirmed the presence of the organic shell around the nanoparticles, as it has been demonstrated by TEM.
FTIR characterization of the nanoparticles before and after gelatin coating was performed. The FTIR spectra obtained from the different MSN@Gel materials evidenced the presence of gelatin by the appearance of new bands in the region between 1200 and 1700 cm−1 (Fig. S3, ESI†). More specifically, the bands at 1633 and 1559 cm−1 could be attributed to the vibrations of amide I (νCO strain) and amide II (δNH bending and νCN strain), respectively. The amide III band (in-phase combination of νCN strain and δNH bending) was represented by a set of three weak signals centered at 1240 cm−1, characteristic of gelatin.51
The amount of organic matter present in the different MSN@Gel nanosystems was determined by TGA-DTA analysis (Table 1). The results show a percentage by weight of organic matter of approximately 30% in the materials with gelatin, more than five times higher than the organic content of the gelatin-free materials, which also confirmed the success of the coating process.
The amount of drug (LVX or AC) loaded into the different compartments of the nanosystems was determined by chemical microanalysis using the percentages of C and N. The obtained results showed that the LVX amount loaded inside the mesopores was 3.0 ± 0.2%, the amount of LVX embedded in the gelatin compartment was 3.5 ± 0.9%, and the amount of AC incorporated in the gelatin shell was 2.9 ± 0.8%. In addition, the successful incorporation of the drug into the mesopores of the nanoparticles was confirmed by FTIR and N2 adsorption porosimetry (Fig. S4 in the ESI†).
Since these MSN@Gel nanomaterials were designed as antimicrobial co-delivery nanosystems for potential biomedical applications, the in vitro stability was evaluated in physiological media. The nanoparticles were suspended in PBS 1× (pH = 7.4) at 37 °C for 96 h, and then the changes produced in the nanosystems were evaluated by TEM. Fig. 2 shows a summary of these results, where although the quasi-spherical morphology of the nanosystems remained constant after 96 h of incubation, a partial loss of the mesostructural order was observed in all materials analyzed (Fig. 2C and G).52 Partial dissolution of silica is consistent with the leaching of silicon into the medium as determined by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES), yielding values of 31.4 ± 0.1 μg mg−1 and 31.8 ± 0.1 μg mg−1 for MSN-L@GelL and MSN-L@GelAC samples, respectively. These results indicate that the