DOI:
10.1039/D4NA00818A
(Paper)
Nanoscale Adv., 2025,
7, 1048-1060
Platinum nanoparticles wrapped in carbon-dot-films as oxygen reduction reaction catalysts prepared by solution plasma sputtering†
Received
1st October 2024
, Accepted 13th December 2024
First published on 24th December 2024
Abstract
Fuel cells have become increasingly important in recent years because of their high energy efficiency and low environmental impact. However, key challenges remain in the widespread adoption of fuel-cell vehicles, including reducing Pt usage in catalysts and improving their durability. In this study, a high-performance Pt@carbon-dot-film core–shell catalyst was successfully synthesized using a nonequilibrium reaction field, i.e., solution plasma (SP) process, by adjusting the electrolyte pH. Four pH solutions (pH = 4.4, 7, 8, and 11) were employed as the discharge liquid environment for the SP process. The catalyst synthesized in the pH = 8 solution exhibited a mass activity of approximately 500 mA mg−1, which was twice as high as that of the commercial Pt/C catalyst (256 mA mg−1) with the same loading amount. The onset and half-wave potentials were 0.99 and 0.89 V, respectively, both of which exceeded those of commercial Pt/C catalysts (0.95 and 0.86 V, respectively). Furthermore, the enhanced catalytic performance corresponded to the Pt/C bonding between Pt and the carbon shell generated during the SP process.
Introduction
Over half of the world's energy is generated from non-renewable and unsustainable fossil fuels, including oil, coal, and natural gas.1,2 However, the incomplete combustion of these fuels causes severe environmental problems, including the well-known greenhouse effect, extreme climate, acid rain, and rising sea levels, and these problems adversely affect the health of all living things on Earth.3,4 Researchers worldwide are seeking clean energy alternatives to reduce reliance on traditional fossil fuels and address these challenges.5–8 One promising alternative is the proton-exchange membrane fuel cell (PEMFC), also known as the hydrogen–oxygen fuel cell. In recent years, PEMFCs have gained importance because of their high energy efficiency and low environmental impact, making them a crucial component in realizing a society that utilizes hydrogen energy.8–12
In the working process of the hydrogen–oxygen fuel cell, hydrogen is absorbed on the surface of the anode and oxidized into protons. Further, oxygen is reduced into oxygenated species on the cathode (oxygen reduction reaction, ORR) using a catalyst, which is then combined with protons passing through the Nafion membrane to form water.13–15 However, the reaction rate on the cathode is low, which is an important obstacle in the development of the PEMFCs.16–19 The anode reaction rate is 5–6 orders of magnitude higher than that in the cathode.20 This indicates that hydrogen can provide numerous electrons, whereas the oxygen on the cathode can only accept a small amount because of the low reduction reaction rate. Therefore, a significant overpotential and a lower electromotive force than the theoretical value always exist during the working process of PEMFCs, which makes ORR a rate-determining step. Additionally, the high cost of catalysts is another limiting factor in the application of fuel cells because most of the catalytic metals are precious metals.21–23 In general, Pt loaded on carbon support is a prevalent commercial fuel cell catalyst.24 However, owing to the harsh working environment and longtime usage conditions of PEMFCs, Pt nanoparticles are very easily dissolved and agglomerated, and their Ostwald ripening can significantly reduce their catalytic activity and durability.25–27
Numerous researchers have reported various effective methods for optimizing the performance of Pt-based catalysts.28–35 A well-known effective method is particle engineering, i.e., downsizing the Pt nanoparticles or preparing the Pt-related alloy with transition metals, such as iron, cobalt, and nickel, or rare earth metals. The former works by increasing the electrochemical active surface area (ECSA), thereby providing high-density active sites28 to exhibit a certain facet of Pt crystals with high activity,29 while the latter usually leads to the reduction of the Pt amount and adjustment of the electronic structure.30,31 Another method is to load Pt onto carbon materials with a characteristic structure, which will enhance their catalytic activity and durability. For instance, Roudbari et al.32 synthesized nitrogen-functionalized carbon nanotubes as a support for Pt catalysts, which increased their fuel cell power and enhanced their durability. Similarly, Ruiz-Camacho et al.33 used carbon Vulcan, graphene sheets, and a mixture of carbon Vulcan and graphene sheets as supports and found that the mixture support exhibited the best ORR performance.
Recently, the wrapping of a few layers of graphene or a few layers of N-doped graphene on Pt nanoparticles has been known as an effective method for ORR catalyst enhancement, although the deposited carbon on Pt nanoparticles typically inhibits catalysis. This indicates that the regulated graphene with the dopants can promote the ORR activity. Li et al.34 investigated the Pt–graphene core–shell structure catalyst and found that adjusting the heat treatment temperature to 700 °C resulted in a high mass activity (MA), 190 mA mg−1 (that of commercial Pt/C was 130 mA mg−1 in their work). Oluf et al.35 showed FeC with few layers of graphene. Park et al.36 showed a high MA (730 mA per mg-Pt) and high ECSA (41.2 m2 g−1) for PtFe nanoparticles with a shell of a few layers of N-doped graphene. Moreover, the durability of the catalyst was drastically improved.
The active site can be the neighbor Pt and/or C atoms, forming Pt–C and/or Pt–N bonds at the interface between the core and shell. This is because the activity can be shown through the wrapping of a few layers of graphene and/or a few layers of N-doped graphene. However, it can be inferred that even these shells sacrifice ORR activity. Therefore, if the carbon shell can be made thinner and the shell is a discontinuous membrane structure, such as single- or double-layer carbon dot films, rather than normal graphene, it is hypothesized that a Pt-only core can significantly improve the MA. Yu Wang et al.37 synthesized graphene-wrapped FePt nanoparticles anchored on N-doped hierarchically porous graphitic carbon nanoflakes and got higher onset potential (1.01 V) and better durability than commercial Pt/C for ORR. Rui-Min Sun et al.38 showed the well entrapped platinum–iron nanoparticles on three-dimensional nitrogen-doped ordered mesoporous carbon catalyst has good performance both in ORR and zinc-air battery.
Although traditional methods, such as sputtering,39–41 ball milling,42–44 and wet-reduction processes,45–47 are typically used to synthesize nanoparticles, most of these methods have a problem with thermal processes at high temperatures. In particular, when synthesizing carbon shells, the general method is to heat-treat nanoparticles coated with organic materials or organic films to leave graphene or N-doped graphene on the nanoparticles. However, in thermal processes, not only do the nanoparticles aggregate and become coarse, it is also difficult to control the thickness and structure of the films.
In a solution plasma (SP) process, the structure of the carbon film can be varied by changing the type of organic compound contained in the solution. Furthermore, this SP process is an electron-active process and can be a room-temperature process with the standard of gas temperature; thus, there is no aggregation or coarsening of nanoparticles. Our research group has successfully synthesized nitrogen-doped graphene,48 carbon dots,49 and carbon shells50 using the SP process. In this study, sodium citrate, which has an acyclic structure, was applied as the carbon-based raw material, rather than the six- and five-membered ring organic compounds used in graphene synthesis by the SP process. Resultantly, the carbon crystallites obtained will be small, and it is expected that the carbon dots will be stacked in a disordered manner. Finally, the Pt–C bond is expected to increase, and the spatial density as a thin film will decrease; thus, the ORR activity will increase.
