
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFn14-targeting, NIR-II responsive nanomaterials for enhanced radiotherapy against glioblastomas†
Wei
Fu‡
 ab,
Qing
Liang‡
ab,
Yuxi
Ma‡
ab,
Shiqiong
Lei
c,
Ruiqi
Li
ab,
Xin
Zheng
ab,
Lian
Chen
ab,
Jiayuan
Chen
ab,
Xing
Cai
ab,
Xiaofang
Dai
ab,
Hongwei
Duan
de,
Wenshan
He
*c and
Jinghua
Ren
*abf
ab,
Qing
Liang‡
ab,
Yuxi
Ma‡
ab,
Shiqiong
Lei
c,
Ruiqi
Li
ab,
Xin
Zheng
ab,
Lian
Chen
ab,
Jiayuan
Chen
ab,
Xing
Cai
ab,
Xiaofang
Dai
ab,
Hongwei
Duan
de,
Wenshan
He
*c and
Jinghua
Ren
*abf
aCancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. E-mail: jhrenmed@hust.edu.cn
bHubei Key Laboratory of Precision Radiation Oncology, Wuhan 430022, China
cDepartment of Breast and Thyroid Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. E-mail: hewenshan@hust.edu.cn
dSchool of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637457, Singapore
eLee Kong Chian School of Medicine, Nanyang Technological University, Singapore 636921, Singapore
fShenzhen Huazhong University of Science and Technology Research Institute, Shenzhen 518057, China
First published on 11th March 2025
Abstract
Radiotherapy is a common treatment option for patients with glioblastoma multiforme. However, tumor heterogeneity causes varying responses to radiation among different tumor subpopulations. Cancer cells that endure radiotherapy exhibit radioresistance, resulting in the ineffectiveness of radiation therapy and eventual tumor relapse. In this study, we discovered that the fibroblast growth factor-inducible 14 (Fn14)-positive tumor cells were enriched in tumor residual foci after radiation, ultimately leading to treatment failure. Fn14-expressing glioma cells survived ionizing radiation through preferential activation of DNA damage checkpoint response. We have thus engineered an Fn14-targeting and NIR-II responsive plasmonic gold nanosystem named Fn14-AuNPs, which can precisely internalize into Fn14-overexpressed glioma cells and have an excellent BBB-crossing capability. As gold nanoparticles, by inhibition of DNA repair processes and induction of G2/M cells cycle arrest, Fn14-AuNPs nanoparticles improved the radiosensitivity of tumor cells. Meanwhile, Fn14-AuNPs induced localized heat under NIR-II photoirradiation, thus impeding RT-induced DNA damage checkpoint response. This versatile nanosensitizer, combined with NIR-II laser photoirradiation, can eradicate radioresistant subpopulations of glioblastoma and improve the therapeutic effect of radiotherapy. This finding presents an effective radiosensitization strategy by targeting radioresistant subpopulations, which can efficiently overcome the constraints imposed in clinical radiotherapy and offer a hopeful avenue to enhance the treatment effectivity of radiotherapy in glioblastoma.
1. Introduction
Glioblastoma multiforme (GBM) is recognized as the most aggressive and primary malignant brain tumor in adult patients.1,2 Despite undergoing standard-of-care therapies like surgery, radiotherapy, and chemotherapy, patients with GBM have a median overall survival of only 12 to 15 months, with a 5-year survival rate of merely 4.7%.1–4 The restricted efficacy of current therapies is partially attributable to their heterogeneous nature.5–9 GBM cells with distinct genotypes and phenotypes may harbor divergent signaling pathways in response to treatment, enabling certain cells to adapt and resist treatment. Rational therapeutic combinations based on an in-depth understanding of the relevant subpopulations associated with therapy resistance are potentially of great utility in glioma therapy.Radiation therapy, as an integral component of GBM treatment, has been validated to enhance overall disease control.10,11 Ionizing Radiation (IR) causes DNA damage through direct ionization and reactive oxygen molecular products, which in turn destroys tumor cells and impedes cell proliferation.11 In spite of the progress made in modern radiation therapy techniques, local recurrence resulting from radiation-resistant tumor cells remains a barrier to the long-term survival of GBM patients.12 Previous studies mainly ascribed the mechanism of radiation resistance to DNA damage repair and hypoxia.13–15 Given that GBM is recognized as a highly heterogeneous disease, tumor cells with low radiosensitivity could potentially trigger local recurrence, ultimately resulting in the failure of radiotherapy.16 Therefore, selectively eliminating the tumor cells with reduced radiosensitivity at conventional radiation therapy doses could be crucial in overcoming the resurgence of GBM.
In this study, we found that high expression of fibroblast growth factor-inducible 14 (Fn14) in glioblastoma tissue was associated with poor progression-free survival (PFS) using the datasets of the Cancer Genome Atlas (TCGA). Fn14, also known as TNFRSF12A, has been identified as a member of the tumor necrosis factor receptor superfamily. It is a type I transmembrane protein composed of 129 amino acid residues.17–19 The binding of Fn14 with its ligand TWEAK may facilitate cellular activities including proliferation, invasion, angiogenesis and inflammation. Here, we showed that the Fn14+ subpopulation in glioblastoma was resistant to radiotherapy by preferential activation of DNA damage checkpoint response and induction of G2/M cells. Our results further demonstrated that mild hyperthermia hindered DNA damage repair. Thus, we speculated that mild hyperthermia in combination with radiotherapy may represent a therapeutic approach for GBM through targeting Fn14+ cells.
Previous studies have shown that nanoparticles can enhance the radiosensitivity of tumor cells by several crucial processes, including enzyme-like catalytic activity, the production of reactive oxygen species (ROS), induction of cell cycle arrest, disruption of radiation-induced DNA damage repair, and inhibition of hypoxia.20,21 Here, we constructed the Fn14-targeting and near-infrared (NIR)-II-activated nanosystem that can sensitize the Fn14-positive GBM cells to radiotherapy through impairment of DNA repair and induction of cell-cycle arrest. In line with our previous work,22 we synthesized plasmonic gold nanoparticles (AuNPs) using a tailored approach involving the dopamine-mediated reduction of gold precursors. Delivering nanoparticles to the brain is challenging owing to the existence of the blood–brain barrier (BBB).23 Under normal physiological conditions, substances can pass through the BBB by: (i) passive diffusion, (ii) transport via carriers, (iii) transcytosis mediated by adsorption, (iv) transport mediated by receptors, and (v) efflux pumps. A previous study showed that AuNPs can traverse the BBB via passive diffusion, owing to their small size (≤60 nm).24 Therefore, AuNPs have an excellent capacity to penetrate the BBB. Besides, the hyperbranched AuNPs exhibit localized surface plasmon resonance (LSPR) across a wide range of wavelengths, extending from the visible to the NIR spectral range. Upon the attachment of the D-enantiomeric peptide ligand to Fn14, AuNPs transform into the photonic and Fn14-targeting nanomaterial, Fn14-AuNPs. Relying on the disruption of DNA repair pathways, the increase of G2/M cells and its NIR-II photothermal effect, Fn14-AuNPs enhanced the radiation damage effect under conventional radiation doses on GBM and selectively eliminate tumor cells in residual GBM sites, striving to suppress tumor development and prolong its long-term survival.
2. Experimental section
2.1 Reagents and antibodies
The enantiomeric peptide ligand of Fn14 (D-FNB, sequenced as DCDHDPDRDEDVDDDVDEDLDYDSDTDVDFDGDH) was synthesized by Bankpeptide (Hefei, China). In this research, multiple primary antibodies were applied, including anti-γ-H2AX (rabbit, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000, Abcam, ab81299), anti-CD266 (Fn14, TWEAK Receptor) monoclonal antibody (ITEM-4) (mouse, 1:250, Abcam, ab109365), anti-AKT monoclonal antibody (rabbit, 1:1000, Abcam, ab314110), anti-phospho-AKT (Ser473) monoclonal antibody (rabbit, 1:1000, Abcam, ab81283), anti-PI3 Kinase p110 beta monoclonal antibody (rabbit, 1:1000, Abcam, ab302958), anti-DNA-PKcs polyclonal antibody (rabbit, 1:1000, Sigma, SAB4502385), anti-phospho-DNA-PKcs (Ser2056) polyclonal antibody (rabbit, 1:1000, Sigma, SAB4504169) and anti-GAPDH (rabbit, 1:5000, Abcam, ab8245).