hydrogel gelatin coating allows the penetration of the aqueous medium into the inorganic core matrix, which is essential for the diffusional release of LVX.38 In the case of the gelatin hydrogel coating, TEM images after staining with PTA reveal that it remained after 96 h (Fig. 2D and H). These findings are in line with the use of a non-acidic pH (pH = 7.4) and with the absence of proteolytic enzymes in the incubation medium, which would cause the gelatin degradation.53–55 The colloidal stability in PBS 1× was verified by DLS measurements, indicating similar hydrodynamic size distributions centered at ca. 250 nm, as displayed in Fig. 2. Additionally, ζ-potential measurements were carried out in PBS 0.1×, and the results show a reduction towards less negative values after 96 h of incubation, which could be attributed to the adsorption of phosphate anions from PBS on the nanoparticle surface, as it has been previously reported for diverse nanomaterials.56 The colloidal stability of the nanoparticles was also evaluated in cell culture medium, namely, Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) (DMEM + 10% FBS). Hydrodynamic size distributions by intensity were measured by DLS of the MSN-L@GelL and MSN-L@GelAC nanosystems at different time periods in DMEM + 10% FCS medium (Fig. S5, in the ESI†). Initially, nanoparticles showed a hydrodynamic size centred at 245 nm, which barely differed from those obtained in water. However, after 96 h in the cell culture medium, the mean size of the particles suspended in DMEM + 10% FCS experienced a noticeable decrease, reaching values of 192 nm and 170 nm for MSN-L@GelL and MSN-L@GelAC samples, respectively. These results suggest a specific interaction between the FCS constituents and the nanoparticles. Several serum proteins, such as albumin, immunoglobulin, and fibrinogen, could be adsorbed on nanoparticles, enhancing the colloidal stability via a steric mechanism, as reported in the literature.57
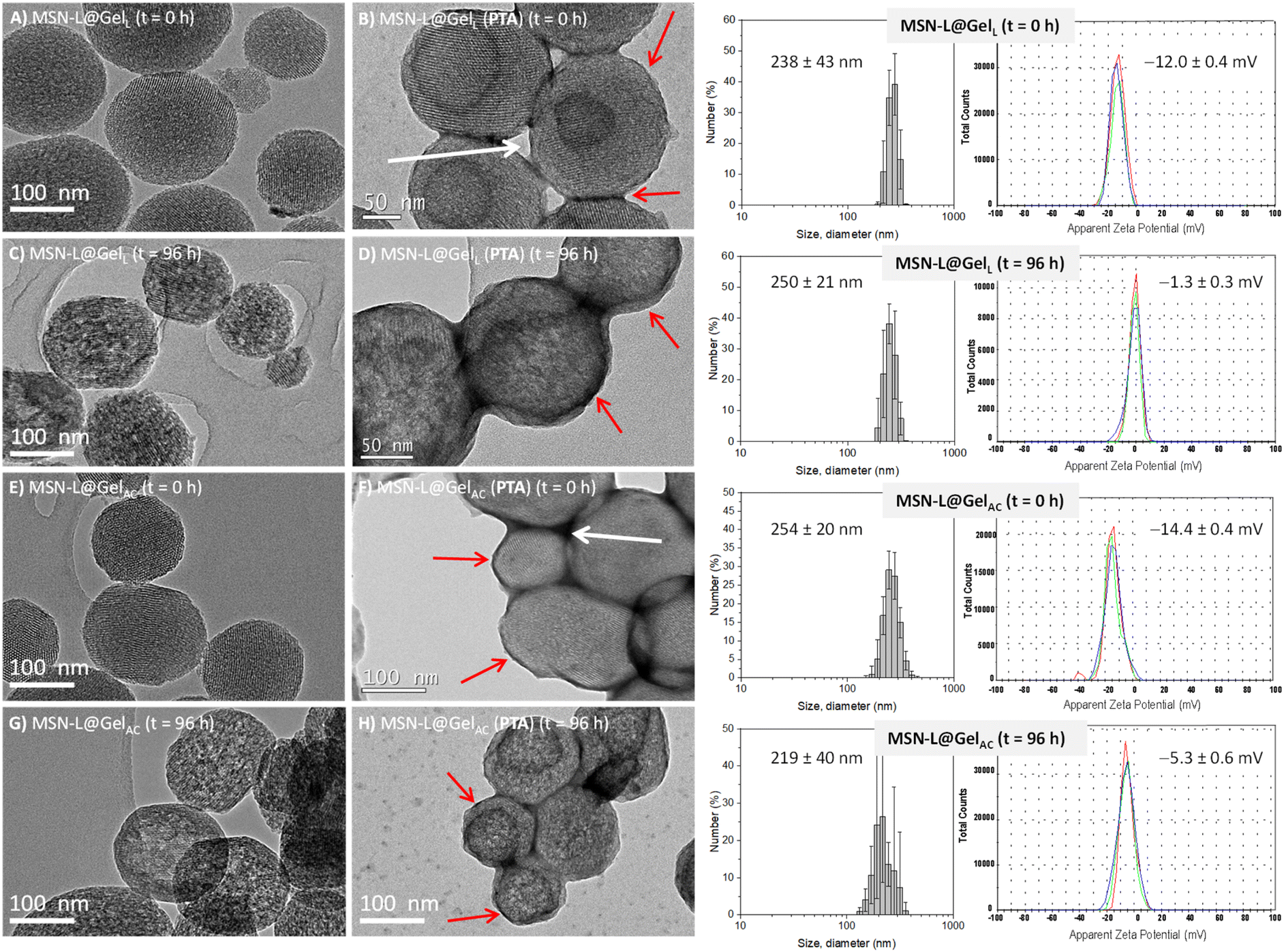 | ||
| Fig. 2 Left: TEM images of the MSN@Gel nanosystems before (t = 0 h) and after (t = 96 h) incubation under physiological conditions (PBS 1×, pH = 7.4 at 37 °C). The study was performed to verify the mesoporous structure of the unstained and 1% PTA-stained nanoparticles, as well as to observe the external gelatin coating due to the presence of organic matter. The gelatin coating is observed as a darker area around the nanoparticle (pointed with red arrows). Interparticle voids (empty or gelatin-filled) are marked with white arrows. Right: Hydrodynamic size distributions by number measured by DLS and ζ-potential distributions of MSN@Gel nanosystems before (t = 0 h) and after (t = 96 h) incubation under physiological conditions (PBS 1×, pH = 7.4 at 37 °C). | ||
