
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integration of focal adhesion morphogenesis and polarity by DOCK5 promotes YAP/TAZ-driven drug resistance in TNBC†
Patricia
Pascual-Vargas
 *,
Mar
Arias-Garcia
,
Theodoros I.
Roumeliotis
,
Jyoti S.
Choudhary
and
Chris
Bakal
*
*,
Mar
Arias-Garcia
,
Theodoros I.
Roumeliotis
,
Jyoti S.
Choudhary
and
Chris
Bakal
*
Chester Beatty Laboratories, Division of Cancer Biology, Institute of Cancer Research, 237 Fulham Road, London SW3 6JB, UK. E-mail: patricia.pascualvargas@icr.ac.uk; chris.bakal@icr.ac.uk
First published on 12th May 2025
Abstract
YAP and TAZ are transcriptional co-activators that are inhibited by sequestration in the cytoplasm. Cellular signalling pathways integrate soluble, mechanical (cytoskeleton, adhesion), and geometric (cell size, morphology) cues to regulate the translocation of YAP/TAZ to the nucleus. In triple-negative breast cancer (TNBC) cells, both signalling and morphogenesis are frequently rewired, leading to increased YAP/TAZ translocation, which drives proliferation, invasion, and drug resistance. However, whether this increased YAP/TAZ translocation is due to alterations in upstream signalling events or changes in cell morphology remains unclear. To gain insight into YAP/TAZ regulation in TNBC cells, we performed multiplexed quantitative genetic screens for YAP/TAZ localisation and cell shape, enabling us to determine whether changes in YAP/TAZ localisation following gene knockdown could be explained by alterations in cell morphology. These screens revealed that the focal adhesion (FA)-associated RhoGEF DOCK5 is essential for YAP/TAZ nuclear localisation in TNBC cells. DOCK5-defective cells exhibit defects in FA morphogenesis and fail to generate a stable, polarised leading edge, which we propose contributes to impaired YAP/TAZ translocation. Mechanistically, we implicate DOCK5's ability to act as a RacGEF and as a scaffold for NCK/AKT as key to its role in FA morphogenesis. Importantly, DOCK5 is essential for promoting the resistance of LM2 cells to the clinically used MEK inhibitor Binimetinib. Taken together, our findings suggest that DOCK5's role in TNBC cell shape determination drives YAP/TAZ upregulation and drug resistance.
Introduction
A major question in cell biology is how cells sense changes in cell and tissue morphology to regulate processes such as proliferation, migration, and differentiation. Signalling pathways act as sensors of size and shape by integrating aspects of cell/tissue geometry (‘rulers’), morphological dynamics (‘timers’), and protein/lipid concentration (‘osmometers’), thereby regulating transcriptional, translational, and post-translational processes.1–5 However, there remains a poor understanding of how components of these pathways function in cellular space over different time scales to coordinate cell behaviour with changes in shape and size.First identified in mutagenesis screens that led to tissue overgrowth6,7 the Hippo-YAP signalling pathway has emerged as a conserved regulator of transcription in response to changes in cell morphology and size.3,8–10 For example, in normal epithelial tissues, injury, damage, and noxious stimuli—such as disruption of cell–cell adhesion and increases in cell-ECM adhesion10 (i.e. at the leading edge of tissue wounds)—trigger the translocation of the transcriptional co-activators YAP1 and TAZ from the cytoplasm to the nucleus, a process referred to as YAP/TAZ ‘activation’. Activated YAP/TAZ act in concert with transcription factors to regulate cell morphogenesis, invasion, and extracellular matrix (ECM) remodelling, thereby mediating tissue repair and regeneration.3,5,8–15 We have previously demonstrated that the dynamics of focal adhesion (FA) assembly and disassembly couple the duration of cell-ECM adhesion to YAP/TAZ translocation, effectively timing a potential repair response.10
Increased YAP/TAZ translocation is frequently observed in many cancer subtypes, especially in triple negative breast cancer (TNBC) cells,16–18 promoting a number of cancer relevant behaviours such as survival, proliferation, drug resistance, the epithelial-to-mesenchymal transition (EMT), migration, and invasion.3,4,14,19,20 ECM remodelling driven by YAP/TAZ activity not only enhances cancer cell migration but also modifies the tumour microenvironment to support metastatic niche formation.4,14,19,20 Moreover, the evolved drug resistance of many cancers is due to dysregulation of YAP/TAZ activity.21–28 The mechanisms that lead to increased YAP/TAZ translocation in cancers, and especially triple negative breast cancers (TNBC) are very poorly understood. Notably, activating mutations in YAP and TAZ are relatively rare,4,17,29,30 suggesting that cancer cells have evolved mechanisms to rewire the regulation of YAP/TAZ translocation dynamics in response to stimuli, rather than constitutively driving nuclear import. Given that YAP/TAZ translocation is highly sensitive to cell shape, size, adhesion, and cytoskeletal organisation, differences in these factors between normal and cancer cells may be key drivers of increased YAP/TAZ activity.
Rho family GTPases, such as CDC42, Rac1/2/3, and RhoA/C are master regulators of cell and tissue morphogenesis.31,32 As such their actions have been heavily implicated in the regulation of YAP/TAZ nuclear translocation in both normal and cancer cells.33–37 Rho GTPases appear to act both ‘indirectly’ by changing the organization of size, shape, adhesion, or cytoskeletal structures to regulate YAP/TAZ translocation, or ‘directly’ by recruiting and activating the downstream kinases such as p21-activated kinases (PAKs) whose actions converge on YAP/TAZ regulators such as LATS kinases.3,4,13,14 We have proposed a model whereby the activity of Rho GTP Exchange Factors (RhoGEFs), such as ARHGEF7/βPIX, act as Focal Adhesions in both ‘indirect’ and ‘direct’ means to regulate YAP/TAZ translocation in normal epithelia during migration.10 Specifically, during the initial formation of nascent adhesions at the leading edge of spreading or migrating cells, ARHGEF7 functions as a CDC42 GEF and PAK1 activator, triggering transient YAP/TAZ translocation. Simultaneously, ARHGEF7 promotes FA maturation via RacGEF leading to further recruitment of ARHGEF7 and other RhoGEFs to growing FAs, sustaining and amplifying YAP/TAZ nuclear import. FA maturation also facilitates actomyosin contractility via RhoA, further enhancing YAP/TAZ translocation. Ultimately, actomyosin contractility and microtubule recruitment promote FA disassembly,10,38 terminating YAP/TAZ translocation signals. Thus, RhoGEF/Rho signalling provides a mechanism by which YAP/TAZ translocation is directly coupled to FA morphogenesis and leading-edge dynamics.
While we demonstrated FA-ARHGEF7 activity as a regulator of YAP/TAZ translocation in normal breast epithelial cells, we found little evidence of this mechanism in TNBC breast cancer cells10 suggesting that TNBC cells have different mechanisms to regulate YAP/TAZ translocation.
There are likely multiple ways in which RhoGEFs and/or Rho GTPase Activating Proteins (RhoGAPs) drive YAP/TAZ translocation in cancer. As essential and conserved regulators of morphogenesis, cytoskeletal organisation, and adhesion, their dysregulation in cancer cells could have profound effects on YAP/TAZ translocation rates. Notably, in cases where YAP/TAZ upregulation drives drug resistance, increased YAP/TAZ activation is almost always coincident with changes in cell shape and actin reorganisation.22,24,28,39–42 Moreover, we recently demonstrated that in breast cancer cells, YAP/TAZ translocation rates are regulated by signalling pathways such as RAS/MAPK, ensuring that nuclear YAP/TAZ levels remain robust to changes in cell size.1 Since dysregulation of RAS/MAPK signalling is a common event in cancer, it could affect the relationship between YAP/TAZ and shape and size. While the mechanisms underlying this sensing remain to be elucidated, Rho-family GTPases, as conserved regulators of signalling in response to geometric and mechanical cues, are likely to be involved in coupling size to YAP/TAZ translocation.
To elucidate the basis for increased YAP/TAZ translocation in drug-resistant metastatic breast cancer LM2 cells, we performed multiplexed high-throughput genetic screens, systematically knocking down the majority of human RhoGEFs and RhoGAPs while simultaneously measuring YAP/TAZ nuclear localisation. To determine whether differences in YAP/TAZ translocation could be attributed to changes in cell shape, we also assessed single-cell morphology. These screens identified the focal adhesion RacGEF DOCK5 (dedicator of cytokinesis 5) as essential for YAP/TAZ translocation in drug-resistant metastatic breast cancer cells. The morphology of DOCK5-deficient cells was phenotypically similar to CDC42- and YAP1-depleted cells, suggesting a shared pathway. Indeed, DOCK5 exhibited synthetic interactions with both CDC42 and YAP1. Chemical perturbation indicated that DOCK5's RacGEF activity contributes to YAP/TAZ translocation, but we also found evidence for a non-catalytic role of DOCK5 in YAP/TAZ regulation.
We further demonstrated that DOCK5 is essential for TNBC cell migration and 3D invasion. Migration defects in DOCK5-deficient cells were partially attributable to FA morphogenesis defects and an inability to maintain stable protrusions. Proteome-wide mass spectrometry analysis revealed that DOCK5 depletion leads to significant decreases in GSK3β protein levels, which stabilises the polarised leading edge in migrating cells. We propose that following the initiation of a leading edge by CDC42, DOCK5 is recruited to nascent FAs, where it acts as a RacGEF and a non-catalytic activator of AKT. This stabilises FAs, reinforcing positive feedback through further DOCK5 and GEF recruitment while promoting GSK3β phosphorylation to maintain the leading edge. These feedback loops ensure the formation of a single leading edge, akin to symmetry breaking in yeast. The formation of a FA-rich leading edge promotes YAP/TAZ translocation in a manner dependent on CDC42, DOCK5, AKT, and RhoA activity, effectively ‘timing’ YAP/TAZ translocation with leading-edge dynamics. Crucially, as DOCK5 is essential for LM2 cell resistance to the MEK inhibitor Binimetinib, this suggests that a polarised migratory morphology itself is a driver of drug resistance.
Results
DOCK5 depletion lowers YAP/TAZ nuclear translocation
LM2 cells are a metastatic derivative of MDA-MB-231 TNBC cells.43 Using an antibody (63.7 sc-101199, Santa Cruz Biotechnology) that recognises both YAP1 and TAZ in immunofluorescence (IF) and western blot, we detected YAP/TAZ proteins predominantly in the nucleus of proliferating LM2 cells (Fig. S1, ESI†).44 In fact, compared to the TNBC cell lines hs578t and SUM159, and the non-metastatic mammary carcinoma cell line T47D, LM2 cells had much higher nuclear YAP/TAZ at sub-confluent densities (Fig. S2A, ESI†). Increasing the cell density of LM2 cell populations to high levels (2.0 × 105 cells per ml, 6000 cells per well) reduced YAP/TAZ nuclear translocation in LM2 cells, demonstrating that YAP/TAZ in LM2 cells was partially sensitive to cell numbers as observed in normal tissues10,45 (Fig. S2A, ESI†). High basal levels of nuclear YAP/TAZ in sub-confluent LM2 cells also contrasted markedly to those observed in MCF10A normal breast epithelial cells (Fig. S2A, ESI†).10 Throughout this study we used LM2 cells at low density (3.3 × 104 cells per ml, 1000 cells per well). The higher baseline levels of nuclear YAP/TAZ in sub-confluent metastatic LM2 cells correlate with observations that YAP/TAZ are drivers of metastasis11,46–48To identify regulators of Rho-family GTPases that contribute to the regulation of YAP/TAZ dynamics in TNBC cells, we systematically depleted 82 RhoGEFs, 67 GAPs and 19 Rho GTPases in LM2 cells, and quantified the ratio of nuclear to cytoplasmic YAP/TAZ as a proxy for YAP/TAZ activation.3,10 LATS1, YAP1 and ECT2 siRNAs were used as positive controls:44 siLATS1/2 for our ability to assess YAP/TAZ localisation, as YAP/TAZ nuclear localisation is expected to increase in the absence of LATS1/2;49 siYAP1 for our ability to detect YAP/TAZ protein; and siECT2 to assess transfection efficiency as ECT2 depletion results in multinucleate cells.50,51
Knocking down individual Rho-family GTPases had distinct effects on YAP/TAZ nuclear translocation. Specifically, knockdown of CDC42, and RHOA resulted in a significant drop in nuclear YAP/TAZ (Fig. 1A). Knockdown of either RAC1 or RAC3 led to statistically insignificant decreases in nuclear YAP/TAZ, while knockdown of RAC2 led to a moderate decrease (Fig. 1A). Thus, the basal levels of YAP/TAZ nuclear translocation observed in LM2 TNBC cells are partially due to the activation of CDC42 and RHOA. However, it is possible that that RAC1 and RAC2, but likely not RAC3,52 promote YAP/TAZ nuclear translocation in LM2 cells in a partially redundant manner.53
 | ||
| Fig. 1 DOCK5 regulates YAP/TAZ localisation. (A) Graph depicting Z-scores of log YAP/TAZ ratios following knockdown of Rho GTPases. siLATS1/2 included as controls for YAP/TAZ translocation. mean ± s.d (n = 3–4 wells per condition, 1000 cells per well, one-way ANOVA to mock). Z-Scores were then calculated per screen using the average and standard deviation of all mock transfected cells. Each well was assigned a QMS consisting of the Z-score for each shape (B) Z-scores per well for YAP/TAZ ratio for OTP siRNA screen in LM2. Above 1.5 (red dotted line) are siRNAs which are hits for high YAP/TAZ ratio and thus can be considered inhibitors of YAP/TAZ. Below −1.5 (red dotted line) are siRNAs which are hits for low YAP/TAZ ratio and thus can be considered YAP/TAZ activators. LATS1/2 and YAP siRNAs included as controls for high and low YAP/TAZ ratios are shown. Four individual replicates for siDOCK5 are shown. (C) Representative images showing YAP/TAZ staining for mock transfected and siDOCK5 transfected LM2 cells (40×). Arrows point to cells with high YAP/TAZ ratio (mock) and low YAP/TAZ ratio (siDOCK5). Scale bar is 20 μm. Graphs showing quantification of YAP/TAZ ratio, and whole cell YAP/TAZ levels in mock versus siDOCK5. siYAP + siTAZ show YAP/TAZ antibody is working appropriately. mean ± s.d (n = 3) independent biological repeats separate from screen data, dots represent individual wells, Student's t-test (left) and ANOVA (right). (D) Validation of siDOCK5 resulting in a decrease in YAP/TAZ ratio via subcellular fractionation. Representative western blot of subcellular fractions for mock and siDOCK5. Nuc: nuclear fraction. Mem: membrane fraction. Cyt: cytoplasm fraction. Blotted for DOCK5, YAP and TAZ. Same amounts of protein loaded (6.5 ng). Quantification of ratio of YAP/TAZ in the nucleus to the cytoplasm fraction from western blots of subcellular fractionations. mean ± s.d (n = 2). Student's t-test. Representative distribution of YAP/TAZ in all fractions with and without DOCK5. (E) Graph depicting Z-scores of log YAP/TAZ ratios following knockdown of Rho GTPases and DOCK5 in OTP screens in MDA-MB-231, SUM159 and hs578t. Mean ± s.d (n = 3–4 wells per condition, 1000 cells per well, one-way ANOVA to mock per cell line). Representative images for DOCK5 knockdown and mock transfection in each cell line. Scale bar is 50 μm. (F) Immunofluorescence images and quantification of YAP/TAZ localisation in mock or siDOCK5 transfected cells treated with DMSO or DOCK5 inhibitor C21. Arrows indicate representative cells for each condition. Mean ± s.d (n = 9 wells/condition, 1000 cells per well, one-way ANOVA). Significance for all panels: * P < 0.05, ** P < 0.001 *** P < 0.001, **** P < 0.0001. Imaged at 40×. Scale bar is 20 μm. | ||
To identify RhoGEFs that might be responsible for basal activation of YAP/TAZ in LM2 cells, we focused on RhoGEF siRNAs that reproducibly decreased nuclear YAP/TAZ levels. DOCK5 siRNA was the most consistent hit for siRNAs that caused low YAP/TAZ ratio (Fig. 1B), suggesting it is required for basal levels of YAP/TAZ activation in LM2 cells. All (4/4) replicates of DOCK5 siRNA were hits in our initial screen (Table S1, ESI†). We performed the same screen using another siRNA library (Dharmacon siGENOME; siG), where again 4 out of 4 replicates of a DOCK5 siRNA pool with distinct sequences from the OTP pool decreased YAP/TAZ nuclear levels (Fig. S2B, Z-scores per well: −1.73, −2.33, −2.77, −0.79, ESI†). Notably, using both the OTP and siGENOME siRNA, depleting DOCK5 consistently reduced YAP/TAZ ratio without changing the total levels of YAP/TAZ (Fig. 1C, D and Fig. S2C, ESI†).
To validate that decreases in YAP/TAZ ratio were due specifically to DOCK5 depletion, we quantified mRNA expression levels following OTP and siGENOME knockdown of DOCK5 by quantitative RT-PCR and found that DOCK5 mRNA expression levels were significantly reduced using either pool (Fig. S2D, ESI†). We also tested the ability of each individual sequence (4 siRNAs per pool) from the two different siRNA pools (OTP and siG) to knockdown DOCK5 mRNA and found that all 8 siRNAs did so effectively (Fig. S1E, ESI†). Further, 7/8 single siRNAs significantly reduced YAP/TAZ ratio and none but siG3 reduced total YAP/TAZ levels (Fig. S2C, ESI†). Because it is virtually impossible that 7/8 DOCK5 siRNAs would reduce YAP/TAZ translocation through any other manner than by depleting DOCK5 mRNA and protein these data prove that DOCK5 promotes YAP/TAZ nuclear translocation in LM2 cells.
To corroborate our immunofluorescence imaging-based observations that DOCK5 depletion affects YAP/TAZ nuclear translocation, we performed subcellular fractionation of mock and DOCK5 depleted LM2 cells, and used the same antibody (sc-101199) used for immunofluorescence to probe for YAP and TAZ protein. In DOCK5 depleted cells the cytoplasmic fraction had significantly higher levels of both YAP and TAZ, compared to the same fraction from mock transfected cells. Indeed, we quantified the ratios of nuclear to cytoplasmic YAP and TAZ individually, and together (YAP/TAZ), all of which were significantly lower (Fig. 1D). These results show that DOCK5 depletion results in decreases of both nuclear YAP and TAZ. Importantly, depletion of DOCK5 appears to limit translocation without affecting YAP/TAZ stability.
To understand whether the effect of DOCK5 depletion is specific to LM2 cells, or common across TNBC cell lines, we performed the same RNAi screens as for LM2, in the parental MDA-MB-231 cell line as well as two weakly metastatic TNBC cell lines: SUM159 and hs578t. We found that DOCK5 depletion also decreased YAP/TAZ nuclear translocation in both screens (siGENOME and OTP) using the parental cell MDA-MB-231 (Fig. S3, ESI†). Further, this decrease was also observed with RHOA siRNA in MDA-MB-231(Fig. 1E). DOCK5 was not a hit for YAP/TAZ translocation in SUM159 and hs578t cells (OTP, Fig. 1E). This suggests that DOCK5 plays a role in more strongly metastatic MDA-MB-231 and LM2 TNBC subtypes, but less so in weakly metastatic SUM159 and hs578t lines.54
To test the role of DOCK5 RAC GEF activity on YAP/TAZ translocation we used C21, an inhibitor which specifically blocks DOCK5's DHR2 catalytic domain by hindering the interaction between DOCK5 and RAC1.55–57 In particular, C21 has been demonstrated not be a general RAC inhibitor as it does not affect the exchange activity of TRIO, a RacGEF unrelated to DOCK5.55 Treatment with C21 decreased YAP/TAZ by the same amount as DOCK5 siRNA (Fig. 1F). Thus DOCK5 catalytic activity on RAC-type GTPases is an essential component of how DOCK5 promotes YAP/TAZ nuclear translocation.
DOCK5 contributes to proliferation and resistance to MEK inhibitors
In many cell types, YAP/TAZ acts as a transcriptional co-activator essential for survival and/or proliferation.28,49,58–63 To determine if DOCK5, Rho GTPases, and basal YAP/TAZ activity contribute to the survival of LM2 TNBC cells we performed growth assays over 160 hours. We normalized confluency values per condition to that of mock transfected cells per well and compared fold change to mock at 50% confluency (Methods). DOCK5 depleted cells proliferated at a slower rate as there were significantly less cells (68% less) following 60 hours post-transfection of DOCK5 siRNA where the mock population had reached 50% confluency (Fig. 2A). Furthermore, DOCK5 depleted cells took 32 hours more than mock transfected cells to reach confluence (Fig. 2E and Fig. S4A, ESI†). Cells where CDC42 was depleted by siRNA also proliferated at a slower rate than mock transfected cells (Fig. 2A, B and Fig. S4B, ESI†). But cells depleted of RHOA, RAC1, YAP, or TAZ proliferated at the same rate as mock transfected cells (Fig. 2A and Fig. S4C–F, ESI†). However, the proliferation of cells simultaneously depleted of both YAP1 and TAZ was significantly impaired (Fig. 2A and Fig. S4G, ESI†). We propose that DOCK5 and CDC42 promote proliferation of LM2 cells at least in part by promoting the nuclear translocation of both YAP1 and TAZ, though we cannot exclude the possibility that DOCK5 and/or CDC42 regulate proliferation independently of YAP/TAZ activation. | ||
| Fig. 2 DOCK5 contributes to growth and resistance to MEK inhibitors. (A) Fold change to control mock transfected wells at 50% confluency for growth curves (Fig. 2B–D and Fig. S4, ESI†). Each data point is from one well. mean ± s.d, Student's t-test. (B)–(D) Growth curves for (B) siDOCK5, siCDC42, siDOCK5 + siCDC42, (C) siDOCK5, siRAC1, siDOCK5 + siRAC1, (D) siDOCK5, siRHOA, siDOCK5 + siRHOA transfected cells. Averages per well are shown. (E), (G) and (H) Growth curves and (J) representative images at 50% confluency for mock and siDOCK5 transfected cells treated with DMSO (closed circle, square) and 10 μM final MEKi Binimetinib (open circle, square) for (E) MDA-MB-231, (G) A375P, (H) LM2. (F) and (I) Fold change to control mock transfected wells at 100% and 50% confluency respectively for LM2, MDA-MB-231, and A375 mock and siDOCK5 transfected cells treated with DMSO or Binimetinib. Each data point is from one well. mean ± s.d. Scale bar is 400 μm. All experiments performed in triplicate, representative experiment shown with n = 3 wells per condition. One-way ANOVA. For all panels: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. (K) Data plotted from Depmap website based on dependency of breast cancer cell lines to KRAS or DOCK5 knockdown. A CERES (computational method to estimate gene-dependency levels from CRISPR-Cas9 essentiality screens)64 a lower score indicates that cell line will be more dependent on that gene. | ||
To determine if DOCK5 regulates proliferation in a fashion dependent on Rho GTPases, we assessed whether DOCK5 depletion is synthetically lethal with depletion of RAC1, RHOA, and CDC42. Indeed, DOCK5 and CDC42 exhibit synthetic lethality (Fig. 2B), such that at 156 hours when mock transfected cells were confluent, CDC42 and DOCK5 depleted cells were at 32% confluency. In contrast, depletion of either RAC1 or RHOA with DOCK5 partially rescued the lethality of DOCK5 depletion (Fig. 2C and D respectively). We conclude that DOCK5 exhibits genetic interactions with CDC42, RAC1, and RHOA; a negative/aggravating interaction with CDC42 and positive/alleviating interactions with RAC1 and RHOA. Positive/alleviating interactions are symptomatic of “within pathway” interactions,65–67 which is consistent with the fact that DOCK5 activates RAC GTPases.56,68–70 That DOCK5 has a negative/aggravating interaction with CDC42 suggests they function independently of each other – i.e. in parallel pathways, and is consistent with the idea that DOCK5 is unlikely to be a CDC42 GEF in LM2 cells.55,57,71
LM2 cells harbour mutant KRAS (G13D),72 which activates the kinases MEK and ERK, driving proliferation and survival.73,74 However, while MEK inhibition (MEKi) effectively kills some cancer cells such as melanoma cells, breast cancer cells are often resistant to MEKi.75,76 Because MEKi resistance in melanoma, pancreatic and non-small lung cancer (NSLC) is often due to YAP/TAZ activation,25,26,77,78 we assessed whether the MEKi resistance of LM2 cells was due to DOCK5 and/or YAP/TAZ activity. To test this hypothesis, we first treated LM2, MDA-MB-231 (from which LM2 cells are derived), and A375p melanoma cells (which harbour activated mutant BRAF) with the MEKi Binimetinib. MEKi significantly inhibited survival in MDA-MB-231 (Fig. 2E and F) and A375p (Fig. 2F and G) cells, but only had a modest effect on the survival of LM2 cells (Fig. 2F and H). DOCK5 depletion did not have any effect on the survival or proliferation of MDA-MB-231 or A375p cells in the absence or presence of MEKi (Fig. 2F, I and J). However, depletion of DOCK5 and MEKi treatment in LM2 cells led to a synergistic effect on proliferation or survival where confluency stalled at 10% for the whole duration at the assay (Fig. 2H–J). Thus, MEKi aggravates DOCK5 depletion suggesting that ERK and DOCK5 are functioning in compensatory pathways, and DOCK5 is essential for resistance of LM2 cells to MEK inhibition.
Supporting the idea that a chemical-genetic interaction between YAP/TAZ and RAS-ERK signalling exists, mining of the DEPMAP database reveals that CRISPR-mediated depletion of DOCK5 and KRAS affects cell viability in a significantly similar manner across a large panel of cell lines (Fig. 2K); 0.4 Pearson correlation between effects of KRAS and DOCK5 depletion across cell lines as determined by DEPMAP profiling.64 These data support the idea that both DOCK5-YAP1 and RAS-ERK pathways signalling promote survival in breast cancer cells, and that while inhibition of one axis can blunt survival, simultaneous inhibition of both is lethal.
DOCK5 depletion reduces 3D invasion
To determine if DOCK5 contributes to the ability of LM2 cells to invade 3D tissues, we quantified the extent to which DOCK5 depleted cells invade rat tail collagen I (Col-I) gels. Cells typically invaded from the bottom of the plate into depths of 30 μM in the collagen gel within 24 hours (Fig. 3A and B). We quantified cell invasion by calculating the invasion index, defined as the number of cells at various planes within the gel, divided by the total number of cells in all planes plus the bottom plane. In a 2.0 mg ml−1 rat-tail Col-I gel, DOCK5 depletion significantly reduced invasion (Fig. 3C). Further, depleting TAZ but not YAP significantly reduced invasion, by the same amount as depleting DOCK5. The similarity in the phenotypes following DOCK5 and TAZ depletion strongly suggests that DOCK5 regulates TAZ as well as YAP translocation to promote invasion. | ||
| Fig. 3 DOCK5 depletion reduces 3D invasion. (A) Representative 2-photon images of LM2 cells (GFP) stained for DNA (blue) mixed with rat-tail collagen I at 2 mg ml−1 (white) at the bottom of the plate (0 μm plane) and the middle (30 μm plane). (B) Representative confocal spinning disk images (12 fields per well) for knockdowns shown in (C) at 0 μm and 30 μm. (C) Graph shows quantification of invasion index for knockdowns in rat-tail collagen I at 2 mg ml−1. Invasion index is indicative of the number of cells in middle planes divided by total number of cells in all planes quantified, including bottom plane. Mean ± sd (n > 3 wells per condition, 1000 cells per well. ANOVA, **** P < 0.0001). (D) Invasion index of mock versus siDOCK5 transfected cells at collagen concentrations: 1.6 mg ml−1, 3.8 mg ml−1, and 4.8 mg ml−1. Mean ± sd (n > 3 wells per condition, 1000 cells per well, ANOVA, ** P < 0.01). Scale bars, 100 μm. (E) Representative images of mock transfected cells and siDOCK5 transfected cell spheroids 48 hours post transfection and pre 100% Matrigel (left), and one (middle) and six (right) days post 100% Matrigel addition. Scale bar is 200 μm. (F) Graph shows quantification of area of Matrigel in arbitrary units over 8 days post adding 100% Matrigel to the spheroids. Mock transfected cells in black, siDOCK5 transfected cells in red. Each line represents one well. n = 3 wells. Statistical significance obtained by performing student t-test on row means (* P = 0.0150). | ||
Because collagen pore size has been shown to be inversely proportional to collagen concentration,79 we used collagen gels of different concentrations as a way of varying pore size. Depleting DOCK5 consistently reduced invasion across a Col-I density range of 1.6 to 4.8 mg ml−1 compared to mock transfected cells (Fig. 3D). Overall, these data suggest that DOCK5 is required for LM2 single-cell invasion in rat tail collagen matrices independently of pore size.
To explore whether DOCK5 is required for invasion of cells in multicellular bodies – i.e. in tumour models, we generated spheroids and quantified cell invasion into 100% Matrigel (Methods). Mock transfected LM2 cells invaded into the gel progressively, while cells lacking DOCK5 did not invade at all (Fig. 3E and F). Overall, this suggests that LM2 cells require DOCK5 to invade into 3D structures.
DOCK5 depletion results in polarity and adhesion defects
Changes in YAP/TAZ localization following systematic RNAi of RhoGEFs could be due to changes in cell/membrane morphology, size, cytoskeletal organization, or adhesion.10 Alternatively, it is formally possible differences in YAP/TAZ translocation could be due to alteration in signalling that is independent of these factors. To differentiate between these two possibilities we quantified the cell shape of single LM2 cells following depletion of 149 RhoGEF/GAPs and 19 Rho GTPases to generate quantitative morphological signatures or “QMSs”.80 In this case, QMSs describe the percentage of cells with a particular shape in an RNAi-treated population, and thus account for the phenotypic heterogeneity observed in different cell lines.81 To generate QMSs, we measured 127 morphological and texture features for single cells in the screens, and trained linear classifiers to identify five visually distinctive reference shapes. The percentage of cells binned into each sub-population is a QMS for that specific RNAi (Methods). The five visually distinct shapes we used to characterize populations are: (1) ‘spindly’ – elongated cells with typically two protrusions; (2) ‘large round’ – spread cells of a large area which are often circular; (3) ‘triangular’ – cells with three distinct protrusions; (4) ‘fan’ – asymmetrically-shaped cells with nucleus to one side; and (5) ‘small round’ – cells which have a small area and high roundness (Fig. 4A and B). LM2 cells are highly heterogeneous, and in a mock-transfected population 9.4% cells are classified as spindly, 0.9% as large round, 60.4% as triangular, 3.7% as fan, and 25.5% as small round (Fig. 4C). Depletion of different RhoGEF and RhoGAP genes by siRNA resulted in significant changes in the number of cells in each sub-population. For example, depletion of FAM13A resulted in a significant increase in spindly and triangular cells, and a significant decrease in large cells with: 35.3% of cells being classified as spindly, 0.3% as large, 42.1% as triangular, 1.3% as fan and 21.1% as small round (Fig. 4C). | ||
| Fig. 4 Shape classification and relationship with YAP/TAZ. (A) Heat map and dendrogram of the features describing each shape and which Columbus software used to classify cells into the five visually distinctive reference shapes, as well as their contribution, using single cell data. Yellow and blue represent a high and low score respectively in arbitrary units. (B) Images from one field of view from mock transfected well with cells coloured by their classified shape by software showing highly heterogenous population, and (C) the percentage of cells classified into each shape in one sample mock transfected well and for an siRNA which was a top hits for spindly shape, depicting a rearrangement in the shapes seen for mock-transfected cells. Mean per well ± sd are shown (n > 3, 1000 cells per well, student's t-test on row for each shape between siFAM13A and mock-transfected cells, * P < 0.05, ** P < 0.01). (D) Graph depicting Z-score of log10 ratio of nuclear to ring region YAP/TAZ for single LM2 cells following transfection with siRNA which results in the biggest enrichment for each particular shape: siECT2 for large, siDOCK11 for fan, siARHGAP5 for small round, siDOCK10 for triangular, siFAM13A for spindly. Mean ± sd are shown. Data taken from at least 5 fields of view from 2 wells. (E) Images of population from which single cell data shown in (D) were taken. Arrows indicate examples of nuclear YAP/TAZ for each shape. Images stained for YAP/TAZ (red) and F-actin (green). Scale bar is 50 μm. | ||
We also developed a classifier for ‘normal’ cells (Methods, Fig. S5, ESI†). This allowed us to identify siRNAs which had significant effects on cell shape based on whether they significantly depleted the percentage of normal cells compared to mock (Z-score equal to or below −1 for normal classifier). In essence we could thus describe the penetrance of each genetic perturbation.81 89 siRNAs resulted in significant depletion of normal cells; of which 79 were RhoGEF/GAPs, 7 were Rho GTPases, and 3 (siLATS1, siLATS2 and siYAP1) were controls for YAP/TAZ translocation.
To determine if cell shape changes regulate YAP/TAZ in LM2 cells, for example following depletion of RhoGEFs or RhoGAPs, we next asked whether exemplars of different cell shapes that were used to derive classifiers had predictable variability in YAP/TAZ nuclear translocation. Effectively asking the question, does shape predict YAP/TAZ dynamics? Indeed, different classifier shapes had significantly different levels of nuclear YAP/TAZ ranging from high nuclear YAP/TAZ levels in polarised “fan” cells with broad lamellipodia to lower levels in “spindly” unpolarised cells (Fig. 4D and E). Thus as in other cell types,3,4,10,14,44 in LM2 cells morphology can largely predict YAP/TAZ translocation dynamics.
To identify how different RhoGEFs, RhoGAPs, and Rho GTPases, and in particular DOCK5, regulate LM2 cell shape specifically, we performed hierarchical clustering on 5-shape QMSs for shape hits (<−1 Z-score for normal classifier) in the LM2 OTP screen, and defined “phenoclusters” as genes grouped at the highest node of clustering with a Pearson Correlation Coefficient (PCC) of >0.73 (Fig. 5A and Table S2, ESI†). This resulted in 10 multigene clusters and 1 singleton formed by mock transfected LM2 cells.
 | ||
| Fig. 5 DOCK5 regulates shape. (A) Clustering of high-dimensional quantitative signatures based on the effect of RhoGEF/GAPs and Rho GTPase siRNAs on the TNBC cell line LM2. Columns are Z-score values of the percentage of cells classified into that shape when said siRNA is depleted. Red and blue indicate high and low values respectively. Clustering performed using Pearson correlation. 10 clusters and a singleton were identified and defined as genes grouped at the highest node of clustering with a value of uncentered Pearson correlation coefficient above 0.73. Numbering indicates clusters and images are representative for mock and siRNAs which enrich for a particular shape in example clusters. Staining shows DNA (blue), tubulin (yellow), F-actin (green) and YAP/TAZ (red). Scale bar is 50 μm. (B) Representative images for a mock “normal” and an siDOCK5 transfected cell imaged on SoRa. Staining shows Paxillin (green), α-tubulin (red), F-actin (blue). Square represents zoom in below. Scale bar is 10 μm, zoom in 5 μm. (C) Clustering of QMS for siRNAs shown as well as C21 treatment at a Pearson correlation of 0.74. Images for siRAC2, C21, siLATS1, and siYAP are shown, stained for DNA (blue), tubulin (yellow), F-actin (green) and YAP/TAZ (red). Scale bar is 50 μm. | ||
The QMS of DOCK5 siRNA belonged to cluster 8 which is characterized by increases in fan, spindly, and triangular shapes (Fig. 5A). Cells following DOCK5 depletion also closely resembles spindle/unpolarised shapes found in cluster 7 following depletion of the canonical and conserved polarity regulator CDC4233,82–85 (Fig. 5A) and cluster 9 – which is characterised by very large flat cells, such as following depletion of YAP/TAZ. Importantly, the relatively high similarity in morphology following depletion of DOCK5, CDC42 and YAP/TAZ supports the idea that DOCK5 and CDC42 regulate YAP/TAZ translocation.
We next asked whether the QMS of C21 treated cells is similar to that of DOCK5, YAP1, or LATS1 depleted cells. Because C21 inhibits GEF activity of DOCK5 towards RAC-type GTPases, we compared the DOCK5 QMS with QMSs following knockdown of CDC42, RAC1, RAC2, and RHOA. Hierarchical clustering of QMSs revealed that the QMS of C21 is similar to that of RAC2 and CDC42 depleted cells (Pearson correlation coefficient (PCC) > 0.77), but only partially similar to the QMS of DOCK5 and YAP1 depleted cells (PCC > 0.40) (Fig. 5C). Thus, DOCK5 depletion is not phenotypically identical in terms of shape to disruption of DOCK5 GEF activity and suggests that DOCK5 mediated regulation of shape occurs both via GEF mediated regulation of RAC-type GTPase (i.e. RAC1 and/or RAC2), as well as by acting as a scaffold.
DOCK5 depletion alters the levels of polarity proteins
To investigate how depletion of DOCK5 may affect cell shape, and YAP/TAZ localisation, we performed mass spectrometry to determine total protein levels in mock and siDOCK5 transfected LM2, MDA-MB-231, and hs578t cells. Protein samples were processed 48 hours after transfection and labelled with TMT-10plex (Tandem Mass Tag) multiplex reagents before being submitted to LC–MS/MS (liquid chromatography mass spectrometry; Methods). Protein abundance values obtained after analysis of mass spectra, scaled within cell lines, and the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 ratio values of siDOCK5 versus mock-transfected lines were calculated. DOCK5 protein was depleted by approximately 78% by siRNA (Fig. 6A and Table S3, ESI†). Further there were no changes in protein levels of DOCK5's closest homolog, DOCK186 (Fig. 6B), suggesting that the DOCK5 siRNA is not targeting DOCK1. In addition, there were no changes in the protein levels of RhoGEFs which clustered with DOCK5 by their QMS. Thus, DOCK5 siRNA has specific on-target effects. While LM2, MDA-MB-231, and hs578t could be distinguished by their proteome, DOCK5 depletion had similar effects on polarity proteins across cell lines (Fig. 6A, C and Table S3, ESI†).
2 ratio values of siDOCK5 versus mock-transfected lines were calculated. DOCK5 protein was depleted by approximately 78% by siRNA (Fig. 6A and Table S3, ESI†). Further there were no changes in protein levels of DOCK5's closest homolog, DOCK186 (Fig. 6B), suggesting that the DOCK5 siRNA is not targeting DOCK1. In addition, there were no changes in the protein levels of RhoGEFs which clustered with DOCK5 by their QMS. Thus, DOCK5 siRNA has specific on-target effects. While LM2, MDA-MB-231, and hs578t could be distinguished by their proteome, DOCK5 depletion had similar effects on polarity proteins across cell lines (Fig. 6A, C and Table S3, ESI†).
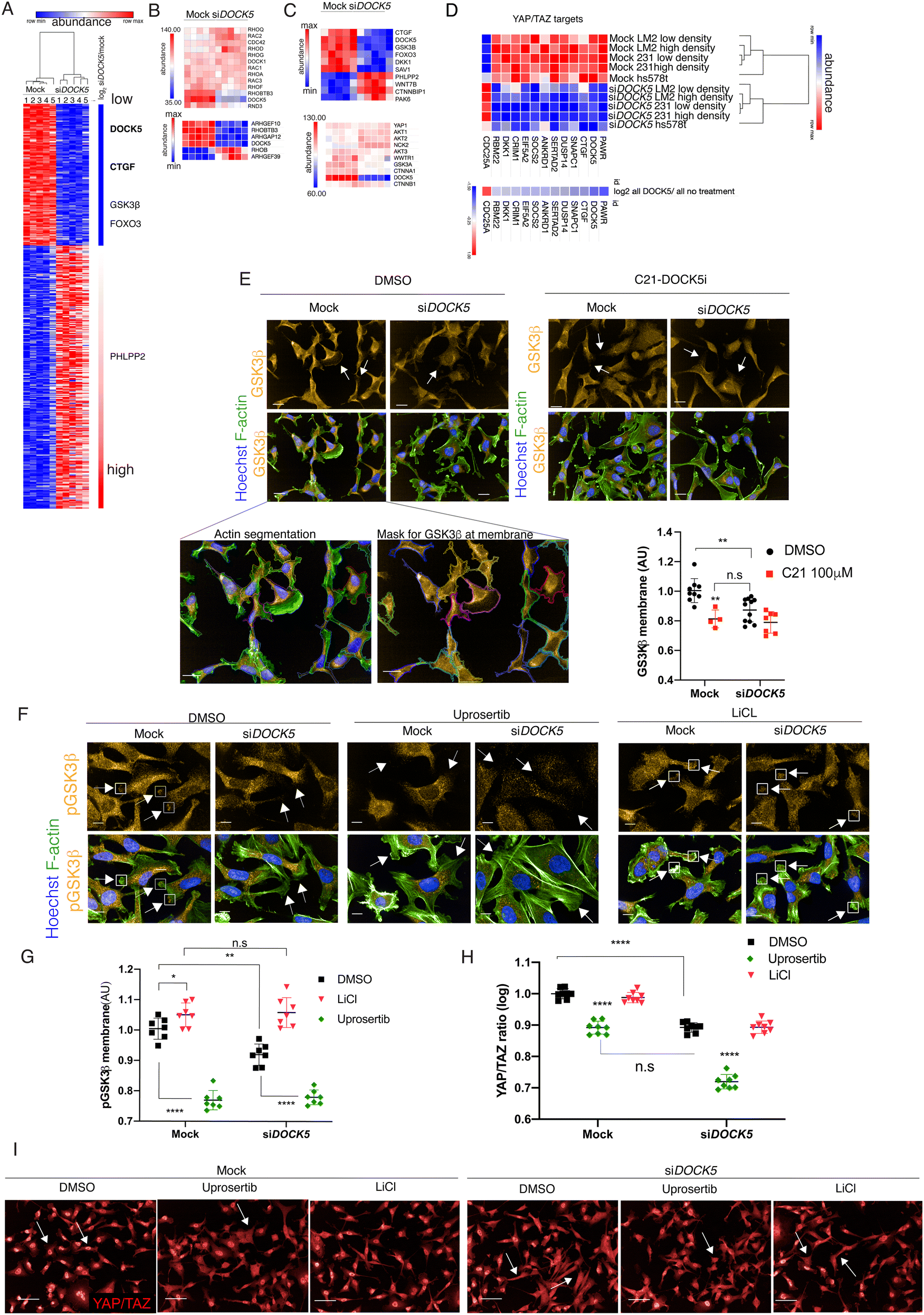 | ||
| Fig. 6 DOCK5 depletion alters the levels of polarity proteins. (A) Heat map of proteins significantly up or downregulated following DOCK5 knockdown via siRNA in LM2,1,2 MDA-MB-2313,4 and hs578t.5 Significance based on log2 ratios for protein levels of siDOCK5 v mock being above 0.5 and below −0.5 and permutation FDR < 0.05 (t-test). Red and blue indicate high and low protein abundance levels (row max to min respectively). Proteins up and downregulated based on log2 value. Proteins involved in regulating polarity and AKT signaling pathways shown are shown as well as CTGF. YAP/TAZ targets shown in bold (B) top protein abundance levels of Rho GTPases and DOCK1 that were unchanged with siDOCK5 abundance levels for comparison. Bottom protein levels of Rho GTPases and RhoGEF/GAPs which were significantly upregulated or downregulated upon DOCK5 knockdown. Row max (red) to min (blue). (C) Top: Heat map of protein levels of proteins regulating polarity that were significantly up and down regulated. Values normalised to cell line. Row max (red) to min (blue). Bottom: Abundance levels of proteins of interest that were not significantly up or downregulated, with siDOCK5 abundance levels for comparison. (D) Heat map of proteins significantly up or downregulated following DOCK5 knockdown via siRNA in TNBC cell lines which have been shown to be YAP/TAZ target genes in MDA-MB-231. Red and blue indicate high and low protein abundance levels (row max to min respectively). Log2 value below. (E) Representative images and immunofluorescence quantification of GSK3β at the membrane in mock and siDOCK5 transfected cells in DMSO or DOCK5 inhibitor C21 Mean per well ± sd (n > 3 wells/condition, 1000 cells per well; ANOVA). White arrows indicate GSK3β at the membrane (mock) and lack thereof (siDOCK5 and C21). GSK3β imaged at 40×, scale bar, 20 μm. Inserts show actin segmentation and mask used to quantify GSK3β at the membrane. (F) Representative images and (G) immunofluorescence quantification of pGSK3β at the membrane in mock and siDOCK5 transfected cells treated with DMSO (black), 5 mM LiCl (pink) or 2 μM final Uprosertib (green). Mean per well ± sd are shown (n > 3, 1000 cells per well, 2 ways ANOVA, Tukey's multiple comparison's test * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). White arrows indicate pGSK3β at the membrane (mock and LiCl) and lack thereof (siDOCK5 and Uprosertib). pGSK3β imaged at 60× with water objective (NA = 1.2). Scale bar, 10 μm. (H) Immunofluorescence quantifications of YAP/TAZ ratio of mock and siDOCK5 transfected cells treated with DMSO (black), 5 mM LiCl (pink) or 2 μM final Uprosertib (green). Mean per well ± sd are shown (n > 3, 1000 cells per well, 2 ways ANOVA, Tukey's multiple comparison's test * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). (I) Immunofluorescence images of YAP/TAZ for mock and siDOCK5 transfected cells treated with DMSO (black), 5 mM LiCl (pink) or 2 μM final Uprosertib quantified in (H). Imaged at 20×. Scale bar, 50 μm. | ||
Protein levels of connective tissue growth factor (CTGF), which are upregulated by YAP/TAZ-TEAD activity15,87 were significantly downregulated in DOCK5 depleted cells (log2 ratio: −0.77695) (Fig. 6A and Table S3, ESI†); supporting the idea that depleting DOCK5 renders YAP/TAZ cytoplasmic and decreases YAP/TAZ transcriptional activity. Notably, total YAP1 and TAZ (encoded by the WWTR1 gene) protein levels remained unchanged (Fig. 6C), matching our immunofluorescence data and confirming that DOCK5 regulates YAP/TAZ localisation rather than YAP/TAZ levels. In addition to CTGF, the protein levels of 10 established YAP/TAZ target genes87 were also significantly downregulated following DOCK5 knockdown in LM2 and MDA-MB-231 (Fig. 6D and Table S3, ESI†).
We performed hierarchical clustering on the abundance values of proteins which were significantly upregulated (40% increase) or downregulated (40% decrease) upon DOCK5 depletion (log2 ratios above 0.5 and below −0.5 and permutation FDR < 0.05 (false discovery rate) (t-test)). DOCK5 depletion resulted in changes in the levels of AKT kinase effectors, AKT regulators, and proteins implicated in the establishment of front-rear polarity. Specifically, the protein levels of the AKT effectors GSK3β (log2 ratio: −0.58753) and FOXO3A (log2 ratio: −0.59131) were downregulated (Fig. 6A and C). GSK3β plays a role in the establishment of polarity where it has been described to be phosphorylated and inhibited at the leading edge of migrating cells in a mechanism dependent on integrin mediated activation of CDC42-PAR6-PKCζ.83,88,89 Phosphorylated GSK3β promotes polarity by capturing/stabilizing microtubules (MTs) at the leading edge of migrating cells. Interestingly although it did not make the log2 ratio cut-off of −0.5 with a −0.46log2 value, levels of the polarity regulator PAR6b were also substantially decreased (27%) in DOCK5 depleted cells (p = 0.0002, q = 0.0008). Finally, PHLPP2 which inhibits AKT90 and aPKCζ,91 was upregulated (log2 ratio: 0.855787) (Fig. 6A and Table S3, ESI†). PHLPP2 upregulation has been shown to inhibit the establishment of polarity.91 Thus decreased levels of polarity proteins broadly correlated with the cellular unpolarized morphology of DOCK5 depleted cells.
We used quantitative single-cell imaging approaches to quantify the subcellular localisation of GSK3β at the tips of cell protrusions (Fig. 6D). DOCK5 depleted cells had significantly reduced levels of GSK3β at the edge of protrusions compared to control cells (Fig. 6E). Total GSK3β levels were also reduced by C21 treatment, demonstrating that DOCK5's exchange activity on RAC-type GTPases is essential to maintain GSK3β levels in polarised protrusions. We propose that the inability of DOCK5 depleted cells to recruit GSK3β to the membrane leads to defects in the maintenance of polarity.
DOCK5 has been shown in osteoclasts to promote serine 9 (Ser9) phosphorylation downstream of AKT activation in both RAC dependent and independent ways that lead to MT stabilisation.92,93 To investigate whether DOCK5 similarly regulates the levels of phosphorylated GSK3β at the leading edge of LM2 cells to promote MT stability and the maintenance of cell polarity, we quantified the levels of phosphorylated GSK3β in LM2 cells at membrane regions following DOCK5 depletion and/or treatment with the AKT inhibitor Uprosertib (final concentration 2 μM). We confirmed that Uprosertib indeed inhibits AKT by measuring FOXO3A nuclear translocation (Fig. S6A and B, ESI†).94,95 We also used LiCl (final concentration 5 mM) as a positive control of our ability to measure GSK3β phosphorylation (pGSK3β). As expected, LiCl caused an increase in pGSK3β levels in protrusions in mock-transfected cells due to disruption of the negative feedback on AKT-mediated Ser9 phosphorylation by GSK3β kinase activity.96 Moreover, LiCl led to a significant increase in whole cell β-catenin levels via immunofluorescence (Fig. S6C and D, ESI†).97–100
DOCK5 depleted cells significantly decreased pGSK3β at the leading edge,101 while AKT inhibition resulted in an even greater decrease in pGSK3β levels at the protrusive edges in both mock and siDOCK5 transfected cells (Fig. 6F and G). Strikingly, LiCl treatment rescued the phenotype of pGSK3β levels in DOCK5 depleted cells resulting in the same levels of pGSK3β in the protrusions of LiCl treated siDOCK5 and mock transfected cells (Fig. 6F and G). The ability of LiCl to restore pGSK3β levels in DOCK5 depleted cells is likely because LiCl blocks GSK3β's ability to inhibit AKT mediated phosphorylation of Ser 9 independently of DOCK5.96
Taken together these data support a mechanism by which DOCK5 acts as a RAC GEF to promote phosphorylation of GSK3β at the leading edge by AKT and other kinases. Combined with prior observations that DOCK5 acts to recruit AKT to different subcellular milieus,92,93 we propose that recruited AKT is likely activated downstream of RAC to promote GSK3β Ser9 phosphorylation. We propose that in the absence of DOCK5 mediated phosphorylation, GSK3β and other polarity proteins become destabilized.
To test whether YAP/TAZ translocation to the cytoplasm could be due to decreases in the levels of phosphorylated GSK3β at the membrane, and decreased AKT signalling, we quantified YAP/TAZ translocation following inhibition of AKT or GSK3β kinase activity, in both mock and siDOCK5 treated cells. Inhibiting AKT directly with Uprosertib resulted in the same decrease in YAP/TAZ ratio in mock transfected cells similar to depleting DOCK5 by RNAi (Fig. 6H and I). Furthermore, treating DOCK5 depleted cells with Uprosertib decreased the ratio significantly compared to mock transfected cells (Fig. 6H and I). LiCl however had no effect on YAP/TAZ. Elevating pGSK3β levels in control cells at the membrane alone does not further increase YAP/TAZ translocation. Hence, we propose that YAP/TAZ translocation is regulated by AKT in LM2 cells.
DOCK5 is required for the maintenance of polarity during wound healing
Given the significant reduction of GSK3β following DOCK5 depletion, and the role of GSK3β in front-rear polarity,83,89 we hypothesised that DOCK5 contributes to the establishment of front-rear polarity in migrating cells. To test this, we performed wound healing assays, which are tests of polarity initiation and maintenance, 2D migration speed, and migration persistence (Methods). We also examined whether YAP/TAZ activation may be important for polarity establishment and migration. In these assays, we depleted DOCK5, YAP1, TAZ, and YAP1 and TAZ in combination. Depleting DOCK5, TAZ, and YAP1 + TAZ significantly reduced migration rates but depleting YAP1 did not (Fig. 7A and B). Therefore, TAZ appears to be driving migration, as well as invasion (Fig. 3C) in LM2 cells, instead of YAP1. | ||
| Fig. 7 DOCK5 is required for the maintenance of polarity during wound healing. (A) Brightfield images at 0, 10 and 16 hours post creating scratch wound for mock, siDOCK5, siYAP, siTAZ, and siYAP + siTAZ transfected cells. Dotted black line indicates the starting line from where cells migrated over time. Scale bar, 100 μm. (B) Graph shows migration rates (μm/hour) (n = 5 wells per condition, with at least 2 biological repeats). Values were normalised to average of mock for each biological repeat. Mean ± sd, one-way ANOVA to mock. *** P < 0.001, **** P < 0.0001 (C) and (D) images depicting Golgi immunofluorescence stain and Hoechst, wound is shown with white dotted line. (C) Angle measuring relative to wound is depicted, example of 90 degree angle and orientation towards the wound. Scale bar is 50 μm. (E) Percentage frequency of cells with Golgis orientated 0, 30, 60, 90, 120, 150, and 180 degrees relative to the wound depicted as a bar chart. Percentages for mock and siDOCK5 transfected cells at 90 and 180 degrees indicated. | ||
The establishment and maintenance of polarity during wound healing is characterised by the reorientation of the Golgi, and microtubule organising centre (MTOC), toward the wound area and direction of migration.101 Therefore to determine if DOCK5 contributes to the initiation and/or establishment of leading edge polarity, we quantified the orientation of the Golgi following wounding, in cells at the edge of the wound.88,102 Specifically, we quantified the angle of the Golgi oriented to the wound such that a 90 degree angle indicates the Golgi is facing toward the wound and front-rear polarity has been established (Fig. 7C and D). Mock transfected populations had a high frequency of cells with Golgi directly facing the wound (65.8%) (Fig. 7E). In contrast, DOCK5 depleted populations had a higher frequency of Golgi oriented parallel to the wound (180 degrees) and/or that were poorly polarised (35.4% to 17.7% respectively) (Fig. 7E). YAP1 and TAZ combined depletion resulted in the condition with the least percentage of cells displaying front-rear polarity (with Golgi oriented 90 degrees to the wound) and the highest percentage of cells with Golgi oriented 30 degrees relative to the wound, suggesting these cells are unable to integrate cues from the microenvironment for directional motility (Fig. 7E). Taken together, these data support the idea that DOCK5 promotes the formation of polarised leading-edge protrusions. In addition, because combined inhibition of YAP/TAZ is likely to have deleterious effects on various aspects of cell behaviour, such as cytoskeletal reorganisation required for polarity, the data suggest that YAP/TAZ can be both regulated by DOCK5-mediated polarisation, and also regulate polarisation itself.
DOCK5 promotes the maintenance of polarity and alters FA dynamics.
We and others have demonstrated that DOCK5 localises in close proximity to FAs peripherally to Paxillin, but co-localises with Paxillin in the absence of GIT2.69,103 In line with these data, we observed that overexpressed DOCK5-YFP is also localised in puncta at the very edge of the lamellipodial protrusions in TNBC cells. DOCK5 bordered the forward-facing side of Paxillin-positive FAs at the leading edge (Fig. 8A). Notably, the rear facing side of Paxillin-positive FAs at the leading edge was often bordered by polymerised alpha-tubulin (Fig. 8A). | ||
| Fig. 8 DOCK5 promotes the maintenance of polarity and alters FA dynamics. (A) Overexpressed DOCK5-tagged YFP localises to focal adhesions (paxillin). Cells stained for paxillin (red), tubulin (blue). Scale bar is 5 μm. (B) Sequences of zoomed in sections of mock, and siDOCK5 transfected LM2-CAAXGFP cells over time. Frames are every 3 minutes. Arrows indicate an example of an adhesion to follow. Dotted line represents the edge of the cell at the beginning of each sequence of frames shown. Adhesion signal represents Talin mApple intensity: red more intense, blue less intense. Scale bar is 2 μm. (C) Representative images of how Focal Adhesion Analysis Server (FAAS)104,105 picks up adhesions on mock and siDOCK5 transfected cells. Scale bar is 10 μm. (D) Focal adhesions tracked over time in same cells as in (C). Blue to red indicates adhesions which have been present for less, and longer amounts of time respectively. (E) and (F) Cumming estimation plots of Hedge's g for comparison between (E) assembly and disassembly rates, and (F) assembly phase length and disassembly phase length of adhesions in mock versus siDOCK5 transfected cells. N shows number of adhesions quantified, data from 4 cells. Raw data is plotted in the top axes, while mean difference is plotted in lower axes as a bootstrap sampling distribution. Mean differences are shown as dots, 95% confidence intervals are shown by the ends of the vertical error bars. Generated using Ho et al., 2019106 website. Effect size legend |0.0–0.2| no effect (blank), |0.2–0.5| small (S), |0.5–0.8| medium (M), |0.8–1.2| large (L), |1.2–2.0| very large (VL). (E) Unpaired Hedge's g between mock and siDOCK5 for assembly and disassembly rates are 1.65 (VL) (95% CI 1.33, 1.96), and 1.28 (VL) (95% CI 1.07, 1.50) respectively. Two-sided P values of Mann–Whitney tests are 3.64 × 10−15, and 1.04 × 10−15 for assembly and disassembly rates respectively. (F) Unpaired Hedge's g between mock and siDOCK5 for assembly phase length and disassembly phase length are 0.101 (95% CI −0.199, 0.405), and −0.00986 (95% CI −0.38, 0.155) respectively. Two-sided P values of Mann–Whitney tests are 0.602, and 0.437 for assembly and disassembly phase lengths respectively. N number is individual adhesions from 4 cells, filtered by them being present for a minimum of 5 frames (phase length 5) and mean axial ratio less than 3. Unpaired Mann–Whitney test, P < 0.001. | ||
To determine if DOCK5 has a role in FA dynamics in LM2 cells which may explain the polarity defects, we used total internal reflection fluorescence (TIRF) microscopy to visualise the recruitment of Talin-mApple to FAs in migrating mock and DOCK5 depleted cells. In migrating mock transfected LM2 cells, small Talin-positive FAs were established 0–4 μm from the leading edge in the lamellipodia of migrating cells (Fig. 8B and C, adhesions coloured by FAAS software; Methods, Movie S1, ESI†). These FAs then enlarged/matured and turned over in the same region of attachment to the ECM, and FAs ere not stable in the lamella and/or cell body as the cell moved over this region (Fig. 8D – dynamics, Movie S1, ESI†).
While migrating mock cells established, and maintained, a polarised leading edge which is the site of both FA formation and turnover (Fig. 8C and Movie S1, ESI†), the changes in FA dynamics in DOCK5 depleted cells coincided with a failure of these cells to maintain polarity (Fig. 8D and Movie S2, ESI†). Once established, the leading edge in randomly migrating control cells was maintained for approximately 120 min. In DOCK5 depleted cells there was rarely only one leading edge at any given time but rather many protrusions lasting approximately 40 minutes on average (Movie S2, ESI†). Thus DOCK5 depleted cells were capable of initial establishing polarised protrusions, but the protrusions were not stable. These data demonstrate that DOCK5 depleted cells initiated the formation of a polarised leading edge but fail to maintain it.
We quantified the assembly and disassembly dynamics of individual FAs in mock and DOCK5 depleted cells. We used a Cumming estimation plot to demonstrate the mean difference in the assembly and disassembly rates and phase lengths. Effect sizes based on Cohen's initial suggestions107,108 indicate that values of 0.2 to 0.5 are small, 0.5 to 0.8 are medium, 0.8 to 1.2 are large, and 1.2 to 2.0 are very large effect sizes. In DOCK5 depleted cells, FAs have significantly accelerated assembly and disassembly rates compared to control FAs (Fig. 8E, orange unpaired Hedges’ g of 1.65; red unpaired Hedges’ g of 1.28). But disassembly phase lengths were largely unaffected (Fig. 8F, Hedge's g of 0.101 and −0.0986 for assembly and disassembly phase lengths respectively). Despite accelerated disassembly rates, FAs in DOCK5 depleted cells often failed to completely turnover (Fig. 8B–D) and DOCK5 depleted cells shift their mass over FA structures that have not turned-over completely as they migrate such that FAs appear to be near the nucleus (Fig. 8D and Movie S2, ESI†).
The most parsimonious explanation for these observations is that in the absence of DOCK5, nascent FAs can form, but are also highly unstable and exhibit fast shrinkage rates, potentially because actin-mediated force maturation is also impaired in DOCK5 depleted cells.109 Meaning, FAs in DOCK5 depleted cells are poorly coupled to RHOA-dependent actomyosin activity and do not mature properly. However, the absence of actomyosin engagement at FAs in DOCK5 depleted cells also means that turnover of existing FA structures is also impaired, and thus FAs are “left behind” as the cell moves over them (Fig. 8D). This model is consistent with the observation that lamellipodia are highly unstable in DOCK5 depleted cells (Movie S2, ESI†).
CDC42 and RHOA depletion affect FA morphogenesis in metastatic cells
We investigated whether the defects in FA morphogenesis following DOCK5 depletion could be attributed to decreased DOCK5-mediated RAC activation, or whether DOCK5 regulates FA morphogenesis in RAC-independent ways. We first knocked down RAC1, RHOA, and CDC42 and measured Talin-mApple dynamics in migrating LM2 cells (Fig. 9A). We also measured FA dynamics following treatment with Uprosertib to determine the effect of AKT signalling on FA morphogenesis. | ||
| Fig. 9 CDC42 and RHOA depletion affect FA morphogenesis in metastatic cells. (A) Representative images of LM2-CAAXGFP cells transfected with Talin mApple and siDOCK5, siCDC42, siRHOA, siRAC1, mock, and treated with the AKT inhibitor Uprosertib. Left and right panels per condition show FAAS picking up the adhesions and adhesions tracked over time for the same cell. (B)–(D) Cumming estimation plots of Hedge's g for comparison between (B) assembly and (C) disassembly rates, as well as (D) stability phase length of adhesions in mock versus siDOCK5, siCDC42, siRHOA, siRAC1 transfected cells, and Uprosertib treated cells. n shows number of adhesions. Data is taken from 3 cells per condition. Raw data is plotted in the upper axes, while mean difference is plotted in lower axes as a bootstrap sampling distribution. Mean differences are shown as dots, 95% confidence intervals are shown by the ends of the vertical error bars. Generated using Ho et al., 2019106 website. Effect size legend |0.0–0.2| no effect (blank), |0.2–0.5| small (S), |0.5–0.8| medium (M), |0.8–1.2| large (L), |1.2–2.0| very large (VL). (B) Unpaired Hedge's g for assembly rates between mock and conditions: siDOCK5: 0.375 (S) (95% CI −0.0214, 0.776); siCDC42: 0.851 (L) (95% CI −0.499, 1.19); siRHOA: 0.106 (95% CI −0.229, 0.436); siRAC1: 0.0311 (95% CI −0.332, 0.387); Uprosertib: 0.105 (95% CI −0.276, 0.479). Two-sided P values of Mann–Whitney tests are 0.242, 2.090 × 10−5, 0.922, 0.963, 0.808 respectively. (C) Unpaired Hedge's g for disassembly rates between mock and conditions: siDOCK5: 0.574 (M) (95% CI 0.0979, 1.04); siCDC42: 0.321 (S) (95% CI −0.0767, 0.715); siRHOA: 0.376 (S) (95% CI 0.0138, 0.732); siRAC1: 0.145 (95% CI −0.252, 0.558); Uprosertib: 0.467 (S) (95% CI 0.0641, 0.887). Two-sided P values of Mann–Whitney tests are 0.0363, 0.1840, 0.0580, 0.4030, 0.0652 respectively. (D) Unpaired Hedge's g for stability phase lengths between mock and conditions: siDOCK5: −0.507 (M) (95% CI −1.20, 0.494); siCDC42: 0.167 (95% CI −0.464, 0.856); siRHOA: −0.395 (S) (95% CI −1.03, 0.517); siRAC1: −0.299 (S) (95% CI −0.827, 0.259); Uprosertib: −0.456 (S) (95% CI −1.04, 0.393). Two-sided P values of Mann–Whitney tests are 0.0602, 0.6390, 0.06080, 0.4590, 0.0476 respectively. (E) Histogram of assembly phase lengths for all conditions (top) and unpaired Hedge's g for assembly rate phase lengths between mock and conditions (bottom): siDOCK5: 0.307 (S) (95% CI −0.0508, 0.662); siCDC42: 0.256 (S) (95% CI −0.0605, 0.536); siRHOA: −0.0537 (95% CI −0.35, 0.247); siRAC1: 0.336 (S) (95% CI 0.0297, 0.621); Uprosertib: −0.187 (95% CI −0.471, 0.148). Two-sided P values of Mann–Whitney tests are 0.140, 0.154, 0.471, 0.237, 0.390 respectively. Mock n = 83, siDOCK5 n = 56, siCDC42 n = 74, siRHOA n = 83, siRAC1 n = 67, Uprosertib n = 61. (F) Histograms of disassembly phase lengths (top) and unpaired Hedge's g for disassembly rate phase lengths between mock and conditions (bottom): siDOCK5: −0.12 (95% CI −0.439, 0.39); siCDC42: 0.106 (95% CI −0.224, 0.483); siRHOA: −0.255 (S) (95% CI −0.505, 0.0726); siRAC1: −0.251 (S) (95% CI −0.494, 0.14); Uprosertib: 0.037 (95% CI −0.285, 0.456). Two-sided P values of Mann–Whitney tests are 0.550, 0.633, 0.270, 0.435, 0.818 respectively. Mock n = 92, siDOCK5 n = 35, siCDC42 n = 50, siRHOA n = 64, siRAC1 n = 42, Uprosertib n = 50. (G) Summary of results and how each siRNA and treatment is affecting adhesion life cycle. Not to scale. (H) Cartoon summarising our data where CDC42 inhibits FA growth and turnover, RAC1 promotes FA growth, and RHOA and AKT promote engagement of FAs with actomyosin leading to FA maturation (stability) as well as normal turnover. | ||
Depleting CDC42 significantly accelerated assembly rates (Hedges’ g: 0.851) in a similar manner to DOCK5 depletion, and large FAs formed rapidly (Fig. 9B) (Movie S3, ESI†). However, all other aspects of FA morphogenesis including disassembly rates (Fig. 9C), stability (Fig. 9D), the length of assembly (Fig. 9E), and disassembly phase length (Fig. 9F) were largely unaffected.
In contrast, RAC1 depletion increased the length of the assembly phase (Fig. 9E); consistent with the role of RAC1 in promoting FA assembly110 (Movie S4, ESI†). RHOA depletion resulted in a significant increase in FA disassembly rates (Fig. 9C), in line with the idea that in the absence of RHOA-mediated actomyosin engagement, FAs fail to mature (Movie S5, ESI†). Uprosertib treatment resulted in similar effects as RHOA inhibition, as FAs had high disassembly rates, indicative of unstable adhesions (Fig. 9C) (Movie S6, ESI†). However, the effect on FA morphogenesis based on all other metrics following depletion of RAC1 and RHOA, or following Uprosertib treatment, was relatively mild.
Taken together these data suggest that CDC42 inhibits FA growth and turnover, RAC1 promotes FA growth, while RHOA and AKT promote engagement of FAs with actomyosin leading to FA maturation (stability) as well as normal turnover (Fig. 9H). The phenotype observed following DOCK5 depletion encompasses all those observed following depletion of Rho GTPases. This suggests that DOCK5 has much broader effects on FA morphogenesis than any single Rho GTPase alone.
DOCK5 and CDC42 stabilise polarity
We next sought to explore the idea that DOCK5 acts to modulate a CDC42-GSK3β axis that stabilises MTs at the leading edge of protrusive cells to reinforce the establishment of polarity. Cells were plated on fibronectin (FN) and fixed after 3 hours. After 3 hours, spreading LM2 cells establish a single leading edge adhered to the matrix by stable FAs, and thus have an asymmetric morphology (Fig. 10). In regions with large FAs, MTs were typically extended into the protrusions (Fig. 10 zoomed insert, arrows) supporting the idea that MTs are captured at the cortex in regions with active FA morphogenesis. | ||
| Fig. 10 DOCK5 and CDC42 stabilise polarity. Images taken on SoRA for mock, siDOCK5, siCDC42, siRAC1+ siRAC2, siRHOA, and treated with FAKi, and Blebbistatin. Stained for DNA, Paxillin, α-tubulin, F-actin. Merge 2D projection and zoomed section: Paxillin (blue), α-tubulin (red), F-actin (green). Merge 3D projection: Paxillin (red), DNA (blue), F-actin (green). Scale bar = 10 μm. | ||
After 3 hours of spreading, DOCK5 depleted cells established multiple smaller protrusions, leading to asymmetric shapes (Fig. 10). Paxillin-positive FAs in DOCK5 depleted cells were very small and appeared diffusely spread in the leading edge (Fig. 10 zoomed insert). Consistent with previous reports demonstrating a role of DOCK5 in promoting MT stability,92,93 there were dramatically fewer MT filaments in the periphery of DOCK5 depleted cells, and MTs appear thin and poorly organized compared to those of mock transfected cells. In DOCK5 depleted cells MTs appear organised largely as bundles around the nucleus. MT bundles are often hallmarks of stabilised, old MTs that exhibit low instability and are not captured at the cortex.111,112 Thus we conclude DOCK5 is essential for MT capture at the cortex in protrusions.
CDC42 depletion led to a partially similar phenotype as that of DOCK5, as cells formed expanded lamellipodia that nearly encompassed the entire circumference of the cells, and adopted a rounded, symmetric shape (Fig. 10). However, Paxillin-positive FAs in CDC42 depleted cells appeared globular and poorly formed – consistent with live cell imaging data suggesting CDC42 inhibits FA assembly (Fig. 10). In contrast, cells depleted of RHOA, or RAC1 + RAC2 all formed polarised lamellipodia with multiple large FAs (Fig. 10), hallmarks of defective FA maturation and/or turnover, but not in MT organization or establishment of polarity. Treatment of cells with either a focal adhesion kinase inhibitor (FAKi), which prevents focal adhesion turnover; or Blebbistatin, a contractility inhibitor, did not inhibit the formation of polarised lammelipodia, although no FAs formed in Blebbistatin treated cells (Fig. 10). We conclude DOCK5 promotes the stability of polarised leading edge by acting at FAs to promote CDC42-mediated MT capture at the cortex.
DOCK5 promotes spreading following MT depolymerisation
DOCK5 has been shown to promote MT stability downstream of GSK3β phosphorylation.92,93 To investigate the relationship between MTs and DOCK5 at the leading edge, we performed assays to analyse cell morphology following washout of the MT-depolymerization agent nocodazole (NZ) in media containing either: NZ, the MT-stabilising agent taxol (TX) or DMSO. Following 4 hours of NZ both mock and DOCK5 depleted cells showed decreased spread area, adopted a rounded morphology and actin intensity at the membrane was significantly decreased (Fig. 11A–C). However, while in control cells MT staining was expectedly diffuse, in DOCK5 depleted cells tubulin was localised into large high-intensity “bundles” which was reflected by significant increases in the SER Ridge Feature (texture feature) (Fig. 11D). These data show that MTs are differentially affected by MT depolymerising agents in control vs DOCK5 depleted cells, and we propose that depletion of DOCK5 leads to MT bundling and stability because MTs are not captured at the cell cortex.111,112 | ||
| Fig. 11 DOCK5 promotes spreading following MT depolymerisation. (A) Immunofluorescence images (maximum projections) of mock or siDOCK5 transfected cells treated just with DMSO or treated with nocodazole (NZ) for 4 hours and then washed out in either media containing DMSO, NZ, or taxol (TX). Stained for DNA (blue), tubulin (yellow), and F-actin (green). Scale bar is 10 μm. Imaged at 60× with water objective (NA = 1.2). Images shown with same intensity settings. White box represents zoomed in sections below. (B)–(D) Quantifications of (A) showing (B) cell area, (C) actin intensity at the membrane, and (D) whole cell SER Ridge tubulin (texture). Mean per well ± sd are shown (n > 3 wells, 1000 cells per well, 2 ways ANOVA, Tukey's multiple comparison's test * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). Significance between Mock and siDOCK5 for each condition shown with no bars. Significance between mock NZ (black) and siDOCK5 NZ (red) compared to other treatments for same transfection. | ||
Thirty minutes after washout in DMSO containing media, control cells formed abundant filopodial protrusions and increased in cell area (Fig. 11A and B, p = 0.013) likely as a result of MT outgrowth and RAC1 promoting actin polymerisation.113,114 Indeed, actin intensity at the membrane significantly increased in DMSO media washout denoting actin polymerisation (Fig. 11C). DOCK5 depleted cells also spread and increased in cell area under DMSO containing media washout (Fig. 11B, p = 0.013 for mock, p = 0.0894 for siDOCK5) but had lowered levels of actin at the membrane (Fig. 11C) – supporting the idea that DOCK5 is required for the stability of actin-based protrusions. DOCK5 depleted cells did not display polarised fronts for any condition supporting our hypothesis that DOCK5 is required for maintenance of polarity. Notably washout of NZ in DOCK5 depleted cells affected MT organisation in an identical manner as control – effectively rescuing the effects of DOCK5 depletion on MT organisation following NZ treatment. Therefore, it is unlikely DOCK5 regulates polymerisation of MTs during spreading or migration, but rather regulates MT capture.
Washout in TX containing media resulted in a significant increase in cell area for both mock and DOCK5 depleted cells compared to washout in DMSO alone, effectively accelerating the rate of spreading, though the ability of TX to rescue spreading was blunted in DOCK5 depleted cells (Fig. 11A and B, p = 0.005 for mock, p = 0.9114 for siDOCK5). Accelerated spreading was likely due to increased actin polymerisation, as both mock and DOCK5 depleted cells in TX had increased levels of actin at the membrane compared to DMSO washout alone, and were at levels comparable to controls. Strikingly however, TX resulted in significant increase of MT bundles in DOCK5 treated cells, that were even higher than the DMSO treated DOCK5 depleted cells (Fig. 11D). We propose that the formation of MT bundles in TX washout is due to accelerated MT polymerisation due to TX, but a failure to capture MT plus ends at the leading edge.
Discussion
In both yeast and mammalian cells the recruitment and activation of CDC42 at the membrane serves to seed polarised cortical structures that are enriched complexes and cytoskeletal elements that would otherwise be diffusely or asymmetrically organised in cells.115 During the formation of the bud in Saccharomyces cerevisiae, localised CDC42 activation marks the site of bud assembly at one site on the membrane.116,117 A key aspect of bud morphogenesis are positive feedback loops initiated by CDC42 which increase the local concentration of GTP-bound and active CDC42,118 resulting in one bud site outcompeting all other potential sites for the recruitment of bud assembly.119,120 Indeed in yeast mutants where the reinforcement, but not necessarily the establishment, of polarity is disrupted, multiple bud sites form.120,121In migrating mammalian cells, the establishment of polarity occurs with the coincident reorganisation of the actomyosin cytoskeleton to generate protrusions such as lamella and lamellipodia, and the engagement of the ECM by integrins to form FAs.122,123 Classical studies have shown that CDC42 regulates microtubule stability and establishment of polarity via PAR6 and AKT/PKCζ mediated phosphorylation of GSK3β at the leading edge of migrating cells in a manner that is integrin dependent.83,88,89 Here we propose that DOCK5 acts at FAs in breast cancer cells to establish positive feedback loops which maintain polarity in the presence of stable integrin-ECM attachments and cellular tension. By reinforcing polarity in sub-cellular regions with stable FA attachments, DOCK5 promotes processive migration and 3D invasion (Fig. 12).
 | ||
| Fig. 12 DOCK5 acts at FAs in breast cancer cells to establish positive feedback loops which maintain polarity in the presence of stable integrin-ECM attachments and cellular tension. Schematic of proposed model where in WT conditions DOCK5 gets recruited to FAs and acts as GEF for RAC-type GTPase to promote FA assembly and turnover/maturation in a positive feedback loop. At the same time DOCK5 recruits and activates AKT to FAs near the leading-edge maintaining GSK3β, which is targeted by CDC42-PKCζ for phosphorylation, leading to the maintenance of polarity. Here DOCK5 increases the levels of AKT and thus promotes further GSK3β stability and phosphorylation in another positive feedback loop. Further, actomyosin engagement by FA results in high RHOA and consequently high nuclear YAP/TAZ. Finally, DOCK5 could potentially mature FAs to recruit CDC42 GEFs which can further amplify CDC42 activation at the leading edge. | ||
We and others have previously observed that DOCK5 is recruited to FAs,69,103 and here we show that DOCK5 acts to couple FA morphogenesis and polarity by coordinating multiple GTPase activities. Firstly, DOCK5 acts as a GEF for RAC-type GTPase to promote FA assembly and ultimately maturation/turnover. In the absence of DOCK5, FAs do not increase in size, and the turnover of existing FAs is disrupted (Fig. 8) likely because of a failure of DOCK5-depleted FAs to engage actin at the leading edge.109,124,125 Failure to mature FAs and/or engage actomyosin likely results in decreases in RHOA activation (Fig. 12). Secondly, by recruiting and activating AKT to FAs near the leading edge, DOCK5 maintains GSK3β at the leading edge, which is targeted by CDC42-PKCζ for phosphorylation, leading to the maintenance of polarity. GSK3B phosphorylation leads to stabilization of the leading edge, further CDC42 recruitment and activation, and ultimately the recruitment of MTs which will disassemble FAs. Indeed, a role for DOCK5 recruitment of MTs to the leading edge has previously been established.92,93,126
In cells depleted of DOCK5, CDC42 activation at the membrane and the initial establishment of polarity is initially unaffected. But either C21 treatment or lack of DOCK5 reduces the levels of GSK3β that can be phosphorylated by CDC42-PKCζ, leading to a failure of cells to maintain polarity. CDC42 activation (which is initially independent of DOCK5) at another subcellular site can lead to formation of a new unstable leading edge in other regions of the cells. Indeed, a characteristic of DOCK5 depleted cells is not the ability to form protrusions, but rather to maintain a stable polarised protrusion. As such this phenotype is highly reminiscent of rdi1 and bem1/2 yeast mutants which form numerous unstable buds.
DOCK5 has been previously described to recruit and activate AKT and promote GSK3β phosphorylation through both RAC-dependent and independent ways.92,93 In particular, DOCK5's C-terminus has been shown to recruit NCK/AKT complexes to promote GSK3β phosphorylation in ways that are independent of catalytic activation of RAC-type GTPase through its DHR2/GEF domain.92,93 But our observation that the DOCK5 GEF inhibitor C21 mimics the effect of both DOCK5 siRNA and AKT inhibition by Uprosertib on GSK3β localisation at the membrane, and YAP/TAZ translocation, strongly suggests that DOCK5 mediated activation of RAC-type GTPases via its DH2/GEF is required for GSK3β phosphorylation and localisation at the leading edge. One model which resolves these previously contradictory observations is that activated DOCK5 exists in a conformation that recruits NCK and AKT to its C-terminus independent of it GEF domain; but also functions as a GEF to promote RAC-mediated FA assembly – which recruits more DOCK5/NCK/AKT to FAs in a positive feedback loop. Thus, inhibition of RAC by C21 breaks this loop and lowers AKT activation. Our observation that AKT inhibition by Uprosertib mimics different aspects of DOCK5 depletion on FA morphogenesis in fact suggests that AKT activity itself promotes FA morphogenesis and is involved in this positive feedback loop (Fig. 12).
We and others have previously demonstrated that YAP/TAZ nuclear translocation is dependent on cell shape and geometry.3,4,10,14,44 our data suggest that decreases in YAP/TAZ nuclear translocation in DOCK5 depleted cells are due to the inability of DOCK5 depleted cells to maintain a polarised, adherent morphology leading to low YAP/TAZ. Mechanistically this can be explained by the inability of cells to assemble stable a polarized leading edge and mature FAs. This leads to both defects in assembly of complexes at FA which promote YAP/TAZ activation – i.e. via signalling pathways such as PAK210 or FAK-PI3K127 and/or the failure of FAs in DOCK5 depleted cells to engage actin and upregulate actomyosin contractility122,123 which promotes YAP/TAZ activation.128 Indeed, that both CDC42 and RHOA depletion phenocopy DOCK5 depletion in terms of decreased nuclear YAP/TAZ, they act downstream of FAs to promote YAP/TAZ translocation (Fig. 1A, 9B and 10).
Finally, we cannot dismiss the potential changes in Hippo and/or Wnt signalling pathways that may occur following DOCK5 depletion which could alter YAP/TAZ translocation.14,129 Indeed, our proteomic data reveals downregulation of the Hippo component SAV, and of the Wnt signalling inhibitor DKK1 (Fig. 6C) in DOCK5 depleted cells. Moreover, the ligand Wnt7b was increased following DOCK5 depletion. As both DKK1 and SAV are YAP/TAZ targets87 and DKK1 has been shown to increase as a result of YAP overexpression,130 this suggests that their downregulation is likely a result of decreased nuclear YAP/TAZ.
It is tempting to speculate that sustained YAP/TAZ nuclear translocation, and resulting capabilities such as drug resistance, may be a consequence of cancer cells adopting stable polarised migratory forms. This model would contrast the classical idea that drug resistance and migration are coincident events, and/or that YAP/TAZ itself drives migration. Indeed, studies in melanoma cells have shown that YAP/TAZ upregulation which promotes drug resistance is caused by shape changes.22,24,40,41 Moreover, a number of recent studies have shown that EMT in breast cancer, which is characterized by morphological changes and increased invasive capabilities, also leads to drug resistant cells.131 Thus, shape changes may be causal to many aspects of TNBC, and suggests that therapeutic intervention could occur by preventing cells from altering their shape.
Materials and methods
Cell culture
LM2s (subpopulation 4172 from MDA-MB-231) were obtained from Joan Massagué (Sloan Kettering Institute, New York),43 and stably express a triple-fusion protein reporter construct encoding herpes simplex virus thymidine kinase 1, green fluorescent protein (GFP) and firefly luciferase (Minn et al., 2005). MDA-MB-231 cells were obtained from Janine Erler (University of Copenhagen, Denmark), while SUM159 and hs578t were a gift from Rachel Natrajan (Institute of Cancer Research, London). Cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin in T75 falcons, at 37 °C and supplemented with 5% CO2 in humidified incubators. MCF10A were obtained from ATCC and were grown in DMEM/F12 supplemented with 5% horse serum, 10 μg ml−1 insulin, 20 ng ml−1 epidermal growth factor, 100 ng ml−1 cholera toxin, 500 ng ml−1 hydrocortisone, and 100 mg ml−1 penicillin/streptomyocin in T75 falcons.T47D were obtained from from Rachel Natrajan (ICR), and were grown in Roswell Park Memorial Institute (RPM1)-1640 culture medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin in T75 falcons.
siRNA transfections
All siRNAs were made up to a stock of 20 μM. Briefly, for 384 well plates, siRNA reverse transfections were performed by adding 40 nl of siRNA to each well followed by 5 μl of Opti-MEM® Reduced Serum Media. 5 minutes later 5 μl of mix containing Opti-MEM and RNAimax reagent in a 125:1 ratio were added per well. Plates were spun at 1000 rpm for 1 minute, and incubated at room temperature for 20 minutes to allow siRNA-RNAimax complexes to form. Cells were plated on top in 30 μl of DMEM (10% FBS, 1% penicillin/streptomyocin), resulting in a total volume of 40 μl per well: 40 nl siRNA (final concentration of 20 nM), 5 μl Opti-MEM, 5 μl of Opti-MEM and RNAimax mix (125:1) plus 30 μl of cells. For transfections performed on 96 well plates and 6 well plates, 4 and 64 times the amounts used for 384 well plates were used, respectively. When siRNAs were transfected in combination, the same amount of each siRNA was used. siRNAs and sequences used can be found in Table 1.
| siRNA | Sequences | Catalogue number |
|---|---|---|
| DOCK5 OTP pool | GAGGAGAGAUUGUUAAGUU | # L-018931-00-0005 |
| GAACAUUAAACACAACCUA | ||
| GCAAAUUCAUUCAAAGCAU | ||
| GGGUUGCGAUCUAUAACUA | ||
| DOCK5 OTP pool deconvoluted5–8 | GAGGAGAGAUUGUUAAGUU | # LU-018931-00-0002 |
| GAACAUUAAACACAACCUA | ||
| GCAAAUUCAUUCAAAGCAU | ||
| GGGUUGCGAUCUAUAACUA | ||
| DOCK5 siGENOME pool deconvoluted1–4 | AGAACUAUCUAAUUCGUUG | # MU-018931-01-0002 |
| GUAACGGGAUGCCCAAGGA | ||
| GAGUGGCAGUGAUGGAUAU | ||
| UAUCAUACAUGGGAAGGUG | ||
| YAP OTP pool | GCACCUAUCACUCUCGAGA | # L-012200-00-0005 |
| UGAGAACAAUGACGACCAA | ||
| GGUCAGAGAUACUUCUUAA | ||
| CCACCAAGCUAGAUAAAGA | ||
| WWTR1 (TAZ) OTP pool | CCGCAGGGCUCAUGAGUAU | # L-016083-00-0005 |
| GGACAAACACCCAUGAACA | ||
| AGGAACAAACGUUGACUUA | ||
| CCAAAUCUCGUGAUGAAUC | ||
| LATS1 OTP pool | GGUGAAGUCUGUCUAGCAA | #L-004632-00-0005 |
| UAGCAUGGAUUUCAGUAAU | ||
| GGUAGUUCGUCUAUAUUAU | ||
| GAAUGGUACUGGACAAACU | ||
| LATS2 OTP pool | GCACGCAUUUUACGAAUUC | #L-003865-00-0005 |
| ACACUCACCUCGCCCAAUA | ||
| AAUCAGAUAUUCCUUGUUG | ||
| GAAGUGAACCGGCAAAUGC | ||
| RHOA OTP pool | CGACAGCCCUGAUAGUUUA | # L-003860-00 |
| GACCAAAGAUGGAGUGAGA | ||
| GCAGAGAUAUGGCAAACAG | ||
| GGAAUGAUGAGCACACAAG | ||
| RAC1 OTP pool | GUGAUUUCAUAGCGAGUUU | # L-003560-00 |
| GUAGUUCUCAGAUGCGUAA | ||
| AUGAAAGUGUCACGGGUAA | ||
| GAACUGCUAUUUCCUCUAA | ||
| RAC2 OTP pool | UGACAACUAUUCAGCCAAU | # L-007741-00 |
| CCAAGGAGAUUGACUCGGU | ||
| CCAAGUGGUUCCCAGAAGU | ||
| UGAAAACCGUGUUCGACGA | ||
| RAC3 OTP pool | AAACUGACGUCUUUCUGAU | # L-008836-00 |
| ACAAGAAGCUGGCACCCAU | ||
| GAAGACAUGCUUGCUGAUC | ||
| CGUGAUGGUGGACGGGAAA | ||
| CDC42 OTP pool | CGGAAUAUGUACCGACUGU | # L-005057-00 |
| GCAGUCACAGUUAUGAUUG | ||
| GAUGACCCCUCUACUAUUG | ||
| CUGCAGGGCAAGAGGAUUA |
Plasmid transfections
For transfections with Talin mApple and DOCK5-GFP, Effectene Transfection Reagent (Qiagen) was used according to manufacturer's protocol. Cells were plated at a density of 1.0 × 106 cells per ml in 1 ml in a 6 well plate. Then, 2.56 ng of plasmid were combined with 150 μl of EC buffer and vortexed followed by addition of 8 μl of Enhancer and vortexing. After 10 minutes 5 μl of Effectene were added, mix was vortexed, 10 minute later 1 ml of DMEM was added and mixture was dispensed dropwise per well. If siRNA was used as well, cells were reverse transfected as usual. Experiments were performed 48 hours later.Generation of CAAX-GFP tagged cell line
CAAX-GFP tag was a kind gift from Nicola Ferrari (previously Institute of Cancer Research, currently Astex Pharmaceuticals, UK), who generated it in HEK-293 cells using a second-generation lentiviral vector (plasmid EGFP-CAAX pCSII-IRES2-hygro). Cells were plated at a density of 6.4 × 104 cells per ml in a 6 well plate. Cells were transfected 24 hours later using a 1:1 dilution of lentivirus containing the CAAX-GFP tag solution and DMEM per well according to the manufacturers protocol. Transfected cells were selected with Hygromycin B (Invitrogen, cat#10687-010) at a final concentration of 500 μg ml−1 (elucidated from carrying out a concentration curve) 72 hours post transfection. CAAX tag did not affect YAP/TAZ ratio or cell morphology.
Drug treatments
Drugs were usually added on top of media already present. Appropriate amounts of DMSO or H2O depending on what the drug was dissolved in were added to mock transfected cells. A list of drugs used can be found in Table 2. Drugs were added for a period of 4 hours, unless other stated, so as to eliminate any cell cycle effects.| Drug | Inhibitor type/target | Company and catalogue number | Final concentration |
|---|---|---|---|
| PF-573228 | FAK inhibitor | Tocris Biosciences, #3239; CAS 869288-64-2 | 2 μM10 |
| C21 | DOCK5 inhibitor | Alfa Aesar, #H60031.MD; CAS 54129-15-6 | 100 μM |
| Blebbistatin | Myosin II light chain | Sigma, #B0560; CAS 856925-71-8 | 10 μM |
| Binimetinib | MEK inhibitor | Selleckchem, 10 μM #S707 | 10 μM132 |
| Uprosertib (GSK2141795) | AKT inhibitor | 2 μM | |
| Taxol (Paclitaxel) | Microtubule stabiliser | Sigma, #T7191 | 10 μM |
| Nocodazole | Microtubule depolymeriser | Sigma, #M1404 | 10 μM |
| LiCl | GSK3 inhibitor | Wako Chemicals | 5 mM92 |
For the nocodazole and taxol experiment, cells were mock and siDOCK5 reverse transfected as described in RNAi screening methodology for 48 hours. Then, cells were trypsinised and plated at a density of 1500 cells per well and allowed to settle for 20 hours. Microtubule depolymerisation was induced by nocodazole at a final concentration of 10 μM for 4 hours. Washout was performed for 30 minutes in either DMSO, 10 μM nocodazole, or 10 μM taxol – containing DMEM as indicated in Fig. 11.
RNAi screening methodology
40 nl (0.08 pmol) of siRNA from an ONTARGETPlus human siRNA RhoGEF/GAP and Rho GTPase library (stock concentration of 20 μM, Dharmacon) were arrayed using the acoustic liquid handler Echo 550 prior to transfection and kept at −80 °C. All siRNAs were arrayed in duplicate per plate and each plate was arrayed in duplicate resulting in a total of 4 plates per library. Pre-stamped siRNA plates were thawed at room temperature for 30 minutes prior to use. 5 μl of Opti-MEM® Reduced Serum Media were then added per well followed by 5 μl of RNAiMax reagent (Invitrogen) and Opti-MEM® mix in a 1:125 ratio. Plates were spun down at 1000 rpm for 1 min and incubated at room temperature for 20 min. Cells (LM2 and MDA-MB-231) were then seeded at a concentration of 33000 cells per ml (1000 cells per well ∼1.5 million cells per screen, 4 wells per siRNA, 4000 cells per siRNA per screen) in 30 μl DMEM supplemented with 12.8% FBS, 1.28% penicillin/streptomyocin (10% FBS and 1% penicillin/streptomyocin final) to account for the 10 μl of mix already present in the well, resulting in a total volume of 40 μl per well. Automated handling of plates on the Cell::Explorer robot station was carried out using the PlateWorks software (PerkinElmer). Plates were fixed 48 hours later with 8% warm formaldehyde in PBS (final concentration 4%) for 15 minutes and permeabilised with 0.1% Triton-X-100 in PBS for 10 minutes. Cells were then blocked in 2% BSA in PBS for 2 hours. All antibody incubations were performed in solution containing 0.5% BSA, 0.01% Triton-X-100 dissolved in PBS in a 1:1000 ratio, with the first primary antibody being added overnight at 4 °C and the rest being incubated at room temperature for 1.5 hours. Plates were washed 3× with PBS between every step using a Microplate Washer (BioTek). Antibodies were added sequentially to prevent cross reactivity. Antibodies used were: mouse YAP/TAZ (Santa Cruz Biotechnologies, cat#sc-101199), rat α-tubulin (Bio-Rad, cat#MCA77G), AlexaFluor 647 goat anti-mouse (Life Technologies, cat#A21235), AlexaFluor 647 goat anti-rat (Life Technologies, cat#A11077), phalloidin (Invitrogen, cat#A12379). Finally Hoechst (Sigma Aldrich, cat#33258) was added for 15 minutes, and after washing, cells were left in PBS and plates were sealed. Cells were imaged using an automated Opera Quadrupule Enhanced High Sensitivity (QEHS) spinning-disk confocal microscope (PerkinElmer) with 20× air lens. 28 fields of view were imaged per well. Cells were plated at increasing densities on columns 1, 2, 23 and 24 to be able to account for density dependent effects. SUM159 and hs578t were plated at a density of 67000 cells per ml (2000 cells per well) and 50000 cells per ml (1500 cells per well) respectively (∼2.5 million cells per screen, 4 wells per siRNA, 1500 to 2000 cells per well, 6000–8000 cells per siRNA per screen).
Images were processed and analysed using Columbus 2.6.0. Software Platform (PerkinElmer). Briefly, single cells were identified using the nuclear Hoechst signal. α-Tubulin intensity was using to segment the cytoplasm. Cells touching the image border were filtered out. Following this, cell morphology and texture features were extracted from all stains as described extensively in Pascual-Vargas et al., Scientific Data 2017. In the same script, a cell shape linear classifier was manually trained on visually distinctive cells that represented each category (spindly, large round, triangular, fan, and small and round) and included all the features extracted from the stains except those based on YAP/TAZ. YAP/TAZ ratio was calculated as log ratio of nuclear to ring region (perinuclear region) and was not included in the classifier. The percentage of cells classified into each shape or YAP/TAZ ratio was calculated per well and normalized per plate. Z-Scores were then calculated per screen by taking away the average of all mock transfected cells from the RNAi containing well, and dividing by the standard deviation of all mock transfected cells. Each well was assigned a QMS consisting of the Z-score for each shape.
Because we have previously observed that following efficient gene depletion, many cells in a population can appear wild-type,81 which can potentially obscure the presence of phenotypes resulting from gene depletion, we identified and filtered normal cells from our data set in the following way. Briefly, we performed principal component analysis (PCA) on single cell dataset comprised of texture and morphological features of populations which were either mock transfected or transfected with siRNAs which scored the highest for enriching for a particular shape. The data were projected into 2D PC space and each data point (cell) colour coded by the shape of each cell (Fig. S5A, ESI†). We defined the region of shape space where different classified cells overlapped in PC space as normal cells, defined as those whose PC coordinates lay within 1 SD of the mean of PC1 and PC2 for the siRNA transfected population (black box, Fig. S5B, ESI†). We trained the new linear classifier on the cells that fell in this space in the mock transfected population (black box, Fig. S5A, ESI†) and defined this population as ‘normal’. Running the classifiers again on the same data, performing the same PCA analysis on this new dataset and removing cells classified as ‘normal’ resulted in cells classified into the different shapes separating better in PC shape space (Fig. S5C, ESI†). Further analyses are described in the results section.
Quantitative real-time PCR (qRTPCR)
RNA was extracted using phenol:chloroform (TRIzol®, ThermoFisher cat#15596026) and RNAeasy kit (Qiagen cat#74104) according to the manufacturer's protocol. RNA was converted to cDNA using a cDNA conversion kit (Applied Biosystems cat#4387406) according to the manufacturers protocol. qRTPCR was performed on a QuantStudio® Flex Real-Time PCR system, using PCR mastermix SyBR green (Applied Biosystems cat#4309155) and primers in Table 3.| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| DOCK5 | GCTTTGAACTTCAGCTCTGG | CCACATGTCCCGGATTCTAA |
| WWTR1/TAZ | TATGGGACAGTCCGGGAGC | CGAGGCTTGGCTGACAAATC |
| YAP1 | GACTTCCTGAACAGTGTGGA | CAGCCAAAACAGACTCCATG |
| GAPDH | AGATCCCTCCAAAATCAAG | GGCAGAGATGATGACCCTT |
3D collagen invasion assay
We chose rat tail Col-I as our matrix because it is the most abundant type of collagen in mammalian tissues,133–137 and has been shown to form networks with physiological mechanical properties in vitro.138–140Cells were reverse transfected in 6 well plates as previously described. 48 hours later cells were trypsinised and resuspended in a solution containing rat-tail collagen I (Corning cat#354249), ultrafiltered H2O (prepared in house according to standard protocols), DMEM 5× and HEPES (1 M pH 7.5, prepared in house according to standard protocols). Collagen solutions were prepared on ice and 96 well plates were prechilled prior to use. Collagen solutions were made up as in Table 4 starting with HEPES, followed by DMEM 5× with rat-tail collagen being added last. Neutralisation of collagen was tested with pH strips prior to use to ensure a pH of 7.4. DMEM 5× was made up using 12.5 g DMEM powder, 25 ml of 1 M HEPES ph 7.5, 5 g NaHCO3, H2O up to 250 ml and was filtered under sterile conditions before aliquoting into 10 ml batches and freezing them at −20 °C. Cells were spun down in 1.5 ml eppendorfs at 1000 rpm for 4 minutes, and supernatant was removed. Cell pellets were resuspended carefully so as to not introduce bubbles in 500 μl of collagen solution, and 100 μl of cell-collagen mix was dispensed per well of a 96 well plate. Once all conditions were dispensed, plates were spun down for 1 minute at 4 °C to ensure all cells were at the bottom of the plate. Collagen was allowed to polymerise at 37 °C for one hour. Then 50 μl of DMEM were added on top of the collagen gels. 150 μl of PBS were added to all outside wells to prevent gel dehydration and edge effects due to dehydration. Cells were fixed 48 hours later with 50 μl per well of 16% PFA containing Hoechst (1:500) for 2 hours. Gels were then washed with PBS and left in PBS prior to sealing. The Opera QEHS spinning-disk confocal microscope (PerkinElmer) with 20× air lens was used to image these plates. A minimum of three Z stacks were taken per well, spaced 30 μm apart with 0 μm referring to the bottom of the plate and subsequent stacks referring to inside the gel. 20 fields were imaged per well per stack, and were all in the centre of the well to prevent edge effects. Columbus software was used to quantify cell number at every stack, using a filtering threshold method to ensure only cells with a certain nuclear and GFP intensity were counted in each stack (Fig. S7, ESI†).
| Stock | Final concentration | |
|---|---|---|
| Rat-tail collagen I | 10.31 mg ml−1 | 2.0 mg ml−1 |
| UF H2O (cold) | — | remaining |
| DMEM 5× | 5× | 1× |
| HEPES 1 M pH 7.5 | 100% | 2% |
Spheroid invasion assay
Cells were reverse transfected in 96 well ultra-low attachment (ULA) plates as described in the siRNA transfections section. Quantities used per well: 20 μl OPTIMEM + 0.16 μl RNAimax, 20 μl OPTIMEM + 0.16 μl siRNA. Cells were plated at a density of 4.0 × 104 cells per ml in 120 μl, resulting in a final volume of 160 μl per well. Due to their mesenchymal nature, LM2 cells required 2% Matrigel in ultra-low attachment plates to form spheroids, therefore 5 hours later a solution of 2.5% Matrigel in 40 μl of DMEM was added per well to promote spheroid formation resulting in a total volume of 200 μl per well. 48 hours later 180 μl of medium was carefully removed from each well so as not to disturb the spheroid, and replaced with 180 μl of 100% Matrigel. Spheroids were imaged prior to adding 100% Matrigel and immediately after using the Celigo Imaging Cytometer (Nexcelom Bioscience). Spheroids were imaged every day for 8 days, following the protocol outlined in ref. 141. Images were processed using ImageJ. Invasion area was determined by drawing a circle to the edge of where cells invaded and subtracting the initial area of the spheroid at day 0. Proliferation was quantified by drawing a circle around the edge of the area of the spheroid where the spheroid is grey, rather than the edge, so as to exclude any invading cells.Western blotting
Cells were reverse transfected in 6 well plates at a density of 3.3 × 104 cells per ml in a 6 well plate (same density as screens and all other experiments unless otherwise stated) for 48 hours. Cells were harvested by trypsinising, spinning down in 1.5 ml eppendorfs and removing supernatant. Cells were then washed in PBS 3 times. Pellets were then resuspended in 500 μl of solution containing a ratio of 3:1 of water to LDS Sample Buffer (4×) (Invitrogen) with a final concentration of 2 mM DTT. Samples were then boiled at 100C for 10 minutes and spun down for 2 minutes at 13000 rpm. Samples were frozen at −20 °C. Samples were boiled and spun down again prior to use. 16 μl of sample were loaded into 1.0 mm 15 well pre cast Novex 4–20% Tris–glycine gels (Invitrogen #XP04205BOX) and 30 μl into 10 well pre cast gels. Ladder used was Color Protein Standard, Broad Range (11–245 kDa) (New England Biolabs, #P7712S). Lysates were run in 1× Tris–glycine (Thermo) running buffer at 130 V until the bands reached the bottom of the gel. Gels were transferred in transfer buffer at 4 °C at 0.2 A onto a methanol activated polyvinylidene fluoride (PVDF) membrane (Immobilon-FL). Membranes were then stained with Ponceau (Sigma) dissolved in acetic acid to ascertain where the proteins were before cutting the membrane at required places. Membranes were then blocked in either 5% milk (Marvel) or 5% BSA if staining for phospho-proteins in Tris-buffered saline (TBS, prepared in house according to standard protocols) 0.1% Tween20 (TBST) solution for 1 hour at room temperature (RT)on a rolling shaker. Membranes were then washed once in TBST. Primary antibodies were diluted in either 5% milk TBST or 5% BSA TBST solution and incubated at 4 °C overnight on a rolling shaker. 10% glycerol was added to primary antibody solutions to be able to reuse them post storing at −20 °C. Membranes were washed 3 × 15 minutes in TBST. Membranes were then incubated for one hour at RT in either appropriate (mouse or rabbit) Horse radish peroxidase (HRP) – linked secondary antibodies (cell signaling) in TBST for chemiluminescent detection. Membranes were washed 3 × 5 minutes with TBST. Membranes were treated with ECL substrate (Thermo, #32106) and exposed using Azure Biosystems c600 western blot imaging system. Descriptions of antibodies used and dilutions can be found in Table 5. Western Blots were quantified using Image J and normalised to GAPDH unless otherwise stated.
| Target | Antibody and species | Company and cat number | W.B dilution | I.F Dilution |
|---|---|---|---|---|
| YAP/TAZ | Anti-YAP/TAZ (mouse) | Santa Cruz Biotechnologies, cat#A21235 | 1:1000 |
1:1000 |
| α-Tubulin | Anti-α-tubulin (rat) | Bio-Rad, cat#MCA77G | 1:1000 |
|
| GSK3β | Anti-GSK3β (rabbit) | CST, cat#12456T | 1:300 |
|
| pGSK3β | Anti-phospho GSK3β (Ser9) (D85E12) (rabbit) | CST, cat#5558T | 1:400 |
|
| Golgi | Anti-GM180 (mouse) | Abcam, cat #52649 | 1:500 |
|
| Beta-catenin | Anti-beta-catenin (mouse) | BD Transduction Laboratories, cat#610153 | 1:1000 |
|
| FOXO3A | Anti-FOXO3A (75D8) (rabbit) | CST, cat#2497S | 1:200 |
|
| DOCK5 | Anti-DOCK5 (rabbit) | Frank et al., 2017. | 1:500 |
|
| PHH3 | Anti-P-Histone H3 (S10) (mouse) | CST, cat#9706S | 1:1000 |
|
| KIF5B | Anti-KIF5B (rabbit) | Protein Tech, cat #231632-1-AP | 1:1000 |
|
| PCNA | Anti-PCNA (mouse) | Santa Cruz Biotechnology, cat #sc-56 | 1:1000 |
|
| GAPDH | Anti-GAPDH (1D4) (mouse) | Novus, cat#NB300-221 | 1:2000 |
|
| Paxillin | Anti-Paxillin (mouse) | BD transduction labs#610052 | 1:300 |
Subcellular fractionation
Thermo's Subcellular Protein Fractionation Kit for Cultured Cells (#78840) was used according to the manufacturer's protocol to extract cytoplasmic, membrane, nuclear soluble, chromatin-bound and cytoskeletal proteins. Briefly, cells were harvested by trypsining them and centrifuging for 5 minutes. Cell pellets were washed with ice-cold PBS. Cells were counted so as to be able to use the corresponding ratios of cytoplasmic extraction buffer (CEB), membrane extraction buffer (MEB), nuclear extraction buffer (NEB) and pellet extraction buffer (PEB):CEB:MEB:NEB:PEB 200:200:100:100 μl respectively. Fractions were stored at −80 °C.
Protein concentrations of each subcellular fraction were measured using the Pierce BCA Protein Assay kit (Thermo). Samples were diluted in water (Sigma) to the lowest sample concentration to ensure the same amount of protein for each fraction was loaded into the gels. 4 μl of NuPage LDS Sample Buffer (4×) were added to 16 μl of each sample, which were then boiled for 10 minutes and spun down before freezing. Samples were then run as described in western blotting subsection.
Mass spectrometry
LC–MS/MS analysis was performed on the Dionex Ultimate 3000 system coupled with the Q Exactive HF Orbitrap Mass Spectrometer (Thermo Scientific). Each peptide fraction was reconstituted in 40 μl 0.1% formic acid and 7 μl were loaded to the Acclaim PepMap 100, 100 μm × 2 cm C18, 5 μm, 100 Å trapping column at 10 μl min−1 flow rate. The sample was then subjected to a gradient elution on the EASY-Spray C18 capillary column (75 μm × 50 cm, 2 μm) at 45 °C. Mobile phase A was 0.1% formic acid and mobile phase B was 80% acetonitrile, 0.1% formic acid. The gradient separation method at flow rate 250 nl min−1 was as follows: for 90 min gradient from 5%-38% B, for 10 min up to 95% B, for 5 min isocratic at 95% B, re-equilibration to 5% B in 5 min, for 10 min isocratic at 5% B. The top 15 precursor ions between 350–1850 m/z were selected with mass resolution of 120k, AGC 3 × 106 and max IT 50 ms for HCD fragmentation with isolation width 1.0 Th. Collision energy was set at 35% with AGC 1 × 105 and max IT 100 ms at 60k resolution. Targeted precursors were dynamically excluded for 30 seconds.
Abundance values were scaled within each cell line and the log2 ratio of abundance in siDOCK5 containing samples to mock transfected samples was calculated. Log2 ratios above 0.5 and below −0.5 and permutation FDR < 0.05 (t-test) were considered to be proteins which were significantly up (∼40% minimum increase) and down regulated (∼40% minimum decrease) respectively. Statistical analysis of proteomics data was done in the Perseus software.142
Microscopy
| Secondary antibody/dyes | Company | Dilution |
|---|---|---|
| Phalloidin | Invitrogen, cat#A12379 | 1:1000 |
| Hoechst | Sigma Aldrich, cat#33258 | 1:1000 |
| AlexaFluor 647 goat anti-mouse | Life Technologies, cat#A21235 | 1:1000 |
| AlexaFluor 568 goat anti-rat | Life Technologies, cat #A11077 | 1:1000 |
| AlexaFluor 647 goat anti-rabbit | Life Technologies, cat #A21245 | 1:1000 |
| AlexaFluor 568 goat anti-rabbit | Life Technologies, cat #A11036 | 1:1000 |
| Anti-mouse IgG, HRP-linked antibody | CST, cat #7076S | 1:2000 |
| Anti-rabbit IgG, HRP-linked antibody | CST, cat #7074S | 1:2000 |
We displayed the data using estimation statistics using a website generated by Ho et al., 2019,106 as we think that by showing the effect size we can visualise and understand this type of data better.
Wound heal assay
Cells were reverse transfected as previously described and were plated in a 96 well PerkinElmer Cell Carrier Ultra plate at a density of 2.7 × 106 cells per ml. 48 hours later a 10 μM pipette tip was used to scratch the monolayers from top to bottom of the well, resulting in a vertical cell-free gap or wound. Medium was removed and wells were carefully washed with warm PBS so as to remove debris. 100 μl of fresh medium was them added to all wells. Imaging began as soon as the wound was created, using a Basic Confocal Spinning Disk Microscope (3i). Images were taken every 10 minutes for 16 hours. Images were processed using ImageJ. We quantified Golgi orientation in cells that had their Golgi within 180 degrees of the wound and quantified the angle of orientation by drawing a line from the position of the Golgi towards the wound and another parallel to the wound (Fig. 7C and D) as in Zhang et al., 2008.102 A 90 degree angle is indicative of Golgi facing toward the wound and indicative of front-rear polarity.Growth curve assay
Cells were reverse transfected in 6-well plates and plated at a density of 3.3 × 104 cells per ml per well, with three replicates per condition. When drugs were used one plate containing mock transfected cells and DOCK5 siRNA would be treated with DMSO, and another with the drug. Binimetinib (MEKi) and FAKi (PF-573288) were added to a final concentration of 10 μM and 2 μM respectively. Drugs and DMSO were added in 500 μl DMEM 48 hours after transfection. Plates were incubated in the IncuCyte S3 Live-Cell Analysis System (Sartorius) at 37 °C and 5% CO2 and imaged at 6 hour time points for a minimum of 156 hours (6.5 days). A minimum of 9 fields of view were imaged per well. Analysis of confluence over time was carried out using IncuCyte software (Sartorius). To compare confluence between conditions at a point where mock transfected cells reached 50% or 100% confluency, each well was normalized to the mock transfected wells for each plate, prior to statistical analysis. Representative growth curves are shown per plate with the average of three wells per condition.Statistical analyses
Statistical analyses performed for each assay are indicated in text and figure legends. Most analyses were carried out using Prism 8 (GraphPad). PCA analysis was performed in Jupyter Notebooks.143 Hierarchical clustering and heat maps were made with Morpheus Broad Institute Software (https://software.broadinstitute.org/morpheus).144Author contributions
P. P.-V. contributed to experimental design, carried out the experiments, performed image and data analysis, wrote and prepared the manuscript. M. A.-G. optimized RNAi screening methodology, performed experiments, data and image analysis. T. I. R, J. S. C performed mass spectrometry and proteomic analysis. C. B contributed to experimental design, imaging, and manuscript writing and preparation.Data availability
Proteomic data available on PRIDE database: PXD020102. Morphological and YAP/TAZ staining data in LM2 and MDA-MB-231 cells following RhoGEF/GAP knockdown is available from P. Pascual-Vargas, et al., RNAi screens for Rho GTPase regulators of cell shape and YAP/TAZ localisation in triple negative breast cancer., Sci. Data, 2017, 4, 170018.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Nicola Ferrari (Astex Pharmaceuticals, UK) for the CAAX-GFP construct, Aaron Farrugia for 2-photon imaging (ICR, London, UK), and Vicky Bousgouni (Dynamical Cell Systems Team, ICR, London, UK) for assistance with the RNAi library array and helpful comments on the manuscript. We thank Oliver Rocks (Max-Delbrück-Center for Molecular Medicine, Berlin, Germany) for the DOCK5-YFP construct and Steen Hansen (Boston Children's Hospital, Boston, USA) for the DOCK5 antibody. We thank Frankie Butera for helpful comments on the manuscript. This work was funded by a Cancer Research UK and Stand Up to Cancer UK Programme Foundation Award to C. B. (C37275/1A20146). The work of T. I. R and J. S. C was funded by the CRUK Centre grant with reference number C309/A25144.References
- I. Jones, M. Arias-Garcia, P. Pascual-Vargas, M. Beykou, L. Dent and T. P. Chaudhuri, et al., YAP activation is robust to dilution, Mol Omics, 2024, 20(9), 554–569 RSC.
- L. G. Dent, N. Curry, H. Sparks, V. Bousgouni, V. Maioli and S. Kumar, et al., Environmentally dependent and independent control of 3D cell shape, Cell Rep., 2024, 43(5), 114016 CrossRef CAS PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti and M. Cordenonsi, et al., Role of YAP/TAZ in mechanotransduction, Nature, 2011, 474(7350), 179–183 CrossRef CAS PubMed.
- S. Piccolo, S. Dupont and M. Cordenonsi, The Biology of YAP/TAZ: Hippo Signaling and Beyond, Physiol. Rev., 2014, 94(4), 1287–1312 CrossRef CAS PubMed.
- M. Aragona, T. Panciera, A. Manfrin, S. Giulitti, F. Michielin and N. Elvassore, et al., A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors, Cell, 2013, 154(5), 1047–1059 CrossRef CAS PubMed.
- J. Huang, S. Wu, J. Barrera, K. Matthews and D. Pan, The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP, Cell, 2005, 122(3), 421–434 CrossRef CAS PubMed.
- S. Wu, J. Huang, J. Dong and D. Pan, hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts, Cell, 2003, 114(4), 445–456 CrossRef CAS PubMed.
- D. Pan, The Hippo Signaling Pathway in Development and Cancer, Dev. Cell, 2010, 19(4), 491–505 CrossRef CAS PubMed.
- T. Moroishi, C. G. Hansen and K. L. Guan, The emerging roles of YAP and TAZ in cancer, Nat. Rev. Cancer, 2015, 15(2), 73–79 CrossRef CAS PubMed.
- J. E. Sero and C. Bakal, Multiparametric Analysis of Cell Shape Demonstrates that β-PIX Directly Couples YAP Activation to Extracellular Matrix Adhesion, Cell Syst., 2017, 4(1), 84–96.e6 CrossRef CAS PubMed.
- J. M. Lamar, P. Stern, H. Liu, J. W. Schindler, Z. G. Jiang and R. O. Hynes, The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(37), E2441–E2450 CrossRef CAS PubMed.
- F. Nallet-Staub, V. Marsaud, L. Li, C. Gilbert, S. Dodier and V. Bataille, et al., Pro-Invasive Activity of the Hippo Pathway Effectors YAP and TAZ in Cutaneous Melanoma, J. Invest. Dermatol., 2014, 134(1), 123–132 CrossRef CAS PubMed.
- J. E. Sero, H. Z. Sailem, R. C. Ardy, H. Almuttaqi, T. Zhang and C. Bakal, Cell shape and the microenvironment regulate nuclear translocation of NF-κB in breast epithelial and tumor cells, Mol. Syst. Biol., 2015, 11(3), 790 CrossRef PubMed.
- A. Totaro, T. Panciera and S. Piccolo, YAP/TAZ upstream signals and downstream responses, Nat. Cell Biol., 2018, 20(8), 888–899 CrossRef CAS PubMed.
- B. Zhao, X. Ye, J. Yu, L. Li, W. Li and S. Li, et al., TEAD mediates YAP-dependent gene induction and growth control, Genes Dev., 2008, 22(14), 1962–1971 CrossRef CAS PubMed.
- Cosmic. cancer.sanger.ac.uk [Internet]. [cited 2020 Dec 24]. Available from: https://cancer.sanger.ac.uk/cosmic.
- K. F. Harvey, X. Zhang and D. M. Thomas, The Hippo pathway and human cancer, Nat. Rev. Cancer, 2013, 13(4), 246–257 CrossRef CAS PubMed.
- J. G. Tate, S. Bamford, H. C. Jubb, Z. Sondka, D. M. Beare and N. Bindal, et al., COSMIC: the Catalogue Of Somatic Mutations In Cancer, Nucleic Acids Res., 2019, 47(D1), D941–D947 CrossRef CAS PubMed.
- M. Cordenonsi, F. Zanconato, L. Azzolin, M. Forcato, A. Rosato and C. Frasson, et al., The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells, Cell, 2011, 147(4), 759–772 CrossRef CAS PubMed.
- W. Zhao, M. Wang, M. Cai, C. Zhang, Y. Qiu and X. Wang, et al., Transcriptional co-activators YAP/TAZ: Potential therapeutic targets for metastatic breast cancer, Biomed. Pharmacother., 2021, 133, 110956 CrossRef CAS PubMed.
- P. C. Calses, J. J. Crawford, J. R. Lill and A. Dey, Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities, Trends Cancer, 2019, 5(5), 297–307 CrossRef CAS PubMed.
- M. L. Fisher, D. Grun, G. Adhikary, W. Xu and R. L. Eckert, Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells, Oncotarget, 2017, 8(66), 110257–110272 CrossRef PubMed.
- A. Kapoor, W. Yao, H. Ying, S. Hua, A. Liewen and Q. Wang, et al., Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic, Cancer Cell, 2014, 158(1), 185–197 CAS.
- M. H. Kim, J. Kim, H. Hong, S. H. Lee, J. K. Lee and E. Jung, et al., Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation, EMBO J., 2016, 35(5), 462–478 CrossRef CAS PubMed.
- S. Kitajima, H. Asahina, T. Chen, S. Guo, L. G. Quiceno and J. D. Cavanaugh, et al., Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS, Cancer Cell, 2018, 34(3), 439–452.e6 CrossRef CAS PubMed.
- L. Lin, A. J. Sabnis, E. Chan, V. Olivas, L. Cade and E. Pazarentzos, et al., The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies, Nat. Genet., 2015, 47(3), 250–256 CrossRef CAS PubMed.
- C. Nguyen and C. Yi, YAP/TAZ Signaling and Resistance to Cancer Therapy, Trends Cancer, 2019, 5(5), 283–296 CrossRef CAS PubMed.
- D. D. Shao, W. Xue, E. B. Krall, A. Bhutkar, F. Piccioni and X. Wang, et al., KRAS and YAP1 Converge to Regulate EMT and Tumor Survival, Cell, 2014, 158(1), 171–184 CrossRef CAS PubMed.
- Y. Wang, X. Xu, D. Maglic, M. T. Dill, K. Mojumdar and P. K. S. Ng, et al., Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer, Cell Rep., 2018, 25(5), 1304–1317.e5 CrossRef CAS PubMed.
- F. Zanconato, G. Battilana, M. Cordenonsi and S. Piccolo, YAP/TAZ as therapeutic targets in cancer, Curr. Opin. Pharmacol., 2016, 29, 26–33 CrossRef CAS PubMed.
- F. M. Vega and A. J. Ridley, Rho GTPases in cancer cell biology, FEBS Lett., 2008, 582(14), 2093–2101 CrossRef CAS PubMed.
- A. B. Jaffe and A. Hall, RHO GTPASES: Biochemistry and Biology, Annu. Rev. Cell Dev. Biol., 2005, 21(1), 247–269 CrossRef CAS PubMed.
- A. Hall, Small GTP-Binding Proteins and the Regulation of the Actin Cytoskeleton, Annu. Rev. Cell Biol., 1994, 10(1), 31–54 CrossRef CAS PubMed.
- A. J. Ridley, Rho GTPases and cell migration, J. Cell Sci., 2001, 114(Pt 15), 2713–2722 CrossRef CAS PubMed.
- A. J. Ridley, Rho GTPase signalling in cell migration, Curr. Opin. Cell Biol., 2015, 36, 103–112 CrossRef CAS PubMed.
- A. Sadok and C. J. Marshall, Rho GTPases, Small GTPases, 2014, 5(4), e983878 CrossRef PubMed.
- M. L. Gardel, I. C. Schneider, Y. Aratyn-Schaus and C. M. Waterman, Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration, Annu. Rev. Cell Dev. Biol., 2010, 26(1), 315–333 CrossRef CAS PubMed.
- K. Porshneva and G. Montagnac, Mechanotransduction mediated by microtubules, Nat. Mater., 2022, 21(3), 271–272 CrossRef CAS PubMed.
- D. Lai, K. C. Ho, Y. Hao and X. Yang, Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF, Cancer Res., 2011, 71(7), 2728–2738 CrossRef CAS PubMed.
- C. H. Lin, F. A. Pelissier, H. Zhang, J. Lakins, V. M. Weaver and C. Park, et al., Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors, Mol. Biol. Cell, 2015, 26(22), 3946–3953 CrossRef CAS PubMed.
- D. A. Lionarons, D. C. Hancock, S. Rana, P. East, C. Moore and M. M. Murillo, et al., RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance, Cancer Cell, 2019, 36(1), 68–83.e9 CrossRef CAS PubMed.
- F. Zanconato, M. Cordenonsi and S. Piccolo, YAP/TAZ at the roots of cancer, Cancer Cell, 2016, 29(6), 783–803 CrossRef CAS PubMed.
- A. J. Minn, G. P. Gupta, P. M. Siegel, P. D. Bos, W. Shu and D. D. Giri, et al., Genes that mediate breast cancer metastasis to lung, Nature, 2005, 436(7050), 518–524 CrossRef CAS PubMed.
- P. Pascual-Vargas, S. Cooper, J. Sero, V. Bousgouni, M. Arias-Garcia and C. Bakal, RNAi screens for Rho GTPase regulators of cell shape and YAP/TAZ localisation in triple negative breast cancer, Sci. Data, 2017, 4(1), 170018 CrossRef PubMed.
- B. Zhao, X. Wei, W. Li, R. S. Udan, Q. Yang and J. Kim, et al., Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control, Genes Dev., 2007, 21(21), 2747–2761 CrossRef CAS PubMed.
- J. S. A. Warren, Y. Xiao and J. M. Lamar, YAP/TAZ Activation as a Target for Treating Metastatic Cancer, Cancers, 2018, 10(4), 115 CrossRef PubMed.
- B. Zhao, L. Li, L. Wang, C. Y. Wang, J. Yu and K. L. Guan, Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis, Genes Dev., 2012, 26(1), 54–68 CrossRef CAS PubMed.
- Q. Y. Lei, H. Zhang, B. Zhao, Z. Y. Zha, F. Bai and X. H. Pei, et al., TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway, Mol. Cell. Biol., 2008, 28(7), 2426–2436 CrossRef CAS PubMed.
- S. Piccolo, M. Cordenonsi and S. Dupont, Molecular Pathways: YAP and TAZ Take Center Stage in Organ Growth and Tumorigenesis, Clin. Cancer Res., 2013, 19(18), 4925–4930 CrossRef CAS PubMed.
- A. L. Miller and W. M. Bement, Regulation of cytokinesis by Rho GTPase flux, Nat. Cell Biol., 2009, 11(1), 71–77 CrossRef CAS PubMed.
- K. C. Su, T. Takaki and M. Petronczki, Targeting of the RhoGEF Ect2 to the equatorial membrane controls cleavage furrow formation during cytokinesis, Dev. Cell, 2011, 21(6), 1104–1115 CrossRef CAS PubMed.
- A. Hajdo-Milasinović, S. I. J. Ellenbroek, S. van Es, B. van der Vaart and J. G. Collard, Rac1 and Rac3 have opposing functions in cell adhesion and differentiation of neuronal cells, J. Cell Sci., 2007, 120(Pt 4), 555–566 CrossRef PubMed.
- J. W. Jang, M. K. Kim, Y. S. Lee, J. W. Lee, D. M. Kim and S. H. Song, et al., RAC-LATS1/2 signaling regulates YAP activity by switching between the YAP-binding partners TEAD4 and RUNX3, Oncogene, 2017, 36(7), 999–1011 CrossRef CAS PubMed.
- S. Conner, J. R. Guarin, T. T. Le, J. Fatherree, C. Kelley and S. Payne, et al., Cell morphology best predicts tumorigenicity and metastasis in vivo across multiple TNBC cell lines of different metastatic potential, bioRxiv, 2023, preprint, bioRxiv:2023.06.14.544969 DOI:10.1101/2023.06.14.544969.
- Y. Ferrandez, W. Zhang, F. Peurois, L. Akendengué, A. Blangy and M. Zeghouf, et al., Allosteric inhibition of the guanine nucleotide exchange factor DOCK5 by a small molecule, Sci. Rep., 2017, 7(1), 1–13 CrossRef CAS PubMed.
- V. Vives, M. Laurin, G. Cres, P. Larrousse, Z. Morichaud and D. Noel, et al., The Rac1 Exchange Factor Dock5 Is Essential for Bone Resorption by Osteoclasts, J. Bone Miner. Res., 2011, 26(5), 1099–1110 CrossRef CAS PubMed.
- V. Vives, G. Cres, C. Richard, M. Busson, Y. Ferrandez and A. G. Planson, et al., Pharmacological inhibition of Dock5 prevents osteolysis by affecting osteoclast podosome organization while preserving bone formation, Nat. Commun., 2015, 6(1), 6218 CrossRef CAS PubMed.
- S. Di Agostino, G. Sorrentino, E. Ingallina, F. Valenti, M. Ferraiuolo and S. Bicciato, et al., YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins, EMBO Rep., 2016, 17(2), 188–201 CrossRef CAS PubMed.
- C. Lin, E. Yao, K. Zhang, X. Jiang, S. Croll and K. Thompson-Peer, et al., YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis, eLife, 2017, 6, e21130 CrossRef PubMed.
- T. Mizuno, H. Murakami, M. Fujii, F. Ishiguro, I. Tanaka and Y. Kondo, et al., YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes, Oncogene, 2012, 31(49), 5117–5122 CrossRef CAS PubMed.
- J. Rosenbluh, D. Nijhawan, A. G. Cox, X. Li, J. T. Neal and E. J. Schafer, et al., β-Catenin-Driven Cancers Require a YAP1 Transcriptional Complex for Survival and Tumorigenesis, Cell, 2012, 151(7), 1457–1473 CrossRef CAS PubMed.
- K. Schlegelmilch, M. Mohseni, O. Kirak, J. Pruszak, J. R. Rodriguez and D. Zhou, et al., Yap1 Acts Downstream of α-Catenin to Control Epidermal Proliferation, Cell, 2011, 144(5), 782–795 CrossRef CAS PubMed.
- F. X. Yu, Y. Zhang, H. W. Park, J. L. Jewell, Q. Chen and Y. Deng, et al., Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation, Genes Dev., 2013, 27(11), 1223–1232 CrossRef CAS PubMed.
- R. M. Meyers, J. G. Bryan, J. M. McFarland, B. A. Weir, A. E. Sizemore and H. Xu, et al., Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells, Nat. Genet., 2017, 49(12), 1779–1784 CrossRef CAS PubMed.
- E. B. Styles, K. J. Founk, L. A. Zamparo, T. L. Sing, D. Altintas and C. Ribeyre, et al., Exploring Quantitative Yeast Phenomics with Single-Cell Analysis of DNA Damage Foci, Cell Syst., 2016, 3(3), 264–277.e10 CrossRef CAS PubMed.
- A. H. Y. Tong, M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader and N. Pagé, et al., Systematic Genetic Analysis with Ordered Arrays of Yeast Deletion Mutants, Science, 2001, 294(5550), 2364–2368 CrossRef CAS PubMed.
- A. H. Y. Tong, G. Lesage, G. D. Bader, H. Ding, H. Xu and X. Xin, et al., Global Mapping of the Yeast Genetic Interaction Network, Science, 2004, 303(5659), 808–813 CrossRef CAS PubMed.
- M. Laurin and J. F. Cote, Insights into the biological functions of Dock family guanine nucleotide exchange factors, Genes Dev., 2014, 28(6), 533–547 CrossRef CAS PubMed.
- P. M. Müller, J. Rademacher, R. D. Bagshaw, C. Wortmann, C. Barth and J. van Unen, et al., Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions, Nat. Cell Biol., 2020, 22, 498–511 CrossRef PubMed.
- J. F. Côté and K. Vuori, GEF what? Dock180 and related proteins help Rac to polarize cells in new ways, Trends Cell Biol., 2007, 17(8), 383–393 CrossRef PubMed.
- G. Gadea and A. Blangy, Dock-family exchange factors in cell migration and disease, Eur. J. Cell Biol., 2014, 93(10–12), 466–477 CrossRef CAS PubMed.
- B. D. Lehmann, J. A. Bauer, X. Chen, M. E. Sanders, A. B. Chakravarthy and Y. Shyr, et al., Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies, J. Clin. Invest., 2011, 121(7), 2750–2767 CrossRef CAS PubMed.
- S. S. Kumar, T. J. Price, O. Mohyieldin, M. Borg, A. Townsend and J. E. Hardingham, KRAS G13D Mutation and Sensitivity to Cetuximab or Panitumumab in a Colorectal Cancer Cell Line Model, Gastrointest Cancer Res. GCR, 2014, 7(1), 23–26 Search PubMed.
- Y. Tokumaru, M. Oshi, E. Katsuta, L. Yan, V. Satyananda and N. Matsuhashi, et al., KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival, Am. J. Cancer Res., 2020, 10(3), 897–907 CAS.
- J. Huang, Q. Luo, Y. Xiao, H. Li, L. Kong and G. Ren, The implication from RAS/RAF/ERK signaling pathway increased activation in epirubicin treated triple negative breast cancer, Oncotarget, 2017, 8(64), 108249–108260 CrossRef PubMed.
- V. E. van der Noord, R. P. McLaughlin, M. Smid, J. A. Foekens, J. W. M. Martens and Y. Zhang, et al., An increased cell cycle gene network determines MEK and Akt inhibitor double resistance in triple-negative breast cancer, Sci. Rep., 2019, 9, 13308 CrossRef PubMed.
- I. Bado and X. H. F. Zhang, Senesce to Survive: YAP-Mediated Dormancy Escapes EGFR/MEK Inhibition, Cancer Cell, 2020, 37(1), 1–2 CrossRef CAS PubMed.
- G. E. Coggins, A. Farrel, K. S. Rathi, C. M. Hayes, L. Scolaro and J. L. Rokita, et al., YAP1 mediates resistance to MEK1/2 inhibition in neuroblastomas with hyperactivated RAS signalling, Cancer Res., 2019, 79, 6204–6214 CrossRef CAS PubMed.
- J. Sapudom, S. Rubner, S. Martin, T. Kurth, S. Riedel and C. T. Mierke, et al., The phenotype of cancer cell invasion controlled by fibril diameter and pore size of 3D collagen networks, Biomaterials, 2015, 52, 367–375 CrossRef CAS PubMed.
- C. Bakal, J. Aach, G. Church and N. Perrimon, Quantitative Morphological Signatures Define Local Signaling Networks Regulating Cell Morphology, Science, 2007, 316(5832), 1753–1756 CrossRef CAS PubMed.
- Z. Yin, A. Sadok, H. Sailem, A. McCarthy, X. Xia and F. Li, et al., A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes, Nat. Cell Biol., 2013, 15(7), 860–871 CrossRef CAS PubMed.
- J. Cau, Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways, J. Cell Sci., 2005, 118(12), 2579–2587 CrossRef CAS PubMed.
- S. Etienne-Manneville and A. Hall, Cell polarity: Par6, aPKC and cytoskeletal crosstalk, Curr. Opin. Cell Biol., 2003, 15(1), 67–72 CrossRef CAS PubMed.
- D. I. Johnson and J. R. Pringle, Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity, J. Cell Biol., 1990, 111(1), 143–152 CrossRef CAS PubMed.
- L. Stowers, D. Yelon, L. J. Berg and J. Chant, Regulation of the polarization of T cells toward antigen-presenting cells by Ras-related GTPase CDC42, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(11), 5027–5031 CrossRef CAS PubMed.
- G. Gadea and A. Blangy, Dock-family exchange factors in cell migration and disease, Eur. J. Cell Biol., 2014, 93(10–12), 466–477 CrossRef CAS PubMed.
- F. Zanconato, M. Forcato, G. Battilana, L. Azzolin, E. Quaranta and B. Bodega, et al., Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth, Nat. Cell Biol., 2015, 17(9), 1218–1227 CrossRef CAS PubMed.
- S. Etienne-Manneville, Cdc42 – the centre of polarity, J. Cell Sci., 2004, 117(8), 1291–1300 CrossRef CAS PubMed.
- S. Etienne-Manneville and A. Hall, Cdc42 regulates GSK-3b and adenomatous polyposis coli to control cell polarity, 2003, 421, 4.
- J. Brognard, E. Sierecki, T. Gao and A. C. Newton, PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms, Mol. Cell, 2007, 25(6), 917–931 CrossRef CAS PubMed.
- X. Xiong, X. Li, Y. A. Wen and T. Gao, Pleckstrin Homology (PH) Domain Leucine-rich Repeat Protein Phosphatase Controls Cell Polarity by Negatively Regulating the Activity of Atypical Protein Kinase C, J. Biol. Chem., 2016, 291(48), 25167–25178 CrossRef CAS PubMed.
- K. Ogawa, Y. Tanaka, T. Uruno, X. Duan, Y. Harada and F. Sanematsu, et al., DOCK5 functions as a key signaling adaptor that links FcεRI signals to microtubule dynamics during mast cell degranulation, J. Exp. Med., 2014, 211(7), 1407–1419 CrossRef CAS PubMed.
- S. Guimbal, A. Morel, D. Guérit, M. Chardon, A. Blangy and V. Vives, Dock5 is a new regulator of microtubule dynamic instability in osteoclasts, Biol Cell, 2019, 111, 271–283 CrossRef CAS PubMed.
- A. Brunet, J. Park, H. Tran, L. S. Hu, B. A. Hemmings and M. E. Greenberg, Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a), Mol. Cell. Biol., 2001, 21(3), 952–965 CrossRef CAS PubMed.
- B. D. Manning and A. Toker, AKT/PKB Signaling: Navigating the Network, Cell, 2017, 169(3), 381–405 CrossRef CAS PubMed.
- W. T. O’Brien, J. Huang, R. Buccafusca, J. Garskof, A. J. Valvezan and G. T. Berry, et al., Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice, J. Clin. Invest., 2011, 121(9), 3756–3762 CrossRef PubMed.
- E. Erdal, N. Ozturk, T. Cagatay, E. Eksioglu-Demiralp and M. Ozturk, Lithium-mediated downregulation of PKB/Akt and cyclin E with growth inhibition in hepatocellular carcinoma cells, Int. J. Cancer, 2005, 115(6), 903–910 CrossRef CAS PubMed.
- J. Huang, X. Guo, W. Li and H. Zhang, Activation of Wnt/β-catenin signalling via GSK3 inhibitors direct differentiation of human adipose stem cells into functional hepatocytes, Sci. Rep., 2017, 7(1), 1–12 CrossRef PubMed.
- S. B. Schleicher, J. J. Zaborski, R. Riester, N. Zenkner, R. Handgretinger and T. Kluba, et al., Combined application of arsenic trioxide and lithium chloride augments viability reduction and apoptosis induction in human rhabdomyosarcoma cell lines, PLoS One, 2017, 12, e0178857 CrossRef PubMed.
- D. Wu and W. Pan, GSK3: a multifaceted kinase in Wnt signaling, Trends Biochem. Sci., 2010, 35(3), 161–168 CrossRef CAS PubMed.
- S. Etienne-Manneville and A. Hall, Integrin-Mediated Activation of Cdc42 Controls Cell Polarity in Migrating Astrocytes through PKCζ, Cell, 2001, 106(4), 489–498 CrossRef CAS PubMed.
- S. Zhang, K. Schafer-Hales, F. R. Khuri, W. Zhou, P. M. Vertino and A. I. Marcus, The Tumor Suppressor LKB1 Regulates Lung Cancer Cell Polarity by Mediating cdc42 Recruitment and Activity, Cancer Res., 2008, 68(3), 740–748 CrossRef CAS PubMed.
- S. R. Frank, C. P. Köllmann, J. F. van Lidth de Jeude, J. R. Thiagarajah, L. H. Engelholm and M. Frödin, et al., The focal adhesion-associated proteins DOCK5 and GIT2 comprise a rheostat in control of epithelial invasion, Oncogene, 2017, 36(13), 1816–1828 CrossRef CAS PubMed.
- M. E. Berginski, E. A. Vitriol, K. M. Hahn and S. M. Gomez, High-Resolution Quantification of Focal Adhesion Spatiotemporal Dynamics in Living Cells, PLoS One, 2011, 6(7), e22025 CrossRef CAS PubMed.
- M. E. Berginski and S. M. Gomez, The Focal Adhesion Analysis Server: a web tool for analyzing focal adhesion dynamics, F1000Research, 2013, 2, 68 Search PubMed.
- J. Ho, T. Tumkaya, S. Aryal, H. Choi and A. Claridge-Chang, Moving beyond P values: data analysis with estimation graphics, Nat. Methods, 2019, 16(7), 565–566 CrossRef CAS PubMed.
- J. Cohen, Statistical power analysis for the behavioral sciences, Psychology Press, New York, NY, 2nd edn, reprint, 1988. p. 567 Search PubMed.
- S. S. Sawilowsky, New Effect Size Rules of Thumb, J. Mod. Appl. Stat. Methods, 2009, 8(2), 597–599 CrossRef.
- L. B. Case and C. M. Waterman, Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch, Nat. Cell Biol., 2015, 17(8), 955–963 CrossRef CAS PubMed.
- A. M. Pasapera, S. V. Plotnikov, R. S. Fischer, L. B. Case, T. T. Egelhoff and C. M. Waterman, Rac1-Dependent Phosphorylation and Focal Adhesion Recruitment of Myosin IIA Regulates Migration and Mechanosensing, Curr. Biol., 2015, 25(2), 175–186 CrossRef CAS PubMed.
- C. J. Bakal, D. Finan, J. LaRose, C. D. Wells, G. Gish and S. Kulkarni, et al., The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(27), 9529–9534 CrossRef CAS PubMed.
- G. G. Gundersen, Evolutionary conservation of microtubule-capture mechanisms, Nat. Rev. Mol. Cell Biol., 2002, 3(4), 296–304 CrossRef CAS PubMed.
- C. M. Waterman-Storer, R. A. Worthylake, B. P. Liu, K. Burridge and E. D. Salmon, Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts, Nat. Cell Biol., 1999, 1(1), 45–50 CrossRef CAS PubMed.
- T. Wittmann, G. M. Bokoch and C. M. Waterman-Storer, Regulation of leading edge microtubule and actin dynamics downstream of Rac1, J. Cell Biol., 2003, 161(5), 845–851 CrossRef CAS PubMed.
- J. G. Chiou, M. K. Balasubramanian and D. J. Lew, Cell Polarity in Yeast, Annu. Rev. Cell Dev. Biol., 2017,(33), 77–101 CrossRef CAS PubMed.
- J. M. Johnson, M. Jin and D. J. Lew, Symmetry breaking and the establishment of cell polarity in budding yeast, Curr. Opin. Genet. Dev., 2011, 21(6), 740–746 CrossRef CAS PubMed.
- T. J. Richman, M. M. Sawyer and D. I. Johnson, Saccharomyces cerevisiae Cdc42p localizes to cellular membranes and clusters at sites of polarized growth, Eukaryot Cell, 2002, 1(3), 458–468 CrossRef CAS PubMed.
- L. Kozubowski, K. Saito, J. M. Johnson, A. S. Howell, T. R. Zyla and D. J. Lew, Symmetry-breaking polarization driven by a Cdc42p GEF-PAK complex, Curr. Biol., 2008, 18(22), 1719–1726 CrossRef CAS PubMed.
- A. S. Howell, M. Jin, C. F. Wu, T. R. Zyla, T. C. Elston and D. J. Lew, Negative feedback enhances robustness in the yeast polarity establishment circuit, Cell, 2012, 149(2), 322–333 CrossRef CAS PubMed.
- C.-F. Wu, J.-G. Chiou, M. Minakova, B. Woods, D. Tsygankov and T. R. Zyla, et al., Role of competition between polarity sites in establishing a unique front, Elife, 2015, 4, e11611 CrossRef PubMed.
- T. Freisinger, B. Klünder, J. Johnson, N. Müller, G. Pichler and G. Beck, et al., Establishment of a robust single axis of cell polarity by coupling multiple positive feedback loops, Nat. Commun., 2013, 4(1), 1807 CrossRef PubMed.
- S. Etienne-Manneville, Polarity proteins in migration and invasion, Oncogene, 2008, 27(55), 6970–6980 CrossRef CAS PubMed.
- A. J. Ridley, M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg and G. Borisy, et al., Cell Migration: Integrating Signals from Front to Back, Science, 2003, 302(5651), 1704–1709 CrossRef CAS PubMed.
- A. Hall, Rho GTPases and the Actin Cytoskeleton, Science, 1998, 279(5350), 509–514 CrossRef CAS PubMed.
- C. D. Nobes and A. Hall, Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia, Cell, 1995, 81(1), 53–62 CrossRef CAS PubMed.
- M. Okabe, T. Sato, M. Takahashi, A. Honjo, M. Okawa and M. Ishida, et al., Autism Spectrum Disorder- and/or Intellectual Disability-Associated Semaphorin-5A Exploits the Mechanism by Which Dock5 Signalosome Molecules Control Cell Shape, Curr. Issues Mol. Biol., 2024, 46(4), 3092–3107 CrossRef CAS PubMed.
- N. G. Kim and B. M. Gumbiner, Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway, J. Cell Biol., 2015, 210(3), 503–515 CrossRef CAS PubMed.
- F. Calvo, N. Ege, A. Grande-Garcia, S. Hooper, R. P. Jenkins and S. I. Chaudhry, et al., Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts, Nat. Cell Biol., 2013, 15(6), 637–646 CrossRef CAS PubMed.
- L. Azzolin, T. Panciera, S. Soligo, E. Enzo, S. Bicciato and S. Dupont, et al., YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response, Cell, 2014, 158(1), 157–170 CrossRef CAS PubMed.
- H. W. Park, Y. C. Kim, B. Yu, T. Moroishi, J. S. Mo and S. W. Plouffe, et al., Alternative Wnt Signaling Activates YAP/TAZ, Cell, 2015, 162(4), 780–794 CrossRef CAS PubMed.
- A. Dongre and R. A. Weinberg, New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer, Nat. Rev. Mol. Cell Biol., 2019, 20(2), 69–84 CrossRef CAS PubMed.
- K. Jacobsen, J. Bertran-Alamillo, M. A. Molina, C. Teixidó, N. Karachaliou and M. H. Pedersen, et al., Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer, Nat. Commun., 2017, 8, 410 CrossRef PubMed.
- E. Brown, T. McKee, E. diTomaso, A. Pluen, B. Seed and Y. Boucher, et al., Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation, Nat. Med., 2003, 9(6), 796–800 CrossRef CAS PubMed.
- M. Magzoub, S. Jin and A. S. Verkman, Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin, FASEB J., 2008, 22(1), 276–284 CrossRef CAS PubMed.
- A. Oldberg, S. Kalamajski, A. V. Salnikov, L. Stuhr, M. Morgelin and R. K. Reed, et al., Collagen-binding proteoglycan fibromodulin can determine stroma matrix structure and fluid balance in experimental carcinoma, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(35), 13966–13971 CrossRef CAS PubMed.
- F. Sabeh, I. Ota, K. Holmbeck, H. Birkedal-Hansen, P. Soloway and M. Balbin, et al., Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP, J. Cell Biol., 2004, 167(4), 769–781 CrossRef CAS PubMed.
- F. Sabeh, R. Shimizu-Hirota and S. J. Weiss, Protease-dependent versus – independent cancer cell invasion programs: three-dimensional amoeboid movement revisited, J. Cell Biol., 2009, 185(1), 11–19 CrossRef CAS PubMed.
- J. Sapudom, L. Kalbitzer, X. Wu, S. Martin, K. Kroy and T. Pompe, Fibril bending stiffness of 3D collagen matrices instructs spreading and clustering of invasive and non-invasive breast cancer cells, Biomaterials, 2019, 193, 47–57 CrossRef CAS PubMed.
- D. R. Stamov and T. Pompe, Structure and function of ECM-inspired composite collagen type I scaffolds, Soft Matter, 2012, 8(40), 10200 RSC.
- K. Wolf, I. Mazo, H. Leung, K. Engelke, A. Y. Strongin and E. B. Bröcker, et al., Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis, J. Cell Biol., 2003, 160, 267–277 CrossRef CAS PubMed.
- M. Vinci, C. Box and S. A. Eccles, Three-Dimensional (3D) Tumor Spheroid Invasion Assay, J. Visualized Exp., 2015,(99), e52686 Search PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein and T. Geiger, et al., The Perseus computational platform for comprehensive analysis of (prote)omics data, Nat. Methods, 2016, 13(9), 731–740 CrossRef CAS PubMed.
- T. Kluyver, B. Ragan-Kelley, F. Pérez, M. Bussonnier, J. Frederic and J. Hamrick, et al., Jupyter Notebooks—a publishing format for reproducible computational workflows, in Positioning and Power in Academic Publishing: Players, Agents and Agendas, 2016, pp. 87–90 Search PubMed.
- https://software.broadinstitute.org/morpheus .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mo00154k |
| This journal is © The Royal Society of Chemistry 2025 |