:5000, Abcam, ab81299), anti-CD266 (Fn14, TWEAK Receptor) monoclonal antibody (ITEM-4) (mouse, 1:250, Abcam, ab109365), anti-AKT monoclonal antibody (rabbit, 1:1000, Abcam, ab314110), anti-phospho-AKT (Ser473) monoclonal antibody (rabbit, 1:1000, Abcam, ab81283), anti-PI3 Kinase p110 beta monoclonal antibody (rabbit, 1:1000, Abcam, ab302958), anti-DNA-PKcs polyclonal antibody (rabbit, 1:1000, Sigma, SAB4502385), anti-phospho-DNA-PKcs (Ser2056) polyclonal antibody (rabbit, 1:1000, Sigma, SAB4504169) and anti-GAPDH (rabbit, 1:5000, Abcam, ab8245).
2.2 Synthesis of Fn14-AuNPs
Synthesis of Fn14-AuNPs was schematically shown in Fig. 2a. Briefly, 1.8 ml dopamine at 4 mg ml−1 was incorporated into a mixture containing 27 ml Tris/HCl buffer (pH 8.5, 10 mM), 6 ml H2O, 0.9 ml PVP (2 mg ml−1, 10 kD) and 3 ml ethanol. Before adding 379 μl of HAuCl4 (24.085 mM), the mixture was stirred forcefully for 3 minutes. Then, the supernatant was abandoned and 3 ml bicine buffer (pH 8.5, 10 mM) was added to the rest. Next, 150 μl NH2-PEG-NH2 (5kD, 10 mg ml−1) was incorporated to react with succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate through sustainably stirring for 24 h under room temperature, which further connected to polypeptide D-FNB with cysteine terminal. After centrifugation (8000 rcf) for 5 min, the precipitation was dispersed to PBS.2.3 The TCGA analysis
From the TCGA database, 153 tumor samples in the glioblastoma datasets were obtained, accessible viahttps://portal.gdc.cancer.gov/repository. The obtained datasets were converted into transcripts per million values. Survival analyses were performed by the Cox proportional-hazards model. The hazard ratio was assessed relative to the lowest-risk group using a two-sided Wald test. The split Fn14 expression among GBM patients was established at 20%, with PFS ranging from 0 to 50 months and disease-specific survival (DSS) spanning 0 to 80 months.2.4 Cell culture and viability assay
The glioblastoma-derived human cell (U251 cell) was sourced from the National Collection of Authenticated Cell Cultures in Shanghai, China. The mouse-derived microglial cell (BV2 cell) was provided by Xiaorong Dong from Huazhong University of Science and Technology. The brain-derived endothelialcell.3 (bEnd.3 cell) was purchased from National Collection of Authenticated Cell Cultures (JSCALL, Shanghai, JSY-CC1889). As described previously, Fn14 overexpression (Fn14OE) U251 cells were constructed by using the lentiviral system.25 Cells were cultured with DMEM (GIBCO, USA) containing 10% fetal bovine serum (GIBCO, USA) and 5% streptomycin/penicillin, and maintained at 37 °C in a 5% CO2 atmosphere. Cell proliferation was evaluated by using Cell Counting Kit-8 (Beyotime, C0037, China). Cells were seeded at a density of 6 × 103 in 96-well plates and cultured for 24, 48, and 72 h respectively. Subsequently, 8 μl per well assay solution was added to 96 well, then incubated for 1 h. The measurement of absorbance at 450 nm was carried out using a Multimode Plate Reader (EnSpire® 2300, USA).2.5 Western blot analysis
Protein was extracted from U251 or Fn14OE U251 cells with RIPA buffer (Beyotime, P0013B) and separated by 10% SDS-PAGE gels. Subsequently, the protein-containing gels were transferred to polyvinylidene difluoride (PVDF, Bio-rad) membranes. Primary antibodies were mentioned below: anti-CD266 (Fn14, 1:250), anti-AKT (1:1000), anti-phospho-AKT (Ser473) (1:1000), anti-PI3K (1:1000), anti-DNA-PKcs (1:1000), anti-phospho-DNA-PKcs (Ser2056) (1:1000) and anti-GAPDH (1:5000).
2.6 Colony formation assay
U251-parental or U251-Fn14OE cells were planted in six-well plates at densities of 500, 1000, 2000, 4000, and 6000 per well, and then exposed to irradiation at doses of 0, 2, 4, 6, and 8 Gy using 6 MV X-rays at a rate of 600 cGy min−1 with the Trilogy System Linear Accelerator. After being exposed to IR, cells were cultured for approximately two weeks. Then, the cells were fixed using 4% paraformaldehyde (Beyotime, P0099) and stained using the crystal violet staining solution (Beyotime, C0121). The colonies with more than 50 cells were counted. Plating efficiency (%) = (number of colonies formed/number of cells plated) × 100. As previously described,26 the single-hit multi-target model was applied to compute the surviving fraction. To confirm the repeatability of the experiment, we verified our findings through three separate experiments.2.7 γ-H2AX assay
Cells were fixed using 4% paraformaldehyde, permeabilized using 0.2% Triton X-100 (Beyotime, P0096) and blocked using 5% BSA (Beyotime, ST023) at 0.5, 4 and 24 h after irradiation, respectively. The cells were then incubated overnight at 4 °C with anti-γ-H2AX (rabbit, Abcam, ab81299, 1:5000). The following day, cells were treated with a fluorescent secondary antibody (Abcam, ab150083, 1:1000) and subsequently stained with DAPI (1 mg ml−1) (Beyotime, C1005). γ-H2AX foci were detected using an excitation wavelength of 652 nm and an emission wavelength of 668 nm. In addition, images were captured at a high magnification (63× objective) with a laser confocal microscope (Nikon EZ-C1 Si, Japan). To define a focus and filter out noise, we established a brightness threshold of 100/255 on the original image, and the minimum spot size was set to 7 pixels. The quantifications of mean γ-H2AX foci numbers per cell were estimated from 3 different fields of view.
2.8 Real-time quantitative polymerase chain reaction (qRT-PCR)
To investigate the possibility that irradiation may upregulate the expression of Fn14 in GBM cells, U251 cells at a density of 0.5 × 105 per well were planted in six-well plates and cultured overnight and physically irradiated at 5, 10 Gy. Then, total RNA was isolated from cells on days 5 and 10 after irradiation via the RNA Extraction Kit (Omega, USA). The total RNA was subsequently reverse-transcribed into cDNA through HiScript III-RT SuperMix (Vazyme, China). qRT-PCR was conducted using ChamQ SYBR qPCR Master Mix (Vazyme, China). By the ΔΔCt method, the relative levels of mRNA for the target gene were computed. The primers for Fn14 are: sense 5′-CCA AGC TCC TCC AAC CAC AA-3′ and anti-sense 5′-TGG GGC CTA GTG TCA AGT CT-3′. GAPDH primers: sense strand 5′-ATC CCA TCA CCA TCT TCC-3′, anti-sense strand 5′-ATG ACC CTT TTG GCT CCC-3′.2.9 Cytotoxicity measurement
To enable cell attachment, Fn14OE U251 cells (2× 105 per well) were planted onto a 24-well micro-plate overnight. Subsequently, the initial culture media was substituted with fresh DMEM solutions containing Fn14-AuNPs (150 μg ml−1). The next day, cells were then separately irradiated via a 1064 nm laser (0.33 W cm−2, 15 min, controlled temperature at about 42.5 °C and 48 °C). To visually observe the killing effects, cells were stained with the live/dead staining kit (Beyotime, C2030S) for 30 min, washed with PBS three times and then imaged using a laser confocal microscope.