3.2. Drug release assays
To investigate the release mechanism of the drug molecules incorporated in the gelatin coating, fluorescein was used as a model molecule (sample MSN@GelF), and in vitro release assays were performed as described above. The use of this fluorescent molecule allowed elucidation of the release mechanism of the hydrogel coating. The amount of fluorescein loaded into the external gelatin shell was 4.3 ± 0.9%, as determined by chemical microanalysis. Fig. 3A shows the fraction of fluorescein released versus time. The amount of fluorescein released at each time was normalized to the total concentration released at infinite time (W0). The experimental data were fitted to the Korsmeyer–Peppas or “Power Law” equation (eqn (3)):58| Wt/W0 = KK–Ptn | (3) |
 | ||
| Fig. 3 Representation of the released fraction of fluorescein (A) in MSN@GelF and LVX (B) in the different MSN@Gel nanosystems depending on time. Data are presented as mean ± σ (***P < 0.001 in all cases compared to the MSN control). Experiments were performed in triplicate. | ||
This kinetic model was chosen because it is the most widely used to study drug release from hydrogels,59,60 since eqn (3) provides a lot of information with the value of n being related to the release mechanism involved.58,61 In the case of matrices with spherical geometry, n < 0.43 indicates a quasi-Fickian diffusion mechanism and n = 0.43 indicates a Fickian diffusion. This means that the drug can diffuse through the hydrogel matrix because it is much smaller than the mesh size and diffuses rapidly. If n > 0.85, this is called Case II transport (zero-order kinetics), and the release is due to the swelling of the system. The values of 0.43 < n < 0.85 correspond to anomalous (non-Fickian) transport, and the release is due to a combination of diffusion and swelling of the hydrogel.
As shown in Fig. 3A, the value of n = 0.22 corresponds to a quasi-Fickian diffusion mechanism, indicating that the fluorescein model molecule can diffuse through the gelatin hydrogel matrix in which it is embedded to the release medium, with a rate constant of 0.06 h−n. The release profiles of LVX from the different nanosystems are shown in Fig. 3B, and the results derived from fitting the experimental data to eqn (3) are displayed in Table 2.
| Nanosystem | Maximum released LVX (μg mg−1) | R 2 | Kinetic constant KK–P × 102 (h−n) | Release exponent n | Diffusion mechanism |
|---|---|---|---|---|---|
| MSN-L | 10.6 ± 0.6 | 0.97 | 6 ± 1 | 0.41 ± 0.04 | Fickian |
| MSN-L@Gel | 5.1 ± 0.3 | 0.89 | 10 ± 1 | 0.14 ± 0.03 | Quasi-Fickian |
| MSN-L@GelL | 8.5 ± 0.1 | 0.87 | 14 ± 3 | 0.14 ± 0.02 | Quasi-Fickian |
| MSN-L@GelAC | 0.1 ± 0.01 | 0.83 | 0.3 ± 0.1 | 0.13 ± 0.04 | Quasi-Fickian |
| MSN-L@GelL-AC | 2.3 ± 0.07 | 0.87 | 1.9 ± 0.3 | 0.16 ± 0.04 | Quasi-Fickian |
It can be observed that LVX release from the MSN-L sample obeys a Fickian diffusion mechanism, with a value of n close to 0.43 and a kinetic constant KK–P = 0.06 h−n. However, MSN@Gel samples show n values in the range of 0.13 to 0.16, which would support a quasi-Fickian diffusion mechanism, which is in good agreement with the results derived from the fluorescein release from the hydrogel gelatin coating. In addition, the maximum amount of LVX released (ca. 11 μg mg−1) was achieved for the gelatin-free nanosystem. This fact could be ascribed to a higher drug retention degree due to the gelatin hydrogel coating in MSN@Gel samples, which would be acting as a diffusion barrier for the LVX loaded into the mesopores of the inorganic matrix. Finally, the samples incorporating LVX in the gelatin coating show the highest maximum amount of drug release due to the easy penetration of the aqueous medium into the polymeric compartment, in good agreement with the results derived from fluorescein model release assays. Finally, it should be noted that there is a sustained LVX release over time from all nanosystems, a consequence of the well-established strong attractive interactions between the LVX molecules and the silanol (–SiOH) groups present in the silica matrix.62,63
3.3. Antibiofilm efficacy tests
Antimicrobial efficacy tests were performed by placing different concentrations of nanosystems in mature E. coli biofilms. To evaluate the biofilm matrix-disruptive effect of AC, confocal laser scanning microscopy studies were performed by staining the protective mucopolysaccharide biofilm matrix with calcofluorine (blue) and the live/dead bacteria with the LIVE/DEAD® BacLight™ reagent, which stains dead (red) and live (green) bacteria. Moreover, quantitative studies of the antimicrobial efficacy of the nanosystems were performed by counting CFU mL−1, following the procedure described above. | ||
| Fig. 4 Confocal laser scanning microscopy images of E. coli biofilms after treatment with (A) 10 μg mL−1 and (B) 50 μg mL−1 of the MSN@Gel materials and their respective fluorescence intensity quantifications of eight images (C and D). Two columns are shown for each sample. On the left, the mucopolysaccharide matrix (blue), live bacteria (green) and dead bacteria (red) can be observed. On the right, only the matrix (blue) is shown to highlight the disaggregating effect on the biofilm matrix. Data are represented as mean ± σ (*P < 0.05, compared to control, **P < 0.01, compared to control, ***P < 0.001, compared to control). The experiment was performed in triplicate. | ||
 | ||
| Fig. 5 Relative percentage of bacterial growth obtained from in vitro antibiofilm activity of the different MSN@Gel materials on mature E. coli biofilms, at concentrations of 10 and 50 μg mL−1. The upper chart is an extension of the percentages up to 1%. Data are represented as mean ± σ (***P < 0.001, compared to control). The experiment was performed in quadruplicate. | ||
3.4. In vitro cell biocompatibility tests
Since the whole nanosystem (MSN-L@Gel-L-AC) provided the best antimicrobial effect against E. coli biofilm, its in vitro biocompatibility was evaluated, which is an essential study before any in vivo testing. Therefore, preliminary in vitro cellular cytotoxicity and cell cycle assays were performed in MC3T3-E1 preosteoblastic cells, using the MSN@Gel nanosystems at different concentrations (10, 25, 50 and 75 μg mL−1). Overall, the results indicate good biocompatibility, showing a small dose-dependent decrease in cell viability at 24 h, which is recovered after 72 h of testing. The decrease in cellular viability when cells are treated with LVX has already been described in the literature.64 In fact, previous results from our research group reported a notable reduction in cell viability upon exposure to similar doses of pure silica MSNs carrying LVX.38 In contrast, in the present work, the reduction in cell viability is noticeably less pronounced, which could be attributed to the dual effect of the gelatin coating, which hinders the initial LVX burst release effect and produces a more sustained release of the antibiotic (Fig. 3B), and the AC, which may protect cells from oxidative damage.31To reinforce the biocompatibility tests, a cell cycle study was performed to evaluate possible changes in the different phases of the cell cycle after treatment with the nanosystem. The results indicated that the process of duplication of the genetic material (G1 and S phases), the preparatory phase to division (G2 phase) and cell division (M phase) were not affected after contact with the MSN-L@GelL-AC nanomaterial, since no changes were observed at any of the concentrations used compared to the control (Fig. 6B). These results confirm the high biocompatibility of this gelatin-containing nanosystem, which exhibits high efficacy against mature E. coli biofilms.
 | ||
| Fig. 6 Cell biocompatibility assays in the MC3T3-E1 cell line with different concentrations of the MSN-L@GelL-AC sample. MC3T3-E1 cells were treated with different concentrations of the MSN-L@GelL-AC sample (10, 25, 50 and 75 μg mL−1) and analyzed for (A) cell viability at 24 h and 72 h, and (B) cell cycle was measured after 24 h of treatment by flow cytometry. The panel shows representative cell cycle distributions. The cell cycle phases are defined as the G0/G1 phase (Quiescence/Gap 1), the S phase (Synthesis) and the G2/M phase (Gap 2 and Mitosis). The bottom right panel shows quantification of percentages of cells in each cell-cycle phase. Data are presented as mean ± SD from three independent experiments (*P < 0.05, compared to control; ***P < 0.001, compared to control). | ||
4 Conclusions
Biocompatible nanosystems with combined mucolytic and antibiotic activity based on mesoporous silica-based nanoparticles were developed, which showed high efficiency against mature Escherichia coli biofilms. The appropriate design of the nanocarrier allows loading the antimicrobial agents into different well-defined compartments. In this way, the antibiotic levofloxacin was loaded into the mesoporous silica structure, while N-acetylcysteine, as a mucolytic agent, was embedded in the outer gelatin layer. The sequential co-delivery of antimicrobial agents from the compartmented nanocarrier allows the release of N-acetylcysteine at the earliest stage, followed by the sustained release of levofloxacin. This release behavior produces an enhanced antimicrobial effect, allowing for approximately 99.8% reduction of mature E. coli biofilms. Fine-tuning the co-delivery of drugs from silica-based mesoporous nanocarriers could lead to the development of efficient therapies to treat infections associated with mature E. coli biofilms while reducing the risk of antimicrobial resistance development.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Anna Aguilar-Colomer: methodology, validation, formal analysis, investigation, data curation, writing – original draft, visualization; Carla Jiménez-Jiménez: methodology, validation, formal analysis, investigation, data curation, writing – original draft, visualization; Blanca González: methodology, formal analysis, investigation, writing – original draft, writing – review & editing, visualization, supervision; Jaime Esteban: conceptualization, methodology, data curation, writing – original draft, writing – review & editing, visualization, supervision; María Vallet-Regí: resources, supervision, researcher responsible, project administration, funding acquisition; Montserrat Colilla: conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration; Isabel Izquierdo-Barba: conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Spanish Government, Ministerio de Ciencia e Innovación through project PID2020-117091RB-I00 (NANONICHE), the European Research Council ERC-2015-AdG (VERDI) grant No. 694160 and the Fundación Ramón Areces (FD5/22_01, Nano4Infection) for their support.References
- G. Sharma, S. Sharma, P. Sharma P, D. Chandola, S. Dang, S. Gupta and R. Gabrani, J. Appl. Microbiol., 2016, 121, 309–319 CrossRef CAS PubMed.
- P. N. Danese, L. A. Pratt and R. Kolter, J. Bacteriol., 2000, 182, 3593–3596 CrossRef CAS PubMed.
- J. W. Costerton, Z. Lewandowski, D. E. Caldwell, D. R. Korber and H. M. Lappin-Scott, Annu. Rev. Microbiol., 1995, 49, 711–745 CrossRef CAS PubMed.
- H. C. Flemming, E. D. van Hullebusch, T. R. Neu, P. H. Nielsen, T. Seviour, P. Stoodley, J. Wingender and S. Wuertz, Nat. Rev. Microbiol., 2023, 21, 70–86 CrossRef CAS PubMed.
- O. Y. A. Costa, J. M. Raaijmakers and E. E. Kuramae, Front. Microbiol., 2018, 9, 1636 CrossRef PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- R. T. Sturbelle, L. F. de Avila, T. B. Roos, J. L. Borchardt, R. da Conceição, O. A. Dellagostin and F. P. Leite, Vet. Microbiol., 2015, 180, 245–252 CrossRef CAS PubMed.
- N. Høiby, T. Bjarnsholt, M. Givskov, S. Molin and O. Ciofu, Int. J. Antimicrob. Agents, 2010, 35, 322–332 CrossRef PubMed.
- J. F. González, M. M. Hahn and J. S. Gunn, Pathog. Dis., 2018, 76, 1–7 Search PubMed.
- V. Ballén, V. Cepas, C. Ratia, Y. Gabasa and S. M. Soto, Microorganisms, 2022, 10, 1103 CrossRef PubMed.
- S. M. Ribeiro, M. R. Felício, E. V. Boas, S. Gonçalves, F. F Costa, R. P. Samy, N. C. Santos and O. L. Franco, Pharmacol. Ther., 2016, 160, 133–144 CrossRef CAS.
- L. Lu, W. Hu, Z. Tian, D. Yuan, G. Yi, Y. Zhou, Q. Cheng, J. Zhu and M. Li, Chin. Med., 2019, 14, 11 CrossRef.
- Y. G. Kim, J. H. Lee, G. Gwon, S. I. Kim, J. G. Park and J. Lee, Sci. Rep., 2016, 6, 36377 CrossRef CAS.
- J. Sun, J. P. Marais, C. Khoo, K. LaPlante, R. M. Vejborg, M. Givskov, T. Tolker-Nielsen, N. P. Seeram and D. C. Rowley, J. Funct. Foods, 2015, 17, 235–242 CrossRef CAS PubMed.
- Y. B. Bai, M. Y. Shi, W. W. Wang, L.-Y. Wu, Y.-T. Bai, B. Li, X.-Z. Zhou and J.-Y. Zhang, Front. Microbiol., 2022, 13, 1003692 CrossRef PubMed.
- O. Fleitas Martínez, M. H. Cardoso, S. M. Ribeiro and O. L. Franco, Front. Cell. Infect. Microbiol., 2019, 9, 74 CrossRef PubMed.
- L. Y. Peng, M. Yuan, Z. Q. Cui, Z.-M. Wu, Z.-J. Yu, K. Song, B. Tang and B.-D. Fu, Microb. Pathog., 2018, 119, 54–59 CrossRef CAS PubMed.
- S. G Sanmukh, J. Admella, L. Moya-Andérico, T. Fehér, B. V. Arévalo-Jaimes, N. Blanco-Cabra and E. Torrents, Cells, 2023, 12, 344 CrossRef.
- B. C. Sanchez, E. R. Heckmann, S. I. Green, J. R. Clark, H. B. Kaplan, R. F. Ramig, C. Hines-Munson, F. Skelton, B. W. Trautner and A. W. Maresso, Front. Microbiol., 2022, 13, 796132 CrossRef PubMed.
- Y. Gu, Y. Xu, J. Xu, X. Yu, X. Huang, G. Liu and X. Liu, Appl. Microbiol. Biotechnol., 2019, 103, 315–326 CrossRef CAS.
- A. Fontanot, I. Ellinger, W. W. J. Unger and J. P. Hays, Antibiotics, 2024, 13, 343 CrossRef CAS PubMed.
- G. Batoni, G. Maisetta and S. Esin, Biochim. Biophys. Acta, 2016, 1858, 1044–1060 CrossRef CAS PubMed.
- R. M. Pinto, F. A. Soares, S. Reis, C. Nunes and P. Van Dijck, Front. Microbiol., 2022, 11, 952 CrossRef PubMed.
- S. Wang, Y. Zhao, A. P. Breslawec, Z. Deng, L. L. Kuperma and Q. Yu, NPJ Biofilms Microbiomes, 2023, 9, 63 CrossRef PubMed.
- R. Torelli, M. Cacaci, M. Papi, F. Paroni Sterbini, C. Martini, B. Posteraro, V. Palmieri, M. De Spirito, M. Sanguinetti and F. Bugli, Colloids Surf., B, 2017, 158, 349–355 CrossRef CAS PubMed.
- G. V. Tetz, N. K. Artemenko and V. V. Tetz, Antimicrob. Agents Chemother., 2009, 53, 1204–1209 CrossRef CAS PubMed.
- C. Cheng, L. Du, J. Yu, Q. Lu, Y. He and T. Ran, Pathol. Res. Pract., 2015, 211, 982–988 CrossRef CAS PubMed.
- Y. Zhang, Y. Fu, J. Yu, Q. Ai, J. Li and N. Peng, J. Infect. Chemother., 2015, 21, 808–815 CrossRef CAS PubMed.
- A. Pani, V. Lucini, S. Dugnani and F. Scaglione, Int. J. Antimicrob. Agents, 2022, 59, 106529 CrossRef CAS PubMed.
- B. Leite, F. Gomes, P. Teixeira, C. Souza, E. Pizzolitto and R. Oliveira, Enferm. Infecc. Microbiol. Clín., 2013, 31, 655–659 CrossRef PubMed.
- B. Pedre, U. Barayeu, D. Ezeriņa and T. P. Dick, Pharmacol. Ther., 2021, 228, 107916 CrossRef CAS PubMed.
- S. Aslam, B. W. Trautner, V. Ramanathan and R. O. Darouiche, Antimicrob. Agents Chemother., 2007, 54, 1556–1558 CrossRef.
- S. Pollini, S. Boncompagni, T. Di Maggio, V. Di Pilato, T. Spanu, B. Fiori, F. Blasi, S. Aliberti, F. Sergio, G. M. Rossolini and L. Pallecchi, J. Antimicrob. Chemother., 2018, 73, 2388–2395 CrossRef CAS PubMed.
- A. Manoharan, S. Ognenovska, D. Paino, G. Whiteley, T. Glasbey, F. H. Kriel, J. Farrell, K. H. Moore, J. Manos and T. Das, Antibiotics, 2021, 10, 900 CrossRef CAS PubMed.
- A. Marchese, M. Bozzolasco, L. Gualco, E. A. Debbia, G. C. Schito and A. M. Schito, Int. J. Antimicrob. Agents, 2003, 22, 95–100 CrossRef PubMed.
- Y. Samuni, S. Goldstein, O. M. Dean and M. Berk, Biochim. Biophys. Acta, 2013, 1830, 4117–4129 CrossRef CAS.
- Z. Wang, X. Liu, Y. Duan and Y. Huang, Biomaterials, 2022, 280, 121249 CrossRef CAS PubMed.
- A. Aguilar-Colomer, M. Colilla, I. Izquierdo-Barba, C. Jiménez-Jiménez, I. Mahillo, J. Esteban and M. Vallet-Regí, Microporous Mesoporous Mater., 2020, 311, 110681 CrossRef PubMed.
- M. Colilla and M. Vallet-Regí, Chem. Mater., 2023, 35, 8788–8805 CrossRef CAS PubMed.
- S. Medaglia, I. Otri, A. Bernardos, M. D. Marcos, E. Aznar, F. Sancenón and R. Martínez-Máñez, Int. J. Pharm., 2024, 654, 123947 CrossRef CAS PubMed.
- C. Carucci, J. L. Pablos, J. A. Romero-Antolín, B. González, M. Colilla, I. Izquierdo Barba, A. Salis, M. Monduzzi and M. Vallet-Regí, Microporous Mesoporous Mater., 2024, 363, 112810 CrossRef CAS.
- J. J. Aguilera-Correa, M. Gisbert-Garzarán, A. Mediero A, R. A. Carias-Cálix, C. Jiménez-Jiménez, J. Esteban and M. Vallet-Regí, Acta Biomater., 2022, 137, 218–237 CrossRef CAS PubMed.
- E. Álvarez, M. Estévez, C. Jiménez-Jiménez, M. Colilla, I. Izquierdo-Barba, B. González and M. Vallet-Regí, Acta Biomater., 2021, 136, 570–581 CrossRef PubMed.
- A. García, B. González, C. Harvey, I. Izquierdo-Barba and M. Vallet-Regí, Microporous Mesoporous Mater., 2021, 328, 111489 CrossRef.
- J. J. Aguilera-Correa, M. Gisbert-Garzarán, A. Mediero, M. J. Fernández-Aceñero, D. De-Pablo-Velasco, D. Lozano, J. Esteban and M. Vallet-Regí, Acta Biomater., 2022, 154, 608–625 CrossRef CAS PubMed.
- W. Tasia, C. Lei, Y. Cao, Q. Ye, Y. He and C. Xu, Nanoscale, 2020, 12, 2328–2332 RSC.
- M. Martínez-Carmona, D. Lozano, M. Colilla and M. Vallet-Regí, RSC Adv., 2016, 6, 50923–50932 RSC.
- J. Cieslinski, V. S. T. Ribeiro, C. K. Lima, L. Kraft, P. H. Suss and F. F. Tuon, J. Bras. Neurol., 2023, 45, 373–377 Search PubMed.
- B. Herigstad, M. Hamilton and J. Heersink, J. Microbiol. Methods, 2001, 44, 121–129 CrossRef CAS PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- C. Tengroth, U. Gasslander, F. O. Andersson and J. P. Jacobsson, Pharm. Dev. Technol., 2005, 10, 405–412 CrossRef CAS PubMed.
- J. L. Paris, M. Colilla, I. Izquierdo-Barba, M. Manzano and M. Vallet-Regí, J. Mater. Sci., 2017, 52, 8761–8771 CrossRef CAS.
- M. Ozeki, T. Ishii, Y. Hirano and Y. Tabata, J. Drug Target., 2001, 9, 461–471 CrossRef CAS PubMed.
- M. Yamamoto, Y. Ikada and Y. Tabata, J. Biomater. Sci. Polym. Ed., 2001, 12, 77–88 CrossRef CAS PubMed.
- Y. Ikada and Y. Tabata, Adv. Drug Deliv. Rev., 1998, 31, 287–301 CrossRef PubMed.
- K. Afshinnia and M. Baalousha, Sci. Total Environ., 2017, 581–582, 268–276 CrossRef CAS PubMed.
- A. C. Sabuncu, J. Grubbs, S. Qian, T. M. Abdel-Fattah, M. W. Stacey and A. Beskok, Colloids Surf. B Biointerfaces, 2012, 95, 96–102 CrossRef CAS PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- D. Caccavo, Int. J. Pharm., 2019, 560, 175–190 CrossRef CAS PubMed.
- M. Vigata, C. Meinert, D. W. Hutmacher and N. Bock, Pharmaceutics, 2020, 7, 1188 CrossRef PubMed.
- P. L. Ritger and N. A. Peppas, J. Control. Release, 1987, 5, 23–36 CrossRef CAS.
- M. Cicuéndez, I. Izquierdo-Barba, M. T. Portolés and M. Vallet-Regí, Eur. J. Pharm. Biopharm., 2013, 84, 115–124 CrossRef PubMed.
- B. González, M. Colilla, J. Díez, D. Pedraza, M. Guembe, I. Izquierdo-Barba and M. Vallet-Regí, Acta Biomater., 2018, 68, 261–271 CrossRef PubMed.
- L. Wang, Y. Wu, Y. Tan, X. Fei, Y. Deng, H. Cao, B. Chen, H. Wang, J. Magdalou and L. Chen, J. Appl. Toxicol., 2014, 34, 870–877 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00006h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |