
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the dynamics of fungal disease with proteomics
Mariana
Sa
,
Mayara
da Silva
,
Brianna
Ball
and
Jennifer
Geddes-McAlister
 *
*
Molecular and Cellular Biology, University of Guelph, Guelph, Ontario, N1G 2W1, Canada. E-mail: jgeddesm@uoguelph.ca
First published on 20th February 2025
Abstract
The occurrence and distribution of new and re-emerging fungal pathogens, along with rates of antifungal resistance, are rising across the globe, and correspondingly, so are our awareness and call for action to address this public health concern. To effectively detect, monitor, and treat fungal infections, biological insights into the mechanisms that regulate pathogenesis, influence survival, and promote resistance are urgently needed. Mass spectrometry-based proteomics is a high-resolution technique that enables the identification and quantification of proteins across diverse biological systems to better understand the biology driving phenotypes. In this review, we highlight dynamic and innovative applications of proteomics to characterize three critical fungal pathogens (i.e., Candida spp., Cryptococcus spp., and Aspergillus spp.) causing disease in humans. We present strategies to investigate the host–pathogen interface, virulence factor production, and protein-level drivers of antifungal resistance. Through these studies, new opportunities for biomarker development, drug target discovery, and immune system remodeling are discussed, supporting the use of proteomics to combat a plethora of fungal diseases threatening global health.
1. Introduction
Fungal infections are among the most challenging to manage given a limited arsenal of antifungal drugs and close target homology with the human host.1–3 Restricted funding towards fungal disease research, along with limited accessibility to diagnostic tests and antifungal drugs, causes a disproportionate number of deaths in developing countries, presenting critical threats to global management and eradication of fungal diseases.4–6 Fungal diseases, or mycoses, are classified as superficial, cutaneous, subcutaneous, or systemic infections.7 Globally, the rates of invasive fungal infections are rising with over 6.5 M cases reported annually, leading to over 3.8 M deaths.4 Diverse species of Candida, Aspergillus, Cryptococcus, and Pneumocystis are responsible for >90% of invasive mycoses worldwide.8 The modes of infection for the pathogens vary with many invasive fungal infections occurring in response to a disruption in the normal microflora. For example, Candida albicans, which serves as a commensal organism within the human host, transitions to a pathogenic state upon perturbation of the microflora.9 Similarly, a shift in host immune status towards an immunocompromised state can increase host susceptibility to infection from fungi, such as Aspergillus fumigatus and Cryptococcus neoformans.10It is postulated that fungi evolved within environmental niches independent of human infections and to cause disease within humans, fungi must produce factors to overcome host defenses. For instance, high thermotolerance, an ability to invade the human host, mechanisms for digestion and absorption of human tissue, and tolerance to the human immune system.11 Therefore, changes in host immune status and the production of virulence factors by the fungi are key drivers of disease. Critically, a growing population of immunocompromised individuals corresponding with increased prevalence of immunotherapy, immunosuppression, co-infections, and aging support the need for integrated approaches to study and understand the complex interactions between a host and fungal pathogen during disease. Moreover, human interference, such as global transport that facilitates the spread of potential pathogens to new geographical ranges, agricultural fungicide applications that contribute to the antimicrobial resistance crises, and climate change that select for thermotolerant fungi, fosters the development of these opportunistic pathogens.12–14 To raise awareness about fungal diseases, in 2022, the World Health Organization published its first-ever ranking of priority fungal pathogens to attract attention and strengthen the global response to infections.15,16 This list, termed the Fungal Priority Pathogens List, names fungi of critical or high importance, including C. neoformans, Candida auris, C. albicans, and A. fumigatus. To define new strategies to disarm fungal pathogens, combat fungal infections, and overcome disease, in this review, we focus on the application of proteomics to explore these globally important fungal pathogens.
2. Mass spectrometry-based proteomics
Mass spectrometry-based proteomics encompasses the study of proteins within a given biological system using a combination of analytical and computational techniques with important applications to study infectious diseases, drug discovery, and host–pathogen interactions.17–21 Proteomics enables the identification and quantification of proteins within a cellular and extracellular context, interactions across proteins and within complexes, and modifications that influence protein structure and function. The field of proteomics is broadly defined by top-down approaches, which include the analysis of intact proteins for detection of protein complexes and proteoforms22 and bottom-up approaches, which encompass a discovery-driven approach using peptides to identify proteins and their modifications.23 Additionally, targeted proteomics detects and quantifies predefined peptides within complex mixtures from diverse applications, including biomarker discovery.24 Measurement of proteins or peptides begins with sample separation by high performance liquid chromatography followed by detection and measurement of ions on a high-resolution mass spectrometer. Within this review, bottom-up proteomics experiments are highlighted.For the measurement of peptides, data-dependent acquisition (DDA), which performs selection of the top-N most abundant ions from a survey scan of sequential fragmentation, has been the traditional approach.25 However, recent instrumentation and computational advances have introduced data-independent acquisition (DIA), which fragments all peptides within a cycling mass-to-charge (m/z) window over the entire m/z range, for the identification of peptides.26 For protein quantification, a range of chemical, metabolic, or label-free quantification (LFQ) methods exist.27–29 For instance, metabolic labeling includes SILAC (stable isotope labeling with amino acids in cell culture) to incorporate a label at the cellular or organismal level30 and chemical labelling with tandem mass tags supports multiplexing and normalization across large sample sets.31 For LFQ methods, additional sample handling is not required, and quantification is performed computationally based on relative intensities. Proteomics also enables detection and localization of post-translational modifications, such as phosphorylation, acetylation, ubiquitination, and glycosylation, to provide further insight into protein structure, function, and regulation.32 Finally, proteomics can capture protein–protein interactions and protein complex formation through affinity purification and subcellular localization assays,33,34 protein correlation profiling,35 proximity-based labeling techniques,36,37 and imaging.38 Protein identification is performed using software tools, such as MaxQuant39 and Fragpipe,40 which map peptides to proteome FASTA files from available databases (e.g., UniProt). The output files are analyzed using statistical testing and visualization tools, such as Perseus41 and R programming, to provide tangible information for the identified proteins. Together, proteomics measures and defines regulatory mechanisms associated with protein production across diverse biological systems.
3. Candida spp.
Candida spp. is a polyphyletic group of fungi belonging to the ascomycete yeasts, being commonly found within the commensal flora of the host skin microbiome and gastrointestinal tract with detection in up to 60% of the human population.42,43 Critically, however, dysbiosis, including changes to host immunocompetency, triggers a morphological switch leading to candidiasis and accounting for over 70% of invasive fungal infections.9 Such infections present challenges for rapid and reliable diagnostics and are attributed to mortality rates exceeding 50%.44 The most isolated commensal and pathogenic species of Candida from humans include C. albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis, Candida lusitania, and Candida krusei.43 The transition from commensal to pathogenic yeast is the foundation of many leading-edge studies exploring the relationships between Candida spp. and the human host.45Candidiasis is a broad term referring to infections of the skin, mucosal membranes, and deep organs caused by Candida spp. Invasive candidiasis refers to bloodstream infections (i.e., candidemia) and deep infections, such as intra-abdominal abscesses, peritonitis (i.e., inflammation of the peritoneum, the tissue that covers the inner wall of the abdomen and abdominal organs), or osteomyelitis (i.e., infection of the bones)42 (Fig. 1). C. albicans is the most common fungal species causing disease in both adult and pediatric populations through the production of virulence factors that are critical for fungal survival, growth, and establishment of infections. For instance, secreted aspartyl proteinases, surface adhesins and biofilm-associated proteins (e.g., agglutinin-like sequence family), phospholipases, and the ability to form hyphae are amongst the most well-studied and critical virulence factors produced by the pathogen.46–48
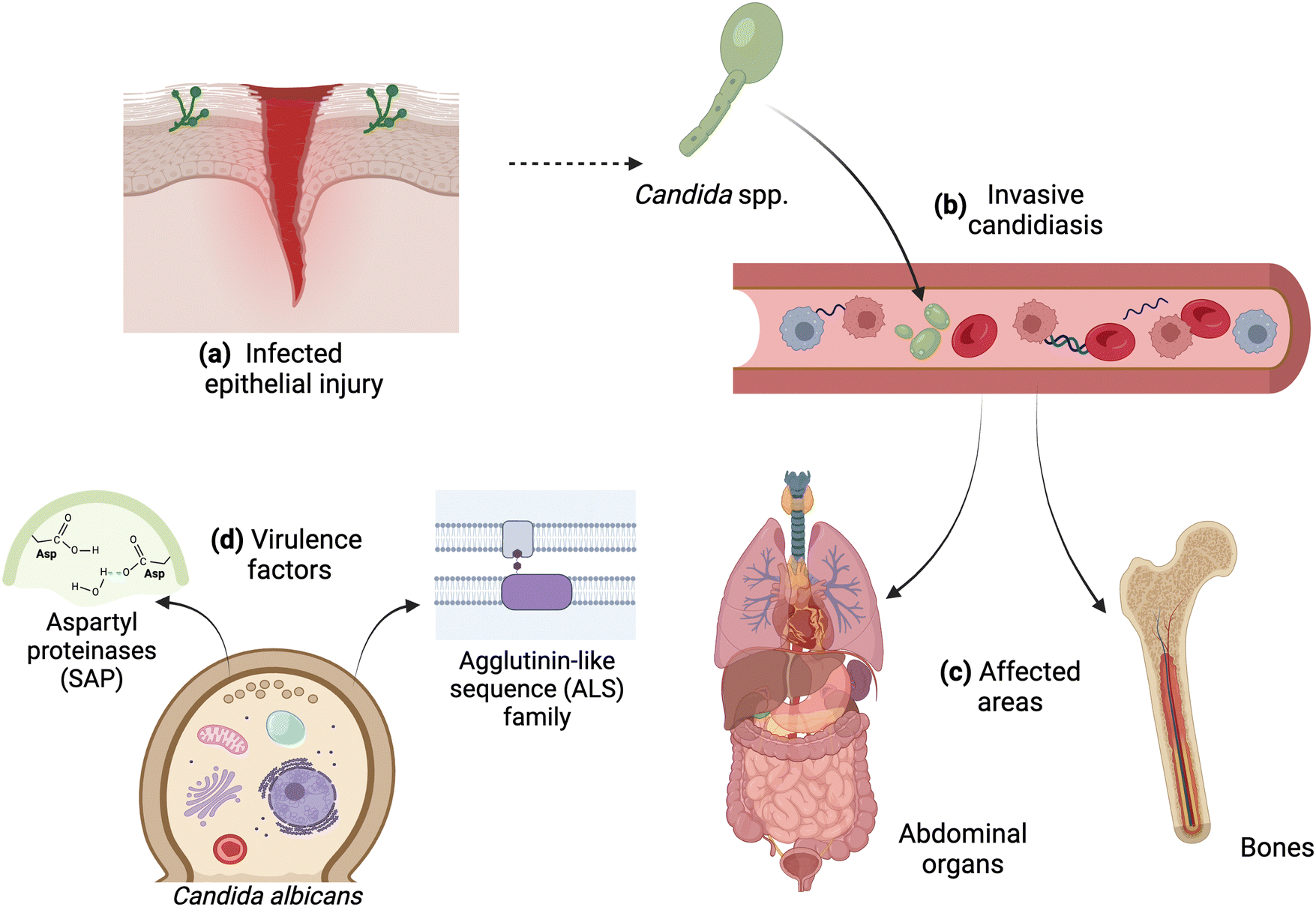 | ||
| Fig. 1 Invasive candidiasis and virulence factors of C. albicans. (a) Infected epithelium upon injury; (b) infection of the bloodstream caused by C. albicans that can affect (c) abdominal organs and bones. (d) Virulence factors produced by the fungi, including secreted aspartyl proteinases (SAP) and surface adhesins, such as those of the agglutinin-like sequence (ALS) family. Image created with https://BioRender.com. | ||
Proteomics provides a quantifiable strategy to characterize morphological changes of Candida spp. that occur during adaptation of the microorganism under different environmental conditions. Proteins with altered abundance profiles under evaluated conditions may present as drivers of fungal pathogenicity, providing new insight into regulatory mechanisms, virulence determinants, antifungal resistance, and the interaction between host and pathogen during infection. An overview of the proteomics approaches used within each of these studies towards Candida spp. shows the diversity of technical options available (Table 1). For example, proteomics investigated the interface of fungal cells and the host environment by measuring surface-exposed proteins collected from C. glabrata, C. parapsilosis, and C. tropicalis under growth conditions of artificial media mimicking the host's saliva, urine, and vaginal space compared to rich media.49 Patterns of protein abundance across five categories, including (i) typical cell wall proteins and secreted proteins, (ii) stress response proteins, (iii) atypical cell wall proteins (i.e., moonlighting proteins), (iv) ribosomal and nuclear proteins, and (v) proteins of unknown function, were defined for each strain. Proteins associated with cell wall maintenance and fungal pathogenesis were identified with elevated abundance under infection-mimicking conditions. Specifically, three moonlighting cell wall proteins were common across the three Candida spp., Pdc11 (pyruvate decarboxylase), Eno1 (enolase), and Tdh3 (glyceraldehyde-3-phosphate dehydrogenase), and exclusive moonlighting proteins identified one common protein across the strains, Mp65 (mannoprotein). A complementary study profiled the surfaceome of extracellular vesicles given their role in communication between a pathogen and host during infection produced by the same non-albicans Candida species.50 Across the strains, a comparison between protein content and phospholipid content correlated, with C. parapsilosis displaying the highest levels. However, these values did not correlate with the average extracellular vesicle size, with C. glabrata displaying the largest vesicles. Proteomics profiling defined diverse extracellular vesicle surface profiles across the strains, including identification of membrane-associated transporters, glycoproteins and enzymes involved in cell wall organization, and cytoplasmic proteins with possible moonlighting roles during infection. Notably, two proteins were common across all three Candida strains, the cell wall protein, Scw4, and an alcohol dehydrogenase, Adh1. The findings highlight strain specific proteome remodeling under altered growth conditions and the complexity of protein exposure at the fungal cell surface or within the extracellular environment to putatively modulate the host immune response to infection.
| Pathogen | Cell type | Environment | Technique | Quant. | Instrument | Mode | Grad. | Ref. |
|---|---|---|---|---|---|---|---|---|
| N/A = not available. Quant = quantification method. Mode = data acquisition mode. Gradient = liquid chromatography gradient length. DDA = data dependent acquisition. DIA = data independent acquisition. TOF = time of flight. TMT = tandem mass tags. LFQ = label-free quantification. | ||||||||
| C. glabrata | Cell surface | Altered culture medium | Cell surface shaving with trypsin | N/A | HCDUltra ETDII ion-trap | DDA | 60 min | Karkowska-Kuleta et al., 201949 |
| C. parapsilosis | ||||||||
| C. tropicalis | ||||||||
| C. glabrata | Extracellular vesicle cell surface | RPMI 1640 media | Cell surface shaving with trypsin | N/A | HCDUltra ETDII ion-trap | DDA | N/A | Karkowska-Kuleta et al., 201949 |
| C. parapsilosis | ||||||||
| C. tropicalis | ||||||||
| C. albicans | Fungal cell | H2O2 or acetic acid treatment | Trypsin digestion | LFQ | Oribtrap Q Exactive Plus | DIA | 120 min | Amador-Garcia et al., 202151 |
| DDA | 60 min | |||||||
| Synthetic heavy-labeled peptides | N/A | QTRAP 5500 | Targeted | 30 min | ||||
| C. albicans | Biofilms | RPMI 1640 media | Trypsin digestion | SWATH spectral library | Triple-TOF 5600 | DDA | 90 min | Abdulghani et al., 202252 |
| C. albicans (clinical isolates) | Fungal cell wall | Caspofungin (+/−) | Trypsin digestion | N/A | Oribtrap Q Exactive Plus | DDA | N/A | Buda De Cesare et al., 202253 |
| C. albicans (clinical isolates) | Fungal cell wall | Fluconazole (+/−) | Trypsin digestion | TMT | Oribtrap Q Exactive Plus | DDA | 24 min | Song et al., 202254 |
Another proteomics study explored the proteome remodeling of C. albicans during transition from a commensal to a pathogenic state initiated by chemical exposure (i.e., H2O2 and acetic acid).51 Using a data-independent acquisition approach for mass spectrometry measurements combined with library-based searching, the authors quantified over 2000 fungal proteins, with increases in protein abundance detected under H2O2 treatment compared to decreased protein abundance profiles under acetic acid treatment. Based on Gene Ontology, proteins with increased abundance upon H2O2 treatment were involved in oxidative stress response, proteasome-dependent catabolism, and protein folding. Specifically, Prn1, a protein similar to pirins and lacking functional knowledge, showed important roles in response to oxidative stress. Proteins with lower abundance upon H2O2 treatment were associated with the respiratory chain and cell wall, as well as ATP synthesis. Upon acetic acid treatment, opposite findings were reported with proteins involved in oxidative response to stress and heat shock proteins showing decreased abundance, along with reduced abundance of proteins of amino acid biosynthesis, protein folding, and rRNA processing. Both treatment conditions demonstrated a modulation of fungal cell apoptosis. These discovery-based findings were coupled with targeted proteomics using selected reaction monitoring (SRM) to detect 32 C. albicans proteins relevant to yeast apoptosis. Comparison of the DIA and SRM data showed comparable patterns of protein abundance changes upon H2O2 and acetic acid treatments. Further experimentation identified an oxidoreductase (Oye32), which plays a role in acetic acid and amphotericin B responses, that correlated with the fungal apoptotic state, supporting a novel role as a putative apoptotic biomarker of fungal stress. Additionally, proteomics profiling of morphological and architectural feature disruption of C. albicans was explored through biofilm growth.52 The authors defined 64 proteins with significant changes in abundance; 31 proteins showed increased abundance and 33 showed decreased abundance. Functional annotation using the Candida Genome Database, UniProt, and the Saccharomyces Genome Database defined higher abundance proteins associated with fungal metabolism, transcription, RNA processing, translation, PTM, proteolysis, transport, stress response, and cell wall composition. Proteins with decreased abundance were associated with common functions to those listed above, including fungal metabolism, cell wall, stress response, RNA processing, translation, PTM, proteolysis, and transport, as well as new categories, such as signal transduction, chromatin remodeling, and DNA repair. The proteomics data were complemented with qRT-PCR analysis of select genes involved in biofilm modulation with only an acyl-CoA desaturase (Ole1) showing differential abundance at the protein level, correlating with transcript expression. Finally, mitochondrial membrane proteins were associated with biofilm formation but no evidence of differential abundance at the protein level was observed. Together, this study detected proteins involved in C. albicans biofilm formation with putative connections to new strategies to combat fungal biofilms upon target disruption; however, further evaluation is needed.
With an emphasis on antifungal resistance, quantitative proteomics provides insight into fungal responses to drug treatment, along with potential mechanisms contributing to resistance. For instance, comparative proteomics of the fungal cell surface in echinocandin-resistant versus -susceptible C. albicans strains in the presence and absence of caspofungin, demonstrated remodeling of cell wall organization and maintenance and changes in cell wall architecture.53 Notably, 30 proteins exclusively identified in the resistant isolates in the absence of caspofungin showed increased abundance and association with the fungal cell wall, as well as cytoplasmic and plasma membrane proteins (potential contaminants). Conversely, in the presence of caspofungin, a decrease in the abundance of proteins associated with host defense and fungal pathogenesis was detected in both resistant and susceptible strains. Specifically, two glycosylphosphatidylinositol (GPI)-anchored proteins (Pga52 and Pga31) showed higher abundance in the resistant isolate in the presence and absence of caspofungin, indicating a baseline and elevated change in protein production upon treatment. For markers of echinocandin resistance, a priority list of 11 proteins, including a GPI-anchored protein (Pga10), with stable differences between drug-resistant and -susceptible strains was defined. Another study explored fluconazole antifungal resistance in clinically isolated C. albicans strains from an immunocompromised individual. Using quantitative proteomics, the study reported enrichment analyses by Gene Ontology and KEGG, functionally annotated and characterized reduced glycolysis, metabolic, and oxidative stress responses in the fluconazole resistant strains, emphasizing the role of proteins in resistance.54 Within the study, protein abundance of common azole resistance determinants was measured with only Cdr1, belonging to the ABC drug efflux transporters, being significantly higher upon a comparison of strains. Given its role in drug transport, it was no surprise that Cdr1 also showed increased production across the isolates upon previous fluconazole exposure. Together, through the described studies, proteomics provided new biological insights into mechanisms of fungal pathogenies, modulation of the host, and antifungal resistance for diverse Candida spp.
4. Cryptococcus spp.
Belonging to the Basidiomycota phylum, the yeast genus Cryptococcus is primarily of environmental origin and is commonly associated with soil, decaying wood, and bird feces.55,56 However, species like C. neoformans and C. gattii can infect humans and cause cryptococcosis, a globally distributed life-threatening disease. Currently, cryptococcosis affects approximately 194![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people annually contributing to 147000 deaths, a mortality rate of almost 80%.4,57 For C. neoformans, the main etiological agent of cryptococcosis, infection is initiated upon the inhalation of desiccated yeast cells or basidiospores followed by colonization of the lungs and, depending on the individual's immune status, dissemination throughout the body via the bloodstream, eventually crossing the blood–brain barrier (BBB) and invasion of the central nervous system (CNS) (Fig. 2). Common clinical manifestations of cryptococcosis in immunocompromised individuals include cryptococcal pneumonia (pulmonary infection), cryptococcemia (blood infection), and cryptococcal meningitis or meningoencephalitis (CNS infection), all resulting from unrestricted fungal growth.58 In contrast, immunocompetent individuals can mount a protective inflammatory response, resulting in the containment of the fungus and reduced fungal replication numbers.59
000 people annually contributing to 147000 deaths, a mortality rate of almost 80%.4,57 For C. neoformans, the main etiological agent of cryptococcosis, infection is initiated upon the inhalation of desiccated yeast cells or basidiospores followed by colonization of the lungs and, depending on the individual's immune status, dissemination throughout the body via the bloodstream, eventually crossing the blood–brain barrier (BBB) and invasion of the central nervous system (CNS) (Fig. 2). Common clinical manifestations of cryptococcosis in immunocompromised individuals include cryptococcal pneumonia (pulmonary infection), cryptococcemia (blood infection), and cryptococcal meningitis or meningoencephalitis (CNS infection), all resulting from unrestricted fungal growth.58 In contrast, immunocompetent individuals can mount a protective inflammatory response, resulting in the containment of the fungus and reduced fungal replication numbers.59
 | ||
| Fig. 2 C. neoformans infection cycle and common clinical manifestations. (a) Environmental sources of cryptococcal dried cells or spores; (b) common manifestations of cryptococcal infection within immunocompetent and immunocompromised individuals; (c) virulence factors produced by Cryptococcus spp. Image created with https://BioRender.com. | ||
To better understand mechanisms regulating infection from both the host and pathogen perspectives, mass spectrometry-based proteomics profiling is a powerful tool for such endeavors. An overview of the proteomics approaches used within each of these studies of Cryptococcus spp. highlights the diversity of technical approaches available (Table 2). For example, a study investigated C. neoformans response to copper-induced-reactive oxygen species stress of the two primary copper detoxifying proteins, copper-sequestering metallothionein (CMT1, CMT2).60 In this study, a proteomic comparison of the double knockout strain to untreated, copper-replete, and toxic copper levels supplemented with reactive oxygen species scavenger conditions, revealed that copper-induced reactive oxygen species decreased the abundance of fungal proteins involved in protein synthesis and increased the abundance of proteins associated with degradation processes. Specifically, copper-induced reactive oxygen species were associated with proteins involved in the ubiquitin ligase complex and proteasome pathway. The discovery-based proteomic profiling was complemented by targeted parallel reaction monitoring for 37 select proteins to confirm detection and abundance; all but two proteins were commonly differentially produced. Moreover, inhibition of the proteasome pathway partially alleviated copper toxicity in fungal cells. Another study explored the connection between fungal virulence and proteasome function through proteomic profiling of the C. neoformans cAMP/Protein Kinase A (PKA) pathway.61 Here, 3222 proteins were identified with 302 proteins being common between a Pka1-regulated C. neoformans strain under pka1 induction or suppression. A STRING analysis of differentially produced proteins identified the ubiquitin-proteasome pathway as a potential fungal pharmacological target due to its ability to control protein turnover and protein aggregations. These data were combined with the connection of PKA towards polysaccharide capsule production. Next, the anticancer drug and proteasome inhibitor, bortezomib, was investigated as a novel drug repurposing strategy revealing C. neoformans sensitivity to treatment. Another study by the same group explored the effect of PKA regulation on the secretome of C. neoformans.62 The study identified regulated virulence-associated proteins in the C. neoformans secretome, including Cig1, Aph1 (acid phosphatase), alpha-amylase, glyoxal oxidase, and a novel protein (CNAG_05312), and aligned protein production with transcript expression. Next, a targeted proteomics approach by multiple reaction monitoring towards these proteins within bronchoalveolar lavage and blood from a murine cryptococcal infection quantified putative diagnostic biomarkers. Ultimately, Cig1, glyoxal oxidase, and CNAG_05312 were detected and quantified within the blood.62 Another study explored the potential of biomarkers from both the host and pathogen infection from the spleen for the detection and monitoring of fungal disease.63 The authors used quantitative proteomics to map signatures of protein production across a temporal cryptococcal infection within a murine model and defined changes in fungi-specific protein responses over time. From the host perspective, four host proteins with known roles in immune response (i.e., metaxin 2, cathelicidin antimicrobial peptide, heat shock protein 90, and complement C3) showed differential production between uninfected and infected samples and across time points of infection, which aligned with additional putative infection-associated biomarkers (e.g., haptoglobin and glutathione peroxidase). From the pathogen perspective, we identified key virulence-associated proteins (i.e., cAMP-dependent protein kinase regulatory subunit, CipC, alpha-amylase, and urease) across time points and proposed novel signatures of disease (e.g., FK506-binding protein and carbonic anhydrase).
| Pathogen | Cell type | Environment | Technique | Quant. | Instrument | Mode | Grad. | Ref. |
|---|---|---|---|---|---|---|---|---|
| N/A = not available. Quant = quantification method. Mode = data acquisition mode. Gradient = liquid chromatography gradient length. DDA = data dependent acquisition. DIA = data independent acquisition. LFQ = label-free quantification. TOF = time of flight. TMT = tandem mass tags. TIMS = trapped ion mobility spectrometry. YPD = yeast peptone dextrose. iTRAQ = isobaric tags for relative and absolute quantification. DMEM = Dulbecco's Modified Eagle's Medium. FBS = fetal bovine serum. | ||||||||
| C. neoformans | Fungal cell | CuSO4; N-acetylcysteine | Trypsin digestion | iTRAQ | Orbitrap Q Exactive Plus | DDA | 120 min | Sun et al., 202160 |
| N/A | Targeted | |||||||
| C. neoformans | Fungal cell | YPD | Trypsin digestion | Dimethyl labeling | LTQ Orbitrap Velos | DDA | N/A | Geddeset al., 201661 |
| C. neoformans | Secreted | Minimal media (+/− galactose) | Trypsin digestion | Dimethyl labeling | LTQ Orbitrap Velos | DDA | 90 min | Geddeset al., 201562 |
| C. neoformans | Secreted | Murine blood; murine bronchoalveolar lavage | Synthetic heavy-labeled peptides | N/A | 6460 Triple Quadrupole | Targeted | 15 min | Geddeset al., 201562 |
| C. neoformans | Tissue | Murine spleen | Trypsin digestion | TMT | Exploris 480 Orbitrap | DDA | 21 min | Muselius et al. 202363 |
| C. neoformans | Extracellular vesicles | Minimal media | Trypsin digestion | N/A | Orbitrap LTQ XL | DDA | 100 min | Rodrigues et al., 200864 |
| C. neoformans, C. deneoformans, C. deuterogattii | Extracellular vesicles | YPD | LysC/trypsin digestion | iBAQ | Orbitrap Q Exactive Plus | DDA | 250 min | Rizzo et al., 202165 |
| C. neoformans | Biofilms; planktonic cells | YPD; minimal media | Trypsin digestion | Spectral counting | Orbitrap LTQ XL | DDA | Offline (300+ min) | Santi et al., 201466 |
| C. neoformans | Biofilms; planktonic cells | YPD; minimal media | Trypsin digestion | Spectral counting | Orbitrap LTQ XL | DDA | Offline (300+ min) | Santi et al., 202467 |
| C. gattii | ||||||||
| C. neoformans | Macrophage co-cultures | DMEM + FBS | LysC/trypsin digestion | LFQ | Orbitrap Exploris 240 | DDA | 240 min | Sukumaran & Ball et al., 202268 |
| C. neoformans | Macrophage co-cultures | DMEM + FBS | LysC/trypsin digestion | LFQ | Hybrid TIMS-quadrupole TOF | DDA | 44 min | Ball et al., 202469 |
| DIA | ||||||||
Proteomic studies also provide insight into virulence-associated structures, including biofilms and extracellular vesicles. For instance, cryptococcal extracellular vesicles were the first fungal-derived extracellular vesicles identified and profiled at the protein level.64 Specifically, researchers observed serological activity specific to extracellular vesicle-associated proteins derived from patients with cryptococcosis. An in-depth analysis of the fungal extracellular vesicles identified 76 proteins with an abundance of proteins correlating to capsule structures. Of these proteins, chaperones, heat shock proteins, superoxide dismutase, signal transduction regulators, antioxidant and cytosolic proteins, as well as enzymes were identified, along with 27 previously reported as vesicular proteins in mammalian exosomes. Another more recent proteomic study identified a core Cryptococcus spp. extracellular vesicle proteome conserved across diverse fungal species.65 Notably, the researchers used intensity-based absolute quantification (IBAQ) to rank the prevalence of C. neoformans proteins across the samples coupled to gene expression levels by RNA-Seq to calculate enrichment values. The authors applied a similar approach to assess extracellular vesicle protein cargo in C. deneoformans and C. deuterogattii with 17 proteins shared across the cryptococcal strains, including chitin deactelylase and glyoxal oxidase. Moreover, the study identified packaged immunoreactive proteins (e.g., mannoproteins) and protective antigens on the surface of the extracellular vesicles resembling the spike complexes on a viral envelope. These findings prompted an investigation into the durability of a cryptococcal extracellular vesicle-based vaccine strategy. Immunization with extracellular vesicles obtained from an acapsular strain provided significant protection from cryptococcal infections.
Cryptococcal biofilms pose a significant threat to the treatment of fungal infections due to the production of antifungal-resistant fungal structures (i.e., cryptococcomas) that lead to persistent lung and brain infections.70 An early comparative proteomic analysis on planktonic and biofilm C. neoformans cells identified 1939 proteins common to the different growth states with <7% unique to the biofilms.66 Proteins with 2-fold higher production during biofilm growth were associated with oxidation–reduction, proteolysis, and stress response (e.g., catalase and heat shock proteins). Notably, 33 proteins were classified as hypothetical with functional annotation defining roles in fungal metabolism, biosynthesis, and replication and transcription, for example. These findings demonstrate the diversity of biological processes associated with cellular remodeling during biofilm formation. A more recent study went beyond a single cryptococcal species to compare biofilm proteome remodeling between C. neoformans and C. gattii.67 This study identified 1819 proteins with >78% commonality between the strains to reveal a conserved Cryptococcus spp. biofilm strategy. The fungal biofilms support an adherent lifestyle by a decreased production of glycolytic proteins, such as glucose-6-phosphate isomerase and malate dehydrogenase, in exchange for increased production of proteins within metabolic pathways associated with energy acquisition and reoxidation, including succinyl-CoA synthetase and cytochrome C oxidase. However, species-specific signatures were also observed, with C. gattii biofilms featuring increased production of proteins related to the electron transport chain, DNA binding, and transcription compared to elevated abundance of proteins related to oxidoreductase, catabolic process, and protein folding in C. neoformans biofilms. These findings highlight the complexity of biofilm structures and define strain specific remodeling to support biofilm development.
Another set of studies used quantitative proteomics to explore the dynamics of cross-kingdom interactions of C. neoformans, a bacterial pathogen (i.e., Klebsiella pneumoniae), and macrophages.68 This study explored the evolution of host and pathogen responses over time and proteome adaptations elicited during coinfection through identification of 2292 host proteins, 128 fungal proteins, and 163 bacterial proteins. The authors observed distinct host and fungal proteome responses due to initial fungal infection followed by a state of dormancy between the species occurring at a later-stage infection time point. These findings provide molecular evidence at the protein level of a characteristic of C. neoformans intracellular adaptation techniques in response to macrophage stress.71 Interestingly, upon co-infection with K. pneumonia, this stabilization was disrupted by virulence-associated fungal and bacterial proteins, including the fungal virulence determinants, catalase and melanin. Ultimately resulting in host dysbiosis, observed through the dramatic increase in tumor necrosis factor α, suggesting a specific immune response tailored to bacterial coinfection. In a second study, a comparison of data-dependent acquisition to data-independent acquisition for cross-kingdom protein identification revealed a significant increase in fungal protein identifications using the data-independent acquisition approach.69 Specifically, a 19%, 55%, and 125% increase in protein identifications for the host, C. neoformans, and K. pneumoniae, respectively, was reported upon DIA measurements. Interestingly, the newly detected fungal and bacterial proteins displayed known and putative roles in virulence, suggesting potential anti-virulence targets. Biological characterization of a previously undetected infection-associated fungal protein, CNAG_05997, revealed roles in fungal growth and thermotolerance, polysaccharide capsule and melanin production, and macrophage infectivity. Together, proteomics has explored and revealed diagnostic and therapeutic potential against cryptococcal infections, strategies for fungal recognition and immune system evasion, and regulatory mechanisms driving pathogenesis.
5. Aspergillus spp.
Aspergillus belongs to the Ascomycota yeasts, comprising a diverse group of species based on morphological, physiological and phylogenetic characteristics. These are saprotrophic fungi found in hospitals, gardens, and fields with essential roles in carbon and nitrogen recycling.72 The most relative species of for human disease is A. fumigatus, which is responsible for 90% of invasive aspergillosis, causing persistent pneumonia, sinusitis that progresses through tissues and brain abscesses in neutropenic patients, and in patients with phagocytic defects, such as chronic granulomatous disease.73,74 Additional pathogenic species include Aspergillus flavus, Aspergillus niger, and Aspergillus terreus. Infection with Aspergillus spp. is potentially fatal in immunosuppressed individuals due to poor susceptibility to antifungal drugs, and a correlation with harmful allergic reactions75 (Fig. 3). Aspergillosis encompasses a range of infections typically caused by A. fumigatus, including allergic bronchopulmonary aspergillosis (i.e., a fungal infection of the lung secondary to a hypersensitivity reaction to antigens of the fungi), chronic pulmonary aspergillosis (i.e., a hypersensitive lung condition primarily affecting patients with asthma and Cystic fibrosis), and invasive aspergillosis.75–77 Invasive aspergillosis is the most severe form of pulmonary aspergillosis, with a mortality rate exceeding 50%; however, combined, these infections account for over 5 M cases of aspergillosis each year.4 | ||
| Fig. 3 Aspergillosis and virulence factors Aspergillus spp. (a) Conidiophores liberating conidia in the air; (b) inhaled conidia disseminate to the pulmonary alveoli; (c) examples of critical virulence factors that activate the immune system of the host. IL = interleukin, IFN = interferon, TNF = tumor necrosis factor, T = T cell, Th = T helper cell, LPS = lipopolysaccharide, GM-CSF = granulocyte macrophage colony stimulating factor. Image created with https://BioRender.com. | ||
Applications of mass spectrometry-based proteomics towards profiling of Aspergillus spp. have revealed important biological insights into diverse pathogenic processes. An overview of the proteomics approaches used within each of these studies of Aspergillus spp. highlights the diversity of technical approaches available (Table 3). For example, a reference proteome map of macrophage phagolysosomes exposed to A. fumigatus conidia from melanin-producing or -non-producing strains identified 2421 murine phagolysosomal proteins and 65 A. fumigatus proteins.78 Notably, 95% of detected proteins were common across the A. fumigatus strains, suggesting few unique proteins drive differential responses and/or quantitative differences prevail. Proteins exclusive to the melanin-producing fungal strains were identified, including catalase, drug response and mitochondrial unfolded protein response elements, and glyceraldehyde-3-phosphate dehydrogenase. Conversely, fungal proteins enriched from the melanin-lacking strain within the phagolysosome included those induced upon oxidative stress or immune cell association, such as a GTPase regulating vesicular transport, RNA helicase, alcohol dehydrogenase, and a transaldolase. For the host, proteins associated with diverse regulatory processes, including phagolysosome acidification (e.g., Rab5), endocytic trafficking, signaling pathways, and proteases (e.g., cathepsin Z) were impacted by the fungus and confirmed by antibody detection. Another study developed the most extensive cell wall proteome map of resting conidia from A. fumigatus using isolation of conidial cell wall proteins by hydrogen–fluoride–pyridine extraction and trypsin shaving coupled to mass spectrometry-based proteomics profiling.79 The hydrogen–fluoride–pyridine method permitted identification of cell wall associated proteins, including GPI-anchored proteins, whereas trypsin shaving identified surface-exposed proteins. In total, 148 fungal proteins were identified with 116 proteins exclusive to the hydrogen–fluoride–pyridine method, 48 proteins exclusive to the trypsin-shaving method, and 15 proteins shared across the approaches. At the intersection of the two methods, RodA, a surface hydrophobin, was the most abundant protein, along with an uncharacterized conidial cell wall protein A (CcpA). Further investigation into CcpA revealed a role in masking the fungal conidia from immune cell recognition through suppressed neutrophil and dendritic cell activation.
| Pathogen | Cell type | Environment | Technique | Quant. | Instrument | Mode | Grad. | Ref. |
|---|---|---|---|---|---|---|---|---|
| N/A = not available. Quant = quantification method. Mode = data acquisition mode. Gradient = liquid chromatography gradient length. DDA = data dependent acquisition. LFQ = label-free quantification. DMEM = Dulbecco's Modified Eagle's Medium. FBS = fetal bovine serum. HF = hydrogen-fluoride-pyridine. | ||||||||
| A. fumigatus | Conidia | Aspergillus minimal media; | Trypsin digestion | LFQ | Orbitrap Q Exactive Plus | DDA | 135 min | Schmidt et al., 201878 |
| Phagolysosome | DMEM + FBS | 360 min | ||||||
| A. fumigatus | Conidial surface | Aspergillus minimal media | Trypsin shaving | N/A | LTQ Orbitrap Velos | DDA | 120 min | Voltersen et al., 201879 |
| HF-pyridine-extraction | QExactive HF Orbitrap | 90 min | ||||||
| A. fumigatus | Conidial surface | Potato dextrose agar | Trypsin shaving | LFQ | Orbitrap Q Exactive HF | DDA | 90 min | Pinzan et al., 202480 |
| A. oerlinghausenensis | ||||||||
| A. fischeri | Macrophage co-culture | DMEM + FBS | Trypsin digestion | LTQ Oribtrap Velos | 80 min | |||
| A. lentulus | ||||||||
| A. fumigatus | Conidial surface; extracellular | Potato dextrose agar | Trypsin shaving; trypsin digestion | DDA | LTQ Orbitrap Velos | DDA | N/A | Venugopalan et al., 202381 |
| A. flavus (clinical isolates) | ||||||||
Other studies explored the conidial surfaceome of diverse pathogenic and non-pathogenic Aspergillus spp. to define proteins exclusive to each species.80 The study identified 1097 conidial surface proteins across four Aspergillus strains, including A. fumigatus, Aspergillus oerlinghausenensis, Aspergillus lentulus, and Aspergillus fischeri, with 75 proteins hared across all strains and 62 unique to A. fumigatus. The majority of these exclusive proteins are associated with cell wall modification, metabolism, cell signalling, and secondary metabolite biosynthesis, as well as unknown functions. Complementary genetic analyses of the protein-encoding genes determined distinguishing characteristics across species. These included altered susceptibility to macrophage and epithelial cells, and modified regulation of host proinflammatory cytokine levels. Another study focused on the proteome mapping of conidial surface-associated and extracellular proteins during the early stages of fungal growth and identified proteins crucial for establishing infection.81 In this study, a comparison of clinical strains identified 116 and 122 proteins in A. flavus and A. fumigatus, respectively, with common protein classes defined, including cell wall modifying enzymes (e.g., chitinase), proteases (e.g., carboxypeptidase), and antioxidant enzymes (e.g., catalase). Additionally, an analysis of the exoproteome identified 239 and 221 proteins in A. flavus and A. fumigatus, respectively, with mutual enrichment of enzymes acting upon cell wall polysaccharides. Next, a comparison of the conidial surface proteins and exoproteins within each strain identified 97 proteins common for A. flavus, including alkaline protease, an allergen was more abundant on the surface than in the extracellular environment. Moreover, 85 proteins were common between the conidial surface proteins and exoproteins for A. fumigatus. Importantly, species-specific protein signatures were defined, including the enrichment of immunoreactive and pathogenicity-related proteins from A. fumigatus compared to the enrichment of enzymes associated with cell wall organization and binding from A. flavus. Study validation included correlative analysis between exoprotein abundance transcript expression for six genes with differential abundance or detection between the strains. Together, these proteomics studies converge on an investigation of Aspergillus spp. conidia and its diverse and critical roles in modulating fungal pathogenicity and the host immune response.
6. Conclusion and future directions
Fungal pathogens represent substantial threats to global human health. Challenges with diagnostics, prognostics, and therapeutic options, combined with the emergence of new pathogens and rising rates of antifungal resistance emphasize an urgent need for improvements. Strategies include expanding our knowledge of mechanisms of pathogenesis and interactions with the environment, including approaches used by the pathogen to survive and proliferate within the host. Mass spectrometry-based proteomics is a powerful, high-resolution technique used to investigate protein-level drivers of fungal pathogenesis, host–pathogen interactions, and mechanisms of antifungal resistance. Presently, the full promise of proteins for new diagnostic and prognostic methods applied within the clinic, and the confirmation of safe and effective novel antifungals towards druggable targets revealed through proteomics, is yet realized. However, with improved technological (e.g., mass spectrometry instrumentation) and computational strategies (e.g., advanced bioinformatics combined with artificial intelligence), the immense potential of proteomics towards fungal research is being revealed through diverse applications at an unprecedented rate. For instance, throughout this review, we highlighted common mechanisms of pathogenesis used by the diverse fungal pathogens in preparation for infection (e.g., nutrient limited media) or within the presence of host cells (e.g., macrophage). These include the production of enzymes for target degradation or cell wall manipulation, such as chitinase and catalase, or the common production of proteins associated with stress response, including oxidoreductases. Moreover, throughout these studies, fungal proteins with known and anticipated roles in virulence mechanisms (e.g., conidial surface-associated proteins, extracellular proteins, biofilm formation) were detected and support further investigation into genetic deletion strains for assessment as putative novel antifungal targets. Lastly, the detection of fungal proteins within host environments, including spleen tissue, blood, and brochoalveolar lavage, warrant investigation as biomarkers of infection for diagnostics and prognostics.Another aspect of comparison and innovation is the diverse technological approaches used to conduct the studies outlined in this review. For example, the use of orbitrap and time-of-flight technologies support identification of diverse proteins based on ion fragmentation and detection. Additionally, differences in labeling techniques, liquid chromatography gradient lengths, and data acquisition methods introduce opportunities for optimization across the studies. Moreover, several studies validate DDA datasets using targeted proteomics strategies for increased sensitivity of detection in clinically relevant matrices. Further, only two highlighted studies applied DIA methods to study fungal pathogenesis but given the increased depth of coverage of the pathogen proteome within complex backgrounds using this approach, integration across future studies may uncover new mechanisms of action used by the pathogen to modulate the immune system or reveal previously undiscovered infection-associated fungal proteins with putative roles as antifungal targets. Overall, the studies presented herein demonstrate the power and potential of proteomics to uncover new biological roles and targets to better understand fungal pathogens and the diseases they cause. Next steps include complementing these proteomics discoveries with biological validation for translational applications. Such translational avenues for fungal disease discovery include moving the information gleaned from in vitro studies focusing on lab-associated and clinical isolates into clinical settings for improved treatment strategies. These include prevention of infections by targeting fungal virulence factors that cause disease, diagnosis of fungal infections upon initial exposure, and monitoring treatment efficacy to disrupt the evolution of antifungal resistance.
Author contributions
M. S., M. dS., and J. G.-M. conceptualized the topics. M. S., M. dS., and B. B. generated figures and wrote the first draft. J. G.-M. edited figures and text for final submission. All authors have reviewed and approve of the final version.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
Funding is provided to J. G.-M. from the University of Guelph, Canadian Institutes of Health Research (Project Grant), New Frontiers Research Fund, and the Canada Research Chairs program. Funding is provided to M. S., M. dS., B. B. from the Natural Sciences and Engineering Research Council of Canada Collaborative Research and Training Experience Program – the Evolution of Fungal Pathogens, and to B. B. from NSERC Canadian Graduate Scholarship – Doctoral. The authors thank members of the Geddes-McAlister lab for their insightful discussions during the writing of this review.References
- F. Bongomin, S. Gago, R. Oladele and D. Denning, J. Fungi, 2017, 3, 57 CrossRef.
- J. Geddes-McAlister and R. S. Shapiro, Ann. N. Y. Acad. Sci., 2019, 1435, 57–78 CrossRef.
- M. C. Fisher, A. Alastruey-Izquierdo, J. Berman, T. Bicanic, E. M. Bignell, P. Bowyer, M. Bromley, R. Brüggemann, G. Garber, O. A. Cornely, S. J. Gurr, T. S. Harrison, E. Kuijper, J. Rhodes, D. C. Sheppard, A. Warris, P. L. White, J. Xu, B. Zwaan and P. E. Verweij, Nat. Rev. Microbiol., 2022, 20, 557–571 CrossRef CAS.
- D. W. Denning, Lancet Infect. Dis., 2024, 24, e428–e438 CrossRef.
- A. Fausto, M. L. Rodrigues and C. Coelho, Front. Microbiol., 2019, 10, 1–5 CrossRef.
- L. C. Horianopoulos, E. Gluck-Thaler, I. Benoit Gelber, L. E. Cowen, J. Geddes-McAlister, C. R. Landry, I. S. Schwartz, J. A. Scott, A. Sellam, D. C. Sheppard, T. Spribille, R. Subramaniam, A. K. Walker, S. D. Harris, R. S. Shapiro and A. C. Gerstein, Can. J. Microbiol., 2021, 67, 13–22 CrossRef CAS.
- T. J. Walsh and D. M. Dixon, in Medical Microbiology, 4th edn, 1996, vol. 75 Search PubMed.
- W. Fang, J. Wu, M. Cheng, X. Zhu, M. Du, C. Chen, W. Liao, K. Zhi and W. Pan, J. Biomed. Sci., 2023, 30, 42 CrossRef CAS.
- I. D. Jacobsen, Curr. Clin. Microbiol. Rep., 2023, 10, 55–65 CrossRef PubMed.
- J. R. Köhler, A. Casadevall and J. Perfect, Cold Spring Harbor Perspect. Med., 2014, 5, a019273 CrossRef.
- A. Rokas, Nat. Microbiol., 2022, 7, 607–619 CrossRef CAS PubMed.
- M. Woods, J. A. McAlister and J. Geddes-McAlister, Wiley Interdiscip. Rev.: Mech. Dis., 2023, 15, e1610 CrossRef.
- M. A. Garcia-Solache and A. Casadevall, mBio, 2010, 1, 1–3 CrossRef.
- A. Casadevall, D. P. Kontoyiannis and V. Robert, mBio, 2019, 10, e01397 Search PubMed.
- M. C. Fisher and D. W. Denning, Nat. Rev. Microbiol., 2023, 21, 211–212 Search PubMed.
- World Health Organization, WHO fungal priority pathogens list to guide research, development and public health action, 2022 Search PubMed.
- R. Aebersold and M. Mann, Nature, 2016, 537, 347–355 CrossRef CAS.
- J. Geddes-McAlister, F. Roux-Dalvai and A. Droit, in Genetics and Evolution of Infectious Diseases, Elsevier, 2024, pp. 465–492 Search PubMed.
- J. Geddes-McAlister, T. Rizakos and B. Muselius, in Detection and Analysis of Microorganisms by Mass Spectrometry, Royal Society of Chemistry, 2023, pp. 215–233 Search PubMed.
- F. Meissner, J. Geddes-McAlister, M. Mann and M. Bantscheff, Nat. Rev. Drug Discovery, 2022, 21, 637–654 CrossRef CAS.
- A. Sukumaran, J. Coish, J. Yeung, B. Muselius, M. Gadjeva, A. MacNeil and J. Geddes-McAlister, J. Leukocyte Biol., 2019, 106, 1221–1232 CrossRef CAS PubMed.
- T. K. Toby, L. Fornelli and N. L. Kelleher, Annu. Rev. Anal. Chem., 2016, 9, 499–519 CrossRef CAS PubMed.
- L. C. Gillet, A. Leitner and R. Aebersold, Annu. Rev. Anal. Chem., 2016, 9, 449–472 CrossRef.
- R. Aebersold, A. Bensimon, B. C. Collins, C. Ludwig and E. Sabido, Proteomics, 2016, 16, 2065–2067 CrossRef CAS PubMed.
- L. C. Gillet, P. Navarro, S. Tate, H. Röst, N. Selevsek, L. Reiter, R. Bonner and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, O111.016717 CrossRef PubMed.
- A. Hu, W. S. Noble and A. Wolf-Yadlin, F1000Res., 2016, 5, 419 Search PubMed.
- J. Derks, A. Leduc, G. Wallmann, R. G. Huffman, M. Willetts, S. Khan, H. Specht, M. Ralser, V. Demichev and N. Slavov, Nat. Biotechnol., 2023, 41, 50–59 CrossRef CAS PubMed.
- A. S. Welter, M. Gerwien, R. Kerridge, K. M. Alp, P. Mertins and M. Selbach, Mol. Cell. Proteomics, 2024, 23, 100839 CrossRef CAS PubMed.
- S. Nahnsen, C. Bielow, K. Reinert and O. Kohlbacher, Mol. Cell. Proteomics, 2013, 12, 549–556 CrossRef CAS.
- S.-E. Ong, Mol. Cell. Proteomics, 2002, 1, 376–386 CrossRef CAS.
- A. Thompson, J. Schäfer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann and C. Hamon, Anal. Chem., 2003, 75, 1895–1904 CrossRef CAS PubMed.
- M. Larance and A. I. Lamond, Nat. Rev. Mol. Cell Biol., 2015, 16, 269–280 CrossRef CAS PubMed.
- A. J. Groen, G. Sancho-Andrés, L. M. Breckels, L. Gatto, F. Aniento and K. S. Lilley, J. Proteome Res., 2014, 13, 763–776 CrossRef CAS PubMed.
- T. P. J. Dunkley, R. Watson, J. L. Griffin, P. Dupree and K. S. Lilley, Mol. Cell. Proteomics, 2004, 3, 1128–1134 CrossRef CAS.
- U. K. Aryal, Z. McBride, D. Chen, J. Xie and D. B. Szymanski, J. Proteomics, 2017, 166, 8–18 CrossRef CAS PubMed.
- A. Mair, S.-L. Xu, T. C. Branon, A. Y. Ting and D. C. Bergmann, eLife, 2019, 8, e47864 CrossRef CAS PubMed.
- T.-W. Kim, C. H. Park, C.-C. Hsu, Y.-W. Kim, Y.-W. Ko, Z. Zhang, J.-Y. Zhu, Y.-C. Hsiao, T. Branon, K. Kaasik, E. Saldivar, K. Li, A. Pasha, N. J. Provart, A. L. Burlingame, S.-L. Xu, A. Y. Ting and Z.-Y. Wang, Plant Cell, 2023, 35, 975–993 CrossRef PubMed.
- B. A. Boughton, D. Thinagaran, D. Sarabia, A. Bacic and U. Roessner, Phytochem. Rev., 2016, 15, 445–488 CrossRef CAS.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- F. Yu, G. C. Teo, A. T. Kong, K. Fröhlich, G. X. Li, V. Demichev and A. I. Nesvizhskii, Nat. Commun., 2023, 14, 4154 CrossRef CAS.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS.
- P. G. Pappas, M. S. Lionakis, M. C. Arendrup, L. Ostrosky-Zeichner and B. J. Kullberg, Nat. Rev. Dis. Primers, 2018, 4, 18026 CrossRef PubMed.
- J. C. O. Sardi, L. Scorzoni, T. Bernardi, A. M. Fusco-Almeida and M. J. S. Mendes Giannini, J. Med. Microbiol., 2013, 62, 10–24 CrossRef CAS.
- H. M. Arnold, S. T. Micek, A. F. Shorr, M. D. Zilberberg, A. J. Labelle, S. Kothari and M. H. Kollef, Pharmacotherapy, 2010, 30, 361–368 CrossRef.
- D. R. Silva, J. de, C. O. Sardi, I. A. Freires, A. C. B. Silva and P. L. Rosalen, Eur. J. Pharmacol., 2019, 842, 64–69 CrossRef CAS.
- L. Mathé and P. Van Dijck, Curr. Genet., 2013, 59, 251–264 Search PubMed.
- N. A. R. Gow, F. L. van de Veerdonk, A. J. P. Brown and M. G. Netea, Nat. Rev. Microbiol., 2012, 10, 112–122 CrossRef CAS PubMed.
- W. Aoki, N. Kitahara, N. Miura, H. Morisaka, Y. Yamamoto, K. Kuroda and M. Ueda, PLoS One, 2012, 7, e32513 CrossRef CAS.