2.10 Cell cycle analysis
Fn14OE U251 cells were treated with Fn14-AuNPs (150 μg ml−1) for 24 h, followed by exposure to 4 Gy irradiation. The following day, the cells were fixed using 4% polyformaldehyde and stained using propidium iodide (20 μg ml−1) containing RNase A (0.2 mg ml−1) and 0.1% Triton X-100. Cell cycle analysis was conducted using FACS. ModFit LT software was utilized for analyzing the acquired data.2.11 In vitro BBB permeability analyses
In vitro BBB models were established using a 24-well transwell culture system as previously described.27 Briefly, bEnd.3 cells (1 × 105 per well) were seeded in the upper chamber with a pore diameter of 0.4 μm. The cells were cultured for 7–9 days to form an intact monolayer. The integrity of the BBB model was monitored by measuring transendothelial or transepithelial electrical resistance (TEER). Fn14OE glioma cells (5 × 104 per well) were seeded in the lower chamber one day before Fn14-AuNPs treatment. Subsequently, 100 μl (150 μg ml−1) AuNPs and Fn14-AuNPs were added to the apical upper chambers of the BBB models. Approximately 6 h after incubation, cells from lower chamber were detached from each well with trypsin and collected. The penetration across BBB and cellular uptake of Fn14-AuNPs were measured by transmission electron microscopy and inductively coupled plasma mass spectrometry (ICP-MS) analysis. For ICP-MS analysis, the trypsinized cells were then digested separately with aqua regia and analyzed for Au content.2.12 The biodistribution and biosafety evaluation of nanoparticles
AuPB (100 μl per mouse, 150 μg ml−1) and Fn14-AuPB (100 μl per mouse, 150 μg ml−1) were intravenously injected into the orthotopic U251-bearing BALB/c nude mice. Major organs, including the heart, spleen, liver, lung, kidney and cancerous brain, were collected at 24 h post-injection. ICP-MS was conducted to quantify the Au content of different organs. To validate the retention ability of Fn14-AuPB in vivo, we also injected AuPB (100 μl per mouse, 150 μg ml−1) and Fn14-AuPB (100 μl per mouse, 150 μg ml−1) through the tail vein on orthotopic U251 GBM-bearing mice. We collected their tumor tissues for ICP-MS analysis on days 1, 5, 10 and 15 after Fn14-AuPB injection.To assess the biosafety of Fn14-AuPB for their potential application in vivo, the injection of PBS, AuPB (100 μl per mouse, 150 μg ml−1) and Fn14-AuPB (100 μl per mouse, 150 μg ml−1) was conducted through the tail vein. The weights of mice were recorded every two days after intravenous injection. Moreover, after 14 days, mice were sacrificed for blood biochemistry, blood routine tests and histology analysis of major organs like the heart, spleen, liver, lung, kidney and brain via H&E staining. The serum, which was separated from blood samples by centrifuging at 1000 rcf for 10 min, was used for the blood biochemistry analysis for alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and creatine (CR). The whole blood was used for blood routine examination.
2.13 Tumor models and treatment
Female BALB/c nude mice with 5–6 weeks, were sourced from Changzhou Cavens Laboratory Animal Co., Ltd. The protocol for the animal study received approval from the Animal Ethics Committee at Huazhong University of Science and Technology (IACUC Number 3066). To construct the orthotopic U251-Luc or Fn14OE U251-Luc glioblastoma tumor-bearing mice models, 8 μl PBS containing 5 × 105 cancer cells was delivered into the mice's right caudate nucleus. Afterward, the tumor growth in mice was observed through bioluminescence imaging (Bruker, Ettlingen, Germany).For in vivo therapy, the mice were anesthetized intraperitoneally (xylazine 7.5 mg kg−1 + zoletil 40 mg kg−1) and injected intravenously with AuNPs or Fn14-AuNPs (150 μg ml−1) at day 20 after tumor inoculation. NIR-II irradiation (0.33 W cm−2, 6 min) was given on days 21, 22, 23, 24 and 25 after implantation. In the X-ray irradiation groups, mice were exposed to whole brain irradiation (2 Gy) within 10 min after mild photothermal therapy. Fractionated radiotherapy (10 Gy in total, 2 Gy per day for 5 days) was given on days 21, 22, 23, 24 and 25 after tumor implantation via 6 MV X-rays (source skin distance: 100 cm; radiation field: 40 × 4 cm; 600 cGy min−1, Trilogy System Linear Accelerator). The progression of GBM was monitored by bioluminescent imaging (intraperitoneal injection of 20 mg per ml D-luciferin) via in vivo MS FX PRO Imager. The mice were followed for survival and euthanized when neurological symptoms became evident, as described previously.28–30
2.14 Tissue immunohistochemical (IHC) and immunofluorescence staining
Histopathology test using Hematoxylin and Eosin Staining Kit (H&E staining Kit, Beyotime, C0105) staining was conducted on the heart, liver, kidney, brain, spleen and lung tissues of treated mice, as previously described.31 Murine glioma specimens for immunofluorescence on histological sections were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% BSA. Then the specimens were incubated with anti-mouse CD266 (1:100 dilution in 0.5% BSA/PBS) at 4 °C overnight while avoiding drying. Sections were washed three times using PBS, and followed by the application of secondary fluorescent antibodies. The slides were subjected to three PBS washes, stained with 200 μl of DAPI (1 mg ml−1) for 15 minutes and observed through a confocal microscope (Nikon EZ-C1 Si, Japan). The images of the slices were taken via the Image-Pro Plus 6.0.
2.15 Statistical analysis
Each experiment was conducted three times. The data were presented as the standard error of the mean (SEM) or as the mean ± standard deviation (SD). A one-way ANOVA was employed for multiple comparisons, while the chi-square test was utilized for analyzing clinical characteristics. Statistical significance was established at *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.3. Results and discussion
3.1 Synthesis and characterization of Fn14-AuNPs
Tumor heterogeneity is a substantial obstacle in traditional GBM treatment including radiotherapy.16 Thus, identifying and targeting glioma cell subpopulations associated with radioresistance can help improve the efficacy of radiotherapy in GBM. By analyzing the TCGA database of GBM patients, we discovered that high Fn14 expression group (Fn14hi group) had shorter PFS and DSS in GBM patients, compared with low expression group (Fn14lo group) (PFS:Fn14hi 5.03 months vs. Fn14lo 7.97 months, p = 0.034; DSS:Fn14hi 14.2 months vs. Fn14lo 16 months, p = 0.046) (Fig. S1a and b†). Together, these results suggested that elevated Fn14 expression was correlated with poor clinical outcomes in GBM patients. Fn14, part of the tumor necrosis factor receptor (TNFR) superfamily, is involved in tumor growth and treatment resistance.32–35 To explore the role of Fn14 in GBM radiotherapy, we overexpressed Fn14 (Fn14OE) in GBM U251 cells (Fig. 1a) and evaluated whether its overexpression enhanced the proliferation capacity of cells. The CCK-8 assay results displayed that there was no notable difference between the Ctrl and Fn14OE groups, indicating that Fn14 overexpression in GBM cells did not intensify cell proliferative capacity (Fig. S1c†). Subsequently, we employed colony formation assays to assess the reproductive death of glioma with different Fn14 expressions after treatment with IR. Fn14OE glioma cells were more resistant to IR treatment than their parental cells (Fig. 1b). To further elucidate the mechanism behind the survival of Fn14OE glioma cells, we investigated the DNA damage dynamics of glioma cells in response to irradiation by γ-H2AX foci assay.36 The γ-H2AX foci assay revealed that the basal level of γ-H2AX foci in Fn14OE glioma cells was lower than their parental U251 cells (Fig. 1c and d), which suggested a blunted early response to the induction of DNA double-strand breaks (DSBs). It should be noted that an accelerated descent of γ-H2AX foci in Fn14OE glioma cells in the subsequent post-incubation period (up to 24 h). In contrast, the sustained presence of γ-H2AX foci in parental U251 cells following irradiation indicated that these cells were relatively sensitive to DNA damage. | ||
| Fig. 1 High expression of Fn14 is correlated with radioresistance in glioblastoma. (a) Western blot for total Fn14 in U251 cells and Fn14OE U251 cells. (b) Clonogenic cell survival curve of U251 cells and Fn14OE U251 cells following different doses of radiation. (c) and (d) Fluorescence staining images and quantification of γ-H2AX foci in U251 cells and Fn14OE U251 cells under radiation (IR: 4 Gy) (Scale bar: 20 μm (upper row) and 10 μm (lower row). (e) Western blot analysis of PI3K, AKT, p-AKT, DNA-PKcs and p-DNA-PKcs in U251 cells and Fn14OE U251 cells after irradiation (IR: 4 Gy). (f) Fluorescence staining images of Fn14 in the brain tissues from irradiated-mice at different time points (day 20, 30 and 40 post-implantation of GBM; scale bar: 100 μm and 50 μm). (g) The mean fluorescence intensity of Fn14 was measured. Data are presented as mean ± SD, with n = 3 per group. Statistical significance is indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Previous evidence suggested that Fn14 positively correlated with the activation of AKT signaling in GBM specimens.37 Moreover, the abnormal activation of the AKT signaling can promote DNA DSB repair, leading to the enhanced radioresistance of GBM cells.38 Hence, we hypothesized that Fn14 reduced the radiosensitivity of GBM cells by aberrant AKT signaling activation. Consistent with our hypothesis, expression levels of both phosphorylated AKT (p-AKT) and phosphorylated DNA-PKcs (p-DNA-PKcs) were elevated in Fn14OE U251 compared to those in parental U251 cells (Fig. 1e). It is likely that Fn14 overexpression could aberrantly activate DNA damage checkpoint response, resulting in radioresistance of GBM. To explore the dynamics of tumor size with different Fn14 expression in response to irradiation, we constructed orthotopic xenograft GBM mice models by intracranially injecting a 9:1 mixture of U251-parental and U251-Fn14OE cells (Fig. S1d†). The tumor-bearing mice underwent fractionated radiotherapy (10 Gy in 5 fractions). Bioluminescent imaging demonstrated a substantial reduction in tumor size on day 30 after implantation, and a subsequent increase in volume on day 40 after implantation after IR (Fig. S1e and f†). In addition, we found that radiation treatment substantially prolonged the survival of glioma-bearing mice by 38.5% (Fig. S1g†). Immunofluorescence staining revealed a dramatically increased proposition of Fn14+ glioma in residual tumors after irradiation (up to 40.0%) (Fig. 1f and g). Furthermore, over 51.4% of tumor cells were Fn14+ cells within the residual foci in mice treated with irradiation. Overall, these data indicated that Fn14+ GBM cells were the predominant population in residual sites after radiotherapy.
