
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and in vitro and ex vivo characterization of a twin nanoparticulate system to enhance ocular absorption and prolong retention of dexamethasone in the eye: from lab to pilot scale optimization†
Muhammad
Sarfraz‡
 *ab,
Goutam
Behl‡
a,
Sweta
Rani
a,
Niall
O'Reilly
a,
Peter
McLoughlin
a,
Orla
O'Donovan
a,
Alison L.
Reynolds
cd,
John
Lynch
a and
Laurence
Fitzhenry
*a
*ab,
Goutam
Behl‡
a,
Sweta
Rani
a,
Niall
O'Reilly
a,
Peter
McLoughlin
a,
Orla
O'Donovan
a,
Alison L.
Reynolds
cd,
John
Lynch
a and
Laurence
Fitzhenry
*a
aOcular Therapeutics Research Group (OTRG), Pharmaceutical & Molecular Biotechnology Research Centre (PMBRC), South East Technological University, Waterford X91 KOEK, Waterford, Ireland. E-mail: muhammad.sarfraz@setu.ie; laurence.fitzhenry@setu.ie
bFaculty of Pharmacy, The University of Lahore, Lahore 56400, Pakistan
cUCD School of Veterinary Medicine, Dublin, Ireland
dUCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin (UCD), Dublin D04 V1W8, Ireland
First published on 23rd April 2025
Abstract
Conventional eye drops show low bioavailability (below 20%) due to the eye's inherent tissue barriers and unique microenvironment. Recent advancements in pharmaceutical nanotechnology have explored various nanoparticle systems, such as micelles, liposomes, and nanoemulsions, to enhance corneal permeation and prolong drug retention. In this study, we propose a twin nanoparticulate system, combining the advantages of two nanoparticles to improve drug targeting and therapeutic efficacy. A dexamethasone-loaded liposome–microemulsion (LME) twin nanoparticulate system was developed using high-pressure homogenization and successfully scaled up. Both liposomes and microemulsions were of similar size (∼60 nm) and displayed uniform distribution (polydispersity index < 0.2) upon combination. The final formulation was hypo-osmolar (osmolality < 100 mOsm per Kg), making it ideal for dry eye relief. Drug release was extended for up to 8 h, following a non-Fickian diffusion pattern. The LME formulation, tested under different conditions (2–8 °C and 25 °C with 60% relative humidity), was found to be stable for 6 months. It showed no cytotoxicity in human corneal epithelial cells up to 10 μM drug concentration. Fluorescence microscopy revealed rapid nanoparticle uptake by cells within 5 minutes. Human corneal epithelial cells showed a marked reduction in inflammatory biomarkers (IL-6, IL-8, and TNF-α) after drug-loaded LME treatments, compared to the control. Corneal tissue imaging confirmed prolonged retention of nanoparticles within the tissue. A whole eye ex vivo permeation study demonstrated higher drug concentrations in the aqueous humour of LME drug-treated rabbit eyes compared to a reference product. This twin nanoparticulate system, loaded with dexamethasone, offers a promising next-generation treatment for dry eye disease (DED).
Introduction
Recent data reveal that approximately 344 million people are affected with dry eye disease (DED) worldwide.1 The associated market is expected to reach $7.49 billion in 2024 and is predicted to increase to $13 billion by 2032.2 DED is a chronic condition that significantly decreases quality of life by causing ocular surface pain and irritation, grittiness and scratchiness, burning and stinging.3 The pathogenesis of this disease is multi-factorial but centres on poor quality or reduced tear volume.3 Advanced age, sex, prolonged use of devices with screens (e.g. tablets and mobile phones) and environmental factors contribute to DED.4 DED can be debilitating, with severe cases leading to visual impairment. Despite the prevalence of this disease, there are relatively few successful treatments available on the market.Topical drug instillation to the eye is the most convenient and applicable approach for anterior segment eye conditions such as DED. However, since merely 5% of the instilled drug is absorbed through the ocular surface,5 optimising such treatments is challenging. Factors that cause potential issues include the innate ocular barriers and microenvironment (osmolarity, pH, tear enzymes, etc.) nasolacrimal secretion, protein binding, enzymatic degradation, or metabolism by protease, and esterase enzyme, Fig. 1.6
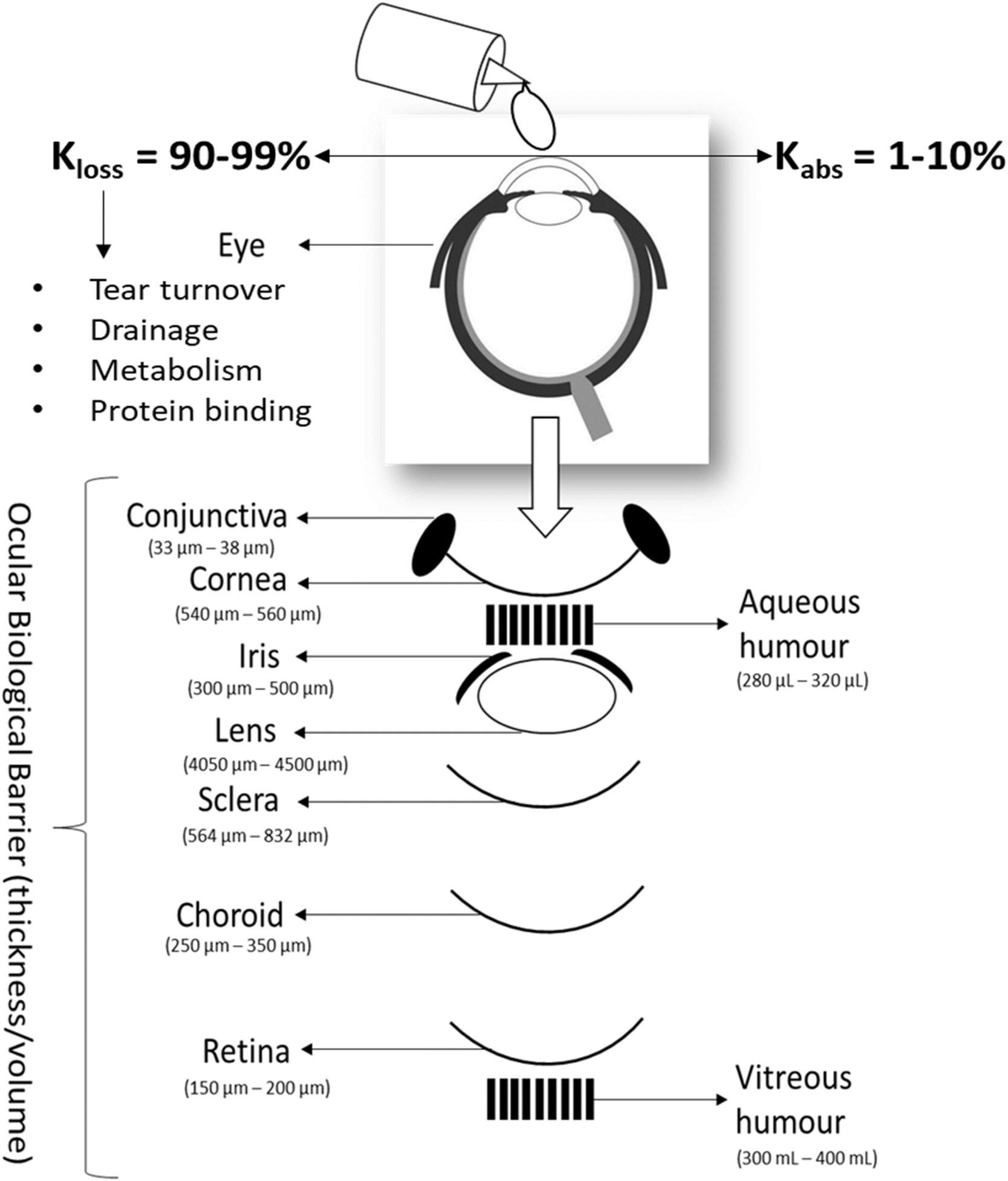 | ||
| Fig. 1 Ocular biological barriers for topical eye drops. | ||
The tear film serves as the first line of defence against pathogens while also acting as a barrier to administered drugs. It maintains eye hydration with a volume of about 3–10 μL and is produced at a rate of 1 μL per minute under normal conditions. Following the administration of a drug solution, the tear turnover rate increases, causing rapid clearance of the drug from the eye within 15–30 seconds.5 The tear film is approximately 8 μm thick and composed of three basic layers: lipid, aqueous, and mucin.7 The lipid layer (40–160 nm) is the first barrier between the eye and the environment.8 Lipids in this layer originate from meibum; an oily, lipid-enriched secretion produced by meibomian glands of human eyelids.9 It is composed of non-polar and polar lipids. The non-polar lipids are the major contributor (82%) to the formation of upper lipid layer of the tear film, consist of wax esters, cholesteryl esters, diesters, triacylglycerols, and free cholesterol.9 Waxes and triacylglycerol make this layer very hydrophobic. The polar (amphiphilic) layer forms a minor fraction of tear lipids (∼8–18% of tear lipids) consisting of phospholipids (such as phosphatidylethanolamine and phosphatidylcholine), ceramide, cerebrosides, free fatty acids, sphingomyelin and (O-acyl)-ω-hydroxy fatty acids.10 This upper lipid layer of the tear film restricts the solubility and penetration of hydrophilic drugs. A tear film, in a healthy person, has an osmolarity of 296–336 mOsm per L, determined mainly by electrolytes in the aqueous phase.8 Ions such as Na+, Cl−, HCo3−, Ca2+, and K+ as well as the ratio of divalent to monovalent cations provide a buffering capacity to the pH of the tears, thus maintaining tear tonicity. Mucins form the innermost layer of the tear film.11 The non-specific binding of drugs with tear enzymes (e.g., lysozyme), the mucin layer, and proteins (e.g., albumin) prevents them from reaching the underlying cornea and anterior chamber, and the drugs are therefore quickly cleared with each blink.7
Abnormalities in the tear film are commonly associated with dry eye conditions, making it a valuable parameter in both the diagnosis and monitoring of disease progression. Other than the tear film, ocular tissues are the physiological barriers to drug penetration when the drug in a solution form. The human cornea is composed of three cell layers: the lipophilic epithelium, the hydrophilic stroma, and the lipophilic endothelium (in order from anterior to posterior).7 The superficial corneal epithelium makes up six to eight layers of cells which allows the permeation of hydrophilic drugs only.12 The hydrophilic stromal matrix (approximately 80% water content) next to the corneal epithelium has a thickness of approximately 450–500 μm, representing 90% of the corneal thickness, and thus imposes significant limitations on lipophilic drugs due to solubility and partition coefficients. The endothelium is a permeable monolayer of cells, approximately 13 μm thick, that offers minimal resistance to the paracellular transport of drugs.13 Overall, the specific sandwich structure of corneal tissue makes it a unique barrier to most lipophilic and hydrophilic drugs. The alternative pathway for drugs to enter the eye following topical instillation is the non-corneal route consisting of the conjunctiva and sclera.7 The conjunctiva possesses a surface area that is about 17 times larger than that of the cornea. Additionally, the conjunctiva's permeability to hydrophilic drugs is significantly higher, being 17 times greater than that of the corneal epithelium. As a result, hydrophilic drugs and macromolecules tend to be absorbed more readily through the conjunctiva.14 The sclera is an extension of the cornea, composed of collagen and mucopolysaccharides and this structure allows for the easy permeation of hydrophilic molecules.15 In addition, metabolism in the eye is also challenging for some drugs. It has been demonstrated that drugs containing aromatic hydrocarbons are metabolized in the pigmented epithelium and ciliary body into their corresponding epoxides and phenols, or further metabolized by other enzymes present in the eye.16
In summary, the ocular anatomical and physiological barriers result in insufficient corneal permeation and a short residence time for topical drugs, leading to non-linear ocular pharmacokinetics. Over the past two decades, advancements in pharmaceutical nanotechnology have driven researchers to explore various types of nanoparticles to enhance corneal permeation and drug residence time on the eye. Some of the examples of nano carrier systems used for this purpose are micelles, liposomes, nanosuspensions, nano-emulsions, nanogels, nanofibers, microspheres, dendrimers, and nanostructured carriers.17–19 The FDA have approved several nanotechnologies for DED conditions, including Restasis® (nanoemulsion), Cequa® (micelles), Artelac Rebalance® (liposome), and VEVYE (semifluorinated alkanes; water-free technology). However, the approved technologies provide a single solution, either eye comfort (e.g. Artelac Rebalance® contains sodium hyaluronate and vitamin B12 act as a lubricant, non-medicated) or pain and inflammation reduction (e.g. Cequa® contains cyclosporine, medicated).
Each class of nanoparticle exhibits unique physicochemical properties that confer specific advantages for drug delivery, particularly in overcoming the diverse barriers associated with ocular administration. However, it is challenging to designate a single nanoparticle system as universally superior for navigating all obstacles in ocular drug delivery. For example, transparency, low viscosity and thermodynamic stability are the features of an emulsion system that has the potential to incorporate both lipophilic and hydrophilic drugs because of the oil and water balance within the system. Furthermore, the ocular surface penetration-enhancing properties of the microemulsion makes it suitable for most routes of administration including ocular delivery.20 However, the drug is directly exposed to the external environment, and hence, prone to being affected by harsh environmental conditions (low pH, enzymes, etc.). In contrast, drug-specific properties determine whether it is encapsulated in the outer lipid layer or inner aqueous core of the liposome, based on its lipophilic or hydrophilic properties, respectively. Furthermore, liposomes show prolonged retention in the eye due to their slow drainage from the cornea as compared to the free drug.21
A hybrid nanoparticulate system, integrating two or more types of nanoparticles, combines the beneficial properties of each individual system, enhancing therapeutic efficacy and overcoming the limitations of using a single nanoparticle, Table 1.
| Hybrid system type | Role of nanoparticles | Model drug | Composition | Target disease | Ref. |
|---|---|---|---|---|---|
| Hydrogel/nanostructured lipid carrier | • NLC – corneal penetration and sustained drug release | Quercetin/baicalin | • NLC – compritol, Miglyol 812N, baicalin, cremophor EL and soy lecithin | Ocular diseases | 22 and 23 |
| • Hydrogel – prolongs corneal retention time and lowers eye irritation with pH and thermosensitive behaviour | • Hydrogel – carboxymethyl chitosan and poloxamer 407 crosslinked by genipin | ||||
| Liposomes–in situ gel | • Liposome – high elasticity to enhance ocular permeation | Itraconazole | • Liposome – SPC, Chol, tween 80/PL188 | Ocular fungal infection | 24 |
| • In situ gel – ocular adhesion | • In situ gel – chitosan or hyaluronic acid or a combination of both | ||||
| Liposome/HPMC | • Liposome – osmo-protectant | Acetazolamide | • Liposome – PC, Chol, and Vit. E dispersed in borates, trehalose and erythritol solution | Glaucoma | 25 |
| • HPMC – increases viscosity, leading to increased retention time | • HPMC – hydroxypropyl methylcellulose | ||||
| Gelatin nanoparticles–HPMC | • Gelatin – increases viscosity and mucoadhesion | Timolol maleate | Gelatin NP and HPMC | Glaucoma | 26 |
| • HPMC – increases viscosity and eye comfort | |||||
| Niosomes/in situ gel | • Niosomes – enhance drug stability | Itraconazole | • Niosome – Span 60, lipoid S100, and cholesterol | Glaucoma and microbial infection | 27 |
| • In situ gel – increases viscosity and mucoadhesion | • In situ gel–chitosan and hyaluronic acid | ||||
| Micelle/hydrogel | • Micelle – enhances drug solubility | Rapamycin | • Micelle – methoxy poly(ethylene glycol)-poly(ε-caprolactone) | Corneal raft rejection | 28 |
| • Hydrogel – mucoadhesive properties and long-term precorneal retention | • Hydrogel – cationic peptide-based hydrogel | ||||
| Dendrimer hydrogel/PLGA nanoparticles | • Hydrogel – increases permeability | Brimonidine and timolol maleate | • Hydrogel–polyamidoamine | Glaucoma | 29 and 30 |
| • Nanoparticles – prolong residence time | • Nanoparticle – poly(lactic-co-glycolic acid (PLGA) | ||||
| Lipid-polymeric nanoparticles | • Nanoparticle – drug stability | Difluprednate | • Nanoparticle – PLGA | Uveitis | 31 |
| • Lipid – enhances permeation | • Lipid – PC and Chol |
This multifunctional approach holds great promise for next-generation drug delivery systems, particularly in addressing the challenges associated with ocular delivery. In the proposed liposome–emulsion twin system, liposomes will encapsulate dexamethasone (DEXA) that has low aqueous solubility and moderate lipophilicity while also protecting it from enzymatic degradation and harsh external conditions. The ability of the proposed twin system to encapsulate both hydrophilic and lipophilic drugs in the aqueous core or lipid bilayer, respectively, offers flexibility for drug loading and liposomes provide prolonged retention time in ocular tissues, thereby improving drug bioavailability.
Blending DEXA-loaded liposomes with an α-linolenic acid (omega-3 fatty acid)-enriched microemulsion offers additional benefits. Omega-3 fatty acids are known for their anti-inflammatory properties, which can work synergistically with the anti-inflammatory effects of DEXA to enhance therapeutic outcomes. Additionally, the microemulsion improves permeability and drug diffusion, facilitating deeper tissue penetration and broader distribution of the liposome and its drug payload across ocular barriers.
Once inside the cell, the system will release the drug in a controlled manner, alleviating the pain and inflammation associated with DED. Ultimately, the system will break down into its constituent components, such as fatty acids and non-polar lipids, helping to restore the tear film and improve eye comfort.
Experimental
Materials
Dexamethasone (DEXA) base (CAS No. 50-02-2) was purchased from CymitQuímica (Barcelona, Spain). Labrafac™ lipophile WL1349 was generously provided by Gattefossé (Lyon, France). Phospholipon 90 G was purchased from Lipoid AG (Germany). Benzalkonium chloride (BAK) (CAS No. 63449-41-2), coumarin 6 (C6) (CAS No. 38215-36-0), paraformaldehyde (PFA) solution, 4% (UNSPSC Code 41116124), acetonitrile (HPLC grade), acetone (HPLC grade), ethanol (HPLC grade) and dimethyl sulfoxide (DMSO/sterile) (CAS No. 67-68-5) were obtained from Sigma-Aldrich (Ireland). Lipopolysaccharide (LPS) [NP_004130.2 (I465F] from E. coli was purchased from Sigma-Aldrich (UK). Cholesterol (CAS No. 57-88-5), ethylenediaminetetraacetic acid (EDTA), disodium salt dihydrate (CAS No. 6381-92-6), tween 80 (T80) (CAS No. 9005-65-6), and α-linolenic acid (ALA) (CAS No. 463-40-1) were purchased from Fisher Scientific (Ireland). Polyinosinic:polycytidylic acid (Poly I:C) (CAS No. 42424-50-0), Pluronic F-127 (CAS No. 9003-11-6), and polyethylene glycol 400 (PEG 400) (CAS No. 25322-68-3) were purchased from Merckmillipore (Ireland). Hydroxypropyl methylcellulose (HPMC) (Hypromellose/METHOCEL™) (CAS No. 9004-65-3) was obtained from Colorcon (UK). Porcine eyes were obtained from Dawn Meats, Co. Waterford, Ireland. Primary human corneal epithelial cells (PCS-700-010) were obtained from LGC Limited (Middlesex, UK), while immortalized (secondary) human corneal epithelial cells (P10871-IM) were purchased from Innoprot (Innovative Technologies, Derio, Spain). Primary cells were cultured in corneal epithelial cell basal media (PCS-700-030) supplemented with a cell growth kit (PCS-700-040), antibiotics (ATCC PCS-999-002) and Phenol Red (ATCC PCS-999-001). Secondary cells were cultured in IM-ocular epithelial cell medium (P60189).Cell culture grade water/USP sterile water for injection (WFI) (product code 25-055-CM) was used for preparing formulations and for washing cells during passages/treatment.
Note: primary human corneal epithelial cells (P-HCECs) were used for a maximum of 4 passages, while immortalized human corneal epithelial cells (IM-HCECs) were used for a maximum of 10 passages.
Liposome formulation development
Liposomes (LIPs) at the lab scale (10 mL batch size) were developed by the thin film hydration method using soya lecithin (Phospholipon 90 G) and cholesterol as core materials at a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Pluronic F-127 was added at 1% concentration in the developed lab scale batch. All the materials were dissolved in 25 mL of a chloroform:methanol mixture (4:1 ratio) in a 250 mL round bottom flask. A yellowish thin film was obtained after 1 hour under vacuum (rotary evaporator) at 40 °C. The obtained film was rehydrated in water (water for injection, WFI, grade) at 55 °C on an oil bath with magnetic stirring followed by sonication to obtain a nano-sized liposomal dispersion. Drug loaded LIPs (LIP-D) were prepared by the same method as stated above except that the drug was dissolved in the organic solvent mixture along with the lipids.
:1. Pluronic F-127 was added at 1% concentration in the developed lab scale batch. All the materials were dissolved in 25 mL of a chloroform:methanol mixture (4:1 ratio) in a 250 mL round bottom flask. A yellowish thin film was obtained after 1 hour under vacuum (rotary evaporator) at 40 °C. The obtained film was rehydrated in water (water for injection, WFI, grade) at 55 °C on an oil bath with magnetic stirring followed by sonication to obtain a nano-sized liposomal dispersion. Drug loaded LIPs (LIP-D) were prepared by the same method as stated above except that the drug was dissolved in the organic solvent mixture along with the lipids.
The obtained drug loaded formulations were then centrifuged at low speed (3000 rpm for 15 minutes) to remove free DEXA. The drug loaded liposome-containing supernatant was collected and stored at 4 °C until further analysis.
The details of formulation variables optimized during development are given in Table S1 to Table S3 (ESI file†).
Microemulsion (ME) preparation
Mixtures of surfactant (PEG 400) and co-surfactant (Hypromellose) (Smix) were prepared in the weight ratios of 1:1, 2:1, 3:1, 4:1, and 1:2 and used as stock solutions for mixing with oil in different proportions. Microemulsions were prepared by the conventional water titration method. A ternary phase diagram was constructed at different Smix ratios (Fig. S1 and the ESI file†). The Smix (1:1, w/w) was mixed with oil (ALA) in different weight ratios (Oil:Smix), i.e., 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, and 1:3. The composition of the various microemulsions prepared is given in Table S4 to Table S10 in the ESI.†
In brief, ALA, PEG 400 and T80 were mixed at 1:4.5:4.5 v/v in 5 mL of WFI under continues magnetic stirring at 40 °C on a hot-plate for 4 h to obtain a final transparent nano-sized microemulsion.
Preparation and characterization of liposome–microemulsion (LME) blend
Liposomes and microemulsion were blended in the composition given below to achieve 1 mg mL−1 conc. of DEXA in the final LME formulation:
| Microemulsion (ME) volume (mL) = 0.105 × target volume of LME (mL) |
EDTA and hypromellose were also added in the formulations as follows:
| EDTA (g) = 0.0001 × target volume of LME (mL) |
| Hypromellose (Methocel®) (g) = 0.00025 × target volume of LME (mL) |
Note: A volume up to the target volume was adjusted with WFI. The pH was adjusted to 7.0, if required. The obtained formulation was sterilized by vacuum filtration using a 0.2 μm filter through Nalgene™ Rapid-Flow™ Sterile Disposable Filter Units.
The pilot scale batch (1 L) was prepared by the same method described above except that the obtained multilamellar vesicles (MLVs) during rehydration were passed through a high pressure homogenizer (HPH) at 20,0000 psi for 6 minutes (3 cycles) in continuous mode to obtain nano-sized homogeneous liposomal formulation. Stability data are given in the ESI file (Fig. S2†).
Coumarin-6 loaded liposomes were also prepared that were further processed as described above to obtain coumarin-6 loaded LME (LME-C6) (100 μg mL−1).
Dynamic light scattering (DLS, Zetasizer Nano ZS90) was utilized to determine particle size, charge and dispersion. DEXA concentration in liposome was determined by the HPLC method32 following the USP43-NF38 monograph method for DEXA assay and organic impurity profiling. In brief, the liposome was lysed by adding an appropriate amount of methanol and the samples were injected into HPLC for drug quantification using the formula given below:


Drug release study
Drug release was carried out by dialysing a formulation sample containing 1.5 mg of drug against 20 mL release media (7.4 pH PBS supplemented with 0.5% v/v tween 80). A dialysis membrane of 14 kDa (UNSPSC Code: 41123100, Sigma Aldrich, Ireland) was used. About 0.2 mL sample was withdrawn each time and replaced with the same volume of fresh media. An aliquot of 100 μL from the collected sample was diluted with 400 μL of methanol and drug concentration was measured using HPLC as stated in the USP43-NF38 drug (dexamethasone) monograph.32Zero order, 1st order, Higuchi and Korsmeyer–Peppas (KMP) kinetic models were applied to the dissolution profile to observe the drug release behaviour. The R2, adjusted R2 (AdjR2), sum of squares (SS), and akaike information criteria (AIC) were computed to find the ‘best fit’ model based on the highest R2 and the lowest AIC values. The drug release mechanism was explained based on the ‘n’ value of the Krosmeyer–Peppas model (applied to the first 60% of the drug release profile).33
Stability study
An intermediate stability study was conducted at 2–8 °C and 25 °C ± 2 °C/60% ± 5 relative humidity (RH) as per the International Council for Harmonization (ICH) guidelines,34 maintained in a commercial facility of Q1 Scientific Ltd. The samples were tested for particle size and drug entrapment at sampling time points of 0, 1 week, 2 weeks, 1 month, 2 months, 3 months and 6 months by the method described in the “Preparation and characterization of LME blend” section, above.Cell toxicity study
The cytotoxicity study of the liposome (LIP) and LME (with and without drug) was carried out on primary (P) and secondary (immortalized, IM) human corneal epithelial cells (HCECs). The cells with an initial density of 10,000 per well (P-HCECs) and 7000 per well (IM-HCECs) were seeded in a 96-well plate and then cultured for 24 h in their respective corneal epithelial culture medium, as detail in the Material section. The cells were then treated with DEXA (free and encapsulated) in a concentration range of 5–100 μg mL−1. The treated cells were incubated in a humidified environment with 5% CO2 at 37 °C. After 24 h, the cell viability was measured using acid phosphatase assay as described in ref.35. The absorbance (O. D.) was recorded with a microplate reader. The cell viability was calculated by using the following equation:
Note: the free drug was dissolved in DMSO. The final conc. of DMSO in the working samples was below 0.4%.
Cellular internalisation
In vitro cellular uptake of coumarin 6-labelled LME (LME-C6), was examined using an inverted microscope (Olympus CKX 53, UK) equipped with cellSens imaging software. The primary and secondary HCEC cells were seeded in a 24-well plate and incubated with 5% CO2 at 37 °C. After confirmation of 70–80% confluency, the cell medium was replaced with low serum (1% FBS) and left in an incubator overnight. Next morning, the low serum containing cell medium was aspirated off and the cells were washed with PBS twice and then the cells were exposed to LME-C6 (5 μg mL−1) containing growth medium for different time intervals (5 minutes, 15 minutes, 30 minutes, 1 h, and 2 h). After the stated time intervals, the LME-C6 treated serum free medium was removed, and the cells were washed twice with cold PBS and fixed with 4% PFA for 15 minutes. Later, the cells were treated with DAPI stain (4′,6-diamidino-2-phenylindole) (1 μg mL−1) to label the cell nucleus. Next, the cells were washed thrice with cold PBS again and then immediately observed under a microscope and photographed. Coumarin-6 suspension in 0.2% tween 80 was used as a control.36In vitro anti-inflammatory biomarker study
The primary and secondary HCECs were seeded in a 24-well plate at a density of 5× the number of cells used in the 96-well plate (section 2.7). Before starting the treatment, the cells were incubated with low FBS (1%) growth medium to bring them to the same metabolic state. Next, the cells were exposed to lipopolysaccharide (LPS) and polyinosinic:polycytidylic acid (Poly I:C) at concentrations of 10 μg mL−1 and 5 μg mL−1, respectively (non-toxic concentration (cell viability > 80%) of LPS and Poly I:C, Fig. S3†) (ESI file†). After 6 h, the cell media were collected (as a positive control) and stored at −20 °C until further analysis within 48 h. Then, the cells were treated with 10 μM of drug/drug loaded LME containing media for 16 h. Later, the cell supernatant was centrifuged at 5000 rpm for 20 min and stored at −20 °C until further analysis within 48 h. Untreated cells were taken as a negative control group. The expression of IL-6, IL-8, and TNF-α cytokines was tested using an ELISA kit as described in the manufacturer's protocol (https://www.assaygenie.com/).Ex vivo corneal permeation
Ex vivo permeation of the LME in the deeper layer of cornea was visualised by fluorescence microscopy.37 Porcine corneas were obtained from an abattoir ((Dawn Meats) of a local market (Co. Waterford, Ireland) (https://www.dawnmeats.com/) within one hour of slaughter and preserved in a PBS solution containing 1% (v/v) antibiotic. All experiments were conducted within 8 h of the initial slaughter. In brief, a transverse section of the cornea was incubated with coumarin-6 loaded LME (0.05%, wt/vol). Coumarin-6 suspension in 0.5% tween 80 was used as the control experiment. After 4 h of exposure, the cornea was washed with cold PBS thrice and then treated with DAPI stain (1 μg mL−1). After 20 min, the cornea was washed with cold PBS and visualised under a fluorescence microscope.Whole eye ex vivo permeation
An ex vivo whole eye permeation study was carried out using a previously reported method.37 In brief, a receiver chamber was fixed over whole porcine eyes (Dawn Meats, Co. Waterford, Ireland), ensuring coverage of the cornea, with the help of a cling film and cellophane tape. About 500 μL of the formulation (0.1% wt/v; DEXA) and Maxidex were added in separate apical chambers. After 5 h, the eyes were washed with running water and about 100 μL aqueous humour was aspirated using a needle and syringe. The sample was diluted with an equal volume of acetonitrile and analyzed on HPLC as described in the Preparation and characterization of LME blend section, above.Statistical analysis
Technical and biological replicates were performed in triplicate (n = 3). Data are presented as the mean ± standard deviation (mean ± SD). A multiple t-test was conducted for group-to-group comparisons. DDSolver was utilized to analyze in vitro drug release kinetics. Significant differences were reported as follows: 0.1234 (ns), 0.0322 (*), 0.0021 (**), 0.0002 (***), and <0.0001 (****).Results and discussion
Liposome development, characterization and scale-up
Particle size (PS), zeta potential (ZP), polydispersity index (PDI), and osmolality (OSM) were evaluated as quality attributes during the testing of the developed formulations. Throughout the development stages, there were no significant changes observed in PS, ZP, or PDI. An average PS of 60 nm was consistently maintained with a PDI below 0.2, while the ZP remained neutral, i.e., 0 ± 1, at every stage of the development process (Table 2 and Fig. 2). An average EE of 72%, corresponding to a drug concentration of 1.8 mg mL−1, was observed at approximately 2.7% DL in liposomes (LIPs). The drug concentration in LIPs was adjusted to 1 mg mL−1 in the final LME formulation, as detailed in the Materials and methods section.| PS (nm) | ZP (mV) | PDI | OSM (mosmol per Kg) | |
|---|---|---|---|---|
| LIP-B | 61.70 ± 3.24 | 0.56 ± 0.15 | 0.27 ± 0.07 | 150.00 ± 8.89 |
| LIP-D | 58.81 ± 2.87 | 0.70 ± 0.27 | 0.15 ± 0.04 | 63.67 ± 9.50 |
| Emulsion | 59.00 ± 2.73 | −1.67 ± 0.30 | 0.21 ± 0.04 | 466.33 ± 0.30 |
| LME-B | 62.75 ± 2.49 | 0.60 ± 0.14 | 0.16 ± 0.05 | 181.00 ± 8.54 |
| LME-D | 61.20 ± 3.64 | 0.60 ± 0.14 | 0.25 ± 0.04 | 62.33 ± 7.02 |
 | ||
| Fig. 2 Quality attributes of the formulation: (A) particle size and (B) zeta potential. | ||
The osmolarity of the tear film in a healthy eye is estimated to be 290–310 mosmol per Kg, and it increases in cases of severe dry eye.38 It is evident that the hypo-osmolar eye drops are more effective over iso-osmolar or hypertonic products.39 We observed that the LIPs were hypo-osmolar (50–150 mosmol per Kg), whereas the emulsion was hyper-osmolar (>450 t is mosmol per Kg). Following blending, the resulting formulation (LME) became hypo-osmolar (50–200 mosmol per Kg). In general, the osmolality of the blank LME (LME-B) ranged between 150 and 200 mosmol per Kg, whereas the dexamethasone-loaded LME (LME-D) exhibited osmolality below 100 mosmol per Kg. While we didn't find any studies justifying a change in osmolality after drug loading, we anticipate that some lipid may also sediment during centrifugation to eliminate free drug, resulting in a decrease in solute (vesicular particles) in the LME-D.
The transition of liposome–microemulsion formulations from laboratory-scale development to pilot-scale production presented several challenges, particularly in maintaining particle size distribution, drug entrapment efficiency, and formulation stability. At the laboratory scale, key formulation variables such as lipid-to-drug ratios, surfactant composition, and processing parameters were optimized to achieve a homogeneous nanosized dispersion. However, when scaling up the process, issues like particle aggregation, phase separation, and variations in entrapment efficiency were observed, requiring further optimization.
One of the primary challenges during scale-up was maintaining a consistent particle size distribution. In early laboratory-scale batches, the formation of large aggregates and lumps was observed, which was attributed to the sedimentation of excess lipid components, including cholesterol and F-127. This led to a broad particle size distribution, as indicated by high polydispersity index (PDI) values. To address this issue, high-pressure homogenization was applied, with three homogenization cycles, each lasting six minutes. This process significantly reduced the particle size, resulting in a more homogeneous dispersion with a final size below 100 nm and a PDI of less than 0.5; results are presented later. The homogenization process improved lipid bilayer uniformity and prevented lipid sedimentation, leading to a more stable formulation.
Drug entrapment efficiency was another critical aspect that required optimization. In the initial lab-scale formulations, drug loading was inconsistent, likely due to incomplete solubilization of dexamethasone within the lipid bilayer. After applying high-pressure homogenization, drug entrapment efficiency improved due to better lipid–drug interactions, reduced free drug precipitation, and enhanced thermodynamic stability of the vesicles. These observations are consistent with previous reports indicating that homogenization enhances drug incorporation within lipid vesicles and prevents drug leakage.
Microemulsion stability was also a major concern during scale-up. Phase separation was observed in early formulations, particularly when optimizing the surfactant-to-co-surfactant ratio (Smix) and the oil-to-Smix ratio. Changes in particle size were also noticeable, indicating instability in some compositions. To resolve these issues, a ternary phase diagram was constructed to identify the stable microemulsion region. The final selection was based on achieving a particle size of less than 100 nm with no phase separation over time. The optimized microemulsion showed good stability and reproducibility when scaled up.
After optimizing both liposomal and microemulsion formulations individually, the two were blended to create a single-phase formulation. This process resulted in a final optimized particle size of approximately 60 nm with a PDI of less than 0.5. The final formulation demonstrated excellent stability, with no phase separation or aggregation observed over extended storage periods. The successful combination of liposomal and microemulsion components confirmed that the developed system was stable and reproducible at the pilot scale; the results are presented in the Stability study section. An illustration of the twin nanoparticulate system is given in Fig. 3.
 | ||
| Fig. 3 An illustration of a twin nanoparticle-based ophthalmic solution compared with conventional eye drops. | ||
Drug release study
A rapid drug release from LME was observed in the initial hours, with approximately 40% of the drug released in 1.5 h. However, this release pattern in initial hours (2 h) was significantly slower than that of free dexamethasone (DEXA) (p = 0.0224 @ 1 h and p = 0.0411 @ 1.5 h). Following this initial phase, LME showed a sustained release, reaching a maximum of 84.26 ± 7.02% at 12 hours. In contrast, more than 50% of the free drug was released within the first 2 hours, with the remaining drug continuing to diffuse into the outer medium for up to 12 hours (95.06 ± 5.11%), as shown in Fig. 4. | ||
| Fig. 4 Drug release study of dexamethasone loaded liposome–microemulsion (LME-D) and in 0.5% tween 80 containing phosphate buffer, pH 7.2, compared with free dexamethasone (DEXA). | ||
The best-fitting in vitro kinetic model was selected based on parameter values, including R2, AdjR2, SS, and AIC, as calculated in Table 2. However, drug release from the system depends on several factors, including but not limited to the method used to study drug release (e.g., dissolution apparatus and dialysis membrane), dissolution media, agitation, and sampling method. A summary of in vitro kinetic models followed by various types of nanoparticles is provided in Table 3.40–42
| Release models | Equation | Mechanism | Examples |
|---|---|---|---|
| Zero order | Q = Q0 + K0t | Concentration independent release from the matrix | Transdermal slow release matrix, coated, and osmotic systems |
| 1st order | logQ = logQ0 − Kt 2.303 |
Concentration dependent release from the matrix | Porous materials, e.g., calcium carbonate nanoparticles |
| Higuchi |

|
Release after swelling and erosion/degradation of the matrix | Polymeric NPs, e.g., PLGA NPs and lipid based drug delivery systems, e.g., emulsion and liposomes |
| Hixson-Crowell | Q 1/30/Q1/3t = KHCt | Release from systems where there is a change in the surface area and diameter of particles or tablets | Polymeric NPs e.g. PLGA NPs |
| Krosmeyer–Peppas | Q t /Q0 = Knt | Drug release from swellable and non-swellable materials | Polymeric NPs e.g. hydrogels and solid lipid nanoparticles |
| Baker-Lonsdale |

|
Drug release from spherical matrices | Nano/microspheres |
| Weibull | Qt/Q0 = 1 − e−K(t−T) | Behaviour when drug is accumulated in the solution at time t | Nano/micro composites |
| Gompertz |
Q
t
/Qmax = Exp [−αeβlogt] |
Drugs having good solubility and an intermediate release rate | Scaffolds and nanofibers |
| Hopfenberg |

|
Release from polymers that could be degraded or eroded during drug loss, regardless of their shape or dimensions | Polymeric NPs |
| Gallagher Corrigan |
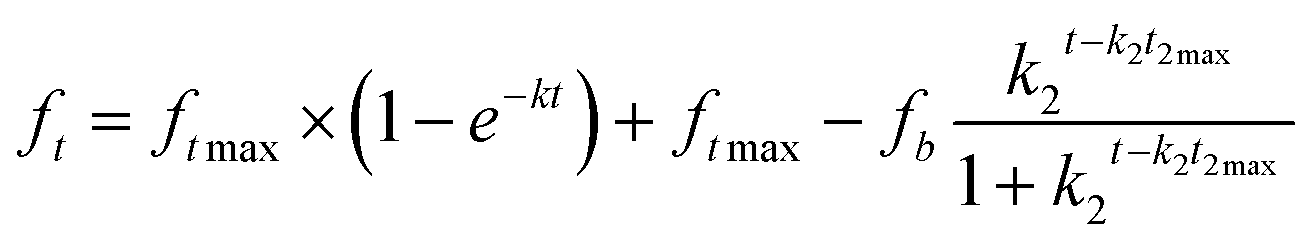
|
Drug diffuses through the interface of the polymer, followed by a second release of the drug entrapped within the polymer structure | Co-polymers/hybrid inorganic–organic system |
It was observed that the drug release from LME followed the KMP model based on R2 and adjR2 > 0.9 and the lowest AIC (12.46). The ‘n’ value confirmed that the drug release pattern was a Non-Fickian diffusion type.33 The drug release under diffusion control is well explained by the KMP model,43Table 4 and our results correlate with previous studies where lecithin-based LIP exhibited the best fit with the same model.44
| Release exponent (n) | Drug transport mechanism | Drug release mechanism |
|---|---|---|
| n = 0.45 | Fickian diffusion | Non swellable matrix diffusion |
| 0.45 < n < 0.89 | Non-Fickian diffusion | For both diffusion and relaxation (erosion) |
| = 0.89 | Case II transport | Zero order release |
| n > 0.89 | Super case II transport | (Relaxation/erosion) |
In vitro kinetic parameters are presented in Table 5.
| Parameter | Zero order | 1st order | Higuchi | KMP | ||||
|---|---|---|---|---|---|---|---|---|
| Free DEXA | LME-D | Free DEXA | LME-D | Free DEXA | LME-D | Free DEXA | LME-D | |
| R 2 | −1.87 | −1.29 | 0.83 | 0.89 | 0.08 | 0.23 | 0.1 | 0.97 |
| AdjR2 | −1.87 | −1.29 | 0.83 | 0.89 | 0.08 | 0.23 | 0.99 | 0.96 |
| SS | 23110.41 | 19447.89 | 1320.58 | 881.71 | 7360.20 | 6483.64 | 8.72 | 24.50 |
| AIC | 112.3932 | 110.47 | 80.18 | 74.96 | 99.66 | 98.16 | 9.18 | 12.47 |
| n | 0.556 | 0.62 | ||||||
Typically, the drug diffuses into the dissolution medium after being released from nanocarriers like LIPs, driven by a concentration gradient under sink conditions.45 We expected a slower and sustained release of DEXA from the LME because of the presence of Pluronic F127 in the carrier system (liposome) that exhibited the slowest dissolution rates and drug release when employed alone or with additives such as methylcellulose 15 cP (MC), and hydroxypropyl methylcellulose 80–120 cP (HPMC).46 On the other hand, ALA, an essential omega-3 fatty acid, plays a crucial role in regulating solubilization dynamics, facilitating membrane permeability, and influencing phase transitions to develop a stable system.47 ALA, an essential omega-3 fatty acid, has gained significant attention in pharmaceutical formulations, particularly for enhancing the solubility, stability, and bioavailability of lipophilic drugs. Given its solubility in surfactants like Tween 80 and its ability to modulate membrane permeability, ALA plays a crucial role in lipid-based drug delivery systems, such as emulsions and self-emulsifying drug delivery systems (SEDDSs).48 One of the key properties of omega-3 fatty acids, including ALA, is their ability to enhance membrane fluidity and permeability. This property can significantly impact drug release and absorption.49
Stability study
To confirm the robustness and reproducibility of the set parameters for product scale up, a pilot scale batch was prepared and stored under pre-defined conditions (2–8 °C and 25 °C ± 2 °C/60% ± 5 relative humidity (RH)).The stability study findings revealed that there were no significant alterations in PS and EE over the 6 months study period under cold (2–8 °C) or ambient (25 °C ± 2 °C) conditions and at 60 ± 5% relative humidity, Fig. 5. All formulations consistently maintained an average PS of 60 nm, with a corresponding 100% EE. It is evident from the literature that lecithin-based liposomes remain stable for at least 3 months in the fridge and at room temperature. The lipid bilayer exhibits different phases depending on the type of lipid. For example, DPPC (dipalmitoylphosphatidylcholine) transitions from the gel phase (Lβ′) to the liquid crystalline phase (Lα′) between 35 and 42 °C. Between 35 and 42 °C, the phospholipid bilayer adopts the Pβ′ or “rippled phase.” The pretransition involves a reorganization of individual lipid molecules within the bilayer. Following the pretransition at 35 °C, numerous conformational changes occur in the lipid molecules, along with alterations in the geometry of the lipid bilayers, resulting in the destabilization of the liposome.50 In this study, we blended the drug-loaded liposome with a microemulsion. Microemulsions, known for their thermodynamic stability51 are anticipated to enhance the overall thermostability of the developed system by altering free energy, surface tension, and interfacial area. It's evident that the lipophilic drug precipitates after leaking from the liposome. Nevertheless, no pellet (free drug) was observed after centrifugation during the stability study. This absence might be attributed to the presence of microemulsion in the surrounding medium, which likely enhanced drug solubilization. It is plausible that the leaked drug remained emulsified within the microemulsion after leakage, thereby improving drug solubility within the system. A zeta potential exceeding +30 mV or falling below −30 mV is deemed a favourable threshold for the stability of colloidal particles, as similarly charged particles repel each other. Despite our system exhibiting a neutral charge, we anticipate that the alteration in viscosity induced by the presence of microemulsion contributed to long-term stability. Enhanced stability may also be attributed to the Brownian movement of particles facilitated by the colloid of liposome vesicles and emulsion droplets. While Brownian motion occurs slowly on a macroscopic scale, it takes place at a significantly faster pace on the nanometer scale.52 Different nanoparticle sizes and nearly neutrally buoyant particle densities are also considered.53
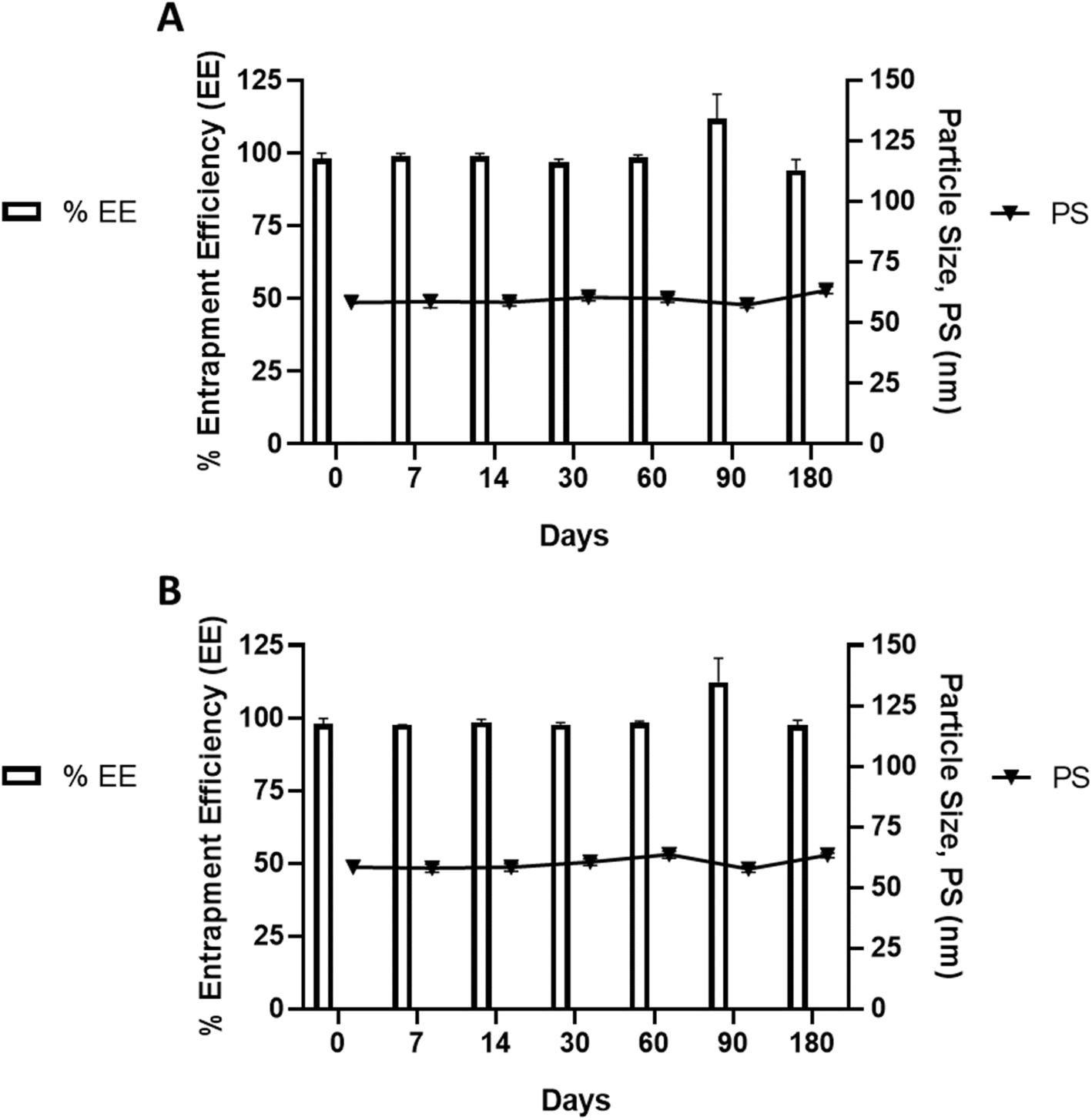 | ||
| Fig. 5 Stability study under controlled temperature and humidity conditions maintained in the commercial facility of Q1 Scientific Ltd. (A) 25 °C ± 2 °C/60% ± 5 relative humidity (RH) (B) 2–8 °C. | ||
Cell toxicity study
Primary cells showed concentration dependent toxicity (Fig. 6). At a concentration of 5 μM DEXA (0.05% DMSO), a cell viability of 83.97 ± 5.25% was observed. However, viability decreased by 27.42% at 100 μM (0.25% DMSO). This decline in cell viability appears to be due to the increase in DMSO concentration. Studies have shown that the acceptable range of DMSO for ocular cells is 0.1% to 1.6%, depending on the cell type.54 We hypothesize that the decrease in cell viability at the higher concentration (100 μM) was due to the elevated DMSO concentration (0.25%), surpassing the tolerable limit. Unfortunately, reducing the DMSO concentration below 0.25% at a drug concentration of 100 μM was impractical due to the drug's solubility limitation in DMSO, which was below 40 mg mL−1. It is noteworthy that the primary cells exhibited a tendency to detach and reattach during regular growth. The product data sheet from the supplier, available online at https://www.atcc.org/products/pcs-700-010, also mentions this characteristic of the cells. We anticipate that this detachment–attachment cycle of cells could contribute to fluctuations in cell viability, particularly during processes such as cell washing and drug treatment, where cells may be lost. No significant change (p > 0.05) in cell viability was observed across various concentrations (ranging from 5 μM to 100 μM) of DEXA-loaded liposomes (LIP-D). Conversely, a significant change in cell viability was evident in cells treated with LME-D. The cell viability decreased from 103.78 ± 2.1% at 5 μM to 56.16 ± 1.68% at 50 μM of LME-D. Subsequently, the cell viability dropped to 18.75 ± 5.94% at 100 μM of LME-D. This decrease in cell viability was higher than that caused by the free drug (DEXA). | ||
| Fig. 6 Cell viability after treatment with DEXA free, DEXA loaded liposome (LIP-D), DEXA loaded LME (LME-D), and blank LME (LME-B). (A) Primary human corneal epithelial cells (P-HCECs) and (B) immortalized human corneal epithelial cells (IM-HCECs). | ||
We conducted a root cause analysis to identify the reason for the decrease in cell viability. Initial findings confirmed that the liposome itself (with or without the drug) was non-toxic. However, the emulsion was found to be highly toxic at a dilution equivalent to 100 μM of LME-D. Next, we tested the components of the emulsion and identified tween 80 as the primary cause for this decrease. To validate our results, we tested tween 80 alone at different concentrations and observed a concentration-dependent decrease in cell viability. Subsequently, we developed a tween 80-free emulsion and a tween 80-free LME, and both formulations were found to be non-toxic. Therefore, tween 80 was confirmed as the cause of the decrease in cell viability (Fig. S4A, S4B and the ESI file†).
Cell toxicity studies were also conducted on IM-HCECs. No significant changes in cell viability were observed with free DEXA and LIP-D at concentrations ranging from 5–100 μM. In contrast, LME-D showed a gradual decrease in cell viability 100.83 ± 3.37% at 5 μM to 49.53 ± 8.9% at 100 μM. These findings were consistent with those observed in primary cell lines mentioned earlier, and an increase in the amount of tween 80 at higher concentration was identified as a potential cause of cell toxicity.
We also prepared drug-loaded liquid microemulsions (LMEs) with and without the preservative benzalkonium chloride (BAK). A notable gradual decrease in cell viability was observed with increasing amounts of BAK, which correlated with higher drug concentrations, Fig. S4C, (ESI file†). Finally, we decided to use preservative free formulations in all further studies.
Cellular internalization
LME loaded with coumarin 6 (C6) was tested on cells to examine uptake. In comparison to free C6, the C6 loaded LME (LME-C6) showed green fluorescence within 5 min on primary cell lines, which was obviously due to fast cell membrane penetration of the developed drug carrier system (LME) (Fig. 7). The fluorescence intensity was persistent during the 2-hour study period, which was evidence of the localization of the nanocarrier within the cells. The cells treated with free C6 started to fluoresce green after 30 minutes, indicating slow penetration of free C6 without a carrier. Moreover, the green fluorescence intensity was apparently higher in primary cells as compared to immortalized cells. The difference in fluorescence intensity of coumarin-6 between primary and immortalized cells likely stems from the distinct endocytic pathways and cellular characteristics of each cell type. Primary cells, retaining natural receptor profiles and active uptake mechanisms, are generally more efficient in nanoparticle internalization compared to immortalized cells, which may exhibit altered or reduced endocytosis due to changes in the membrane structure, receptor expression, and metabolic state. These differences in cellular trafficking contribute to variations in fluorescence intensity observed in different cell types.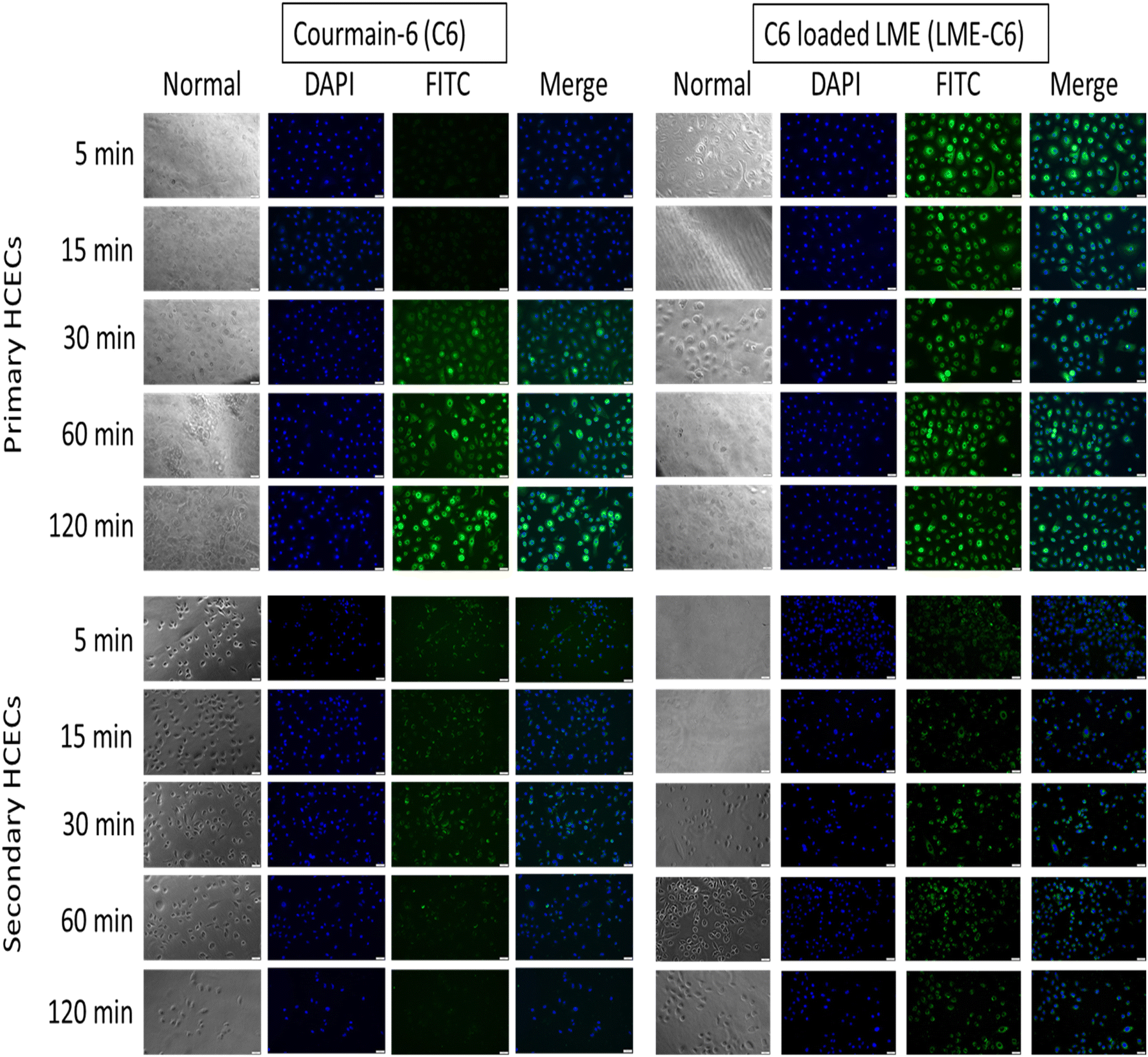 | ||
| Fig. 7 Fluorescence micrographs of cells after treatment with coumarin 6 (C6), a fluorescence dye. | ||
In vitro anti-inflammatory biomarker study
LPS was used to stimulate an inflammatory response in corneal epithelial cells, which were then treated with DEXA in different formulations (Fig. 8). Secreted cytokines interleukin (IL)-6, IL-8, and tumour necrosis factor (TNF)-α were measured in the drug-treated cell media. The LPS-stimulated P-HCECs showed a 72%, 40%, and 45% reduction in IL-6, IL-8, and TNF-α expression, respectively, after LME-D treatment compared to the control (inflamed cells). An average reduction of 17%, 17%, and 26% was also observed in IL-6, IL-8, and TNF-α expression, respectively, after blank LME (without drug) treatment. However, DEXA (free drug) showed a 24% reduction in IL-8 expression only, with no significant change in IL-6 and TNF-α expression. Similarly, the LPS-stimulated IM-HCECs showed an average reduction of 69% and 11% in IL-6 and IL-8 expression, respectively, after LME-D treatment. The blank LME showed a reduction in IL-6 expression only (32%), while the free drug showed a 39% reduction in IL-6 expression only. However, TNF-α was not detected in any of the treated samples. Most probably, the sampling time point (16 h) was not suitable for the detection of the optimum concentration of TNF-α in those samples, as indicated by the very low expression of TNF-α (7.5%) in the cell media 16 h after the removal of LPS-containing media. | ||
| Fig. 8 Effect of different treatments on inflammatory marker expression after stimulation with lipopolysaccharide (LPS)/polyinosinic:polycytidylic acid (P:IC). Cells were treated with LPS)/P:IC for 6 hours, followed by a 16-hour treatment with either dexamethasone (DEXA), DEXA-loaded lipid-based microemulsion (LME-D), or blank LME (LME-B). Expression levels of IL-6, IL-8, and TNF-α were assessed and normalized to the LPS/P:IC-treated group (6 h treatment). A comparison was also made with the control group; initially LPS/P:IC stimulation for 6 hours but no treatment for the following 16 hours (after 16 h). Statistical significance was determined by comparing treatment groups with the inflamed control. | ||
In contrast, the P:IC-stimulated P-HCECs showed elevated expression of all the tested inflammatory biomarkers (IL-6, IL-8, and TNF-α) after treatment with DEXA, LME-D, and LME-B compared to the control (inflamed cells). Similarly, the IL-6 expression was higher than that of the control (inflamed cells) in the P:IC inflamed IM-HCEC media. However, an average reduction of 14%, 21%, and 17% was observed in IL-8 expression after treatment with DEXA, LME-D, and blank LME (LME-B), respectively. However, TNF-α was not detected in inflamed IM-HCEC media after any treatment.
Overall, the expression of IL-6 and IL-8 was higher at 16 h after the aspiration of P:IC-containing media compared to the control (before aspiration of P:IC containing media).
It was inferred from the overall results that P:IC at 5 μg mL−1 concentration was more ‘stressful’ for the cells than the LPS at 10 μg mL−1 concentration, hence producing higher concentration of cytokines in the cell media after 6 h of exposure.
The difference in response to both LPS and P:IC might be due to differences in their target receptors. LPS primarily binds to Toll-like receptor 4 (TLR4), while P:IC binds to Toll-like receptor 3 (TLR3).55 Variations in immune responses were observed in animals following LPS and P:IC treatment, with factors such as timing, administration route, and species playing a role.55,56 A difference in immune response was also observed in human immune cells. IP-10 production from DCs was detected after P:IC treatment at levels similar to those in LPS-stimulated cells. However, unlike LPS, poly I:C did not induce TNFα or IL-6, nor did it stimulate IL-8 production in human DCs.57
Another reason for the reduced response to drug treatment in P:IC treated cells might be that the cells were under greater stress after exposure of P:IC; hence, they continued to produce cytokines even after aspiration of the P:IC containing media, as evidenced by higher cytokine expression at 16 h after inflammation, Fig. 8. Thus, the drug concentration (10 μg mL−1) was not enough to neutralize/reduce cytokine expression in P:IC treated cells compared to LPS treated cells.
A study showed that LPS stimulation led to the rapid production of TNF, while IFN-β mRNA exhibited a strong but short-lived response. In contrast, P:IC triggered a robust and sustained (>12 hours) IFN-β mRNA and protein response.58 Similarly, in our study, we anticipate that P:IC induced a strong and prolonged inflammatory response that was not suppressed by the free drug or formulations.
In this study, we analysed the inflammation inducing potential of LPS and P:IC on primary and secondary HCECs. This study demonstrated that the LPS and P:IC were non-toxic at 10 μg mL−1 and 5 μg mL−1 for both P-HCEC and IM-HCEC cells, respectively, Fig. S3 (ESI file†). However, it was inferred from the presented results that P:IC at 5 μg mL−1 was more stressful for the cells compared to LPS at 10 μg mL−1.
Ex vivo corneal permeation
Fig. 9 shows transverse corneal sections with the upper corneal layer stained with DAPI. In the control experiment no green fluorescence was observed in the lower corneal layer; however intense coumarin-6 green fluorescence was observed in the corneas treated with coumarin-6 loaded LME. Coumarin-6 loaded LME could permeate through the deeper corneal layer with the surface layer clearly marked by DAPI stain. | ||
| Fig. 9 Fluorescence micrographs of the transverse section of the cornea treated with (A) coumarin-6, C6 and (B) C6 loaded LME. | ||
Several researchers have utilized liposomes in ocular therapy to minimize the drug loss associated with traditional eye drops due to rapid turnover of the tears and fast blinking of the eye after the instillation of eye drops.59,60 Our study results were correlated with a published work that suggested that fusogenic liposomes have intrinsic ability to efficiently and rapidly internalize into corneal tissue.61
In this study, we combined drug-loaded liposomes with a microemulsion to create the final formulation with lipid emulsion-like properties (referred to as LME). Lipid emulsions serve as a type of delivery system utilizing lipid/fat as an oil phase stabilized by a surfactant and a co-surfactant. These emulsions have been utilized to improve the ocular bioavailability of drugs by enhancing tissue permeability and prolonging the retention time of the formulation.62 However, the distribution of the drug typically occurs in either the oil or water phase, depending on the drug's physicochemical properties. In other words, the drug is exposed to the outer environment in this type of drug delivery system. In our product design, we encapsulated the drug within the lipid layer in the form of liposomes, which were then blended with the emulsion, aiming for fast penetration and enhanced drug stability (minimizing drug leakage and protecting against enzyme degradation).
The study results demonstrated that the fusogenic nature of the developed nanoparticle (liposome) and prolonged residency due to the presence of microemulsion enabled fast and deep penetration of the LME through the corneal tissue, as evidenced by green fluorescence.
Whole eye ex vivo permeation
A higher permeation of DEXA was observed in the aqueous humour of porcine eyes treated with NPs formulation (0.1% w/v) (135 μg mL−1) as compared to Maxidex (0.1% w/v) (85 μg mL−1) treated group, Fig. 10. Under physiological pH conditions, the corneal surface acquires a negative charge owing to acidic groups like sialic acid residues present on the epithelium's apical surface. Consequently, drug particles with positive charge exhibit a fast penetration rate than the negatively charge or neutral particles.63 However, Lee and Carson's study demonstrated that neutral liposomes showed 100 times higher and sustained release of inulin as compared to positively charged liposomes.64 A significant increase in blinking was also noticed after the instillation of positively charged liposomes as compared to neutral liposomes.65 Although positively charged liposomes were considered safe, but a mild and non-specific type of ocular inflammation was also noticed during the treatment and surface charge was considered the sole cause for this.66 Considering the pros and cons of positively charged particles, we developed a twin model product design that takes advantage of both nanoparticles (liposomes and emulsions), resulting in an overall neutral charge on the surface. | ||
| Fig. 10 Whole eye ex vivo permeation study from porcine cornea. | ||
Conclusion
Our project aims were to develop a nanosized formulation that would have fast penetration power (within 2–5 minutes) and strong mucoadhesive properties to maximize drug localization within the eye, leading to enhanced clinical benefits and the potential for increased patient compliance. Visudyne® and Cyclokat® represent two distinct classes of nanoformulations approved by the FDA: liposomal and emulsion-based, respectively.67 Liposomes are acknowledged for their targeted therapeutic capabilities, prolonged circulation time and ability to enhance the stability of the encapsulated drug. Conversely, emulsions are renowned for their rapid penetration through skin or tissue barriers. Our innovative liposome-microemulsion (LME) system combines the advantageous characteristics of both liposomes and emulsions. By integrating the rapid penetration of emulsions with the prolonged residency of liposomes, our LME system offers dual benefits, resulting in enhanced therapeutic outcomes for dry eye disease. Quality attribute testing of the developed systems has confirmed a nano drug dispersion with an average size of 60 nm, maintaining stability for six months. Cell-based studies have indicated the system's non-toxic nature at lower concentrations and its rapid cell penetration capability, with uptake occurring within 5 minutes. Tissue-based research has showcased robust penetration and adhesion to deep tissues, as evidenced by higher drug concentrations in the aqueous humour compared to the marketed brand (Maxidex) after 5 h of LME-D treatment, indicating prolonged residency within the eye. This comparative ex vivo study suggests the superiority of the developed system over conventional drug suspensions (Maxidex) in terms of targetability, delivering more drug to the target site. The LPS-treated DED cell model further validated the improved therapeutic benefits of the developed system compared to simple drug suspensions. The proposed nanotechnology offers a safe therapeutic option that enhances patient compliance by reducing dose frequency, thanks to its prolonged residency properties.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. S.: conceptualization, methodology, investigation, data curation, writing – original draft, visualization. G. B.: formal analysis, software, validation, writing – review & editing. S. R.: methodology, resources, investigation, writing – review & editing. N. R.: resources, supervision, validation, writing – review & editing. P. M.: project administration, funding acquisition, supervision, writing – review & editing. O. D.: investigation, visualization, writing – review & editing. A. L. R.: validation, supervision, writing – review & editing. J. L.: conceptualization, resources, writing – review & editing. L. F.: conceptualization, project administration, supervision, funding acquisition, writing – review & editing.Conflicts of interest
Dr Laurence Fitzhenry is the lead inventor of a technology presented in a patent titled “MICROEMULSION FOR OPHTHALMIC DRUG DELIVERY” (Patent Number: US 2024/0307306 A1, Published on 19-09-2024), which is directly related to the technology and methods discussed in this study. The patent is owned by South East Technological University (SETU), (formally called the Waterford Institute of Technology) Waterford, Ireland.Acknowledgements
This project was funded by the Enterprise Ireland Commercialization Fund (CF-2020-1511 A).References
- F. Stapleton, F. G. Velez, C. Lau and J. S. Wolffsohn, Ocul. Surf., 2024, 31, 11–20 CrossRef PubMed.
- Fortune Business Insight. Source: https://www.fortunebusinessinsights.com/dry-eye-syndrome-market-102413. Last updated on Sep. 2024.
- M. Uchino and D. A. Schaumberg, Curr. Ophthalmol. Rep., 2013, 1, 51–57 CrossRef PubMed.
- S. Barabino, BMC Ophthalmol., 2022, 22, 85 CrossRef PubMed.
- A. Jünemann, T. Chorągiewicz, M. Ozimek, P. Grieb and R. Rejdak, Ophthalmol. J., 2016, 1, 29–35 CrossRef.
- M. H. Akhter, I. Ahmad, M. Y. Alshahrani, A. I. Al-Harbi, H. Khalilullah, O. Afzal, A. S. A. Altamimi, S. N. M. Najib Ullah, A. Ojha and S. Karim, Gels, 2022, 8(2), 82 CrossRef CAS PubMed.
- M. Mofidfar, B. Abdi, S. Ahadian, E. Mostafavi, T. A. Desai, F. Abbasi, Y. Sun, E. E. Manche, C. N. Ta and C. W. Flowers, Int. J. Pharm., 2021, 607, 120924 CrossRef CAS PubMed.
- S. Masoudi, Exp. Eye Res., 2022, 220, 109101 CrossRef CAS PubMed.
- I. A. Butovich, Exp. Eye Res., 2013, 117, 4–27 CrossRef CAS PubMed.
- S. H. Brown, C. M. Kunnen, E. Duchoslav, N. K. Dolla, M. J. Kelso, E. B. Papas, P. Lazon de la Jara, M. D. Willcox, S. J. Blanksby and T. W. Mitchell, Invest. Ophthalmol. Visual Sci., 2013, 54, 7417–7424 CrossRef CAS PubMed.
- R. R. Hodges and D. A. Dartt, Exp. Eye Res., 2013, 117, 62–78 CrossRef CAS PubMed.
- T. Loftsson and E. Stefánsson, Acta Ophthalmol., 2022, 100, 7–25 CrossRef CAS PubMed.
- S. Gause, K.-H. Hsu, C. Shafor, P. Dixon, K. C. Powell and A. Chauhan, Adv. Colloid Interface Sci., 2016, 233, 139–154 CrossRef CAS PubMed.
- M. A. Watsky, M. M. Jablonski and H. F. Edelhauser, Curr. Eye Res., 1988, 7, 483–486 CrossRef CAS PubMed.
- M. Löscher, C. Seiz, J. Hurst and S. Schnichels, Pharmaceutics, 2022, 14, 134 CrossRef PubMed.
- N. D. Das and H. Shichi, Exp. Eye Res., 1981, 33, 525–533 CrossRef CAS PubMed.
- M. Mofidfar, B. Abdi, S. Ahadian, E. Mostafavi, T. A. Desai, F. Abbasi, Y. Sun, E. E. Manche, C. N. Ta and C. W. Flowers, Int. J. Pharm., 2021, 607, 120924 CrossRef CAS PubMed.
- S. Ahmed, M. M. Amin and S. Sayed, AAPS PharmSciTech, 2023, 24, 66 CrossRef PubMed.
- L.-C. Liu, Y.-H. Chen and D.-W. Lu, Int. J. Mol. Sci., 2023, 24, 15352 CrossRef CAS PubMed.
- H. Y. Karasulu, Expet Opin. Drug Deliv., 2008, 5, 119–135 CrossRef CAS PubMed.
- R. Agarwal, I. Iezhitsa, P. Agarwal, N. A. Abdul Nasir, N. Razali, R. Alyautdin and N. M. Ismail, Drug Delivery, 2016, 23, 1075–1091 CrossRef CAS PubMed.
- Y. Yu, R. Feng, J. Li, Y. Wang, Y. Song, G. Tan, D. Liu, W. Liu, X. Yang, H. Pan and S. Li, Asian J. Pharm. Sci., 2019, 14, 423–434 Search PubMed.
- Y. Yu, S. Xu, S. Yu, J. Li, G. Tan, S. Li and W. Pan, ACS Biomater. Sci. Eng., 2020, 6, 1543–1552 CrossRef CAS PubMed.
- M. M. Badran, A. Alsubaie, M. M. S. Bekhit, A. H. Alomrani and A. Almomen, Gels, 2025, 11, 19 CrossRef CAS PubMed.
- M. Gómez-Ballesteros, J. J. López-Cano, I. Bravo-Osuna, R. Herrero-Vanrell and I. T. Molina-Martínez, Polymers, 2019, 11, 929 CrossRef PubMed.
- S. Esteban-Pérez, V. Andrés-Guerrero, J. J. López-Cano, I. Molina-Martínez, R. Herrero-Vanrell and I. Bravo-Osuna, Pharmaceutics, 2020, 12, 306 CrossRef PubMed.
- M. M. Badran, A. Alsubaie, M. M. Salem Bekhit, A. H. Alomrani, A. Almomen, M. A. Ibrahim and D. H. Alshora, Saudi Pharm. J., 2024, 32, 102208 CrossRef CAS PubMed.
- X. Xu, Y. Wu, R. Gu, Z. Zhang, X. Liu, Y. Hu, X. Li, D. Lin and Z. Bao, Eur. J. Pharm. Biopharm., 2024, 201, 114351 CrossRef CAS PubMed.
- H. Yang, P. Tyagi, R. S. Kadam, C. A. Holden and U. B. Kompella, ACS Nano, 2012, 6, 7595–7606 CrossRef CAS PubMed.
- J. Wang, B. Li, D. Huang, P. Norat, M. Grannonico, R. C. Cooper, Q. Gui, W. Nam Chow, X. Liu and H. Yang, Chem. Eng. J., 2021, 425, 130498 CrossRef CAS PubMed.
- B. Kaviarasi, N. Rajana, Y. S. Pooja, A. N. Rajalakshmi, S. B. Singh and N. K. Mehra, Int. J. Pharm., 2023, 640, 123006 CrossRef CAS PubMed.
- S. Kwende, Analytix Reporter. Downloaded from: https://www.sepscience.com/analytix-dexamethasone-an-hplc-assay-and-impurity-profiling-following-the-usp/, 2021, 19–20.
- R. Kulsoom, M. Sarfraz, A. Afzal, M. Farooq, S. Adnan, M. U. Ashraf and S. A. Khan, Polym. Bull. (Berl.), 2023, 80, 6965–6988 CrossRef CAS PubMed.
- A. Rignall, in ICH Quality Guidelines, 2017, DOI:10.1002/9781118971147.ch1, pp. 3–44.
- T.-T. Yang, P. Sinai and S. R. Kain, Anal. Biochem., 1996, 241, 103–108 CrossRef CAS PubMed.
- X. Miao, Y. Li, I. Wyman, S. M. Y. Lee, D. H. Macartney, Y. Zheng and R. Wang, MedChemComm, 2015, 6, 1370–1374 RSC.
- S. K. Gautam Behl, N. O'reilly, O. O'donovan, P. Mcloughlin, D. Kent and L. Fitzhenry, US Pat. Office, 2024/0307306 A1, Waterford Institute of Technology, Ireland, Assignee, 2024, pp. 1–20 Search PubMed.
- C. G. Wilson, in Specialised Pharmaceutical Formulation: The Science And Technology Of Dosage Forms, ed. G. D. Tovey, The Royal Society of Chemistry, 2022, 10.1039/9781839165603-00001.
- R. M. Dutescu, C. Panfil and N. Schrage, Cornea, 2015, 34, 560–566 CrossRef PubMed.
- P. Trucillo, Processes, 2022, 10, 1094 CrossRef CAS.
- A. Jain and S. K. Jain, Chem. Phys. Lipids, 2016, 201, 28–40 CrossRef CAS PubMed.
- Strategies To Modify The Drug Release From Pharmaceutical Systems, ed. M. L. Bruschi, Woodhead Publishing, 2015, DOI:10.1016/B978-0-08-100092-2.00005-9, pp. 63–86.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- M. T. Vu, N. T. T. Le, T. L.-B. Pham, N. H. Nguyen and D. H. Nguyen, J. Nanomater., 2020, 2020, 8896455 Search PubMed.
- G.-H. Son, B.-J. Lee and C.-W. Cho, J. Pharm. Invest., 2017, 47, 287–296 CrossRef CAS.
- S. D. Desai and J. Blanchard, J. Pharm. Sci., 1998, 87, 226–230 CrossRef CAS PubMed.
- M. Singh, J. Kanoujia, P. Parashar, M. Arya, C. B. Tripathi, V. R. Sinha, S. K. Saraf and S. A. Saraf, Drug Delivery Transl. Res., 2018, 8, 204–225 CrossRef CAS PubMed.
- U. Butt, A. ElShaer, L. A. S. Snyder, A. A. Al-Kinani, A. Le Gresley and R. G. Alany, Nanomaterials, 2018, 8, 51 CrossRef PubMed.
- A. Kar, P. Ghosh, P. Patra, D. S. Chini, A. K. Nath, J. K. Saha and B. Chandra Patra, Clin. Nutr. Open Sci., 2023, 52, 72–86 CrossRef.
- N. T. T. Le, V. D. Cao, T. N. Q. Nguyen, T. T. H. Le, T. T. Tran and T. T. Hoang Thi, Int. J. Mol. Sci., 2019, 20, 4706 CrossRef CAS PubMed.
- E. Ruckenstein, Chem. Phys. Lett., 1978, 57, 517–521 CrossRef CAS.
- O. Gustafsson, S. Gustafsson, L. Manukyan and A. Mihranyan, Membranes, 2018, 8, 90 CrossRef PubMed.
- B. Uma, T. N. Swaminathan, R. Radhakrishnan, D. M. Eckmann and P. S. Ayyaswamy, Phys. Fluids, 2011, 23, 73602–7360215 CrossRef CAS PubMed.
- C. Zhao, B. Lan, J. Hou and L. Cheng, Chin. J. Exp. Ophthalmol., 2015, 33, 216–220 CAS.
- M. Bao, N. Hofsink and T. Plösch, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2021, 322, R99–R111 CrossRef PubMed.
- J. Q. Tran, M. O. Muench, B. Gaillard, O. Darst, M. M. Tomayko and R. P. Jackman, Front. Immunol., 2023, 14, 1281130 CrossRef CAS PubMed.
- A. M. Lundberg, S. K. Drexler, C. Monaco, L. M. Williams, S. M. Sacre, M. Feldmann and B. M. Foxwell, Blood, 2007, 110, 3245–3252 CrossRef CAS PubMed.
- T. Reimer, M. Brcic, M. Schweizer and T. W. Jungi, J. Leukocyte Biol., 2008, 83, 1249–1257 CrossRef CAS PubMed.
- A. Bhattacharjee, P. J. Das, P. Adhikari, D. Marbaniang, P. Pal, S. Ray and B. Mazumder, Eur. J. Ophthalmol., 2018, 29, 113–126 CrossRef PubMed.
- S. Ebrahim, G. A. Peyman and P. J. Lee, Surv. Ophthalmol., 2005, 50, 167–182 CrossRef PubMed.
- Y. T. M, S. S. Tellakula, S. V. Suryavanshi, S. K. G, S. C. Vasudev and S. H. Ranganath, Surv. Ophthalmol., 2023, 5, 6410–6422 Search PubMed.
- R. Tiwari, V. Pandey, S. Asati, V. Soni and D. Jain, Egypt. j. basic appl. sci., 2018, 5, 121–129 Search PubMed.
- A. L. Onugwu, C. S. Nwagwu, O. S. Onugwu, A. C. Echezona, C. P. Agbo, S. A. Ihim, P. Emeh, P. O. Nnamani, A. A. Attama and V. V. Khutoryanskiy, J. Controlled Release, 2023, 354, 465–488 CrossRef CAS PubMed.
- V. H. Lee and L. W. Carson, J. Ocul. Pharmacol., 1986, 2, 353–364 CrossRef CAS PubMed.
- K. Taniguchi, Y. Yamamoto, K. Itakura, H. Miichi and S. Hayashi, J. Pharmacol., 1988, 11, 607–611 CAS.
- N. Mosallaei, T. Banaee, M. Farzadnia, E. Abedini, H. Ashraf and B. Malaekeh-Nikouei, Curr. Eye Res., 2012, 37, 453–456 CrossRef CAS PubMed.
- S. Li, L. Chen and Y. Fu, J. Nanobiotechnol., 2023, 21, 232 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na01086h |
| ‡ Muhammad Sarfraz and Goutam Behl are joint first-authors of this work. |
| This journal is © The Royal Society of Chemistry 2025 |