- J. Karkowska-Kuleta, D. Satala, O. Bochenska, M. Rapala-Kozik and A. Kozik, BMC Microbiol., 2019, 19, 149 CrossRef.
- J. Karkowska-Kuleta, K. Kulig, E. Karnas, E. Zuba-Surma, O. Woznicka, E. Pyza, P. Kuleta, A. Osyczka, M. Rapala-Kozik and A. Kozik, Cells, 2020, 9, 1722 CrossRef CAS.
- A. Amador-García, I. Zapico, A. Borrajo, J. Malmström, L. Monteoliva and C. Gil, mSystems, 2021, 6, 00946 CrossRef PubMed.
- M. Abdulghani, R. Iram, P. Chidrawar, K. Bhosle, R. Kazi, R. Patil, K. Kharat and G. Zore, J. Proteomics, 2022, 265, 104661 CrossRef CAS PubMed.
- G. B. De Cesare, A. Hafez, D. Stead, C. Llorens and C. A. Munro, Virulence, 2022, 13, 1005–1018 CrossRef CAS PubMed.
- N. Song, X. Zhou, D. Li, X. Li and W. Liu, Antimicrob. Agents Chemother., 2022, 66, e0210521 CrossRef PubMed.
- S. E. Kidd, Y. Chow, S. Mak, P. J. Bach, H. Chen, A. O. Hingston, J. W. Kronstad and K. H. Bartlett, Appl. Environ. Microbiol., 2007, 73, 1433–1443 CrossRef CAS PubMed.
- V. Poplin, C. Smith, D. H. Caceres, P. F. Herkert, O. Jegede, G. R. Thompson, J. W. Baddley, I. S. Schwartz, R. Kubat, M. A. Deka, M. Toda, S. R. Lockhart, T. Chiller, F. Hagen and N. C. Bahr, Lancet Microbe, 2024, 100921 Search PubMed.
- R. Rajasingham, N. P. Govender, A. Jordan, A. Loyse, A. Shroufi, D. W. Denning, D. B. Meya, T. M. Chiller and D. R. Boulware, Lancet Infect. Dis., 2022, 22, 1748–1755 CrossRef.
- E. K. Maziarz and J. R. Perfect, Infect. Dis. Clin. North Am., 2016, 30, 179–206 CrossRef.
- J. M. Bednarek and J. C. S. Brown, mBio, 2024, 15, e02155-23 CrossRef PubMed.
- T. Sun, Y. Li, Y. Li, H. Li, Y. Gong, J. Wu, Y. Ning, C. Ding and Y. Xu, Front. Cell. Infect. Microbiol., 2021, 11, 662404 CrossRef CAS PubMed.
- J. M. H. Geddes, M. Caza, D. Croll, N. Stoynov, L. J. Foster and J. W. Kronstad, mBio, 2016, 7, 1–15 Search PubMed.
- J. M. H. Geddes, D. Croll, M. Caza, N. Stoynov, L. J. Foster and J. W. Kronstad, BMC Microbiol., 2015, 15, 1–26 CrossRef.
- B. Muselius, F. Roux-Dalvai, A. Droit and J. Geddes-McAlister, J. Am. Soc. Mass Spectrom., 2023, 34, 1928–1940 CrossRef CAS.
- M. L. Rodrigues, E. S. Nakayasu, D. L. Oliveira, L. Nimrichter, J. D. Nosanchuk, I. C. Almeida and A. Casadevall, Eukaryot Cell, 2008, 7, 58–67 CrossRef CAS PubMed.
- J. Rizzo, S. S. W. Wong, A. D. Gazi, F. Moyrand, T. Chaze, P. Commere, S. Novault, M. Matondo, G. Péhau-Arnaudet, F. C. G. Reis, M. Vos, L. R. Alves, R. C. May, L. Nimrichter, M. L. Rodrigues, V. Aimanianda and G. Janbon, J. Extracell. Vesicles, 2021, 10, e12129 Search PubMed.
- L. Santi, W. O. Beys-da-Silva, M. Berger, D. Calzolari, J. A. Guimarães, J. J. Moresco and J. R. Yates, J. Proteome Res., 2014, 13, 1545–1559 CrossRef CAS.
- L. Santi, M. Berger, J. A. Guimarães, Y. P. Calegari-Alves, M. H. Vainstein, J. R. Yates and W. O. Beys-da-Silva, J. Proteomics, 2024, 290, 105022 CrossRef CAS.
- A. Sukumaran, B. Ball, J. R. Krieger and J. Geddes-McAlister, mBio, 2022, 13, e01687 Search PubMed.
- B. Ball, A. Sukumaran, J. R. Krieger and J. Geddes-McAlister, J. Proteome Res., 2024, 23, 3917–3932 CrossRef CAS PubMed.
- L. Aslanyan, D. Sanchez, S. Valdebenito, E. Eugenin, R. Ramos and L. Martinez, J. Fungi, 2017, 3, 10 CrossRef.
- M. K. Mansour, J. L. Reedy, J. M. Tam and J. M. Vyas, Curr. Fungal Infect. Rep., 2014, 8, 109–115 CrossRef PubMed.
- J.-P. Latgé and G. Chamilos, Clin. Microbiol. Rev., 2019, 33, e00140 CrossRef.
- T. R. T. Dagenais and N. P. Keller, Clin. Microbiol. Rev., 2009, 22, 447–465 CrossRef CAS.
- J.-P. Latgé, Trends Microbiol., 2001, 9, 382–389 CrossRef.
- F. L. van de Veerdonk, M. S. Gresnigt, L. Romani, M. G. Netea and J.-P. Latgé, Nat. Rev. Microbiol., 2017, 15, 661–674 CrossRef CAS.
- J. D. Jenks, H. H. Nam and M. Hoenigl, Mycoses, 2021, 64, 1002–1014 CrossRef PubMed.
- F. El-Baba, Y. Gao and A. O. Soubani, Am. J. Med., 2020, 133, 668–674 CrossRef CAS PubMed.
- H. Schmidt, S. Vlaic, T. Krüger, F. Schmidt, J. Balkenhol, T. Dandekar, R. Guthke, O. Kniemeyer, T. Heinekamp and A. A. Brakhage, Mol. Cell. Proteomics, 2018, 17, 1084–1096 CrossRef CAS.
- V. Voltersen, M. G. Blango, S. Herrmann, F. Schmidt, T. Heinekamp, M. Strassburger, T. Krüger, P. Bacher, J. Lother, E. Weiss, K. Hünniger, H. Liu, P. Hortschansky, A. Scheffold, J. Löffler, S. Krappmann, S. Nietzsche, O. Kurzai, H. Einsele, O. Kniemeyer, S. G. Filler, U. Reichard and A. A. Brakhage, mBio, 2018, 9, e01557 CrossRef CAS.
- C. F. Pinzan, C. Valero, P. A. de Castro, J. L. da Silva, K. Earle, H. Liu, M. A. C. Horta, O. Kniemeyer, T. Krüger, A. Pschibul, D. N. Cömert, T. Heinekamp, A. A. Brakhage, J. L. Steenwyk, M. E. Mead, N. Hermsdorf, S. G. Filler, N. G. da Rosa-Garzon, E. Delbaje, M. J. Bromley, H. Cabral, C. Diehl, C. B. Angeli, G. Palmisano, A. S. Ibrahim, D. C. Rinker, T. J. C. Sauters, K. Steffen, A. Gumilang, A. Rokas, S. Gago, T. F. dos Reis and G. H. Goldman, Nat. Microbiol., 2024, 9, 2710–2726 CrossRef CAS PubMed.
- L. P. Venugopalan, V. Aimanianda, V. P. Namperumalsamy, L. Prajna, D. Kuppamuthu and J. M. Jayapal, Appl. Microbiol. Biotechnol., 2023, 107, 4025–4040 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |