
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface engineering of 3D-printed polylactic acid scaffolds with polydopamine and 4-methoxycinnamic acid–chitosan nanoparticles for bone regeneration†
Abinaya
Shanmugavadivu
and
Nagarajan
Selvamurugan
 *
*
Department of Biotechnology, School of Bioengineering, SRM Institute of Science and Technology, Kattankulathur, Tamil Nadu 603203, India. E-mail: selvamun@srmist.edu.in
First published on 20th January 2025
Abstract
Bone remodeling, a continuous process of resorption and formation, is essential for maintaining skeletal integrity and mineral balance. However, in cases of critical bone defects where the natural bone remodeling capacity is insufficient, medical intervention is necessary. Traditional bone grafts have limitations such as donor site morbidity and availability, driving the search for bioengineered scaffold alternatives. The choice of biomaterial is crucial in scaffold design, as it provides a substrate that supports cell adhesion, proliferation, and differentiation. Poly-lactic acid (PLA) is known for its biocompatibility and biodegradability, but its hydrophobicity hinders cell attachment and tissue regeneration. To enhance PLA's bioactivity, we fabricated 3D-printed PLA scaffolds using fused deposition modelling. They were then surface-treated with NaOH to increase their reactivity, followed by polydopamine (PDA) and 4-methoxycinnamic acid (MCA)-loaded chitosan nanoparticle (nCS) coatings though polyelectrolyte complexation. Even though MCA, a polyphenolic, is known for its therapeutic properties, its osteogenic potential is not yet known. MCA treatment in mouse mesenchymal stem cells (mMSCs) promoted increased levels of Runx2 mRNA, a key bone transcription factor. Due to MCA's hydrophobic nature, nCS were used as carriers. The PLA/PDA/nCS–MCA scaffolds exhibited exceptional compressive strength and bioactivity. Biocompatibility tests confirmed that these scaffolds were non-cytotoxic to mMSCs. Overall, this study highlights the osteogenic potential of MCA and demonstrates the improved biocompatibility, bioactivity, wettability, and cell adhesion properties of the PDA/nCS–MCA-coated PLA scaffolds, positioning it as a promising material for bone tissue regeneration.
1. Introduction
Bone provides structural support and contributes to motor and hematopoietic functions, and internal organ protection. It has a double-layered structure: the outer cortical bone, with 3–5% porosity, constitutes 80% of adult bone mass, while the inner cancellous bone, with 80–90% porosity, accounts for 20% of the total bone mass.1 Bone remodeling is a continuous and dynamic process that balances bone resorption and formation, maintaining skeletal integrity and adapting to mechanical stress. This process is supported by bone's vascularized nature, enabling efficient self-repair without scarring. However, congenital or acquired bone injuries, such as those from aging and osteoporosis, can compromise bone's regenerative abilities, increasing fracture risk. In severe cases involving trauma, infections, inflammation, or tumor resection, lesions may exceed the bone's natural healing capacity, necessitating surgical intervention.2 The limitations of autogenous, allogeneic, and xenogeneic bone grafts can be addressed through advancements in bone tissue engineering (BTE). Bioengineered bone scaffolds aim to replicate the natural properties of bone, enhancing structural support and promoting cell function, offering promising alternatives for patients with critical bone defects.3In BTE, biomaterials, particularly polymers, play a crucial role in designing scaffolds that promote bone regeneration and repair. These materials serve as temporary frameworks that support cell attachment, proliferation, and differentiation, ultimately guiding the formation of new bone tissue.4 Natural polymers, such as collagen and chitosan (CS), are often used due to their biocompatibility and ability to mimic the extracellular matrix (ECM) of bone. However, synthetic polymers like polylactic acid (PLA), polyglycolic acid (PGA), and polycaprolactone (PCL) offer greater control over degradation rates and mechanical properties, making them suitable for creating scaffolds that degrade as new bone forms. Biodegradable polymers are particularly valuable in BTE, as they allow for the gradual transfer of load to the regenerating tissue, reducing the need for additional surgeries.5
PLA is a thermostable aliphatic polyester known for its biocompatibility, biodegradability, and ease of processing. Sourced from renewable materials like corn and sugarcane, PLA degrades into lactic acid, a metabolite that the body can safely eliminate, aligning with the bone regeneration timeline and minimizing long-term complications.6 PLA scaffolds, created using techniques such as 3D printing and electrospinning, offer precise control over architecture, porosity, and mechanical properties, supporting bone growth and cell infiltration.7 Despite their excellent mechanical strength, PLA scaffolds face limitations due to their poor bioactivity. This issue is addressed through various surface modifications, such as roughening, plasma polymerization, ion implantation, and the application of bioactive coatings.8,9 Bioactive coatings in bone scaffolds are surface treatments that enhance scaffold performance by promoting cell attachment, proliferation, and differentiation. Various studies have coated PLA scaffolds with materials like hydroxyapatite, bioactive glass, collagen, CS, and bioactive molecules such as peptides or growth factors, all of which support better integration with bone tissue.10
Polydopamine (PDA) is an adhesive inspired by mussel proteins, known for forming stable coatings that enhance surface bioactivity and cell attachment on scaffolds. PDA mimics mussel-secreted proteins, which function as adhesives in wet environments due to domains rich in 3,4-dihydroxy-L-phenylalanine (DOPA) and lysine amino acids, crucial for adhesion.11 PDA can be polymerized under alkaline conditions followed by catechol oxidation and strong substrate interactions.12
4-Methoxycinnamic acid (MCA) is a commonly found natural phenolic acid with a range of effects, including antibacterial, antifungal, and anti-inflammatory effects, neuroprotective properties, and cancer inhibition.13–15 With a chemical formula of C10H10O3 and a molecular weight of 162.19 g mol−1, MCA features a cinnamic acid backbone with a methoxy group attached to the para position of the phenyl ring. To our knowledge, the osteogenic potential of MCA has not yet been reported, which constitutes the novelty of this study. Also, the hydrophobic nature of MCA may restrict its direct application in the human body, making biomaterial-based delivery systems necessary to enhance the bioavailability of MCA at the bone defect site.16
CS is a linear polysaccharide composed of randomly distributed β-(1→4)-linked D-glucosamine (deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit). CS nanoparticles (nCS) are widely used in drug delivery systems due to their biocompatibility, biodegradability, and ability to provide controlled, sustained release of drugs. They can enhance drug stability, improve solubility, and be engineered for targeted delivery, making them a versatile option for effective therapeutic applications.17 Also, CS has been reported to form stable coatings when PDA is used as a surface anchor forming an intermediate bridging layer between PLA and CS.18
The novelty of this work lies in its first-time evaluation of MCA's ability to upregulate Runx2 mRNA expression, demonstrating its osteogenic potential for BTE applications. Additionally, this study introduces an effective approach for securely anchoring nCS–MCA to 3D-printed PLA scaffolds using a mussel-inspired PDA coating, enhancing the scaffold's surface properties and ensuring the stable immobilization of nCS–MCA for sustained release and improved biomineralization.