To rule out the possibility that radiation may upregulate Fn14 gene expression in GBM cells, we detected the Fn14 expression at different time points after receiving irradiation (Fig. S1h†). However, the results suggested that irradiation had no considerable effect on the expression of Fn14 in U251 cells. Thus, one alternative explanation is that Fn14+ cells enriched in GBM after irradiation are intrinsically resistant to radiotherapy.
The nanoplatform containing an AuNP core and a mesoporous polydopamine (mPDA) shell has been established in our previous work, showing efficient photothermal conversion and peptide cargo loading.22,39,40 Herein, we designed peptide-functionalized AuNPs to target Fn14 positive subpopulation and enhance cellular uptake. As described previously, AuNPs were synthesized using the one-pot seedless and surfactant-free method.41 The D-enantiomeric peptide ligand of Fn14 (D-FNB, sequenced as DCDHDPDRDEDVDDDVDEDLDYDSDTDVDFDGDH) was artificially synthesized and mixed with the AuNPs solution, forming Fn14-AuNPs nanoparticles (Fig. 2a). During the manufacturing process, H2N-PEG-NH2 (MW = 5 kDa) was added to improve the colloidal stability of AuNPs in blood circulation and facilitate the conjugation of D-FNB onto AuNPs. In addition, previous studies showed that PEG surface modification reduced protein corona formation and RES clearance, resulting in superior pharmacokinetic and bioavailability properties.42,43
 | ||
| Fig. 2 Characterization of Fn14-AuNPs. (a) Illustrative diagram for the synthesis of Fn14-AuNPs. (b) and (c) TEM images of Fn14-AuNPs (scale bar: 500 nm and 50 nm). (d) Absorption spectra of Fn14-AuNPs (50 μg ml−1). (e) Laser irradiation at 1064 nm and 1.0 W cm−2 increased the temperature of Fn14-AuNPs solutions at concentrations of 10, 20, and 50 μg ml−1. (f) Laser irradiation using different power intensities (0.3, 0.5 and 1.0 W cm−2) caused temperature increases in the Fn14-AuNPs solution (50 μg ml−1). (g) Heating–cooling curve of Fn14-AuNPs solution (50 μg ml−1) under repeated laser exposure (1.0 W cm−2). | ||
Transmission electron microscopy (TEM) images demonstrated that the structure of Fn14-AuNPs was a hyperbranched and compact framework (Fig. 2b and c). The plasmonic coupling between adjacent branches endowed Fn14-AuNPs absorption across UV-vis-NIR spectra (Fig. 2d). Furthermore, the high absorption and scattering ratio of Fn14-AuNPs, attributed to their hyperbranched structure, led to superior photothermal conversion efficiency. The temperature variations of Fn14-AuNPs solutions were recorded at several concentrations (10, 20 and 50 μg ml−1) with the maximum permission exposure (MPE) of NIR-II laser (1.0 W cm−2, 1064 nm). Within 10 min, the temperature of Fn14-AuNPs (50 μg ml−1) increased rapidly by 17.5 °C (Fig. 2e). It was also exhibited in Fig. 2f that the temperature of Fn14-AuNPs solution (50 μg ml−1) ascended with irradiation time when received to different power intensities (0.33, 0.5 and 1.0 W cm−2) of NIR-II laser. To test the photothermal stability of Fn14-AuNPs, this irradiation was applied to the Fn14-AuNPs solution (50 μg ml−1) for about 10 min (1.0 W cm−2) and repeated for five cycles (Fig. 2g). The peak temperature of Fn14-AuNPs solution in every heating–cooling cycle was retained identically, strongly indicating its photothermal and structural stability.
3.2 Fn14-AuNPs enhanced the radiosensitivity of Fn14OE glioma cells in vitro
The therapeutic efficacy of Fn14-AuNPs will be significantly affected by the tumor targeting. Thus, we first evaluated the ability of Fn14-AuNPs to target Fn14OE glioma cells. Fn14OE U251 cells were co-cultured with Fn14-AuNPs (150 μg ml−1) for 2 h. TEM imaging revealed that Fn14-AuNPs exhibited enhanced cell membrane adherence and internalization compared to the AuNPs group, implying a remarkable capacity for targeting Fn14-AuNPs in vitro (Fig. S2a†). The introduction of nanoparticles with high atomic numbers into tumors has been developed for enhanced radiosensitivity due to their enzyme-like catalytic activity, production of ROS, disruption of radiation-induced DNA damage repair, induction of G2/M cell cycle arrest and inhibition of hypoxia.44,45 To this point, we evaluated the potential radiosensitizing effect of Fn14-AuNPs on Fn14OE U251 cells. As shown in Fig. 3a and b, an increased γ-H2AX foci formation upon IR was noted in Fn14OE cells pre-treated with Fn14-AuNPs when compared to the other groups, indicating an enhanced radiosensitivity associated with Fn14-AuNPs. In line with these results, the colony formation assay also exhibited that Fn14-AuNPs nanoparticles could suppress the Fn14OE U251 cell proliferation (Fig. 3c). Previous studies have shown that gold nanoparticles can promote radiation-induced G2/M arrest, thereby enhancing radiosensitivity.46,47 Therefore, we subsequently assessed the cell cycle distribution with flow cytometry. Compared to Ctrl groups, the G2/M arrest was dramatically induced by pretreatment with Fn14-AuNPs (23.5% of Fn14-AuNPs vs. 18.1% of Ctrl, *p < 0.05) (Fig. 3d and e). Taken together, these results verified the potential of FN14-AuNPs nanoparticles to radiosensitize Fn14OE U251 cells in vitro.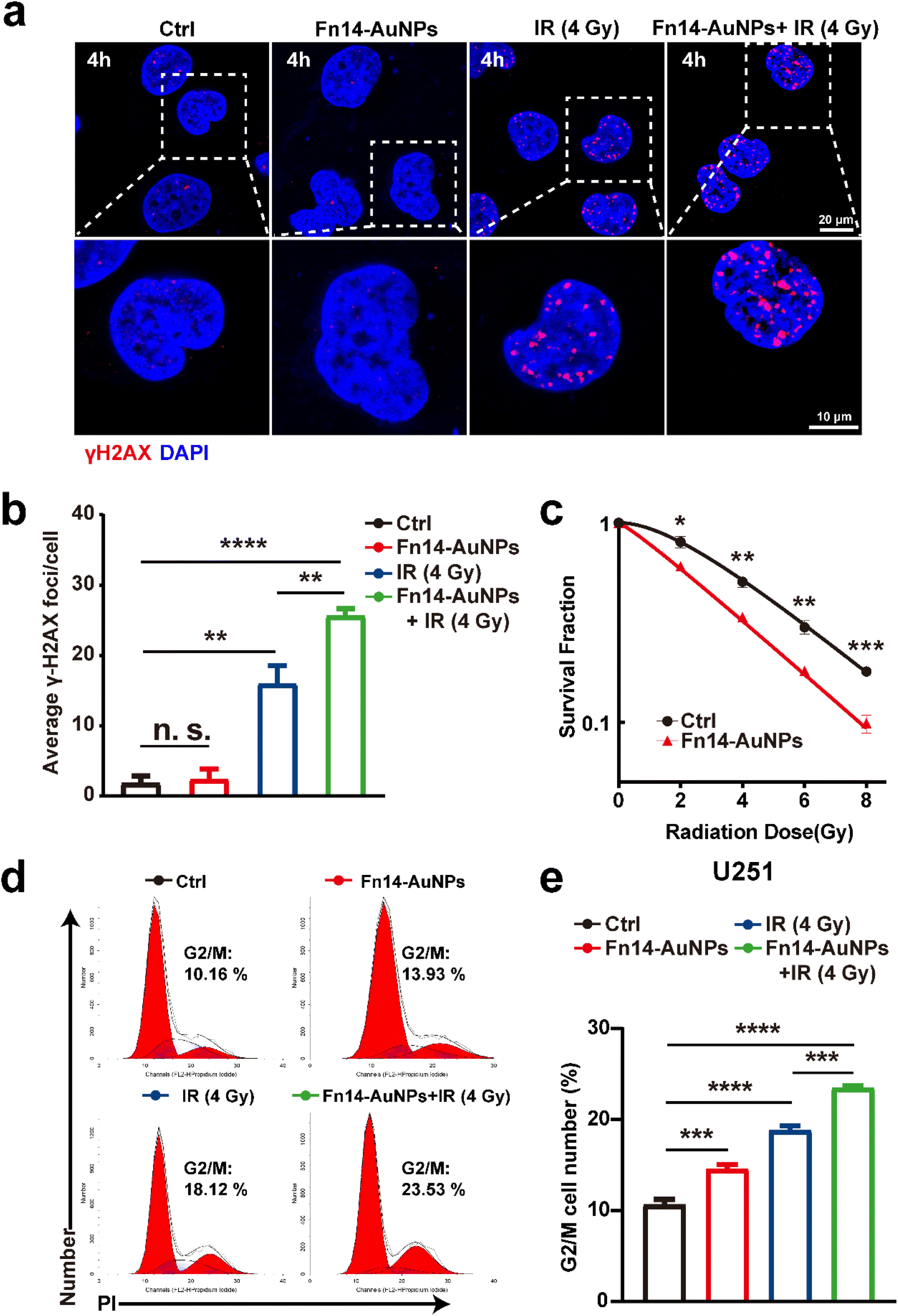 | ||
| Fig. 3 Fn14-AuNPs enhanced the radiosensitivity of Fn14OE glioma cells in vitro. (a) Fluorescence staining images and (b) quantification of γH2AX foci in Fn14OE U251 cells at 4 h after different treatments (Ctrl, Fn14-AuNPs, IR (4 Gy), and Fn14-AuNPs + IR (4 Gy); scale bar: 20 μm (upper row) and 10 μm (lower row). (c) Clonogenic survival assay of Fn14OE U251 cells treated with Fn14-AuNPs. Fn14OE U251 cells were exposed to physical radiation doses of 0 Gy, 2 Gy, 4 Gy, 6 Gy, and 8 Gy, and the cell colonies with ≥50 cells were statistically analyzed after 2 weeks. (d) Representative flow cytometry histogram of cell-cycle analysis in Fn14OE U251 cells after 24 h of indicated treatments: Ctrl, Fn14-AuNPs, IR (4 Gy), and Fn14-AuNPs + IR (4 Gy). (e) Quantification of the cell distribution in the G2/M phase of the cell cycle. Data are presented as mean ± SD, with n = 3 per group. Statistical significance is indicated as follows: n. s. (no significance), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
3.3 Fn14-AuNPs-mediated mPTT further increased the radiosensitivity of Fn14OE glioma cells in vitro
Mild photothermal therapy (mPTT), which converts light energy to mild heat energy, is an emerging strategy that renders cancer cells more vulnerable to treatment modalities including radiotherapy, chemotherapy and immunotherapy.48–50 Lower hyperthermia (<45 °C) can avoid collateral damage to healthy tissues and tumor metastasis resulting from excessive hyperthermia during cancer therapy. Notably, recent research suggested that mPTT can disrupt DNA damage repair pathways by affecting several critical repair mechanisms, including translesion DNA synthesis, nucleotide excision repair,51 base excision repair,52 mismatch repair,53 homologous recombination and non-homologous end joining.54 mPTT is expected to enhance the efficacy of genotoxic therapies such as radiotherapy by preventing the repair of radiation-induced damage.50 Thus, to investigate whether Fn14-mediated mPTT can augment the radiosensitivity of Fn14OE U251 cells, we treated Fn14OE U251 cells with Fn14-AuNPs for 24 h, followed by laser irradiation. An infrared thermal camera was employed to monitor the photothermal temperature in real time. The colony formation assay confirmed that pretreatment with Fn14-AuNPs-based mPTT suppressed cell proliferation (Fig. 4a and b). Similarly, the cell cycle distribution in Fig. 5c and d demonstrated that pretreatment with mPTT significantly induced G2/M arrest and inhibited cell proliferation caused by irradiation (29.9% in the thermoradiotherapy group vs. 17.6% in the radiotherapy-only group, *p < 0.05). The cell viability was 95% in the group treated with mPTT (42.5 °C), whereas the viability of the cells treated with high-temperature PTT (48 °C) decreased to 20.0% (Fig. S2b†). Live/dead co-staining (live cells with AM; dead cells with PI) also demonstrated a comparable pattern (Fig. S2c†). These data indicated that mPTT alone cannot directly kill tumor cells or cause any DNA DSBs. Notably, we observed a significant outcome in the Fn14-AuNPs + mPTT + IR group, where cells were irradiated following hyperthermia (approximately an interval of 10 minutes between the two treatments). And that was a remarkable increase in the level of γ-H2AX in the thermoradiotherapy group (Fig. 4e and f). These results implied that Fn14-AuNPs-based mPTT could efficiently promote radiation-induced DNA DSBs and prevent DNA damage repair, rather than directly eliminate tumor cells. | ||
| Fig. 4 Fn14-AuNPs-mediated mPTT further increased the radiosensitivity of Fn14OE glioma cells in vitro. (a) and (b) representative images of colony formation and colony count of Fn14OE U251 cells in Ctrl, Fn14-AuNPs + mPTT, IR (4 Gy), Fn14-AuNPs + mPTT + IR (4 Gy) groups.(c) and (d) Cell cycle plot and its quantification displaying the percentage of G2/M phase in Fn14OE U251 cells after 24 h of indicated treatments (Ctrl, Fn14-AuNPs + mPTT, IR (4 Gy), Fn14-AuNPs + mPTT + IR (4 Gy)) (e) and (f) fluorescence staining images and quantification of γ-H2AX foci in Fn14OE U251 cells at 4 h after different treatments: Ctrl, Fn14-AuNPs + mPTT, IR (4 Gy), Fn14-AuNPs + mPTT + IR (4 Gy) groups scale bar: 20 μm (upper row) and 10 μm (lower row). Data are presented as mean ± SD, with n = 3 per group. Statistical significance is indicated as follows: n. s. (no significance), **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
 | ||
| Fig. 5 The BBB-crossing ability of Fn14-AuNPs in vitro and in vivo. (a) Schematic representation of the in vitro BBB model. (b) Representative TEM images of the uptake of nanoparticles in Fn14OE U251 cells across different groups (PBS, AuNPs, Fn14-AuNPs, AuNPs + NIR, Fn14-AuNPs + NIR). Scale bar: 5 μm and 500 nm. (c) The intracellular gold accumulation in Fn14-Fn14OE glioma cells after 6 h of different treatments: PBS (Ctrl), AuNPs, Fn14-AuNPs, AuNPs with NIR and Fn14-AuNPs with NIR. (d) The ICP-MS analysis of Fn14-AuNPs accumulation in major organs and tumors from AuNPs and Fn14-AuNPs injected orthotopic tumor-bearing mice in vivo. (e) Retention ability of AuNPs and Fn14-AuNPs in tumor tissues of Fn14OE U251 cells orthotopic tumor-bearing murine model after tail vein injection quantified by ICP-MS at different time points. Data are presented as mean ± SD, with n = 3 per group. Statistical significance is indicated as follows: n. s. (no significance), *p < 0.05, **p < 0.01, ****p < 0.0001. | ||
A study has revealed that applying hyperthermia before radiotherapy can effectively suppress the activation of AKT signaling in GBM cells.15 Moreover, another research has reported that the abnormal activation of the AKT signaling was strongly associated with radioresistance in GBM.38 Based on these, we speculated that mPTT sensitized GBM cells to radiation by suppressing the abnormal activation of the AKT signaling pathway. Consistent with our hypothesis, the Fn14-AuNPs + mPTT + IR group exhibited reduced expression levels of p-AKT and p-DNA-PKcs compared to other IR-treated groups (Fig. S2d†). This indicated that Fn14-AuNPs-mediated mPTT could radiosensitize Fn14OE U251 cells by inhibiting the DNA damage checkpoint response.