This study aims to fabricate 1–2 layers of carbon-dot-films on Pt nanoparticles by the SP sputtering process and show the enhancement effect on the ORR activity of Pt nanoparticles with carbon shells.
Experimental
Chemicals and materials
Sodium citrate (>99.0%), Pt standard ICP solution, commercial Pt/C catalyst (20 and 46 wt%), and Nafion solution (5 wt% in a mixture of lower aliphatic alcohols and water) were purchased from Sigma-Aldrich, Germany. Acetone (99.5%), sodium hydroxide (NaOH, >86%), hydrochloric acid (HCl, 35.0–37.0%), perchloric acid (HClO4, 70%), and isopropanol were purchased from Kanto Chemical Co., Inc., Japan. A Pt electrode (diameter was 0.5 mm, 99.95%) was purchased from Nilaco Co., Japan. Carbon black Vulcan XC 72 was purchased from the Fuel Cell Store USA. Polytetrafluoroethylene (PTFE) membrane filters (pore size 0.1 μm) were purchased from Merck USA.
Preparation of platinum/carbon catalysts by solution plasma sputtering
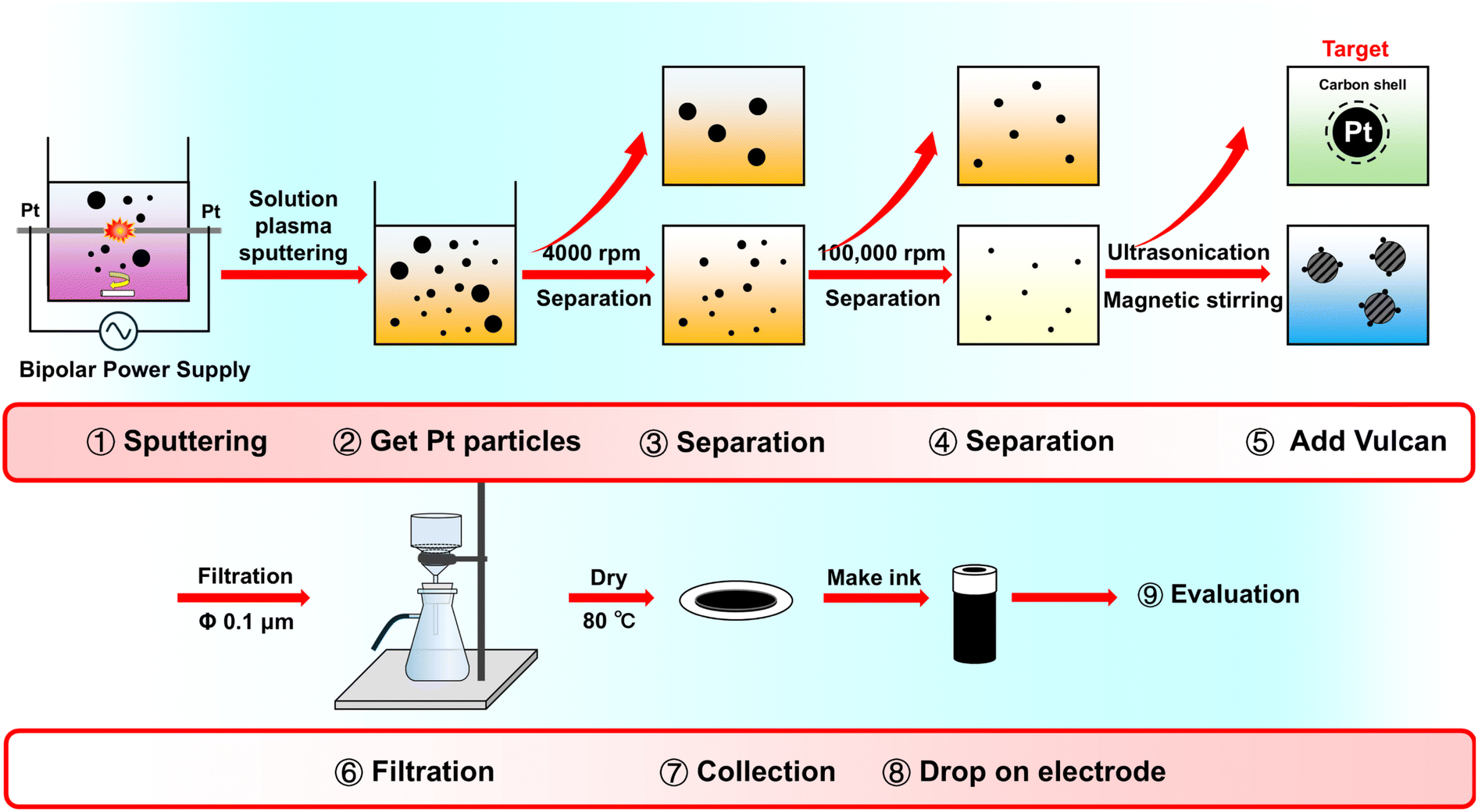
The Pt/C catalysts were synthesized using a nonequilibrium plasma in solution with a pin-to-pin discharge structure, as shown in Fig. 1. A 1.5 mM sodium citrate solution was used as a stabilizer, and NaOH and HCl were added to adjust the pH to 4.4, 7, 8, and 11 for the discharge liquid environment. Pt wires were utilized as discharge electrodes, which were wrapped with ceramic tubes to maintain the same electric field during the discharge process. The electrodes were polished flat before each discharge to prevent tip discharge. A bipolar discharge reaction was conducted using an alternating-current power supply (MPS-R06K01C-WP1-6CH, Kurita, Japan) at a frequency of 30 kHz and a pulse width of 1.5 μs, with a 1.0 mm distance between the electrodes. After a 20 min discharge time, all generated Pt solutions were collected. Thereafter, low-speed centrifugation at 4000 rpm (H-28F, KOKUSAN CORP. Japan) followed by ultracentrifugation at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (GX SERIES himac CS 100GXL, HIMAC CORP., Japan) was performed, and the entire preparation steps are shown in the Fig. 1. In the dispersion step, Vulcan XC 72 was used as carbon support for the synthesized nanoparticles. The process involved ultrasonication and magnetic stirring treatment, after which the resulting mixture was separated and washed with distilled water through filtration using a PTFE 0.1 μm membrane. The final step involved drying in an oven at 80 °C.
000 rpm (GX SERIES himac CS 100GXL, HIMAC CORP., Japan) was performed, and the entire preparation steps are shown in the Fig. 1. In the dispersion step, Vulcan XC 72 was used as carbon support for the synthesized nanoparticles. The process involved ultrasonication and magnetic stirring treatment, after which the resulting mixture was separated and washed with distilled water through filtration using a PTFE 0.1 μm membrane. The final step involved drying in an oven at 80 °C.
 |
| | Fig. 1 Experimental procedures of the Pt/C oxygen reduction reaction (ORR) catalyst by the solution plasma sputtering process. | |
Measurements and characterization
A pH meter (F-72, Horiba Ltd., Japan) was used to confirm the pH of electrolytes. Thermogravimetric analysis (C305759, SHIMADZU CORP., Japan) and inductively coupled plasma atomic emission spectroscopy (SPS-7000; SEIKO CORP., Japan) were employed to measure the percentage of Pt in the catalysts. An ultraviolet-visible (UV-Vis) spectrophotometer (UV-3600, SHIMADZU CORP., Japan) was used to measure the absorbance of the Pt solution. The morphology of the nanosized Pt particles was observed by high-resolution transmission electron microscopy (HR-TEM, JEM-2100F/HK; JEOL Ltd, Japan). The presence of carbon in the product was first confirmed by Raman spectroscopy (Leica DM 2500 M Ren (RL/TL), Renishaw Plc., England). Electron structure and element valence state were characterized by X-ray photoelectron spectrometry (XPS, Thermo Fisher Scientific, ESCALAB 250Xi, USA) using a Mg Kα X-ray source. X-ray absorption fine structure spectroscopy (XAFS, Aichi Synchrotron Radiation Facility, Japan) was conducted to study the fine structure of the synthesized Pt nanoparticles. Optical emission spectroscopy (OES, UV/Vis USB 2000+, Ocean Optics Inc., USA) and an oscilloscope (DLM2024, 2.5 GS/s, 200 MHz, Yokogawa, Japan) were employed to detect generated radicals and current–voltage (I–V) during plasma discharge, respectively.
Electrochemical test
To evaluate the performance of the obtained catalysts for ORR, linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were conducted on a potentiostat (HAG1512m/BP, HOKUTO DENKO CO., Japan). Further, a solution containing 0.1 M HClO4 was used as the electrolyte in the electrochemical measurements. The catalyst ink was composed of 3.5 mg of catalyst, 0.75 mL of isopropanol, 1.75 mL of distilled water, and 20 μL of Nafion (5 wt%). Thereafter, ultrasonic dispersion was carried out to disperse the ink evenly. Finally, a pipette was employed to drop the ink onto the surface of a glassy-carbon rotating disk electrode (HAG1512m/BP, HOKUTO DENKO CO., Japan), and the working electrode was placed on a 600 rpm rotary drying machine. The loading amount of these tested catalysts was 9.18 μg(Pt) cm−2. In the LSV and CV test, a Pt foil and reversible hydrogen electrode were employed as the counter electrode and reference electrode, respectively. Before measurement, 100 sccm N2 was bubbled into the solution for half an hour until it was saturated. Thereafter, the electrode was immersed in the solution for CV testing. The scan range of CV was 0.05–1.2 V, and the scan rate was 50 mV s−1. After the CV test, 100 sccm O2 was flowed into an electrolyte until it was saturated. Afterward, LSV was carried out under 2500, 1600, 900, 400, and 100 rpm from 0.2 to 1.2 V at a scan rate of 10 mV s−1. Further, single cell performance was conducted in this work. Owing to the low percentage of Pt in the prepared catalyst, the loading amount was only 20.4 μg cm−2. For comparison, the loading amount of 46.7% commercial Pt/C was also only 33.0 μg cm−2. The anode was 46.7% commercial Pt/C at the same loading amount (approximately 160 μg cm−2). During the test, the full fuel cell temperature was maintained at 80 °C. H2 was flowed into the anode of the fuel cell, and O2 was induced to the cathode. The catalyst was activated for two hours before the measurement of the I–V curve. CV measurement was conducted at 0.05–0.95 V and 100 mV s−1 in the cathode under N2 atmosphere.
Result and discussion
Solution plasma sputtering process properties
During the SP sputtering process, OES was employed to detect the produced radicals in the plasma, and the OES spectra after normalization are shown in Fig. 2(a). At the beginning stage of discharge, many electrons with high energy came out of the electrode and went into the solution to bombard other species, such as H and C. After the bombardment, several H radicals were produced, particularly in the aqueous solution, which resulted in the high intensity of Hα and Hβ peaks in the OES spectra. Additionally, C2 peaks were found in the OES spectra in these discharge systems, and they may come from the sodium citrate. The C2 peaks in alkaline solutions have much higher intensities than those in the acidic and neutral solutions. The electron temperature can be calculated using eqn (1):51,52| | 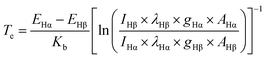 | (1) |
where E is the upper-level energy of the excited state. Kb is the Boltzmann constant (1.38 × 10−23 J K−1). I is the measured peak intensity. λ is the wavelength. E is the energy of the excited state. A is the transition probability of the active species. g is the statistical weight of the upper energy level. In the OES spectrum, the baseline of each spectrum was not a straight line; thus, the gas temperature can be calculated using eqn (2):53| |  | (2) |
λ is the wavelength corresponding to the blackbody radiation peak. The calculation results are presented in Table 1.
 |
| | Fig. 2 (a) Optical emission spectroscopy for the solutions with pH = 4.4, 7, 8, and 11, and (b) the relationship between the power per pulse and peak-ratio of C2 to Hα intensities. | |
Table 1 Power per pulse, electron temperature, gas temperature, and the peak-ratio of C2 to Hα intensities calculated from Fig. 1(a) and S1(b)
| Sample |
Power per pulse (J) |
T
e (K) |
T
g (K) |
C2/Hα |
| pH = 4.4 |
669.18 |
7880 |
5008 |
0.092 |
| pH = 7 |
550.88 |
8729 |
5185 |
0.079 |
| pH = 8 |
761.81 |
12025 |
5197 |
0.162 |
| pH = 11 |
714.54 |
16700 |
5350 |
0.232 |
Fig. S1(a)† shows the relationship between electron temperature and gas temperature. The red dash line represents the equilibrium discharge system, and the ratios of Tg/Te being less than 1 indicates that they are all nonequilibrium discharge systems in this research. As observed in Table 1, the electron and gas temperatures for each condition show that the nonequilibrium state is promoted while the pH value increases from 4.4 to 11. Furthermore, the intensity of C2 indicates that the formation of graphene and carbon can be promoted. The electronic state of C2 has been a point of disagreement for many years. However, in recent years, it has been identified as a singlet biradical, both theoretically and experimentally,54,55 and it has been clarified that it is an intermediate that is essential for the build-up of graphene and C60. Further, this explains why the formation of carbon progressed at pH = 8 and 11, which is consistent with the plasma state. Fig. 2(b) shows the relationship between power per pulse and peak-ratio of C2 to Hα intensities. The power per pulse can be calculated from the I–V curves shown in Fig. S1(b),† and it was determined that the pH = 8 discharge process had the largest pulse power. The intensity of the Hα and C2 peak can be attributed to the electron temperature and gas temperature in the SP sputtering system, respectively. Further, the different peak intensities of Hα and C2 suggest that the discharge processes in these systems were in a nonequilibrium state. Owing to the special structure in the liquid phase, carbon can be generated in the center of the plasma and solution interface, which indicates that in the pH = 8 and 11 solution there may be amorphous carbon or even graphene. The sodium citrate acted not only as stabilizer but also a precursor for carbon generation. Fig. S2(a-1) and (a-2)† showed the optical images for the solution at each pH treated by SP process and the UV-Vis absorbance spectra. The colors of all the solutions after the SP process became brown, and the absorbance spectra showed that the intensity increased with the decreased wavelength because the light scattering occurred because of the produced substances in the solution by the SP process. Fig. S2(b-1) and (b-2)† showed the optical images for the solution separated by 100000 rpm ultracentrifugation. The acidic and neutral solutions became almost transparent, whereas the alkaline solutions became dark yellow. It indicated that the particles in the alkaline solutions were larger than those in the acidic solutions.
Morphology of platinum nanoparticles
Fig. 3 shows the TEM images and histograms of Pt nanoparticle sizes on supporting materials. Fig. 3 ((a-1): pH = 4.4) and ((b-1): pH = 7) show relatively few Pt particles. Contrarily, Fig. 3 ((c-1): pH = 8) and ((d-1): pH = 11) show numerous Pt nanoparticles. Almost all the nanoparticles had diameters of less than 6 nm, as observed from their histograms shown in Fig. 3(a-2)–(d-2). Such distributions occurred because the 100000 rpm ultracentrifugation removed big particles. Fig. S3† shows the image and line profile obtained by Fourier transforming the original TEM image reduce the effect of carbon. Fig. S3(a)† was the original TEM image, Fig. S3(b)† was the image obtained by Fourier transforming the image after removing the spatial frequency of carbon by inverse Fourier transform, and Fig. S3(c)† was the line profile of the contrast from point a to point b on Fig. S3(b).† The average (111) interplanar spacing obtained from the spacing between points a and b was 0.23 nm, which was consistent with the interplanar spacing obtained from XRD (PDF # 87-0636). The graphene wrapping was not observed in the TEM images of the Pt nanoparticles synthesized in solutions with pH = 4.4 and pH = 7. Conversely, when looking at the particles around the edge of the supporting material in the Pt nanoparticles synthesized in the pH = 8 solution, a carbon shell was observed. This is shown in the high-resolution TEM image in Fig. 3(c-3). The shell had an interplanar spacing of 0.34 nm, corresponding to the (002) plane of graphitic carbon.37 The lattice fringes of the carbon film in the TEM image were highlighted with yellow lines. These lattice fringes were extremely short compared to those reported in other studies.56–58 This indicates that the graphitization is incomplete, the diameter of the 2D sheets is small, the number of layers is small, and the layers are disordered. It can be said that the film is composed of 2D-grown carbon dots with a lower degree of development. For the above reasons, the carbon film obtained in this study was defined as a carbon dot film to distinguish it from conventional carbon films (graphite films). Fig. 3(d-3) shows the different TEM views of the synthesized materials in the pH = 11 solution. There were many polymerized carbons. The presence or absence of carbon deposition, a carbon shell, and polymerized carbon were correlated with the intensity of the C2 spectrum observed by OES.
 |
| | Fig. 3 Transmission electron microscopy (TEM) images and particle size histograms of the Pt nanoparticles prepared from solutions with pH values of 4.4, 7, 8, and 11. The TEM images of (a-1)–(d-1) are at the same magnification. (a-2)–(d-2) Are the particle size histograms of Pt nanoparticles in (a-1)–(d-1). (c-3) Is a TEM image of (c-1) taken at a higher magnification from a different field of view, and (d-3) is a TEM image of (d-1) taken at a lower magnification from a different field of view. | |
Carbon structure by Raman spectroscopy
Fig. 4 shows the Raman spectra for Pt nanoparticles prepared by the SP process. These samples for Raman spectroscopy did not include the supporting materials, i.e., the samples included only the particles and carbons prepared by the SP process. The D band was related to the disordered vibration of carbon in sp3, and it typically indicates some defects in the crystal structure or the edge of graphene or a disordered structure. Meanwhile, the G band relates to the vibrations of the in-plane sp2 phonons of carbon atoms in the graphitic planes.48 From the Raman spectra, the spectral D and G bands cannot be seen at pH = 4.4 and 7, and the carbons can be categorized into molecules. At pH = 8 and 11, the spectral D and G band were observed, and the carbons were categorized into inorganic carbons, such as graphite, graphene, and carbon dots. The TEM results showed that at pH = 11, the carbon shape freestanding spheres were the main products, compared with the carbons formed on pt nanoparticles. The Raman spectrum of the Pt nanoparticles prepared at pH = 11 showed that the ratio of IG/ID was over 1. This indicates that the polymerization proceeded. Conversely, the spectrum from the Pt nanoparticles prepared at pH = 8 showed that the ratio of IG/ID was less than 1. This indicates that the diameter of crystal is smaller or the crystal has many defects. In both cases, the length of edges increased. From the TEM images and the Raman spectrum, the one- or two-layer films composed of carbon dots successfully wrapped the Pt nanoparticles.
 |
| | Fig. 4 Raman spectra of nanoparticles obtained using the solution plasma (SP) sputtering process. Some kind of carbon material or carbon material is attached. The samples are not nanoparticles on the carbon such as Vulcan of supporting materials. | |
Platinum chemical bonding state by X-ray photoelectron spectrometry and X-ray absorption fine structure spectroscopy
Fig. 5(a)–(e) and S4† show the XPS Pt 4f and C 1s spectra for the obtained Pt nanoparticles on Vulcan and 20 wt% commercial Pt/C, respectively. The Pt 4f7/2 and Pt 4f5/2 peaks can be deconvoluted into components of Pt(0) and Pt(II). C 1s spectra can be deconvoluted into three or four components: C–C, C–O, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and π–π* shakeup components. The sample synthesized in the pH = 8 solution has the largest Pt(0) content; the Pt(0) contents of the synthesized samples were in the following order from large to small: pH = 8 > pH = 7 > commercial Pt/C > pH = 11 > pH = 4.4. As the free-state Pt typically shows better catalytic performance,59 the pH = 8 sample was expected to perform best in the subsequent test. Further, as shown in Fig. 5(c), the binding energies of Pt(0) and Pt(II) in the pH = 8 sample slightly increased compared with those of the commercial Pt/C, as shown in Fig. 5(e). This indicates that the prepared Pt/C catalysts exhibit different electron structures from the commercial Pt/C. In the C 1s spectra shown in Fig. S4,† there are only three components in the Vulcan, commercial Pt/C, and pH = 4.4 and 7. However, in samples pH = 8 and 11, π–π* bonding structure was found near 291 eV in the catalysts, and these π–π* bonding may be attributed to ordered sp2 structure like graphene.60,61 Additionally, the corresponding atomic percentage of sample pH = 8 is slightly larger than that of sample pH = 11. Finally, the carbon materials on Pt nanoparticles at pH = 8 were closer to carbon dots with two-dimensional (2D) structures and exhibited higher electrical conductivity than amorphous carbons. Its structure might show the high catalytic activity. Fig. 6 shows the results of the XAFS spectroscopy; (a) the extended X-ray absorption fine structure (EXAFS) spectra of the obtained samples, PtO2, and Pt foil at the Pt L3-edge, (b) Fourier transform EXAFS spectra. The peaks were all located between the Pt foil and PtO2 in the Pt L3-edge XAFS spectra and closer to the Pt foil, which indicated that the Pt valence state was between 0 and +4. The fitting results of R space and wavelet transform graphs of all samples were shown in Fig. 6(c)–(i), and the fitting results were listed in Table 2. In the wavelet transform graphs, the bright region near 2.5 nm belonged to Pt–Pt bonding, and the region near 1.5 nm was attributed to Pt–C or Pt–O. the pH = 8 sample exhibited the brightest color in this region. According to the previous Raman, TEM, and OES analysis results, this sample can be fitted into three paths: Pt–O, Pt–C, Pt–Pt, respectively. Other samples can be fitted as only two paths, Pt–O and Pt–Pt. Additionally, the XAFS spectroscopy results proved the existence of graphene shell.
O, and π–π* shakeup components. The sample synthesized in the pH = 8 solution has the largest Pt(0) content; the Pt(0) contents of the synthesized samples were in the following order from large to small: pH = 8 > pH = 7 > commercial Pt/C > pH = 11 > pH = 4.4. As the free-state Pt typically shows better catalytic performance,59 the pH = 8 sample was expected to perform best in the subsequent test. Further, as shown in Fig. 5(c), the binding energies of Pt(0) and Pt(II) in the pH = 8 sample slightly increased compared with those of the commercial Pt/C, as shown in Fig. 5(e). This indicates that the prepared Pt/C catalysts exhibit different electron structures from the commercial Pt/C. In the C 1s spectra shown in Fig. S4,† there are only three components in the Vulcan, commercial Pt/C, and pH = 4.4 and 7. However, in samples pH = 8 and 11, π–π* bonding structure was found near 291 eV in the catalysts, and these π–π* bonding may be attributed to ordered sp2 structure like graphene.60,61 Additionally, the corresponding atomic percentage of sample pH = 8 is slightly larger than that of sample pH = 11. Finally, the carbon materials on Pt nanoparticles at pH = 8 were closer to carbon dots with two-dimensional (2D) structures and exhibited higher electrical conductivity than amorphous carbons. Its structure might show the high catalytic activity. Fig. 6 shows the results of the XAFS spectroscopy; (a) the extended X-ray absorption fine structure (EXAFS) spectra of the obtained samples, PtO2, and Pt foil at the Pt L3-edge, (b) Fourier transform EXAFS spectra. The peaks were all located between the Pt foil and PtO2 in the Pt L3-edge XAFS spectra and closer to the Pt foil, which indicated that the Pt valence state was between 0 and +4. The fitting results of R space and wavelet transform graphs of all samples were shown in Fig. 6(c)–(i), and the fitting results were listed in Table 2. In the wavelet transform graphs, the bright region near 2.5 nm belonged to Pt–Pt bonding, and the region near 1.5 nm was attributed to Pt–C or Pt–O. the pH = 8 sample exhibited the brightest color in this region. According to the previous Raman, TEM, and OES analysis results, this sample can be fitted into three paths: Pt–O, Pt–C, Pt–Pt, respectively. Other samples can be fitted as only two paths, Pt–O and Pt–Pt. Additionally, the XAFS spectroscopy results proved the existence of graphene shell.
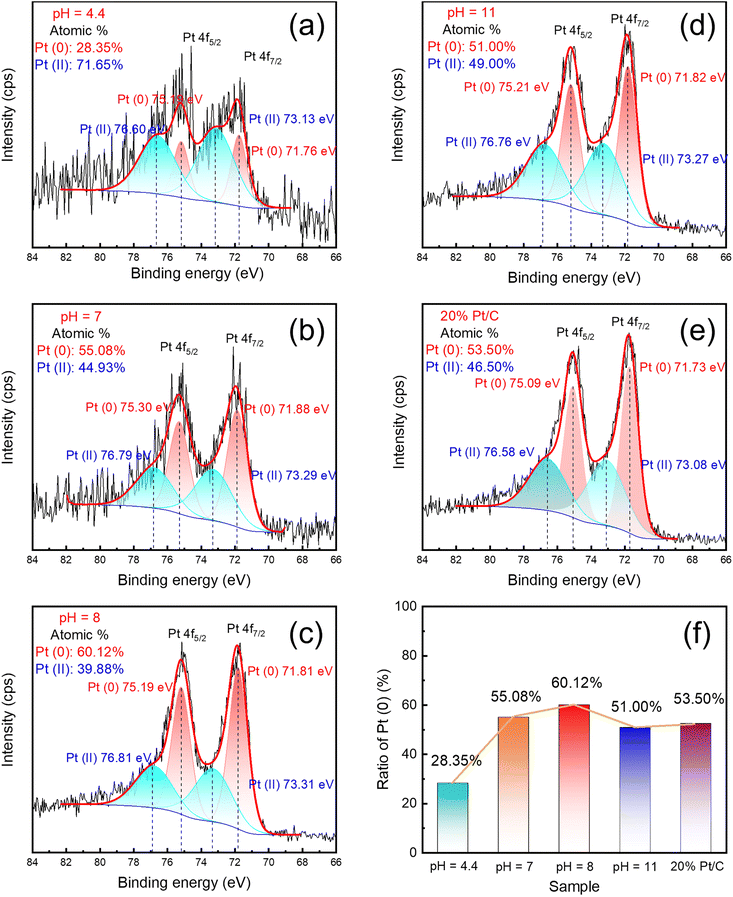 |
| | Fig. 5 X-ray photoelectron spectra of Pt 4f (4f5/2 and 4f7/2) for the nanoparticles prepared from the solutions with (a) pH = 4.4, (b) pH = 7, (c) pH = 8, (d) pH = 11, and (e) 20 wt% commercial Pt/C, and (f) the peak ratios of Pt(0) obtained by the Gaussian deconvolutions. | |
 |
| | Fig. 6 X-ray absorption fine structure spectroscopy; (a) extended X-ray absorption fine structure (EXAFS) spectra of the obtained samples, PtO2, and Pt foil at the Pt L3-edge; (b) Fourier transform EXAFS spectra and (c)–(i) their Fourier fitting results and wavelet transforms. | |
Table 2 Extended X-ray absorption fine structure fitting parameters at the Pt L3-edge for various samples
| Sample |
Shell |
CN |
R (Å) |
ΔE0 (eV) |
σ
2 (Å2) |
|
CN is the coordination number.
R (Å) is the bond distance.
ΔE0 (eV) is the inner potential correction.
σ
2 (Å2) is Debye–Waller factors.
|
| Pt foil |
Pt–Pt1 |
8 (set) |
2.77 |
7.7 |
0.0048 |
| Pt–Pt2 |
6 (set) |
3.91 |
0.0061 |
| PtO2 |
Pt–O |
6 (set) |
2.01 |
8.7 |
0.0033 |
| pH = 4.4 |
Pt–O |
1.6 |
1.95 |
5.1 |
0.0076 |
| Pt–Pt |
6.8 |
2.74 |
0.0076 |
| pH = 7 |
Pt–O |
1.0 |
1.96 |
5.7 |
0.0020 |
| Pt–Pt |
6.5 |
2.74 |
0.0080 |
| pH = 8 |
Pt–O |
1.1 |
1.98 |
6.7 |
0.0080 |
| Pt–C |
1.1 |
2.05 |
0.0020 |
| Pt–Pt |
5.3 |
2.74 |
0.0090 |
| pH = 11 |
Pt–O |
1.8 |
1.99 |
8.2 |
0.0059 |
| Pt–Pt |
5.7 |
2.75 |
0.0083 |
| 20 wt% Pt/C |
Pt–O |
2.1 |
1.99 |
8.5 |
0.0058 |
| Pt–Pt |
5.7 |
2.75 |
0.0082 |
Oxygen reduction reaction performance of the platinum/carbon catalyst
Fig. 7 and Table 3 show the ORR performance of the prepared catalysts. Fig. 7(a) shows the CV curves of the obtained Pt/C catalysts and the commercial Pt/C from 0.05 to 1.2 V in the 0.1 M HClO4 solution with saturated nitrogen. The loading amount of the catalysts was maintained at 9.18 μg cm−2 for better comparison. For commercial Pt/C, the loading amount of 20 μg cm−2 was used, as this was the measurement used by many researchers. The peaks between 0 V and 400 mV were the hydrogen desorption and adsorption regions, as shown in Fig. 7(a). From the hydrogen desorption region, we could calculate the ECSA based on eqn (3):9,11,62,63| |  | (3) |
where QH is the coulombic charge for hydrogen desorption, m is the weight of the Pt loading, and qH (210 μC cm−2) is the charge required to oxidize a monolayer of H2 on the Pt site.11 The sample made in the pH = 8 electrolyte exhibited the largest ECSA at approximately 136.50 m2 g−1, whereas the commercial Pt/C was only 80.95 m2 g−1. This indicated that sample pH = 8 has the highest number of active sites. For sample pH = 4.4 and 7 with very low Pt percentage, maintaining the same loading amount inevitably caused numerous Pt particles to be buried in the carbon support. To evaluate the ORR catalytic performance, the LSV measurement was carried out. Fig. 7(b) shows the LSV curves from 0.2 to 1.2 V. The onset potential of sample pH = 8 was approximately 0.99 V, which was 0.04 V higher than that of the commercial Pt/C at the same loading amount. Additionally, this sample exhibited a more positive half-wave potential than the commercial one. MA typically can be used to judge the performance of catalysts. A high MA indicates that the catalyst can generate large current when the Pt loading amount is low. MA can be calculated using the following equations:64| |  | (4) |
| |  | (5) |
where ik is the kinetic current, and mPt is the loading amount used in the measurement process. id is the diffusion-limiting current, and i is the experimentally measured current. The results showed that the MA of sample pH = 8 was approximately 500 mA mg−1, which was already two times that of commercial Pt/C at the same loading amount. The number of electrons transferred per oxygen molecule during the ORR was calculated using the Koutecky–Levich (K–L) equation as given below:65,66| |  | (6) |
| | | B = 0.62nFCODO2/3v−1/6, | (7) |
where ω is the angular velocity, j is the measured current density, jK and jL are the kinetic and diffusion-limiting current densities, respectively, and n is the number of electrons transferred. F is the Faraday constant (96485 C mol−1), CO is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), DO is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), and v is the kinematic viscosity of the electrolyte (0.01 cm2 s−1). n can be obtained from the slope of the K–L equation. As Fig. 7(c) shows, the red lines represent n = 4, the black line represents n = 2, and all the electron transfer numbers are calculated at 750 mV. The slopes of samples pH = 4.4, 7, 8, and 11 and commercial Pt/C were near the slope n = 4 line, which indicated that all samples were 4 electron pass way reaction. The full cell performance of the prepared Pt/C (sample pH = 8) and commercial (46 wt%) samples were evaluated using the membrane electrode assembly (MEA) measurements. The ink compositions are listed in Table S1.† Table S2† shows the loading amount of Pt at each electrode. In the tested fuel cell, the Pt loading on the anode remained consistent. The loading amount on the pH = 8 cathode was approximately 20.4 μg because of the low percentage of Pt (just approximately 8%), and for comparison, the loading amount of commercial 46 wt% Pt/C should on the same level. The output performance of the fuel cells and CV in nitrogen atmosphere results are shown in Fig. S5.† It shows an oxygen reduction peak at 800 mV and hydrogen under-potential adsorption/desorption peaks at 0–400 mV in the prepared sample. However, there was no peak in the commercial Pt/C. The as-prepared catalyst exhibited larger ECSA and higher output power than commercial Pt/C. Results for the MEA measurement suggested that our prepared catalyst has potential applications in the fuel cells.
 |
| | Fig. 7 Catalytic performance measurements of half-cell (a) cyclic voltammetry curves under N2; (b) linear sweep voltammetry curves under 1600 rpm and O2; (c) Koutecky–Levich plot at 750 mV; (d) mass activity, electrochemical active surface area, onset potential, and half-wave potential of different samples. | |
Table 3 Loading amount and oxygen reduction reaction performance results of all the catalystsa
| Catalyst |
pH = 4.4 |
pH = 7 |
pH = 8 |
pH = 11 |
Commercial 20 wt% Pt/C |
Commercial 20 wt% Pt/C |
|
m
Pt is loading amount; Eonset is onset potential; E1/2 is half-wave potential; MA is mass activity at 0.9 V; ECSA is electrochemical active surface area.
|
| Pt% |
2.3 |
3.7 |
13.1 |
11.0 |
20 |
20 |
|
m
Pt (μg) |
|
|
1.80 |
|
|
3.90 |
|
E
onset (V) |
0.93 |
0.93 |
0.99 |
0.95 |
0.95 |
0.97 |
|
E
1/2 (V) |
0.84 |
0.86 |
0.89 |
0.82 |
0.86 |
0.89 |
| MA (mA mg−1) |
128.0 |
201.3 |
524.1 |
148.5 |
256.0 |
165.8 |
| ECSA (m2 g−1) |
32.75 |
87.76 |
136.50 |
53.47 |
80.95 |
83.20 |
Contribution of platinum/carbon bonding and their formation
The above results show that ORR activity can be improved using single or double layers of carbon shells with a 2D structure, such as carbon dots. The MA (524.11 mA per mg-Pt) was larger than that of the core–shell catalyst, Pt/C,34,67–69 as shown in Fig. S6.† This is thought to be because compared with conventional graphene core–shells, the Pt–C bonds are increased by wrapping the material in layers composed of small 2D materials, such as carbon dots. Further, the disordered structure allows for relatively fast mass transfer to the reaction site. This type of carbon layer is suitable for the ORR catalyst. Fig. 8(a) shows the sputtering process during discharge in one pulse; Fig. 8(b) shows the electron temperature and the gas temperature corresponding to the different pH solutions; Fig. 8(c) illustrates the SP sputtering process for generating the Pt nanoparticles. According to calculation results, the gas temperature and electron temperature gradually increased as the pH increased. A significant amount of C2 was generated at the center of the plasma with the increase in the gas temperature. In the acidic solution, the main species that collided with the metal surface was H+, and in this case, the sputtering rate was low, resulting in a small number and size of particles. In the alkaline solution, the main species that collided with the metal surface was O−, and in this case, the sputtering rate was comparatively high, producing many large particles. In alkaline solutions, when the gas temperature was high, C2 moved to the interface between the plasma and the liquid phase. Owing to the γ effect, the organic groups present in the liquid phase (in this work, the chemical group was citrate ions) underwent dehydrogenation reactions, resulting in the formation of graphene. Conversely, as the electron temperature increased (pH = 8), the electron movement rate was high and the lifetime increased. It bombarded the generated graphene, etched the graphene on the surface of the Pt particles, and formed carbon dots. When the electron temperature further increased (pH = 11), the carbon on the surface of the Pt particles was completely etched, and the carbons were freestanding or adsorbed on the Pt nanoparticles as the bigger particles. The variations in the carbon wrapping layers from pH = 8 and pH = 11 are summarized in Fig. 8(d) and (e). The structure and morphology of the final product were determined by the deposition and etching rates of the carbon produced by the solution plasma. In this experiment, the balance of the pH = 8 solution was suitable for the production of carbon.
 |
| | Fig. 8 (a) Sputtering process during discharge in one pulse; (b) electron temperature and the gas temperature corresponding to the different pH electrolytes; (c) solution plasma sputtering process for generating the Pt nanoparticles; (d) and (e) schematic diagrams of a single Pt particle of the pH = 8 and pH = 11 samples, respectively. | |
Conclusions
We successfully prepared Pt nanoparticles with diameters of less than 6 nm using SP sputtering and separation processes from different sodium citrate aqueous solutions with pH values of 4.4, 7, 8, and 11. The TEM images and EXAFS spectra showed that at pH = 4.4 and 7, the platinum nanoparticles were not wrapped in carbon materials such as graphene and only adsorbed molecular substances were present, as determined by Raman spectroscopy. Conversely, in the pH = 8 sample, the platinum nanoparticles were wrapped in carbon with several layers of graphene. From the Raman spectra, the diameter of the crystals was not very high. This can be identified as a carbon layer comprising a collection of carbon dots. In the pH = 11 sample, the carbon material was in a freestanding state, with no carbon was observed on the Pt nanoparticles. Additionally, the formation of these carbons is reasonable in terms of the balance between carbon formation by C2 in the plasma and carbon etching by H+ and O− ions and the precipitation and morphology of these carbon materials. Electrochemical evaluation showed that the ORR activity at pH = 8 was the highest, and MA was approximately twice as high as that of the commercial 20 wt% Pt/C catalyst at the same loading amount; further, the ECSA was 55.6 m2 g−1 larger. This is thought to be because compared with conventional graphene core–shells, the Pt–C bonds are increased by wrapping the material in layers composed of small 2D materials, such as carbon dots. Further, the disordered structure allows for a relatively fast mass transfer to the reaction site.
Data availability
The authors will supply the relevant data in response to reasonable requests.
Author contributions
Yuanyuan Liu: conceptualization, methodology, data curation, writing original draft. Zhunda Zhu: methodology, formal analysis. Pengfei Wang: methodology. Zhuoya Deng: methodology. Jiangqi Niu: methodology. Yasuyuki Sawada: review & editing, funding acquisition. Nagahiro Saito: visualization, writing-review & editing, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study is partially based on results obtained from a project, JPNP20003, commissioned by the New Energy and Industrial Technology Development Organization (NEDO), Japan. Yuanyuan Liu is also supported by a scholarship from the China Scholarship Council (No. 202106130011).
Notes and references
- K. Vezzùa, A. B. Delpeuch, E. Negroa, S. Polizzi, G. Nawn, F. Bertasi, G. Pagota, K. Artyushkov, P. Atanassova and V. D. Noto, Electrochim. Acta, 2016, 222, 1778–1791 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- M. O'Hare, R. J. Plevin, J. I. Martin, A. D. Jones, A. Kendall and E. Hopson, Environ. Res. Lett., 2009, 2, 024001 CrossRef.
- Y. Zhang, P. Wang, J. Yang, K. Li, X. Long, M. Li, K. Zhang and J. Qiu, Chem. Eng. J., 2020, 401, 126001 CrossRef CAS.
- G.-L. Li, C.-D. Liu, L.-F. Yuan, Q.-M. Wu, W.-W. Chen, G.-C. Cheng, B.-B. Yang and C. Hao, Int. J. Hydrogen Energy, 2018, 43, 1386–1395 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- P. Wang, J. Zhang, Y. Peng, X. Hu, L. Miao and T. Ishizaki, Sustainable Mater. Technol., 2022, 32, e00384 CrossRef CAS.
- W. Li, Z.-Y. Hu, Z. Zhang, P. Wei, J. Zhang, Z. Pu, J. Zhu, D. He, S. Mu and G. V. Van Tendeloo, J. Catal., 2019, 375, 164–170 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- B. P. Vinayan, R. Nagar, N. Rajalakshmi and S. Ramaprabhu, Adv. Funct. Mater., 2012, 22, 3519–3526 CrossRef CAS.
- S. I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef.
- M. Chen, C. Zhao, F. Sun, J. Fan, H. Li and H. Wang, eTransportation, 2020, 5, 100075 CrossRef.
- G. Wang, Y. Yu, H. Liu, C. Gong, S. Wen, X. Wang and Z. Tu, Fuel Process. Technol., 2018, 179, 203–228 CrossRef CAS.
- A. Ozden, S. Shahgaldi, X. Li and F. Hamdullahpur, Prog. Energy Combust. Sci., 2019, 74, 50–102 CrossRef.
- B. Liang, M. Su, Z. Zhao and S. Liang, J. Electroanal. Chem., 2024, 962, 118260 CrossRef CAS.
- H.-Y. Wang, C.-C. Weng and Z.-Y. Yuan, J. Energy Chem., 2021, 56, 470–485 CrossRef CAS.
- H. Zhao, Y. Chen, H. Zuo, L. Li, L. Li, F. Ai, Z. Tang, T. Xing, Y. Zhang, L. Tao, Z. Tian, H. Yang, X. Geng and B. An, Catal. Today, 2024, 438, 114811 CrossRef CAS.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS.
- K. Karan, Electrochem. Commun., 2007, 9, 747–753 CrossRef CAS.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, Z. Wang, Z. Hu, Y. Zhao, H.-W. Li, D. He and S. Mu, Nano Energy, 2021, 88, 106221 CrossRef CAS.
- Z. Song, Y. N. Zhu, H. Liu, M. N. Banis, L. Zhang, J. Li, K. D. Davis, R. Li, T. K. Sham, L. Yang, A. Young, G. A. Botton, L. M. Liu and X. Sun, Small, 2020, 16, 2003096 CrossRef CAS.
- H. Jin, H. Zhou, D. He, Z. Wang, Q. Wu, Q. Liang, S. Liu and S. Mu, Appl. Catal., B, 2019, 250, 143–149 CrossRef CAS.
- H. Lv and S. Mu, Nanoscale, 2014, 6, 5063–5074 RSC.
- E. Antolini, Appl. Catal., B, 2010, 100, 413–426 CrossRef CAS.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- S.-Y. Huang, P. Ganesan and B. N. Popov, Appl. Catal., B, 2010, 96, 224–231 CrossRef CAS.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS.
- K. Kinoshita, J. Electrochem. Soc., 1990, 137, 845–848 CrossRef CAS.
- J. Wang, B. Li, D. Yang, H. Lv and C. Zhang, Electrochim. Acta, 2018, 288, 126–133 CrossRef CAS.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y.-H. Chung, H. Kim, B. S. Mun, K.-S. Lee, N.-S. Lee, S. J. Yoo, D.-H. Lim, K. Kang, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS.
- M. N. Roudbari, R. Ojani and J. B. Raoof, Renewable Energy, 2020, 159, 1015–1028 CrossRef CAS.
- B. Ruiz-Camacho, J. A. Palafox-Segoviano, P. J. Pérez-Díaz and A. Medina-Ramírez, Int. J. Hydrogen Energy, 2021, 46, 26027–26039 CrossRef CAS.
- Z. Li, J. Zou, X. Xi, P. Fan, Y. Zhang, Y. Peng, D. Banham, D. Yang and A. Dong, Adv. Mater., 2022, 34, 2202743 CrossRef CAS PubMed.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- J.-H. Park, N. Saito and M. Kawasumi, Carbon, 2023, 214, 118364 CrossRef CAS.
- Y. Wang, M.-T. Chen, X. Ye, A.-J. Wang, G.-M. Tu, L. Zhang and J.-J. Feng, Appl. Surf. Sci., 2022, 604, 154475 CrossRef CAS.
- R.-M. Sun, R.-Z. Wu, X.-S. Li, J.-J. Feng, L. Zhang and A.-J. Wang, J. Colloid Interface Sci., 2022, 621, 275–284 CrossRef CAS.
- M. Zhu, M. T. Nguyen, W. J. Sim and T. Yonezawa, Adv. Mater., 2022, 3, 8967–8976 RSC.
- S. Hussain, H. Erikson, N. Kongi, M. Merisalu, P. Ritslaid, V. Sammelselg and K. Tammeveski, Int. J. Hydrogen Energy, 2017, 42, 5958–5970 CrossRef CAS.
- S. Hussain, H. Erikson, N. Kongi, A. Tarre, P. Ritslaid, M. Rähn, L. Matisen, M. Merisalu, V. Sammelselg and K. Tammeveski, Int. J. Hydrogen Energy, 2018, 43, 4967–4977 CrossRef CAS.
- T. Tian, S. Zhang, Y. Song, C. Li, X. Zeng, Z. Yang, Q. Ji, X. Zhao and F. Chen, Mater. Lett., 2024, 365, 136468 CrossRef CAS.
- M. Etesami, R. Khezri, A. Abbasi, M. T. Nguyen, T. Yonezawa, S. Kheawhom and A. Somwangthanaroj, J. Alloys Compd., 2022, 922, 166287 CrossRef CAS.
- S. Ratso, A. Zitolo, M. Käärik, M. Merisalu, A. Kikas, V. Kisand, M. Rähn, P. Paiste, J. Leis, V. Sammelselg, S. Holdcroft, F. Jaouen and K. Tammeveski, Renewable Energy, 2021, 167, 800–810 CrossRef CAS.
- S. Maghsoudi, R. Abdullah Mirzaie, M. Ghalkhani and A. Anaraki Firooz, J. Phys. Chem. Solids, 2023, 176, 111269 CrossRef CAS.
- F. T. Angerasa, C.-Y. Chang, E. A. Moges, W.-H. Huang, K. Lakshmanan, Y. Nikodimos, J.-F. Lee, N. G. Habtu, M.-C. Tsai, W.-N. Su and B. J. Hwang, Mater. Today Energy, 2023, 34, 101312 CrossRef CAS.
- Z. X. Liu, B. H. Liu and Z. P. Li, Int. J. Hydrogen Energy, 2014, 39, 5689–5695 CrossRef CAS.
- J. Niu, C. Chokradjaroen and N. Saito, Carbon, 2022, 199, 347–356 CrossRef CAS.
- K. Kim, C. Chokradjaroen and N. Saito, Nano Express, 2020, 1, 020043 CrossRef.
- P. Q. Phan, R. Naraprawatphong, P. Pornaroontham, J. Park, C. Chokradjaroen and N. Saito, Mater. Adv., 2021, 2, 322–335 RSC.
- L. Wang, L. Chen, Z. Yan and W. Fu, Surf. Coat. Technol., 2010, 205, 1651–1658 CrossRef CAS.
- T. Zhan, W. Tu, Y. Cheng, J. Han, B. Su and Y. Cheng, Ceram. Int., 2018, 44, 10402–10411 CrossRef CAS.
- X. C. Y. Huang, K. H. Hu and Z. H. Wei, Sci. Rep., 2016, 6, 37960 CrossRef PubMed.
- K. Miyamoto, S. Narita, Y. Masumoto, T. Hashishin, T. Osawa, M. Kimura, M. Ochiai and M. Uchiyama, Nat. Commun., 2020, 11, 2134 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, W. Wu, P. Su, H. S. Rzepa and P. C. Hiberty, Nat. Chem., 2012, 4, 195–200 CrossRef CAS.
- J.-H. Park, N. Saito and M. Kawasumi, Int. J. Hydrogen Energy, 2024, 72, 642–651 CrossRef CAS.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS.
- J.-H. Jang, A. A. Jeffery, J. Min, N. Jung and S. J. Yoo, Nanoscale, 2021, 13, 15116–15141 RSC.
- C.-T. Shen, K.-W. Wang, C.-J. Tseng, K.-R. Lee and Y.-J. Hsueh, RSC Adv., 2016, 6, 44205–44211 RSC.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard, P. Golob and M. Winter, Appl. Surf. Sci., 2000, 167, 99–106 CrossRef CAS.
- S. Rudi, C. Cui, L. Gan and P. Strasser, Electrocatalysis, 2014, 5, 408–418 CrossRef CAS.
- F.-S. Zheng, S.-H. Liu and C.-W. Kuo, Int. J. Hydrogen Energy, 2016, 41, 2487–2497 CrossRef CAS.
- X. Huang, Z. Zhao, Y. Chen, E. Zhu, M. Li, X. Duan and Y. Huang, Energy Environ. Sci., 2014, 7, 2957–2962 RSC.
- J. Moreira, P. D. Angel, A. L. Ocampo, P. J. Sebastián, J. A. Montoya and R. H. Castellanos, Int. J. Hydrogen Energy, 2004, 29, 915–920 CrossRef CAS.
- W. Chen, J. Kim, S. Sun and S. Chen, J. Phys. Chem. C, 2008, 112, 3891–3898 CrossRef CAS.
- Y. Nie, S. Chen, W. Ding, X. Xie, Y. Zhang and Z. Wei, Chem. Commun., 2014, 50, 15431–15434 RSC.
- M. Sharma, J.-H. Jang, D. Y. Shin, J. A. Kwon, D.-H. Lim, D. Choi, H. Sung, J. Jang, S.-Y. Lee, K. Y. Lee, H.-Y. Park, N. Jung and S. J. Yoo, Energy Environ. Sci., 2019, 12, 2200–2211 RSC.
- J. M. Yoo, H. Shin, D. Y. Chung and Y.-E. Sung, Acc. Chem. Res., 2022, 55, 1278–1289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Current–voltage relationship during discharge; solution photos after SP sputtering process and after ultracentrifugation; absorbance spectra of the solution after ultracentrifugation; the image and line profile obtained by Fourier transforming the original TEM image reduce the effect of carbon; C 1s XPS spectra of Vulcan, pH = 4.4, 7, 8 and 11, and commercial Pt/C (20 wt%) samples; MEA performance of commercial Pt/C (46 wt%) and sample pH = 8; benchmark of different catalyst ORR performance from references. See DOI: https://doi.org/10.1039/d4na00818a |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zhunda
Zhu
a,
Zhunda
Zhu