2. Materials and methods
2.1. Materials
MCA, CS (low molecular weight, 75–85% deacetylated), sodium tripolyphosphate (TPP), MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide], and dopamine hydrochloride were obtained from Sigma-Aldrich, St. Louis, MO, USA. The PLA [poly(lactic acid)] filament, with a thickness of 2.85 mm, was sourced from Solidspace Technology LLP, Nashik, Mumbai, India. Dulbecco's Modified Eagle's Medium (DMEM), trypsin, and phosphate-buffered saline (PBS) were purchased from Lonza, Basel, Switzerland. Fetal bovine serum (FBS) was acquired from Gibco, Gaithersburg, USA. Sodium hydroxide (NaOH), dimethyl sulfoxide (DMSO), acetic acid (AA), ethanol, and other reagents used were of analytical grade.2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (CS:TPP). This process was carried out with continuous stirring at 700 rpm for 1 h to facilitate the ionic cross-linking process and the formation of nCS–MCA, with the pH maintained at 4.5. The nCS–MCA suspension was centrifuged at 15000 rpm for 40 min to separate the nanoparticles from the supernatant.19 The supernatant containing unbound MCA was collected for subsequent quantification to calculate the entrapment efficiency. To ensure the complete removal of DMSO, the nanoparticles were washed repeatedly with a 1% trehalose solution, preserving their structural integrity. They were then centrifuged and lyophilized under vacuum at a pressure below 1 mbar and a temperature of −40 °C to eliminate any residual solvents and obtain a stable nCS–MCA powder. In parallel, blank nCS were prepared using the same ionic gelation method, but without the addition of MCA, to serve as a control in further analyses. The prepared nanoparticles, including nCS, nCS–10 μM MCA, nCS–20 μM MCA, and nCS–40 μM MCA, were analyzed using scanning electron microscopy (SEM), transmission electron microscopy (TEM), dynamic light scattering (DLS), Fourier transform infrared spectroscopy (FT-IR), and X-ray diffraction (XRD).
:5. The spectra were recorded in transmittance mode, scanning across a wavenumber range of 400 to 4000 cm−1.
:1 (CS:TPP). This process was carried out with continuous stirring at 700 rpm for 1 h to facilitate the ionic cross-linking process and the formation of nCS–MCA, with the pH maintained at 4.5. The nCS–MCA suspension was centrifuged at 15000 rpm for 40 min to separate the nanoparticles from the supernatant.19 The supernatant containing unbound MCA was collected for subsequent quantification to calculate the entrapment efficiency. To ensure the complete removal of DMSO, the nanoparticles were washed repeatedly with a 1% trehalose solution, preserving their structural integrity. They were then centrifuged and lyophilized under vacuum at a pressure below 1 mbar and a temperature of −40 °C to eliminate any residual solvents and obtain a stable nCS–MCA powder. In parallel, blank nCS were prepared using the same ionic gelation method, but without the addition of MCA, to serve as a control in further analyses. The prepared nanoparticles, including nCS, nCS–10 μM MCA, nCS–20 μM MCA, and nCS–40 μM MCA, were analyzed using scanning electron microscopy (SEM), transmission electron microscopy (TEM), dynamic light scattering (DLS), Fourier transform infrared spectroscopy (FT-IR), and X-ray diffraction (XRD).
:5. The spectra were recorded in transmittance mode, scanning across a wavenumber range of 400 to 4000 cm−1.
where Tmca is the total amount of MCA that was added and Fmca is the amount of unbound MCA in the supernatant.

2.2.13.1. In vitro biodegradation studies. The scaffolds were incubated in 1× PBS containing lysozyme at a concentration comparable to that found in circulating blood (10
000 U L−1) at 37 °C. After a 28 day incubation period, the scaffolds were frozen at −80 °C overnight and then lyophilized for 12 h. The percentage of degradation was calculated using the formula:where Wo represents the initial weight of the scaffold, and Wt represents the final weight post-incubation.

2.2.13.2. In vitro protein adsorption studies. Scaffolds were initially soaked in 100% ethanol for 30 min and then washed three times with MilliQ water. Following this, they were pre-wetted in 1× PBS for 90 min. After pre-wetting, the scaffolds were incubated in 3 mL of DMEM containing 10% FBS at 37 °C for 1, 6, 12, and 24 h. Upon completion of each incubation period, the scaffolds were carefully removed, blotted to remove excess moisture, and washed three times with PBS to eliminate loosely adhered proteins. The concentration of non-adsorbed proteins in the incubation solution was then measured using the Bradford assay, and the amounts of adsorbed proteins were calculated by subtracting the non-adsorbed protein from the total protein content.
2.2.13.3. In vitro compound release studies. The scaffolds were incubated in 1× PBS at 37 °C for up to 21 days to monitor MCA release. At predetermined intervals, 200 μL of the incubation medium was extracted and replaced with fresh PBS. The MCA concentration was quantified by measuring the absorbance at 288 nm, and a standard curve was used to calculate the drug concentration (20–100 μM). The cumulative release percentage was determined using the formula:
| MCA release% = (amount of MCA released/initial concentration of MCA loaded) × 100 |
To confirm the presence of MCA in the release samples, high-performance liquid chromatography (HPLC) analysis was performed using an Eclipse Plus C18 column (5 μm, 4.6 × 250 mm, Agilent-1260 infinity series). The mobile phase consisted of a 70:30 methanol–water mixture, with a flow rate of 1.00 mL min−1, and detection was carried out at 288 nm. The PBS medium, adjusted to pH 5.5, was used as the blank.
2.2.13.4. In vitro biomineralization studies. Scaffolds were immersed in 1× stimulated body fluid (SBF) and incubated at 37 °C for 7, 14, and 21 days. After each incubation period, the scaffolds were carefully extracted, rinsed three times with deionized water to remove any residual minerals, and lyophilized. The lyophilized scaffolds were then analyzed using SEM and EDS (HSEM – Quanta 200FEG, Netherlands) to assess their biomineralization and the elemental composition of the deposited apatite.
3. Results
3.1. MCA stimulated Runx2 mRNA expression in mMSCs
To assess the osteogenic potential of MCA, the expression level of Runx2 mRNA in mMSCs was examined. Runx2, a master bone transcription factor, governs the differentiation of mesenchymal stem cells into osteoblastic lineage. C3H10T1/2 cells were treated with MCA at varying concentrations, ranging from 5 μM to 60 μM, for a duration of 72 h. Following treatment, total RNA was extracted, and RT-qPCR was performed. The results demonstrated a statistically significant upregulation of Runx2 mRNA expression in 10, 20 and 40 μM MCA-treated cells compared to the control group (p < 0.05). Notably, the highest Runx2 mRNA expression was observed in cells treated with 20 μM MCA. Consequently, subsequent experiments focused on loading concentrations of 10, 20, and 40 μM MCA onto nCS.3.2. Physicochemical characterisation
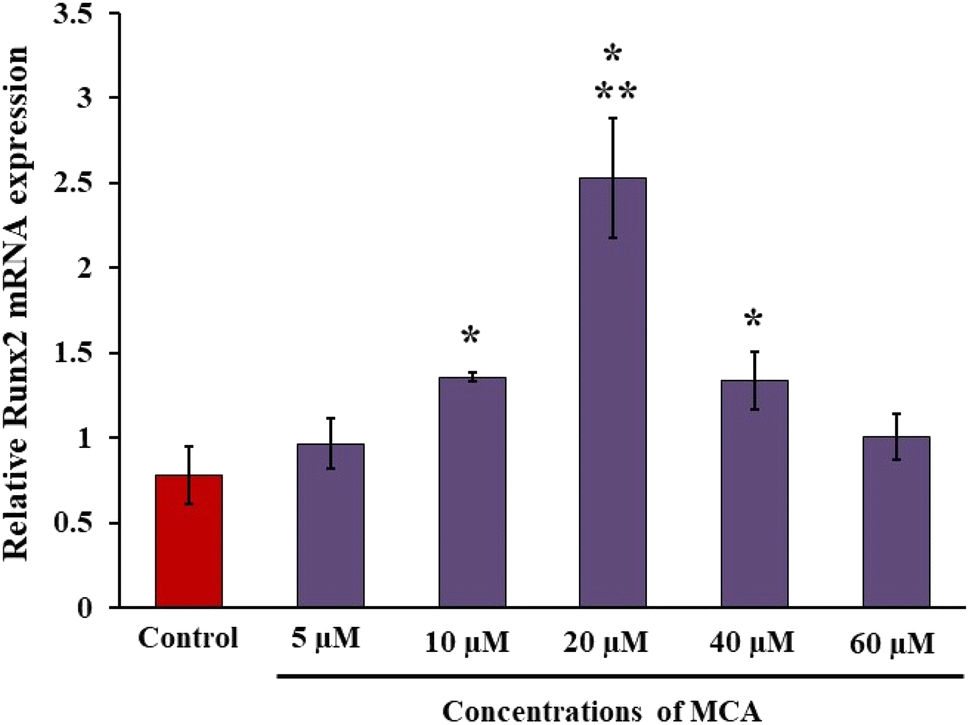 | ||
| Fig. 1 Effect of MCA on osteoblast differentiation. C3H10T1/2 cells were treated with varying concentrations of MCA and cultured for 3 days. Total RNA was isolated, and RT-qPCR was performed to measure the relative mRNA levels of the osteoblast differentiation marker Runx2, normalized to RPL13A. * indicates a significant increase compared to the control (p < 0.05). ** indicates a significant decrease compared to all groups (p < 0.05). | ||
 | ||
| Fig. 2 (A) SEM images of nCS and nCS–MCA with a scale bar of 500 nm. (B) TEM images of nCS and nCS–MCA with a scale bar of 100 nm. (C) SEM images of the PLA/PDA/nCS and PLA/PDA/nCS–MCA scaffolds are presented, with a scale bar of 300 μm. Insets show higher magnification images of the nCS and nCS–MCA distribution on the scaffold surface, with a scale bar of 500 nm. (D and E) DLS results for nCS and nCS–MCA. | ||
The particle size, zeta potential, and polydispersity index (PDI) of nCS and nCS–MCA nanoparticles serve as critical indicators of their physicochemical properties. The zeta potential values, ranging from +38.1 to +42.3 mV, suggested strong colloidal stability (Fig. 2D). This positive charge, primarily contributed by the protonation of CS's amino groups, likely facilitates enhanced surface interactions and uniform nanoparticle dispersion in a suspension. The Z-average size of the nanoparticles ranged from 144.9 to 151.3 nm, with a PDI between 0.293 and 0.34, reflecting consistent nanoparticle formation and a relatively narrow size distribution (Fig. 2E). The entrapment efficiency of nCS was quantified by analyzing the MCA content in the supernatant. The results indicated that MCA was effectively encapsulated within nCS, with an entrapment efficiency ranging from 66.37% to 68.9%.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, respectively.22 The CS spectra showed a broad peak between 3500 and 3000 cm−1, which is attributed to the stretching vibrations of the amine and OH groups in CS. The N–H stretching from amide I and II was observed at 1642 cm−1 and 1554 cm−1, respectively.9 For TPP, the main vibrations observed were PO, PO2 and PO3 stretching at 1212 cm−1, 1148 cm−1 and 1093 cm−1, respectively; and P–O–P antisymmetric stretching at 900 cm−1.23 The PLA spectra revealed characteristic peaks at 3226.91 cm−1 (OH stretching), 2931.80 cm−1 (C–H symmetric valence vibration), and 1749.44 cm−1 (CO valence vibration), with distinct peaks between 1300 and 1100 cm−1 corresponding to C–O–C symmetric valence vibrations.9 The FT-IR spectra of dopamine revealed peaks at 3346 cm−1, 3270 cm−1 and 3050.77 cm−1 corresponding to amine N–H, phenol O–H stretching and aromatic CH stretching, respectively.24 FT-IR of nCS revealed interactions between TPP and the ammonium groups in CS within the nanoparticles. The characteristic peak at 3438 cm−1 shifted to 3320 cm−1 in nCS, signifying hydrogen bonding.19 The spectra of PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds exhibited distinct peaks characteristic of PLA. However, MCA-specific peaks were not observed, which could be attributed to the low concentrations of MCA utilized in the scaffolds.8
O stretching, respectively.22 The CS spectra showed a broad peak between 3500 and 3000 cm−1, which is attributed to the stretching vibrations of the amine and OH groups in CS. The N–H stretching from amide I and II was observed at 1642 cm−1 and 1554 cm−1, respectively.9 For TPP, the main vibrations observed were PO, PO2 and PO3 stretching at 1212 cm−1, 1148 cm−1 and 1093 cm−1, respectively; and P–O–P antisymmetric stretching at 900 cm−1.23 The PLA spectra revealed characteristic peaks at 3226.91 cm−1 (OH stretching), 2931.80 cm−1 (C–H symmetric valence vibration), and 1749.44 cm−1 (CO valence vibration), with distinct peaks between 1300 and 1100 cm−1 corresponding to C–O–C symmetric valence vibrations.9 The FT-IR spectra of dopamine revealed peaks at 3346 cm−1, 3270 cm−1 and 3050.77 cm−1 corresponding to amine N–H, phenol O–H stretching and aromatic CH stretching, respectively.24 FT-IR of nCS revealed interactions between TPP and the ammonium groups in CS within the nanoparticles. The characteristic peak at 3438 cm−1 shifted to 3320 cm−1 in nCS, signifying hydrogen bonding.19 The spectra of PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds exhibited distinct peaks characteristic of PLA. However, MCA-specific peaks were not observed, which could be attributed to the low concentrations of MCA utilized in the scaffolds.8
 | ||
| Fig. 3 (A) FT-IR spectra of CS, TPP, MCA, PDA, nCS, nCS–10 μM MCA, nCS–20 μM MCA, nCS–40 μM MCA, PLA, PLA/PDA, PLA/PDA/nCS, PLA/PDA/nCS–10 μM MCA, PLA/PDA/nCS–20 μM MCA, and PLA/PDA/nCS–40 μM MCA. (B) XRD patterns of CS, TPP, MCA, PDA, PLA, nCS, PLA/PDA, PLA/PDA/nCS, PLA/PDA/nCS–10 μM MCA, PLA/PDA/nCS–20 μM MCA, and PLA/PDA/nCS–40 μM MCA. | ||
XRD analysis was carried out to examine the crystalline nature of the individual components and the biocomposite scaffolds (PLA/PDA, PLA/PDA/nCS and PLA/PDA/nCS–MCA). The XRD spectra of MCA exhibited sharp, intense peaks confirming its crystalline structure (Fig. 3B). The diffraction pattern of TPP and PDA also showed sharp peaks indicating their crystalline nature, whereas the diffraction patterns of nCS and PLA demonstrated semi-crystalline characteristics. The biocomposite scaffolds, PLA/PDA, PLA/PDA/nCS and PLA/PDA/nCS–MCA showed less intense peaks, indicating the semi-crystalline nature of the composites. The degree of crystallinity of the PLA/PDA/nCS scaffold was found to be 27.78%, while the PLA/PDA/nCS–MCA scaffold exhibited a crystallinity of 26.74%. The scaffolds were classified as semi-crystalline, with the inclusion of MCA having no significant effect on the crystallinity, suggesting that MCA incorporation did not alter the crystalline structure of the composite. The compressive strength analysis revealed that the PLA scaffold had a compressive strength of 77.67 MPa, while the PLA/PDA/nCS–MCA scaffold exhibited 83.85 MPa.
 | ||
| Fig. 4 Water contact angle measurement of PLA, PLA (NaOH-treated), PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds. | ||
3.3. In vitro material characterisation
 | ||
| Fig. 5 Biodegradation, protein adsorption, drug release, and HPLC analysis. PLA, PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds were evaluated across several parameters. (A) Biodegradation rates of scaffolds in PBS containing lysozyme over 28 days. * indicates a significant increase compared to PLA (p < 0.05). ** indicates a significant increase compared to PLA/PDA (p < 0.05). (B) Protein adsorption on scaffolds at 1, 6, 12, and 24 h. * indicates a significant increase compared to the respective PLA/PDA scaffolds (p < 0.05). ** indicates a significant increase compared to the respective PLA (p < 0.05). *** indicates a significant increase compared to the respective groups at 1 h (p < 0.05). (C) MCA release from PLA/PDA/nCS–MCA scaffolds over 21 days. (D) HPLC chromatograms confirming the presence of MCA in the released samples, with retention times similar to the MCA standard. | ||
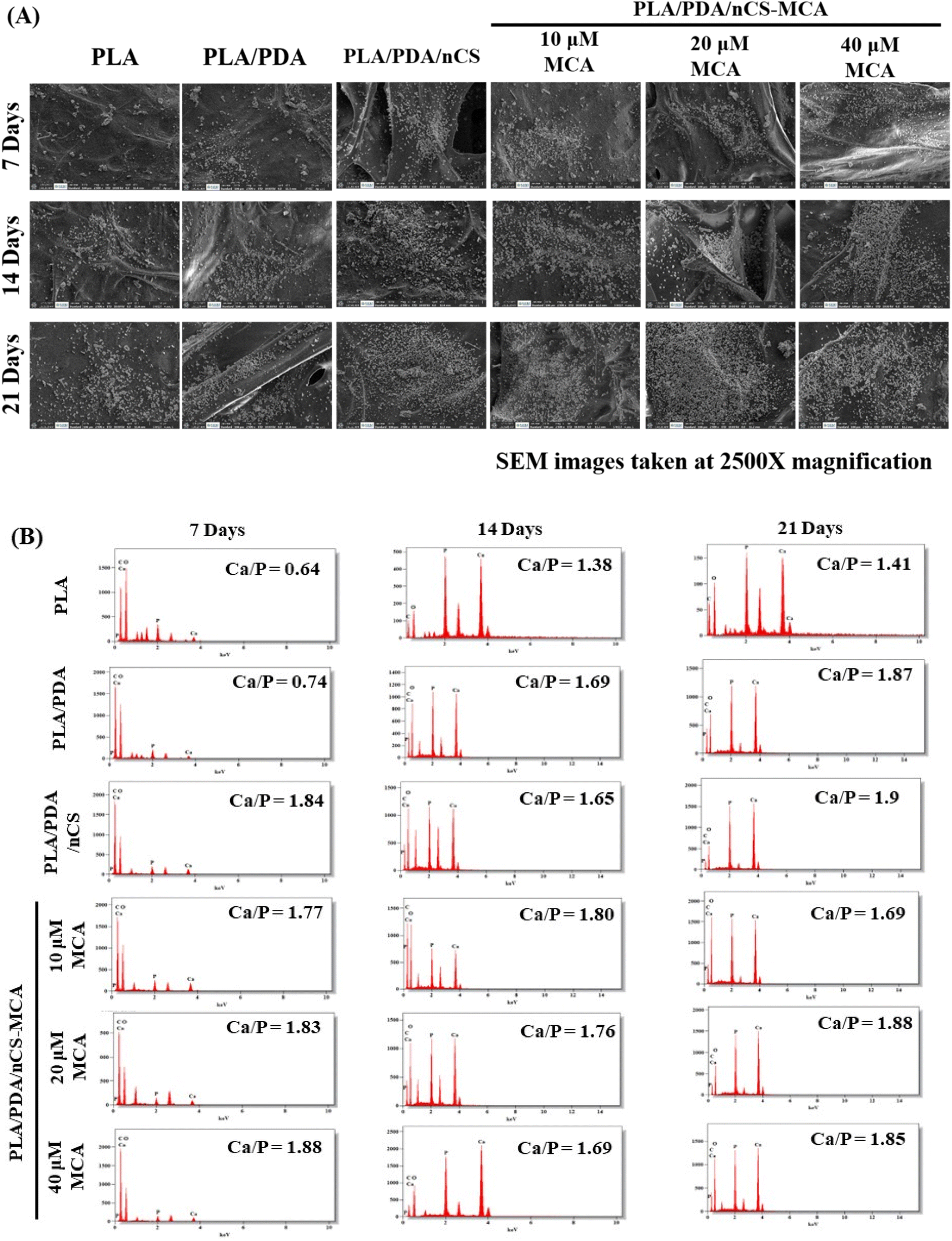 | ||
| Fig. 6 PLA, PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds were assessed for biomineralization in SBF over 7, 14, and 21 days. (A) Mineral deposits on scaffolds observed by SEM analysis at 2500× magnification. (B) EDS spectra confirmed the presence of calcium (Ca), phosphorus (P), carbon (C), and oxygen (O) in the mineralized layers, with the corresponding Ca/P ratios. | ||
3.4. Cytotoxicity, cell viability and cell count studies
The synthesized scaffolds were assessed for cytocompatibility using the MTT assay with mMSCs (Fig. 7A). This assay evaluates cell viability by measuring the conversion of MTT into formazan crystals though mitochondrial succinate dehydrogenase activity. The results demonstrated no significant change in absorbance, indicating that the PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds were non-cytotoxic in mMSCs. | ||
| Fig. 7 Cytotoxicity and cell morphology analysis. mMSCs were seeded onto PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds and cultured for 72 h. (A) MTT assay. (B) The dot plots illustrate the distribution of live cells, dead cells, and cell debris, with the upper left quadrant representing viable cells and the upper right quadrant depicting dead cells. (C and D) Bar graphs showing cell viability and cell number. (E) mMSCs were stained with FDA and examined under a fluorescence microscope. | ||
While the MTT assay provides insight into the metabolic activity of cells, it does not assess membrane integrity. Therefore, cell membrane integrity in response to the scaffolds was further evaluated using a Muse cell analyzer. The results showed no significant change in cell viability or cell number (Fig. 7B–D). All scaffold groups, including PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA, exhibited similar percentages of cell viability and cell counts, indicating that both MCA and the biocomposite scaffolds were non-toxic and do not affect cell proliferation.
FDA staining revealed that mMSCs seeded on PLA/PDA, PLA/PDA/nCS, and PLA/PDA/nCS–MCA scaffolds exhibited well-extended morphology with distinct cytoplasmic extensions that support cell attachment and growth (Fig. 7E). Morphological evaluation at 20× and 40× magnification showed that the addition of MCA in the biocomposite scaffolds did not adversely affect cell morphology, indicating their non-toxic nature. Hence, these results suggested that PLA/PDA/nCS–MCA scaffolds have the potential to accelerate bone regeneration while being non-cytotoxic.
4. Discussion
BTE aims to create scaffolds that mimic the natural ECM and support bone regeneration. Advances in 3D printing, especially FDM, enable precise scaffold design, with PLA being popular due to its biocompatibility and customizable properties.25 However, PLA lacks inherent bioactivity, necessitating surface modifications to enhance osteoconductivity. Incorporating bioactive molecules or growth factors into PLA scaffolds can also make them osteoinductive, enabling the differentiation of MSCs into osteoblasts to support bone regeneration.26 In this work, a feasible and effective method to firmly anchor nCS–MCA onto 3D-printed PLA scaffolds though a mussel-inspired PDA coating was proposed. The nCS were loaded with MCA, a polyphenol whose osteogenic potential was evaluated. The PDA coating, serving as an intermediate bridging layer, offers abundant active sites to couple with nCS–MCA. After physicochemical characterization of the surface-engineered PLA scaffolds, their bioactivity was systematically assessed though various in vitro material characterization and biocompatibility studies.MCA was selected for its potential therapeutic benefits, which are largely attributed to its chemical structure. The biological activity of phytocompounds, including MCA, is significantly influenced by their chemical structure, which impacts solubility, membrane penetration, and enzyme binding. Specifically, for cinnamic acid derivatives, bioactivity is primarily governed by the presence and positioning of hydroxy (–OH) and methoxy (–OCH3) groups on the aromatic ring. The number and placement of these substituents critically define the extent and potency of their biological action. Notably, studies have demonstrated that among phenylpropanoic acids, those with the –OCH3 group in the para position, such as MCA, exhibit pronounced hepatoprotective activity, underscoring their potential for therapeutic applications.27 The evaluation of MCA's osteogenic potential revealed a significant increase in Runx2 mRNA expression in mMSCs, especially at 10, 20, and 40 μM concentrations, which guided the selection of these MCA-loaded nCS concentrations for further experimentation (Fig. 1). Phytocompounds have been reported to achieve peak plasma concentration within a short timeframe and are swiftly eliminated from systemic circulation.28 Therefore, an effective delivery system is needed to prolong their bioavailability and therapeutic effects.
Nano-drug delivery systems have been employed to deliver various drugs, growth factors, and hydrophobic phytocompounds.29,30 Natural polymers, such as CS, are especially beneficial for fabricating these nanoparticles due to their biocompatibility and effectiveness in stabilizing various phytocompounds, by enhancing their solubility and bioavailability.19,31 Nanoparticles offer significant advantages in drug delivery, particularly in enhancing drug stability and bioavailability though controlled release mechanisms.32 SEM and TEM analyses confirmed that both nCS and nCS–MCA nanoparticles possess a spherical morphology (Fig. 2A and B). Zhao et al. reported that spherical nanoparticles had better bioavailability compared to rod-shaped nanoparticles due to their uniform size and shape, which enhanced their ability to be internalized by cells, leading to better cellular uptake and more consistent drug release profiles.33 Hejjaji et al. reported that a zeta potential greater than +30 mV generally indicates lower stability due to reduced electrostatic repulsion, while a zeta potential in the range of +34 to +42 mV is considered optimal for enhanced stability.34 Accordingly, the zeta potentials of nCS and nCS–MCA, ranging from +38 to +42 mV, indicated their excellent colloidal stability (Fig. 2D). Also, the size of nCS and nCS–MCA was observed to be less than 200 nm (Fig. 2E), indicating their potential for enhanced drug release control and improved cellular interactions.31 The entrapment efficiency of nCS–MCA nanoparticles, determined by analysing the MCA content in the supernatant, ranged from 66.37% to 68.9%. Although the direct method of preparation, which involves dispersing the drug solution in a polymeric matrix followed by the addition of a crosslinking agent, is often associated with low entrapment efficiency, this limitation was overcome by optimizing critical parameters such as polymer concentration, CS:TPP mass ratio, pH, stirring conditions, and the addition rate of TPP, as demonstrated in our previous studies.19
The prepared nCS and nCS–MCA were coated on the PLA/PDA scaffold via the immersion technique allowing the PDA to act as a natural crosslinker. Similarly, Salehi et al. fabricated poly(L-lactic acid) scaffolds via freeze-drying and coated them with nCS via the immersion technique.35 The FT-IR spectra of PLA/PDA/nCS–MCA showed no new peaks, indicating no interaction between the components.9 Additionally, the PLA/PDA/nCS–MCA nanocomposite scaffolds maintained their semi-crystalline structure despite the inclusion of the crystalline MCA (Fig. 3).
The hydrophilicity of biomaterials is critical in influencing their interactions with biological systems, such as protein adsorption and cell attachment.20 The surface modification techniques employed, including NaOH treatment, PDA coating and nCS incorporation, substantially improved the hydrophilicity of the scaffolds. The water contact angle measurements demonstrated a marked decrease in hydrophobicity across the modified scaffolds (Fig. 4). The nanocomposite scaffolds, PLA/PDA/nCS and PLA/PDA/nCS–MCA scaffolds exhibited near-zero contact angles, highlighting a significant enhancement in hydrophilic properties. Materials with a water contact angle between 0° and 40° are optimal for bone graft applications,36 supporting the suitability of our PLA/PDA/nCS–MCA scaffolds for BTE applications. Consistent with SEM analysis that confirmed successful nCS–MCA incorporation on the scaffold surfaces (Fig. 2C), the reduced water contact angles further validated this integration. The mechanical strength of a scaffold is crucial for supporting cellular activities like adhesion and proliferation, which influence its overall in vivo performance. It also affects the scaffold's stability under physiological loads and its ability to facilitate mechanotransduction, essential for effective bone regeneration. In this study, the PLA/PDA/nCS–MCA scaffolds showed compression strength values ranging from 77.67 to 83.85 MPa, approaching those of cortical bone. The slight increase in compressive strength of the PLA/PDA/nCS–MCA scaffold can be attributed to the 5-layer coating of PDA and nCS, which enhanced the scaffold's structural integrity and strength through improved material reinforcement and interfacial bonding. The layer-by-layer assembly technique enabled the application of conformal coatings on highly porous 3D templates, effectively transferring microscale mechanical characteristics to larger-scale structures.37
Biodegradation is essential for BTE scaffolds, as it allows for the gradual breakdown of materials to facilitate new bone formation. Scaffolds interact with bodily fluids containing hydrolytic enzymes that accelerate their degradation. In this study, PLA/PDA/nCS and PLA/PDA/nCS–MCA scaffolds exhibited significantly higher degradation rates compared to PLA and PLA/PDA scaffolds, likely due to the increased surface area-to-volume ratio provided by nCS (Fig. 5A). nCS undergo degradation though their β-1,4-glycosidic bond cleavage and N-acetyl linkage hydrolysis in the presence of lysozyme, producing non-toxic oligosaccharides that integrate into ECM components.38 PLA, on the other hand, is degraded via enzyme–PLA complex formation with lysozyme, which cleaves ester bonds and releases by-products.39 Protein adsorption is critical for optimizing cellular interactions. Our findings revealed that PLA/PDA/nCS and PLA/PDA/nCS–MCA scaffolds exhibited higher protein adsorption compared to PLA and PLA/PDA (Fig. 5B). This indicates that the addition of nCS could improve scaffold performance by enhancing cellular responses and facilitating bone regeneration. A consistent and extended drug release profile is essential for bone scaffolds to ensure an effective dosage for bone tissue regeneration. Polyphenols, known for their rapid metabolism, pose a challenge for sustained delivery.9 To overcome this, nCS were employed as a controlled release system for MCA. In vitro studies showed that PLA/PDA/nCS–MCA scaffolds achieve a prolonged and stable release of MCA over 21 days (Fig. 5C).
Biomineralization involves forming new bone by depositing mineral salts on a scaffold's surface when implanted and exposed to body fluids. This process promotes the accumulation of calcium phosphates, such as hydroxyapatite, which interact with the scaffold's organic polymers, enhancing mineralization and supporting bone formation.19 Enhanced biomineralization in the PLA/PDA/nCS and PLA/PDA/nCS–MCA groups can be attributed to the initiation of nucleation sites by the polar groups in nCS (Fig. 6).
Biocompatibility is crucial for the success of biomaterial implants, influencing cell adhesion and long-term viability on the scaffold surface. In this study, the polymers used—PLA, PDA, and CS—demonstrated compatibility with diverse cell types and animal models, making them well-suited for BTE applications.40–42 Evaluating cell attachment, morphology, and distribution on the scaffold provides essential insights into its performance and interactions with cells.43 These characteristics were assessed though various assays, including MTT, Muse cell analysis, and FDA staining. The results confirmed that the PLA/PDA/nCS–MCA nanocomposite scaffolds are biocompatible (Fig. 7), and they do not adversely affect cell proliferation, underscoring their potential for BTE applications.
5. Conclusion
This study highlights the pivotal role of surface engineering in advancing the efficacy of 3D-printed PLA scaffolds for BTE applications. By employing surface modifications such as incorporating PDA, nCS, and MCA, we significantly enhanced the scaffolds' hydrophilicity, biomineralization, and degradation properties. Also, the biocomposite scaffolds were non-cytotoxic to mMSCs. Notably, this study is the first to reveal MCA's osteogenic potential, marking a significant advancement in scaffold development. The inclusion of MCA did not alter any of the material properties of the scaffolds and they remained non-toxic. Future investigations should focus on elucidating the cellular and molecular mechanisms underlying the osteogenic effects of these scaffolds. In vivo evaluations will then be crucial to further assess the scaffolds' bone regenerative potential in a physiological environment.Data availability
The corresponding author will provide the results produced during the current investigation upon reasonable request.Conflicts of interest
There are no conflicts to declare.Abbreviations
| AA | Acetic acid |
| BTE | Bone tissue engineering |
| CS | Chitosan |
| DLA | Dynamic light scattering |
| DMEM | Dulbecco's modified Eagle's medium |
| DMSO | Dimethyl sulfoxide |
| DOPA | 3,4-Dihydroxy-L-phenylalanine |
| ECM | Extracellular matrix |
| EDS | Energy-dispersive X-ray spectroscopy |
| FBS | Fetal bovine serum |
| FDA | Fluorescein diacetate |
| FT-IR | Fourier transform infrared spectroscopy |
| HPLC | High-performance liquid chromatography |
| KBr | Potassium bromide |
| MCA | 4-Methoxycinnamic acid |
| mMSCs | Mouse mesenchymal stem cells |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| NaOH | Sodium hydroxide |
| NCCS | National Centre for Cell Sciences |
| nCS | Chitosan nanoparticles |
| PBS | Phosphate-buffered saline |
| PCL | Polycaprolactone |
| PDA | Polydopamine |
| PGA | Polyglycolic acid |
| PLA | Polylactic acid |
| qPCR | Quantitative polymerase chain reaction |
| SD | Standard deviation |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| TPP | Sodium tripolyphosphate |
| XRD | X-ray diffraction |
References
- A. M. Parfitt, Bone, 2002, 30, 807–809 CrossRef CAS PubMed.
- F. Shang, Y. Yu, S. Liu, L. Ming, Y. Zhang, Z. Zhou, J. Zhao and Y. Jin, Bioact. Mater., 2021, 6, 666–683 CAS.
- N. Xue, X. Ding, R. Huang, R. Jiang, H. Huang, X. Pan, W. Min, J. Chen, J.-A. Duan, P. Liu and Y. Wang, Pharmaceuticals, 2022, 15, 879 CrossRef CAS PubMed.
- H. Qu, H. Fu, Z. Han and Y. Sun, RSC Adv., 2019, 9, 26252–26262 RSC.
- Z. Sheikh, S. Najeeb, Z. Khurshid, V. Verma, H. Rashid and M. Glogauer, Materials, 2015, 8, 5744–5794 CrossRef CAS PubMed.
- M. A. Elsawy, K.-H. Kim, J.-W. Park and A. Deep, Renewable Sustainable Energy Rev., 2017, 79, 1346–1352 CrossRef CAS.
- S. Swetha, K. Balagangadharan, K. Lavanya and N. Selvamurugan, Biotechnol. J., 2021, 16(11), e2100282 CrossRef PubMed.
- B. Ashwin, B. Abinaya, T. P. Prasith, S. V. Chandran, L. R. Yadav, M. Vairamani, S. Patil and N. Selvamurugan, Int. J. Biol. Macromol., 2020, 162, 523–532 CrossRef CAS PubMed.
- R. Bharathi, G. Harini, A. Sankaranarayanan, A. Shanmugavadivu, M. Vairamani and N. Selvamurugan, Int. J. Biol. Macromol., 2023, 253, 127492 CrossRef CAS PubMed.
- A. Dukle and M. R. Sankar, Mater. Today Commun., 2024, 40, 109776 CrossRef CAS.
- L. Jia, F. Han, H. Wang, C. Zhu, Q. Guo, J. Li, Z. Zhao, Q. Zhang, X. Zhu and B. Li, J. Orthop. Transl., 2019, 17, 82–95 Search PubMed.
- L. Li, Y. Li, L. Yang, F. Yu, K. Zhang, J. Jin, J. Shi, L. Zhu, H. Liang, X. Wang and Q. Jiang, Ann. Transl. Med., 2019, 7, 240 CrossRef CAS PubMed.
- Y. Wang, M. Yin, L. Gu, W. Yi, J. Lin, L. Zhang, Q. Wang, Y. Qi, W. Diao, M. Chi, H. Zheng, C. Li and G. Zhao, Int. Immunopharmacol., 2023, 116, 109782 CrossRef CAS PubMed.
- M. Rychlicka, A. Rot and A. Gliszczyńska, Foods, 2021, 10, 1417 CrossRef CAS PubMed.
- S. Gunasekaran, K. Venkatachalam and N. Namasivayam, Environ. Toxicol. Pharmacol., 2015, 40, 966–974 CrossRef CAS PubMed.
- H. S. Han, S. Y. Koo and K. Y. Choi, Bioact. Mater., 2022, 14, 182–205 CAS.
- K. Jafernik, A. Ładniak, E. Blicharska, K. Czarnek, H. Ekiert, A. E. Wiącek and A. Szopa, Molecules, 2023, 28, 1963 CrossRef CAS PubMed.
- B.-B. Wang, Y.-H. Quan, Z.-M. Xu and Q. Zhao, Prog. Org. Coat., 2020, 149, 105967 CrossRef CAS.
- R. S. Leena, M. Vairamani and N. Selvamurugan, Colloids Surf., B, 2017, 158, 308–318 CrossRef CAS PubMed.
- J. Park, S. J. Lee, T. G. Jung, J. H. Lee, W. D. Kim, J. Y. Lee and S. A. Park, Colloids Surf., B, 2021, 199, 111528 CrossRef CAS PubMed.
- X. Ma, G. Wu, F. Dai, D. Li, H. Li, L. Zhang and H. Deng, Carbohydr. Polym., 2021, 251, 117058 CrossRef CAS PubMed.
- W. Ferenc, B. Cristóvão, J. Sarzyński and P. Sadowski, J. Therm. Anal. Calorim., 2012, 110, 739–748 CrossRef CAS.
- F. G. de Carvalho, T. C. Magalhães, N. M. Teixeira, B. L. C. Gondim, H. L. Carlo, R. L. dos Santos, A. R. de Oliveira and Â. M. L. Denadai, Mater. Sci. Eng., C, 2019, 104, 109885 CrossRef CAS PubMed.
- A. Thakur, S. Ranote, D. Kumar, K. K. Bhardwaj, R. Gupta and G. S. Chauhan, ACS Omega, 2018, 3, 7925–7933 CrossRef CAS PubMed.
- B. Zhang, L. Wang, P. Song, X. Pei, H. Sun, L. Wu, C. Zhou, K. Wang, Y. Fan and X. Zhang, Mater. Des., 2021, 201, 109490 CrossRef CAS.
- E. H. Baran and H. Y. Erbil, Colloids Interfaces, 2019, 3, 43 CrossRef CAS.
- R. Rukkumani, K. Aruna, P. Suresh Varma and V. Padmanabhan Menon, J. Med. Food, 2004, 7, 456–461 CrossRef CAS PubMed.
- S. N. B. Selby-Pham, R. B. Miller, K. Howell, F. Dunshea and L. E. Bennett, Sci. Rep., 2017, 7, 1931 CrossRef PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- N. T. Vittala Murthy, S. K. Paul, H. Chauhan and S. Singh, Curr. Drug Delivery, 2022, 19, 182–191 CrossRef PubMed.
- K. Balagangadharan, R. Trivedi, M. Vairamani and N. Selvamurugan, Carbohydr. Polym., 2019, 216, 1–16 CrossRef CAS PubMed.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 4131 CrossRef PubMed.
- E. M. A. Hejjaji, A. M. Smith and G. A. Morris, Int. J. Biol. Macromol., 2018, 120, 1610–1617 CrossRef CAS PubMed.
- M. Salehi, M. Naseri-Nosar, M. Azami, S. J. Nodooshan and J. Arish, Tissue Eng. Regener. Med., 2016, 13, 498–506 CrossRef CAS PubMed.
- N. G. Rim, S. J. Kim, Y. M. Shin, I. Jun, D. W. Lim, J. H. Park and H. Shin, Colloids Surf., B, 2012, 91, 189–197 CrossRef CAS PubMed.
- X. Liu, C. Zhou, Q. Xie, L. Xia, L. Liu, W. Bao, H. Lin, X. Xiong, H. Zhang, Z. Zheng, J. Zhao, W. Liang and J. Transl, Med, 2024, 22, 1001 CAS.
- E. Szymańska and K. Winnicka, Mar. Drugs, 2015, 13, 1819–1846 CrossRef PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- K. Peng, S. Chen, V. Senthooran, X. Hu, Y. Qi, C. Zhang, L. Wu and J. Wang, Int. J. Biol. Macromol., 2024, 279, 135055 CrossRef CAS PubMed.
- J. Miszuk, J. Hu, Z. Wang, O. Onyilagha, H. Younes, C. Hill, A. V. Tivanski, Z. Zhu and H. Sun, J. Biomed. Mater. Res., Part B, 2024, 112(8), e35456 CrossRef CAS PubMed.
- W. Tang, P. Pan, T. Chen, J. Wang, X. Cui, W. Liu and L. Kan, Colloids Surf., B, 2025, 245, 114199 CrossRef CAS PubMed.
- S. Mohammadi, F. Ghasemi, S. A. Alavi G. and E. Alemzadeh, Tissue Cell, 2024, 91, 102536 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00768a |
| This journal is © The Royal Society of Chemistry 2025 |