3.4 The tumor targeting and BBB-crossing capabilities of Fn14-AuNPs in vitro and in vivo
The BBB, a selective barrier formed by endothelial cells, astrocytes, pericytes, and tight junctions also restricts the entry of drugs into the brain parenchyma, eventually leading to the failure of cancer treatments.55,56 To assess the BBB penetration capability of Fn14-AuNPs, we constructed a transwell assay consisting of bEnd.3 cells to mimic the BBB as illustrated in Fig. 5a. The high TEER of approximately 150 Ω cm2 confirmed the integrity of the BBB model for studying Fn14-AuNPs transport57–59 (Fig. S2e†). In this model, the TEM imaging displayed that Fn14-AuNPs exhibited enhanced cell membrane adherence and internalization compared to the AuNPs and PBS groups in Fn14OE glioma cells (Fig. 5b). Moreover, the ICP-MS analysis demonstrated a higher upake of Fn14-AuNPs by Fn14OE glioma cells compared to the AuNPs and PBS groups (Fig. 5c). These results indicated that Fn14-AuNPs had a superior BBB penetration ability in vitro. Previous studies have shown that AuNPs can cross BBB via passive diffusion in Transwell and Microfluidic models in vitro, owing to their small size of less than 50 nm.24,60–62 Therefore, the excellent BBB penetration capability of Fn14-AuNPs may be attributed to their small particle size, which makes it easier for them to traverse the gaps between endothelial cells.To further explore the tumor targeting and BBB-crossing capabilities in vivo, the biodistribution of AuNPs and Fn14-AuNPs was investigated by ICP-MS 24 h after injection of Fn14-AuNPs (150 μg ml−1, 100 μl per mouse) in Fn14OE U251 orthotopic tumor-bearing mice. The accumulation of elemental Au in the brain tumor 24 h after Fn14-AuPB treatment (Fig. 5d) indicated that Fn14-AuNPs also had an excellent BBB penetration ability in vivo. In addition, to evaluate the retention time of AuNPs and Fn14-AuNPs, tumor tissues were harvested from orthotopic Fn14OE U251 tumor-bearing mice at 1, 5, 10, and 15 days following AuNPs and Fn14-AuNPs injection. To evaluate nanoparticle accumulation in tissues, the elemental Au concentration was measured through ICP-MS analysis. As shown in Fig. 5e, the retention time of Fn14-AuNPs or AuNPs in tumors exceeded 15 days, confirming their long-term aggregation at the targeted location following a single dose of Fn14-AuNPs or AuNPs. Overall, these results indicated that Fn14-AuNPs had excellent BBB penetration and tumor-targeting abilities both in vitro and in vivo.
3.5 Biosafety assessment of Fn14-AuNPs
The biosafety of Fn14-AuNPs was assessed both in vitro and in vivo to ascertain their suitability for anticancer therapies. The cytotoxicity of Fn14-AuNPs was evaluated in human GBM cells (Fn14OE U251) and murine microglia cells (BV2) after incubation with Fn14-AuNPs for 24 h by CCK-8 assay. As shown in Fig. S3a and b,† cell viability was maintained at 94% in both Fn14OE U251 and BV2 cells, suggesting that the Fn14-AuNPs had no noticeable cytotoxicity in vitro. To assess the in vivo toxicity of Fn14-AuNPs, mice were divided into PBS group (100 μl per mouse), AuNPs group (100 μl per mouse, 150 μg ml−1), or Fn14-AuNPs group (100 μl per mouse, 150 μg ml−1) (n = 3/group). The weight of mice was measured every two days within 2 weeks. No body weight loss was observed in any of the groups (Fig. S3c†). We further investigated the biocompatibility of AuNPs or Fn14-AuNPs via the H&E staining and blood biochemistry on healthy tissues. Two weeks after the injection, blood chemistry and complete blood count tests were performed. Quantitative analysis of blood tests revealed no notable differences between the Ctrl (PBS) and nanoparticle-treated groups (Fig. S3d and e†). These findings verified that repeated nanoparticle treatments had no harm to the liver, kidney or hematological parameters. Meanwhile, samples from major organs (heart, liver, kidney, spleen and lung) were collected two weeks after injection and subjected to H&E staining. Tissue histopathology indicated that there was no inflammation, ischemia, necrosis, or other histological or architectural irregularities in these organs, indicating that they were not affected by AuNPs or Fn14-AuNPs and maintained physiological condition (Fig. S3f†). These results confirmed the nontoxicity of AuNPs or Fn14-AuNPs in mice.3.6 In vivo Fn14-AuNPs-mediated mPTT suppressed GBM progression
To assess the synergistic efficiency of radiotherapy combined with Fn14-AuNPs + mPTT, orthotopic Fn14OE U251 tumor-bearing mice were randomly allocated into six groups: (i) Ctrl; (ii) Fn14-AuNPs (150 μg ml−1, 100 μl per mouse); (iii) Fn14-AuNPs + mPTT (0.33 W cm−2, 6 min); (iv) IR (10 Gy in total, 2 Gy per day for 5 days); (v) Fn14-AuNPs + IR; (vi) Fn14-AuNPs + mPTT plus IR. The treatment strategy was represented in Fig. 6a. Mice received a dose of Fn14-AuNPs (intravenous injection, i.v) followed by exposure to 1064 nm laser irradiation (0.33 W cm−2, 6 min). During the treatment, the local temperature in tumor-bearing mice administered AuNPs or Fn14-AuNPs was monitored. Within 6 minutes, the temperature in the tumor region injected with Fn14-AuNPs increased quickly from 36.6 °C to 42.5 °C (Fig. 6b). The treatment temperature was sustained at about 42.5 °C. Radiotherapy commenced within 10 minutes following the completion of mPTT. The therapeutic effect was assessed by the tumor growth (bioluminescence signals from Fn14OE U251 cells) and overall survival rates. Tumor progression was considerably inhibited after treatment with IR, Fn14-AuNPs + IR and Fn14-AuNPs + mPTT + IR, as evidenced by lower tumorous luciferase intensity (Fig. 6c and 5e) and H&E images (Fig. 6d). Compared to the other groups, the Fn14-AuNPs + mPTT + IR group achieved the most prominent antitumor effects with no obvious neurological symptoms observed at 50 days after tumor implantation. Consequently, the group of mice treated with Fn14-AuNPs + mPTT + IR exhibited a dramatically prolonged lifespan. Fn14-AuNPs + mPTT + IR extended the median survival of Fn14OE U251-bearing mice to 62 days, a 1.8-fold increase in comparison to the Ctrl group. As shown by Fig. 6f, the other groups without IR including Ctrl, Fn14-AuNPs, and Fn14-AuNPs + mPTT illustrated a limited lifespan, with a median survival of only 34.6 days. IR and Fn14-AuNPs + IR treatment moderately improved survival to 45.8 days, and 53.2 days respectively. In summary, these observations implied that Fn14-AuNPs-mediated mPTT boosted the antitumor therapeutic effect of radiotherapy. | ||
| Fig. 6 In vivo Fn14-AuNPs-mediated mPTT suppressed GBM progression (a) a diagram illustrating the experimental design in vivo. (b) Representative images of the temperature elevation during photothermal therapy (n = 6 per group). (c) Representative bioluminescence assay images of various treatment groups (n = 4 per group). (d) H&E images of mice brain tissues with tumors from various groups were acquired 10 days after tumor implantation (scale bar: 3 mm). (e) The quantitative signal intensity of bioluminescence assay. (f) Kaplan–Meier survival curves for mice intracranially injected with Fn14OE U251 cells and subjected to various treatments (n = 6 per group). Statistical significance was assessed using an unpaired Student's t-test, with results presented as mean ± SD (n. s., no significance, **p < 0.01, ***p < 0.001). | ||
4. Conclusions
In summary, our study revealed that high Fn14 expression correlated with poor clinical outcomes in GBM patients. Furthermore, Fn14+ glioma cells predominated in the residual sites of GBM following radiotherapy, potentially due to the preferential activation of DNA damage checkpoint response. Based on these findings, we developed a photonic and targeted nanoparticle designated as Fn14-AuNPs. The AuNPs core provided the foundation for light-to-heat conversion, while the D-FNB served as the targeting ligand, facilitating the attachment of Fn14-AuNPs to Fn14-expressing glioma cells. Fn14-AuNPs also had the excellent BBB-crossing capabilities and the advantage of sustained retention at targeted sites, along with their established biocompatibility and biosafety. The properties of the gold nanoparticles made Fn14-AuNPs ideal candidates as radiosensitizers. Notably, the therapeutic combination of IR + Fn14-AuNPs and mPTT further boosted the radiosensitivity of Fn14-positive cells because of the mPTT-activated DNA damage checkpoint response. In vivo, utilization of IR + Fn14-AuNPs + mPTT also remarkably suppressed tumor growth. Overall, given the significant role of Fn14 in the radiosensitivity of glioma cells, the combined utilization of IR and Fn14-AuNPs-mediated mPTT is anticipated to be a novel therapeutic approach to enhance radiosensitivity and improve the efficiency of radiotherapy in GBM.Ethical statement
Animal experiments adhered to the ethical guidelines of the Animal Ethics Committee at Tongji Medical College, Huazhong University of Science and Technology (IACUC Number 3066).Consent for publication
The authors agree to publication.Data availability
All data regarding this manuscript are already shown in the graphs of the main paper.Author contributions
The authors confirmed their contribution to the paper as follows: Wei Fu, Jinghua Ren designed the experiments. Qing Liang and Wei Fu performed experiments and collected data. Qing Liang, Wei Fu and Shiqiong Lei, Ruiqi Li, Xin Zheng and Lian Chen contributed to data analysis. Qing Liang, Wei Fu, Yuxi Ma and Jinghua Ren wrote the manuscript. Xiaofang Dai, Hongwei Duan, Wenshan He and Jinghua Ren supervised, directed, and managed the study.Conflicts of interest
There are no competing interests.Acknowledgements
This work was supported by Natural Science Foundation of Hubei Province (No. 2020CFB786), National Natural Science Foundation of China (No. 82072944, 81874084, 82072800 and 82103385), and Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20210324141802006).References
- L. Rong, N. Li and Z. Zhang, J. Exp. Clin. Cancer Res., 2022, 41, 142 CrossRef CAS PubMed.
- C. W. Brennan, R. G. Verhaak, A. McKenna, B. Campos, H. Noushmehr, S. R. Salama, S. Zheng, D. Chakravarty, J. Z. Sanborn, S. H. Berman, R. Beroukhim, B. Bernard, C. J. Wu, G. Genovese, I. Shmulevich, J. Barnholtz-Sloan, L. Zou, R. Vegesna, S. A. Shukla, G. Ciriello, W. K. Yung, W. Zhang, C. Sougnez, T. Mikkelsen, K. Aldape, D. D. Bigner, E. G. Van Meir, M. Prados, A. Sloan, K. L. Black, J. Eschbacher, G. Finocchiaro, W. Friedman, D. W. Andrews, A. Guha, M. Iacocca, B. P. O'Neill, G. Foltz, J. Myers, D. J. Weisenberger, R. Penny, R. Kucherlapati, C. M. Perou, D. N. Hayes, R. Gibbs, M. Marra, G. B. Mills, E. Lander, P. Spellman, R. Wilson, C. Sander, J. Weinstein, M. Meyerson, S. Gabriel, P. W. Laird, D. Haussler, G. Getz and L. Chin, Cell, 2013, 155, 462–477 CrossRef CAS PubMed.
- V. M. Ravi, P. Will, J. Kueckelhaus, N. Sun, K. Joseph, H. Salié, L. Vollmer, U. Kuliesiute, J. von Ehr, J. K. Benotmane, N. Neidert, M. Follo, F. Scherer, J. M. Goeldner, S. P. Behringer, P. Franco, M. Khiat, J. Zhang, U. G. Hofmann, C. Fung, F. L. Ricklefs, K. Lamszus, M. Boerries, M. Ku, J. Beck, R. Sankowski, M. Schwabenland, M. Prinz, U. Schüller, S. Killmer, B. Bengsch, A. K. Walch, D. Delev, O. Schnell and D. H. Heiland, Cancer Cell, 2022, 40, 639–655.e613 CrossRef CAS PubMed.
- K. Yang, Z. Wu, H. Zhang, N. Zhang, W. Wu, Z. Wang, Z. Dai, X. Zhang, L. Zhang, Y. Peng, W. Ye, W. Zeng, Z. Liu and Q. Cheng, Mol. Cancer, 2022, 21, 39 CrossRef CAS PubMed.
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS PubMed.
- T. Kondo, Semin. Cancer Biol., 2022, 82, 176–183 CrossRef CAS PubMed.
- E. Verdugo, I. Puerto and M. Medina, Cancer Commun., 2022, 42, 1083–1111 CrossRef PubMed.
- D. Nehama, A. S. Woodell, S. M. Maingi, S. D. Hingtgen and G. Dotti, Neuro Oncol., 2023, 25, 1551–1562 CrossRef CAS PubMed.
- A. Marusyk, M. Janiszewska and K. Polyak, Cancer Cell, 2020, 37, 471–484 CrossRef CAS PubMed.
- M. J. van den Bent, M. Geurts, P. J. French, M. Smits, D. Capper, J. E. C. Bromberg and S. M. Chang, Lancet, 2023, 402, 1564–1579 CrossRef PubMed.
- Y. Wu, Y. Song, R. Wang and T. Wang, Mol. Cancer, 2023, 22, 96 CrossRef CAS PubMed.
- D. Antoni, L. Feuvret, J. Biau, C. Robert, J. J. Mazeron and G. Noël, Cancer Radiother., 2022, 26, 116–128 CrossRef CAS PubMed.
- L. Barthel, M. Hadamitzky, P. Dammann, M. Schedlowski, U. Sure, B. K. Thakur and S. Hetze, Cancer Metastasis Rev., 2022, 41, 53–75 CrossRef CAS PubMed.
- Y. A. Yabo, S. P. Niclou and A. Golebiewska, Neuro Oncol., 2022, 24, 669–682 CrossRef CAS PubMed.
- J. Man, J. D. Shoemake, T. Ma, A. E. Rizzo, A. R. Godley, Q. Wu, A. M. Mohammadi, S. Bao, J. N. Rich and J. S. Yu, Cancer Res., 2015, 75, 1760–1769 CrossRef CAS PubMed.
- A. Ou, W. K. A. Yung and N. Majd, Int. J. Mol. Sci., 2020, 22, 351 CrossRef PubMed.
- L. C. Burkly, Semin. Immunol., 2014, 26, 229–236 CrossRef CAS PubMed.
- J. G. Perez, N. L. Tran, M. G. Rosenblum, C. S. Schneider, N. P. Connolly, A. J. Kim, G. F. Woodworth and J. A. Winkles, Oncogene, 2016, 35, 2145–2155 CrossRef CAS PubMed.
- H. Wajant, Br. J. Pharmacol., 2013, 170, 748–764 CrossRef CAS PubMed.
- C. Mochizuki, J. Nakamura and M. Nakamura, ACS Omega, 2023, 8, 9569–9582 CrossRef CAS PubMed.
- J. Atkinson, J. Chopin, E. Bezak, H. Le and I. Kempson, Chemphyschem, 2024, 26, e202400764 CrossRef PubMed.
- Q. Liang, J. Chen, S. Hou, D. Li, Y. Zhu, R. Li, L. Chen, J. Li, W. Fu, S. Lei, B. Zhang, X. Zheng, T. Zhang, H. Duan, W. He and J. Ren, Biomaterials, 2023, 300, 122206 CrossRef CAS PubMed.
- S. Zha, H. Liu, H. Li, H. Li, K. L. Wong and A. H. All, ACS Nano, 2024, 18, 1820–1845 CrossRef CAS PubMed.
- Y. Fan, C. Xu, N. Deng, Z. Gao, Z. Jiang, X. Li, Y. Zhou, H. Pei, L. Li and B. Tang, Lab Chip, 2023, 23, 1935–1944 RSC.
- S. Chen, J. Liu, M. Yang, W. Lai, L. Ye, J. Chen, X. Hou, H. Ding, W. Zhang, Y. Wu, X. Liu, S. Huang, X. Yu and D. Xiao, PLoS One, 2015, 10, e0143802 Search PubMed.
- S. Zhang, Y. Hu, Z. Wu, X. Zhou, T. Wu, P. Li, Q. Lian, S. Xu, J. Gu, L. Chen, G. Wu, T. Zhang, J. Tang and J. Xue, Int. J. Radiat. Oncol. Biol. Phys., 2023, 115, 1244–1256 CrossRef PubMed.
- J. H. Pedder, A. M. Sonabend, M. D. Cearns, B. D. Michael, R. Zakaria, A. B. Heimberger, M. D. Jenkinson and D. Dickens, Lancet Neurol., 2025, 24, 246–260 CrossRef CAS PubMed.
- S. Sato, Y. J. Tang, Q. Wei, M. Hirata, A. Weng, I. Han, A. Okawa, S. Takeda, H. Whetstone, P. Nadesan, D. G. Kirsch, J. S. Wunder and B. A. Alman, Cell Rep., 2016, 16, 917–927 CrossRef CAS PubMed.
- S. Liu, F. Yin, W. Fan, S. Wang, X. R. Guo, J. N. Zhang, Z. M. Tian and M. Fan, J. Exp. Clin. Cancer Res., 2012, 31, 52 CrossRef CAS PubMed.
- U. Ahmadov, D. Picard, J. Bartl, M. Silginer, M. Trajkovic-Arsic, N. Qin, L. Blümel, M. Wolter, J. K. M. Lim, D. Pauck, A. M. Winkelkotte, M. Melcher, M. Langini, V. Marquardt, F. Sander, A. Stefanski, S. Steltgens, C. Hassiepen, A. Kaufhold, F. D. Meyer, A. Seibt, L. Kleinesudeik, A. Hain, C. Münk, C. B. Knobbe-Thomsen, A. Schramm, U. Fischer, G. Leprivier, K. Stühler, S. Fulda, J. T. Siveke, F. Distelmaier, A. Borkhardt, M. Weller, P. Roth, G. Reifenberger and M. Remke, Cell Death Dis., 2021, 12, 885 Search PubMed.
- R. Li, L. Chen, Q. Ji, Q. Liang, Y. Zhu, W. Fu, T. Chen, H. Duan, W. He, Z. Xu, X. Dai and J. Ren, Adv. Funct. Mater., 2023, 33, 2213292 CrossRef CAS.
- S. P. Fortin, M. J. Ennis, B. A. Savitch, D. Carpentieri, W. S. McDonough, J. A. Winkles, J. C. Loftus, C. Kingsley, G. Hostetter and N. L. Tran, Mol. Cancer Res., 2009, 7, 1871–1881 CrossRef CAS PubMed.
- C. S. Schneider, J. G. Perez, E. Cheng, C. Zhang, P. Mastorakos, J. Hanes, J. A. Winkles, G. F. Woodworth and A. J. Kim, Biomaterials, 2015, 42, 42–51 Search PubMed.
- D. S. Hersh, B. G. Harder, A. Roos, S. Peng, J. E. Heath, T. Legesse, A. J. Kim, G. F. Woodworth, N. L. Tran and J. A. Winkles, Neuro Oncol., 2018, 20, 1321–1330 CrossRef CAS PubMed.
- J. A. Winkles, N. L. Tran and M. E. Berens, Cancer Lett., 2006, 235, 11–17 CrossRef CAS PubMed.
- M. Gerić, G. Gajski and V. Garaj-Vrhovac, Ecotoxicol. Environ. Saf., 2014, 105, 13–21 CrossRef PubMed.
- M. Martini, M. C. De Santis, L. Braccini, F. Gulluni and E. Hirsch, Ann. Med., 2014, 46, 372–383 Search PubMed.
- M. Toulany, K. J. Lee, K. R. Fattah, Y. F. Lin, B. Fehrenbacher, M. Schaller, B. P. Chen, D. J. Chen and H. P. Rodemann, Mol. Cancer Res., 2012, 10, 945–957 CrossRef CAS PubMed.
- J. Zhou, Y. Jiang, S. Hou, P. K. Upputuri, D. Wu, J. Li, P. Wang, X. Zhen, M. Pramanik, K. Pu and H. Duan, ACS Nano, 2018, 12, 2643–2651 CrossRef CAS PubMed.
- J. Chen, S. Hou, Q. Liang, W. He, R. Li, H. Wang, Y. Zhu, B. Zhang, L. Chen, X. Dai, T. Zhang, J. Ren and H. Duan, ACS Nano, 2022, 16, 2585–2597 CrossRef CAS PubMed.
- L. Huang, J. Xie, Q. Bi, Z. Li, S. Liu, Q. Shen and C. Li, Mol. Pharm., 2017, 14, 1742–1753 CrossRef CAS PubMed.
- L. J. Mah, A. El-Osta and T. C. Karagiannis, Leukemia, 2010, 24, 679–686 CrossRef CAS PubMed.
- A. Kinner, W. Wu, C. Staudt and G. Iliakis, Nucleic Acids Res., 2008, 36, 5678–5694 CrossRef CAS PubMed.
- Y. Zhang, F. Huang, C. Ren, J. Liu, L. Yang, S. Chen, J. Chang, C. Yang, W. Wang, C. Zhang, Q. Liu, X. J. Liang and J. Liu, Adv. Sci., 2019, 6, 1801806 CrossRef PubMed.
- G. Liang, X. Jin, S. Zhang and D. Xing, Biomaterials, 2017, 144, 95–104 CrossRef CAS PubMed.
- C. Ganesh Kumar, Y. Poornachandra and C. Chandrasekhar, Nanoscale, 2018, 10, 19639 RSC.
- W. Roa, X. Zhang, L. Guo, A. Shaw, X. Hu, Y. Xiong, S. Gulavita, S. Patel, X. Sun, J. Chen, R. Moore and J. Z. Xing, Nanotechnology, 2009, 20, 375101 CrossRef PubMed.
- M. Payne, S. H. Bossmann and M. T. Basel, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2020, 12, e1638 Search PubMed.
- Z. Jiang, T. Li, H. Cheng, F. Zhang, X. Yang, S. Wang, J. Zhou and Y. Ding, Asian J. Pharm. Sci., 2021, 16, 738–761 Search PubMed.
- M. R. Horsman and J. Overgaard, Clin. Oncol., 2007, 19, 418–426 CrossRef CAS PubMed.
- J. A. Marteijn, H. Lans, W. Vermeulen and J. H. Hoeijmakers, Nat. Rev. Mol. Cell Biol., 2014, 15, 465–481 CrossRef CAS PubMed.
- D. Fantini, E. Moritz, F. Auvré, R. Amouroux, A. Campalans, B. Epe, A. Bravard and J. P. Radicella, DNA Repair, 2013, 12, 227–237 Search PubMed.
- G. N. Castro, N. Cayado-Gutiérrez, F. C. Zoppino, M. A. Fanelli, F. D. Cuello-Carrión, M. Sottile, S. B. Nadin and D. R. Ciocca, Cell Stress Chaperones, 2015, 20, 253–265 CrossRef CAS PubMed.
- T. Mantso, G. Goussetis, R. Franco, S. Botaitis, A. Pappa and M. Panayiotidis, Semin. Cancer Biol., 2016, 37, 96–105 CrossRef PubMed.
- G. C. Terstappen, A. H. Meyer, R. D. Bell and W. Zhang, Nat. Rev. Drug Discov., 2021, 20, 362–383 Search PubMed.
- D. Wu, Q. Chen, X. Chen, F. Han, Z. Chen and Y. Wang, Signal Transduct. Targeted Ther., 2023, 8, 217 CrossRef PubMed.
- S. Palma-Florez, A. López-Canosa, F. Moralez-Zavala, O. Castaño, M. J. Kogan, J. Samitier, A. Lagunas and M. Mir, J. Nanobiotechnol., 2023, 21, 115 CrossRef CAS PubMed.
- A. Xie, G. Cheng, J. Wu, Z. Li, G. Yu, X. Zhu and T. Chen, Biomaterials, 2025, 312, 122749 CrossRef CAS PubMed.
- T. Xiao, M. He, F. Xu, Y. Fan, B. Jia, M. Shen, H. Wang and X. Shi, ACS Nano, 2021, 15, 20377–20390 CrossRef CAS PubMed.
- G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274–280 CrossRef CAS PubMed.
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 Search PubMed.
- M. Khongkow, T. Yata, S. Boonrungsiman, U. R. Ruktanonchai, D. Graham and K. Namdee, Sci. Rep., 2019, 9, 8278 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Access the Web version on PubMed Central for supplementary resources. See DOI: https://doi.org/10.1039/d4na00788c |
| ‡ Wei Fu, Qing Liang and Yuxi Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |