DOI:
10.1039/D5MH00615E
(Communication)
Mater. Horiz., 2025,
12, 9808-9820
Medium spin FeIII regulating the peroxide selectivity in the heterogeneous oxygen reduction reaction of spin-polarized Fe-TAML complexes
Received
3rd April 2025
, Accepted 7th August 2025
First published on 8th August 2025
Abstract
Hydrogen peroxide (H2O2) can be produced cleanly and sustainably using electrosynthesis from the oxygen reduction reaction (ORR) via a 2-electron pathway, as opposed to the conventional anthraquinone technique. However, the rational design protocols for promoting the peroxide selectivity of ORR catalysts are still inadequate. This work synthesized a series of medium FeIII complexes with tetra-amido macrocyclic ligands (Fe-TAMLs) using a simplified two-step procedure. A relationship between peroxide selectivity and the spin polarization of metal centers in these Fe-TAML complex ORR catalysts is revealed. Using X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES), the spin polarization of metal centers in Fe-TAMLs regulated by the ligand modification was studied. We found that the nitro group-substituted ligand Fe-TAML complex was regulated by its spin-polarization and offered the highest spin state enhancing peroxide selectivity about 55% to 78%. An operando XAS study is employed to verify the stability of the oxidation states and the robustness of the coordination structures of the Fe-TAMLs.
New concepts
Hydrogen peroxide can be produced cleanly and sustainably via electrosynthesis through a 2-electron oxygen reduction reaction (ORR) pathway, offering a greener alternative to the conventional anthraquinone process. However, rational design strategies for enhancing the peroxide selectivity of ORR catalysts remain underdeveloped. In this study, a series of iron complexes with tetra-amido macrocyclic ligands (Fe-TAMLs) were synthesized using a simplified two-step procedure. In these Fe-TAML complex ORR catalysts, a clear relationship between the peroxide selectivity and the spin polarization of metal centers is revealed. Notably, our work provides the first direct evidence that spin-polarized Fe centers govern peroxide selectivity during the ORR. Operando X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES) were employed to confirm the structural stability and electronic modulation of Fe-TAML catalysts under ORR conditions. These analyses validate the role of spin-selective electron transfer, demonstrating how spin states suppress the 4-electron pathway and favor H2O2 formation. Using synchrotron-based XAS and XES, we uncover how spin polarization influences the electronic structure and determines the ORR pathway preference.
|
Introduction
Hydrogen peroxide (H2O2) is widely used as a chemical in bleach and waste-water treatment. To date, over 95% of industrial H2O2 production relies on the anthraquinone process.1 It is imperative to find alternative methods, because this process requires enormous energy costs, uses hazardous reagents, and produces harmful byproducts. Heterogeneous oxygen reduction reaction (ORR) electrosynthesis is a viable method for producing H2O2, in which the reagents for the ORR—water and oxygen gas—are non-toxic and readily available, minimizing the risk during storage and transportation. Single-atom catalysts (SACs) have been extensively studied due to their atom-utilization efficiency. Among those SACs constructed from metal ions and 4-fold coordinating nitrogen atom groups (denoted as M–N4), Fe–N4 has been reported to exhibit the highest ORR activity and optimal binding ability with O2.2–7 Owing to the triplet electronic configuration of O2 molecules, spin polarization may be one of the promising ways to manipulate the catalytic oxygen reduction.3–5 Several researchers have demonstrated manipulating the spin state of reactive centers to improve the selectivity toward a 4-electron pathway by hetero-doping, thereby modifying the affinity of intermediates.6–9 For example, Chen et al. decreased the spin state of Fe-NC through sulfur-doping in the second coordination sphere of Fe-NC, achieving improved selectivity toward the 4-electron pathway.6 Unfortunately, the random seeding of metal ions and/or doping atoms in graphene-based SACs complicates the ability to control their structure as well as manipulate the corresponding electronic configuration.
As compared to random dispersive SACs, molecular metal complexes have attracted increased attention in recent years for the following reasons: (1) their well-defined structures, determined through single crystal X-ray diffraction (SC-XRD), offer extraordinary platforms to study catalytic mechanisms at the atomic scale; (2) the discrete energy level of complexes provides versatile properties by electronic structure manipulation;2 and (3) precisely controlled metal complexes could be achieved through reasonable synthesis protocols for ligand modification. Several molecular complexes, particularly metal macrocyclic complexes like porphyrins,10–22 corroles,23,24 and phthalocyanines,25–34 have been employed as electrocatalysts for the ORR. Inspired by cytochrome P450, a natural enzyme that enables producing hydrogen peroxide through its FeIII–N4 structure,35,36 previous studies on the use of Fe complexes with tetra-amido macrocyclic ligands (Fe-TAMLs) in allylic oxidation reactions,37,38 epoxidation,39 and water oxidation40–42 have demonstrated their potential for oxygen activation. Furthermore, strong σ-donation from TAMLs to the metal has been demonstrated to prevent demetallation and to stabilize the coordination structures of complexes in the case of water oxidation, revealing the feasibility to be applied in aqueous heterogeneous catalysis.40
By considering these approaches for manipulating the corresponding electrocatalytic behaviors, spin polarization could be potentially realized by ligand modification because of significant electron sharing and interactions between metal centers and surrounding ligands. For example, by stabilizing a higher spin state of S = 1 through electron-withdrawing substitution and steric effects, a decrease in covalency between Fe and imido ligands could remarkably promote a hydrogen atom transfer (HAT) reaction.43 Note that the ORR includes two pathways: a 2-electron pathway to produce peroxide and a 4-electron pathway to produce water (or hydroxide in an alkaline environment), and the 2-electron pathway (eqn (1)) is less thermodynamically favorable than the competitive 4-electron pathway (eqn (2)).44 This may explain why many researchers have focused their efforts in enhancing the selectivity of the 4-electron pathway, often overlooking the economic potential of the 2-electron ORR pathway. However, despite numerous studies showcasing the utility of molecular metal complexes for the ORR, their spin-dependent behavior remains unclear and warrants further investigation.
| | | O2 + H2O + 2e− → OOH− + OH−, E0 = 0.695 VRHE | (1) |
| | | O2 + 2H2O + 4e− → 4OH−, E0 = 1.229 VRHE | (2) |
herein, a series of medium Fe
III complexes with tetra-amido macrocyclic ligands (Fe-TAMLs, see
Fig. 1a) were synthesized as aqueous heterogeneous catalysts as a model system to realize the spin-dependent behavior for peroxide selectivity, in which the spin polarization of the reactive metal centers was investigated by introducing nitro or bromo substituents on tetra-amido macrocyclic ligands, which allows evidently revealing a significant correlation between the peroxide selectivity and the spin polarization of the reactive Fe center. It was found that the catalytic properties of Fe-TAMLs were regulated after spin polarization control, in which the nitro group-substituted Fe-TAML presented the highest spin state and greatest enhancement in peroxide selectivity.
 |
| | Fig. 1 Synthesis routes and Fe K-edge XAS of Fe-TAMLs. (a) Scheme of synthesis routes and chemical structures of Fe-TAMLs, including scattering paths in which red and blue double arrows correspond to the Fe–N and Fe–C single scattering paths, respectively. (b) XANES, (inset) the enlargement of the main edges. (c) Phase-corrected EXAFS, the yellow shade corresponds to the Fe–N scattering path, and the gray shade corresponds to Fe–C scattering paths in Fe-TAMLs. | |
Results and discussion
Characterization of Fe-TAML catalysts
The synthesis routes of Fe-TAMLs used in this study and their characterization are summarized in Fig. 1a and Fig. S1–S9. The TAMLs with different substitutions, i.e., ligand-1, ligand-2, and ligand-3, were synthesized following the four-fold condensation reaction between two phenylenediamines and two oxalyl chlorides (two diethyl oxalates for ligand-1).39 Three TAMLs were metallized with FeCl2 to obtain distinctive Fe-TAMLs: Fe-1, Fe-2, and Fe-3, with a yield of nearly 100%. It should be noted that, prior to performing the corresponding ORR catalytic properties of Fe-TAMLs, residual Li ions from n-butyl lithium (nBuLi) were exchanged with tetramethylammonium to eliminate the inactive effects caused by forming clusters with intermediates of the ORR (i.e., OH*–Li+(H2O)x), which could subsequently block the active sites of catalysts and suppress the resulting performance of the ORR.45,46 Pronounced peaks have been found in the negative-ion mode of ESI-MS, suggesting that the complexes are mono-valent (Fig. S7–S9). Fe K-edge X-ray absorption near edge structure (XANES) also provided solid evidence to confirm that the oxidation states of Fe centers are determined to be approximately +3 (Fig. 1b). The redox non-innocent properties of TAMLs are further confirmed and could be explained by the strong σ-donation of four amido-nitrogen to stabilize the Fe center at an oxidation state of +3, even though the TAMLs are metallized with a ferrous precursor.47,48 Moreover, as illustrated in observed edge energies, their oxidation state exhibited an order of Fe-2 < Fe-3 < Fe-1, which can attributed to a divergence in the corresponding electronic configuration caused by substituents, thereby leading to distinct catalytic nature (vide infra). On the other hand, a small feature in the pre-edge region around 7114 eV in all XANES spectra validated the asymmetric geometry of the coordinated environment around Fe centers because of a considerable 3d–4p mixing with a square-pyramid structure of Fe-TAMLs (Fig. 1b). Through a deconvolution into two pseudo-Voigt curves with a Voigt baseline (Fig. S10), the peak feature in the high energy region could correspond to the transition of the 1s → ligand orbital,49 while the low energy one could be attributed to the 3dz2 orbital due to the hybridization with the p orbital (4pz).50 The position of 3dz2 can reveal an order of Fe-1 (7114.65 eV) > Fe-3 (7114.53 eV) > Fe-2 (7114.37 eV), indicating a decrease in 3dz2 energy attributed to the ligand modification through electron-withdrawing group substitution.51
Phase-corrected extended X-ray absorption fine structure spectra (EXAFS) derived from XAS by Fourier transform depicted in Fig. 1c, with two distinct peaks corresponding to the coordination shells of Fe-TAMLs. The first coordination shell at 1.75 Å observed for the Fe-TAMLs (yellow shade in Fig. 1c) represents the scattering path of Fe–N (red double arrow in Fig. 1a) and shows that the ligand modification has no significant influence on the Fe–N distance within the resolution of EXAFS. The second coordination shell at 2.85–2.95 Å (gray shade in Fig. 1c; 2.95, 2.88, and 2.85 Å for complexes 1-H, 1-NO2, and 1-Br) corresponds to Fe–C (blue double arrow in Fig. 1a) and other multiple scattering paths such as Fe–N–C.52 Accordingly, it can be observed that substituting with electron-withdrawing groups alters the position of the 3dz2 state in the order of Fe-1 > Fe-3 > Fe-2, without causing significant changes to the local coordination structures that may be influenced by ligand modifications.53
Electrochemical ORR performance
A series of electrochemical measurements were performed to investigate the ORR capabilities of the Fe-TAML complexes, in which the immobilization of Fe-TAML complexes onto a glassy carbon disk electrode was achieved by using Nafion, maintaining a consistent molar loading density of 6.07 × 10−4 mmol cm−2 across all catalysts. In the cyclic voltammetry (CV) analysis, the Fe-TAML complexes did not exhibit any noticeable peaks within the operating voltage of 0.0–1.0 VRHE in N2-saturated 0.1 M KOH (black curves in Fig. S11). This indicates the absence of any redox reactions involving the Fe-TAMLs under these conditions. However, during CV measurements conducted in an O2-saturated electrolyte (colored curves in Fig. S11), distinct cathodic peaks appeared at around 0.5 VRHE. These additional reduction peaks observed during O2 purging serve as evidence of the Fe-TAMLs' capacity for the ORR. The ORR electrocatalytic activity and peroxide selectivity of the Fe-TAMLs were measured using the rotating ring–disk electrode (RRDE) in an O2-saturated 0.1 M KOH aqueous solution and the corresponding data are summarized in Table 1, where a constant potential of 1.20 VRHE was applied on the ring electrode to probe the generation of peroxide from the disk electrode once the LSV measurement on the disk electrode proceeded. Additionally, two molecular ORR catalysts with the Fe–N4 motif, iron phthalocyanine (FePc) and iron(tetraphenylporphyrinato) chloride (FeTPPCl) are also measured with identical molar loading density of Fe-TAMLs to demonstrate the spin effect on peroxide selectivity (vide infra). Notably, in the upper panel of Fig. 2a, subsequently the ring current could serve as an indicator of the amount of peroxide generated during the ORR. While observing the ring currents within the range of applied disk potentials, Fe-TAMLs followed the order of Fe-2 > Fe-3 > Fe-1, which implies that the electron-withdrawing substituted ligands enhance the rate of peroxide production, with the nitro-substituted complex showing the highest production of peroxide compared to the bromo-substituted one. Additionally, the quantitative peroxide selectivity and the average electron transfer number (n) were determined using the disk and ring currents (Fig. 2b and c). Regarding the peroxide selectivity, Fe-TAMLs exhibited a plateau feature within the operating voltage range of 0.4–0.6 VRHE, and followed the sequence of Fe-2 (75–78%) > Fe-3 (61–65%) > Fe-1 (50–55%). Under biases beyond 0.4 VRHE, there was a decline in the peroxide selectivity as the potential decreased, implying a shift towards the 4-electron pathway branching ratio to generate hydroxide rather than peroxide. Notably, Fe-1 displayed a conspicuous rise in cathodic current compared to the other two complexes when the disk potential was below 0.3 VRHE. The peroxide selectivity exhibits severe decline as observed in Fig. 2b and c beyond an applied disk potential of 0.3 VRHE, corresponding to the favoring reaction towards the 4-electron pathway and explaining that Fe-1 has achieved the highest disk current among Fe-TAML catalysts (Fig. 2a lower panel). Moreover, the peroxide selectivity of Fe-1 attains 0% at a bias potential of 0 VRHE. This phenomenon clearly clarified that the Fe-1 exhibits a significantly different nature regarding the 2-/4-electron pathway, which may be attributed to the discrepancy in 3d energy as illustrated in the XAS results. Compared with several literature reports on ORR catalysts featuring an M–N4 structure for peroxide production (Table S1), Fe-2 demonstrated a competitive advantage in peroxide selectivity. The electrochemical stability test for Fe-2 kept at a constant potential of 0.5 VRHE is carried out over 48 hours, with the results depicted in Fig. S12, demonstrating notable stability of Fe-2 for peroxide selectivity. The disk and ring currents of the RRDE loaded with Fe-2 decay with time, nevertheless, the peroxide selectivity was maintained within 70–80%, demonstrating the stability for peroxide selectivity. Additionally, inductively coupled plasma mass spectrometry (ICP-MS) experiment for Fe-2 showed that the Fe element on the electrode remained about 90% after a 48-hour ORR operation (Fig. S13). Consequently, the findings from electrochemical characterization can reveal evidently the preference for the 2-electron pathway of the oxygen reduction reaction, with the order being Fe-2 > Fe-3 > Fe-1 in terms of the 2-electron pathway (peroxide) selectivity.
Table 1 Summarized results of ORR performances, X-ray spectroscopies, and computations
|
|
V
onset
(VRHE) |
Max. peroxide selectivity (%) |
n
|
Calc. spin statec |
ΔGabsd (eV) |
Charge transfer to OOHe (|e−|) |
|
Applied potential at 0.01 mA cm−2.
Average electron transfer number in the ORR.
Calculated from Fe Kβ XES.
Absorption energy towards OOH.
Based on Bader analyses.
|
| Fe-1 |
0.69 |
55@0.55 VRHE |
2.9 |
2.06 |
−0.064 |
0.72 |
| Fe-2 |
0.70 |
78@0.48 VRHE |
2.4 |
2.17 |
0.205 |
0.45 |
| Fe-3 |
0.70 |
65@0.52 VRHE |
2.7 |
2.11 |
0.011 |
0.65 |
 |
| | Fig. 2 ORR performance of Fe-TAMLs. (a) The RRDE diagram includes the linear sweep voltammetry of the disk electrode at a scan rate of 10 mV s−1 and the ring current responding at a constant potential of 1.20 VRHE. (b) The molar peroxide selectivity and (c) the average number of electron transfers are derived by eqn (4) and (5). (d) Chemical structures of FePc and FeTPPCl. | |
Dynamic spin state through non-resonant X-ray emission spectroscopy (XES)
Beyond local structures, spin polarization, which significantly influences electron population and orbital symmetry, motivates us to explore the dynamic spin state of Fe sites during reactions.54 However, capturing these dynamics remains challenging through both experimental and theoretical methods. While prior studies using in situ Mössbauer spectroscopy6 or ex situ non-resonant Kβ X-ray emission spectroscopy (XES)55 have provided valuable insights into the spin states of Fe–N–C materials, they offer limited information on real-time changes under ORR conditions. To address this gap, we employed operando non-resonant XES to directly probe the dynamic evolution of various Fe sites during oxygen reduction. Operando Fe Kβ XES spectra were obtained using a sophisticated electrochemical cell at several selected applied potentials (Fig. S13). The Kβ1,3 and Kβ′ satellite splitting in the mainline emission reflects the number of unpaired electrons in metal 3d orbitals by the peak area of the Kβ′ peak (located around 7045 eV).56 The heightened presence of unpaired electrons intensifies the 3p–3d interactions, thereby increasing the Kβ′ peak area correlating with the spin state of the emitter.56 Each operando XES depicts the negligible change in Kβ′ areas under ORR conditions, demonstrating the electrochemical performance variations of the Fe-TAMLs within the applied voltage window are independent of the dynamic spin change in Fe centers. Therefore, Fe L3-edge XAS (Fig. S14) and non-resonant Kβ XES (Fig. 3 and Fig. S15) were performed to elucidate the d-electronic structures. The stronger metal–ligand covalency, evidenced by lower absorbance in Fe L3-edge XAS57 and a redshift of the Kβ1,3 peak in Fe XES,58 follows the order Fe-2 > Fe-3 > Fe-1 (Fig. S14a and Fig. 3a), which can be attributed to the strong electron-withdrawing effect of the nitro and bromo substitutions on the ligand enhancing σ-donating ability by depleting the electron density of the resonance structure in the ligand.59 This observation aligns with earlier studies on para-substituted pyridine ligands, where stronger electron-withdrawing substituents were found to increase the covalency between the metal ion and the ligand.60 Because of the 2p3/2 → 3d process of the L3-edge XAS spectra, there are two primary peaks at around 708.3 and 709.7 eV in the Fe L3-edge XAS spectra (Fig. S14a) of the three catalysts attributed to t2g (dxy, dxz, dyz) and eg (dx2–y2, dz2) orbitals. The differences in peak positions, representing the energy gap between the t2g and eg orbitals, follow the order Fe-2 (1.40 eV) < Fe-3 (1.42 eV) < Fe-1 (1.48 eV) (Table S2). Based on the Fe L3-edge XAS spectra and pre-edge fittings in K-edge XANES (Fig. S10, vide supra), electron-withdrawing substituents on the ligands lower the energy level of eg orbitals due to the strong σ-donor characteristics of TAMLs,51,61 leading to a reduced energy gap, which subsequently influences the electron population in the eg and t2g orbitals, ultimately impacting spin polarization.
 |
| | Fig. 3 Fe Kβ XES of Fe-TAMLs (normalized by area). The enlarged XES of (a) Kβ1,3 and (b) Kβ′ peaks. (c) Correlation between the Kβ′ peak area and the spin state. (d) Correlation between the spin state calculated from Fe Kβ XES and the maximum peroxide selectivity in the ORR. | |
To estimate the degree of spin polarization in Fe-TAMLs, further analyses of Fe L3-edge XAS and Kβ XES have been conducted. The electron population ratio at the eg and t2g levels could be extracted from the integral ratio of the two peaks in the L3-edge XAS spectrum.62,63 By deconvoluting into multiple Lorentzian function peaks (peaks 1–4) with an error function as the step function (Fig. S14, all fitting parameters are summarized in Table S3), Fe-TAMLs exhibited an intensity ratio (Ieg/It2g) order of Fe-2 (2.461) < Fe-3 (4.066) < Fe-1 (5.297), implying that Fe-2 has the highest spin state while Fe-1 has the smallest among the three Fe-TAMLs. After normalization by the area of each Kβ XES spectrum, the areas of the Kβ′ peaks (Fig. 3b) exhibited the order of Fe-2 > Fe-3 > Fe-1 and indicated the consistent trend in the spin state observed in the L3-edge XAS. Furthermore, by utilizing the linear relationship between the spin state and Kβ′ peak area with various Fe standards as references (Fig. 3c and Fig. S16),64 the spin states of Fe-TAMLs have been quantified as 2.06 for Fe-1, 2.17 for Fe-2, and 2.11 for Fe-3, indicating that these spin states lie between a quadruplet (S = 1.5) and a sextet (S = 2.5), in agreement with DFT calculations (vide infra). The trend in the spin state among Fe-TAMLs could be elucidated by their energy gap where a smaller gap corresponds to a higher spin state. Fig. S17 and 3d emphasize that the maximum peroxide selectivity of Fe-TAMLs exhibited a negative correlation with Ieg/It2g and a positive correlation with the Kβ′ area. Moreover, FePc and FeTPPCl also exhibited this trend (Fig. 2, 3d). Consequently, Fe L3-edge XAS and Kβ XES confirm a strong correlation between the spin state of the reactive Fe center and the maximum peroxide selectivity, demonstrating that a higher spin state promotes greater peroxide selectivity.
Operando Fe K-edge X-ray absorption spectroscopy
Several studies have pointed out the potential correlation between the dynamic structure variations and the catalytic properties, highlighting the importance of operando studies.65–71 Gu et al. have revealed that FeIII–N–C is more active than FeII–N–C in the CO2 reduction reaction for CO production via the operando X-ray absorption spectroscopy (XAS) technique.72 Wang et al. have also reported that the cobalt complex, [(TPA)CoIII(μ-OH)(μ-O2)CoIII(TPA)](ClO4)3, only exhibits water oxidation activity after transforming into cobalt oxide.73 To verify the robustness of the Fe-TAMLs and exclude the impact of the deformation of catalysts, operando XAS has been conducted to clarify the dynamic variations during the oxygen reduction reaction (Fig. 4 and Fig. S18–S20).74 Due to O2-saturated electrolyte immersion, slight edge shifts towards higher energies are observed for Fe-2 and Fe-3, implying partial oxidation by dissolved O2 (Fig. 4a).75,76 Note that such a partial oxidation phenomenon was absent in the case of Fe-1, indicating a fact that the affinity of Fe-1 toward O2 gas was greatly poor as compared with those of Fe-2 and Fe-3. After applying a potential, Fe-2 and Fe-3 show a negligible edge shift. Nevertheless, Fe-1 exhibits a remarkable edge shift towards lower energy while the applied potential is negative than 0.4 VRHE, implying that partial reduction of the reactive Fe center further leads to the dramatic decay of the peroxide production rate (Fig. 2a upper panel, ring current of RRDE). On the other hand, a negligible change is observed for the Fe–N bond distances in the study of the EXAFS spectra (Fig. S20), which is evidence of the robustness of the coordination structure. Notably, the second coordination shell of all three Fe-TAMLs exhibited a shift towards a larger distance of ∼2.91 Å when immersed in the electrolyte (open-circuit voltage, OCV) and remained unchanged during the ORR course. Even though the oxidation states of Fe-TAMLs were kept at FeIII, the formation of Fe and/or Fe2O3 clusters can be entirely ruled out. The wavelet transform (WT) analyses of metallic Fe and Fe2O3 XAS data show obvious Fe–Fe scattering peaks at 7.9 and 7.0 Å−1, respectively; however, the WT results of Fe-TAMLs operando XAS data (Fig. 4b) reveal no discernible peak at either 7.9 or 7.0 Å−1. This observation indicates that neither demetallation nor aggregation of Fe atoms/ions occurred during the ORR.77,78 Apart from the noticeable decrease in peroxide production below 0.4 VRHE, which could be attributed to the partial reduction of the Fe center in the case of Fe-1, operando XAS and wavelet transform analyses clearly reveal that the variations in ORR performance relative to the applied potential across each Fe-TAML are independent of distortions in Fe coordination structures or changes in Fe oxidation states. The exceptional structural stability of Fe-TAMLs highlights their resilience under the rigorous electrochemical conditions of the ORR.
 |
| | Fig. 4 Wavelet transform analysis of Fe-TAMLs operando Fe K-edge X-ray absorption spectra under ORR conditions and the scheme of the ORR mechanism. (a) Edge shifts (defined by the absorbance of 0.5) under the ORR conditions. (b) Wavelet transform analysis. From bottom to top in each diagram, the contour plots correspond to the Fe2O3 standard, the ORR conditions of the as-prepared electrode, OCV, 0.6, 0.4, 0.2, 0.0 VRHE, and Fe foil standard, respectively (A.P.: as-prepared sample and OCV: open-circuit voltage). | |
Mechanism of spin-selective electron transfer in the ORR
According to the proposed ORR mechanism,79 the Fe–OOH intermediate plays an important role in the branching point of the 2-/4-electron pathway ORR, in other words, the selectivity in the ORR is dependent on the O–O bond cleavage in the OOH group. To address the O–O bond cleavage mechanism, a computational study has been conducted. The selected information from DFT-optimized Fe-TAML structures is listed in Table S4. DFT calculations indicate that the FeIII center in each Fe-TAML is most stable in the medium spin state (S = 1.5). All coordinating N atoms are coplanar in each Fe-TAML, and the slight inconsistency of Fe–N bond lengths in Fe-2 and Fe-3 is owing to the asymmetrical substitution on phenyl rings. According to Table S4, the Fe–N bond lengths exhibited a negligible elongation (<0.02 Å) and an out-of-plane distortion (<0.10 Å, defined by the distance between the Fe center and the N4 plane) after substitution. The results align with EXAFS data (Fig. 1c), indicating that ligand substitution induces no significant structural distortion. Based on the DFT results, the vibrational frequencies of the O–O stretching in Fe–OOH intermediates (S = 1) locate at ∼830 cm−1 (Table 2). After electron injection to form Fe–OOH− intermediates, different degrees of O–O bond weakening are observed, depending on the spin direction of the injected electron. Spin-down electron injection (S = 0.5) causes a redshift of −25 to −137 cm−1, while spin-up one (S = 1.5) results in a redshift of less than 20 cm−1, indicating that spin-down electrons promote a greater O–O bond cleavage than spin-up injection. Moreover, Fe-1 exhibits the largest redshift in O–O stretching frequency by ∼−137 cm−1 among the three Fe-TAMLs, while Fe-2 shows the smallest shift (∼−25 cm−1), implying that O–O bond weakening is most pronounced in Fe-1 after spin-down electron injection. Additionally, according to the results of charge decomposition analysis (CDA, Fig. 5a), both spin-up and spin-down lowest unoccupied molecular orbitals (LUMOs) of all Fe–OOH intermediates show obvious contributions from Fe 3d orbitals along the z-axis (3dz2, 3dxz, or 3dyz). All three spin-up LUMOs include the OOH πh* anti-bonding orbital (parallel to the Fe–O–O plane), however, spin-down LUMOs show the OOH σ* anti-bonding orbital character, except the Fe–OOH intermediate of Fe-2. Noteworthily, the OOH σ* anti-bonding orbital plays a crucial role in the O–O bond cleavage to form the Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediates and undergoes the 4-electron ORR pathway.80 The electron injection into the molecular orbital including the OOH σ* orbital increases the electronic density of the OOH σ* orbital, resulting in the weakening of the O–O bond and promoting the formation of FeO intermediates. In the case of Fe-2 with the highest Fe spin among three Fe-TAMLs, the negligible OOH σ* contribution (<0.5%) in the spin-down LUMO of the Fe–OOH intermediate aligns with its smallest redshift in O–O stretching frequency after spin-down electron injection, demonstrating that Fe-2 has the highest peroxide selectivity due to the negligible electronic injection into OOH σ* during the Fe–OOH intermediate reduction. The additional CDA analysis of Fe–OOH− intermediates also indicates the absence of OOH− σ* in the Fe-2 spin-down highest occupied molecular orbital (HOMO) after a spin-down electron injection (Table S5), further corroborating that O–O bond cleavage is more facilitated in Fe-1 and Fe-3, while it is more hindered in Fe-2. Furthermore, according to the theory of quantum spin exchange interaction (QSEI), a higher spin Fe center promotes parallel-oriented electron transfer through Fe centers owing to the lower repulsion between two electrons with the same spin orientation.81,82 In the three Fe-TAMLs, the lowest peroxide selectivity of Fe-1 could be attributed to its lowest Fe spin, resulting in the least promotion of parallel-oriented (spin-up) electron transfer during the reduction of the Fe–OOH intermediate promoting the O–O bond cleavage (Fig. 5b). On the other hand, the highest Fe spin in Fe-2 prevents the O–O bond cleavage by the highest parallel-oriented electron selectivity, leading to the best peroxide selectivity in the ORR.
O intermediates and undergoes the 4-electron ORR pathway.80 The electron injection into the molecular orbital including the OOH σ* orbital increases the electronic density of the OOH σ* orbital, resulting in the weakening of the O–O bond and promoting the formation of FeO intermediates. In the case of Fe-2 with the highest Fe spin among three Fe-TAMLs, the negligible OOH σ* contribution (<0.5%) in the spin-down LUMO of the Fe–OOH intermediate aligns with its smallest redshift in O–O stretching frequency after spin-down electron injection, demonstrating that Fe-2 has the highest peroxide selectivity due to the negligible electronic injection into OOH σ* during the Fe–OOH intermediate reduction. The additional CDA analysis of Fe–OOH− intermediates also indicates the absence of OOH− σ* in the Fe-2 spin-down highest occupied molecular orbital (HOMO) after a spin-down electron injection (Table S5), further corroborating that O–O bond cleavage is more facilitated in Fe-1 and Fe-3, while it is more hindered in Fe-2. Furthermore, according to the theory of quantum spin exchange interaction (QSEI), a higher spin Fe center promotes parallel-oriented electron transfer through Fe centers owing to the lower repulsion between two electrons with the same spin orientation.81,82 In the three Fe-TAMLs, the lowest peroxide selectivity of Fe-1 could be attributed to its lowest Fe spin, resulting in the least promotion of parallel-oriented (spin-up) electron transfer during the reduction of the Fe–OOH intermediate promoting the O–O bond cleavage (Fig. 5b). On the other hand, the highest Fe spin in Fe-2 prevents the O–O bond cleavage by the highest parallel-oriented electron selectivity, leading to the best peroxide selectivity in the ORR.
Table 2 DFT vibrational frequency shift of O–O stretching after electron injection into Fe–OOH intermediates (S = 1.5 and 0.5 for spin-up and spin-down electron injection, respectively)
|
|
Fe–OOH |
Fe–OOH− |
Δν (cm−1) |
|
S
|
ν(O–O) (cm−1) |
S
|
ν(O–O) (cm−1) |
| Fe-1 |
1 |
836.74 |
1.5 |
819.01 |
−17.73 |
| 0.5 |
699.76 |
−136.98 |
| Fe-2 |
830.10 |
1.5 |
823.10 |
−7.00 |
| 0.5 |
805.04 |
−25.06 |
| Fe-3 |
831.72 |
1.5 |
824.86 |
−6.86 |
| 0.5 |
707.68 |
−124.04 |
 |
| | Fig. 5 LUMO analysis and the spin-selective ORR mechanism. (a) LUMO boundary surfaces of Fe–OOH intermediates. Only orbital contributions (in parentheses) larger than 1% are listed (isovalue: 0.02 e Å−3). (b) Proposed spin-selective ORR mechanism. The red and blue dashed-line arrows indicate the spin-up and spin-down electron transfer, respectively. | |
Additionally, the absorption strength of Fe–OOH is considered as another descriptor of peroxide selectivity in the ORR.83 The absorption strength of Fe–OOH affected by the TAML substitution is also investigated and verified with DFT calculations and Bader analysis (summarized in Table 1). The absorption energies of Fe–OOH derived from DFT calculations are −0.064 eV for Fe-1, 0.205 eV for Fe-2, and 0.011 eV for Fe-3, showing a decreasing trend corresponding to an increase in the electron-withdrawing ability of substituting ligand groups on the aromatic rings. Activation energy calculations for both 2-electron and 4-electron pathways, based on Marcus theory,84,85 demonstrate that ORR selectivity favors the 2-electron pathway even though the 4-electron pathway is thermodynamically more favorable (Fig. S22 and Table S6). Furthermore, the optimal applied potential for peroxide production was observed to be around 0.5 VRHE across all three Fe-TAMLs (Fig. S22(h)). Fig. 6 and Fig. S23 show the DFT-optimized structures and the charge density different (CDD) plots of Fe–OOHs based on the Bader analysis, displaying electron transfer numbers from the Fe center to the OOH adsorbate as 0.72 |e−| for Fe-1, 0.45 |e−| for Fe-2, and 0.65 |e−| for Fe-3, indicating the OOH affinity order as Fe-1 > Fe-3 > Fe-2 due to less charge transfer associated with weaker chemisorption.83 Based on the crucial role of spin-down electron injection in the O–O bond cleavage within Fe–OOH intermediates, the QSEI effect impedes the antiparallel-oriented electron injection into the σ* orbital in the OOH group, constructing a spin-selective channel for electron transfer and lessening the O–O bond cleavage in the OOH group, leading to more release of peroxide ions. Accordingly, both computational thermodynamic and Bader charge analyses explain the substituent effect on peroxide selectivity of the Fe–OOH absorption strength in agreement with the findings of the electrochemical experiments.
 |
| | Fig. 6 (a–c) DFT optimized structures of Fe–OOHs (enlarged) and (d–f) their Bader charge density difference plots. The gray, white, red, blue, and blue-purple spheres represent C, H, O, N, and Fe atoms, respectively. The values correspond to the charge (electron) difference on the atoms. The blue-purple and cyan surfaces represent the accumulation and depletion regions of charges, respectively (isovalue: 0.01 e Å−3). | |
Conclusions
In summary, a series of Fe-TAMLs were synthesized using a simplified two-step procedure and employed as heterogeneous electrocatalysts for the ORR. The Fe center in Fe-TAMLs became more spin-polarized upon the electron-withdrawing nitro or bromo substitution, which strengthened the preference for the 2-electron pathway in the ORR and improved peroxide selectivity. Notably, the nitro-substituted Fe-TAMLs exhibited an increased selectivity towards peroxide from 55% to 78% when compared to the Fe-TAML analogs substituted with any other electro-withdrawing groups. Operando Fe K-edge XAS analysis was performed to evaluate the robustness of the electronic and coordination structures in Fe-TAMLs. The regulation mechanism of peroxide selectivity in the ORR has been explained as the prevention of the antiparallel-oriented electron injection into the OOH σ* orbital which could be attributed to the QSEI effect. Moreover, DFT calculations and Bader analysis also verified the influence of electron-withdrawing group substitution on Fe–OOH absorption strength, corroborating the findings from electrochemical experiments in this work. This study shows that spin-state engineering could be used to regulate the ORR catalytic characteristics of metal complexes, highlighting Fe-TAMLs as viable options for peroxide-selective electrosynthesis catalysts. Our work provides a rational design protocol of molecular metal complex catalysts to enhance peroxide selectivity in the ORR.
Experimental section
General information
All the chemicals were purchased from Acros, Sigma-Aldrich, and Merck, and used without any further purification. The route of synthesis is summarized in Fig. 1a. Ligands were prepared as discussed in a previous report, with modifications.39 All syntheses, except for cation exchange, were carried out under nitrogen, using standard Schlenk-type techniques. The solvents used in the synthesis were purified using a SPBT-1 benchtop solvent purification system (LC Technology Solution, Salisbury, MA, USA). Nuclear magnetic resonance (NMR) spectra were measured using AV-300 NMR equipment (Bruker, Rheinstetten, Germany). High-resolution mass spectra were recorded on a JMS-700 (JEOL, Tokyo, Japan) with the electrospray ionization (ESI) method.
Synthesis of 5,8,13,16-tetrahydrodibenzo[b,h][1,4,7,10]tetraazacyclododecine-6,7,14,15-tetraone (ligand-1)
Benzene-1,2-diamine (0.5 g, 4.6 mmol) and diethyl oxalate (0.67 g, 4.6 mmol) were dissolved in 60 mL of ethanol, followed by the dropwise addition of 8 mL of conc. hydrochloric acid. The mixture was then stirred and heated to flux. After an overnight reaction, a saturated sodium hydrogen carbonate solution was added until the pH of the mixture reached approximately 7. Ethanol was evaporated using a rotary evaporator. The crude product was precipitated using an ice bath, and the remaining solvent was removed by vacuum filtration. The crude product was washed with methanol, ethyl acetate, dichloromethane, and n-hexane; then, the remaining solvent was removed using a high-vacuum system. The yield was 56.4%. 1H NMR (300 MHz, d6-DMSO): δ 11.90 (s, 4 H), 7.06–7.14 (m, 8 H) ppm. 13C NMR (150 MHz, d6-DMSO): δ 155.24, 125.63, 123.05, 115.18 ppm. HRMS (ESI positive mode) [M + Na]+ cacld for C16H12N4O4Na: 347.0751; found 347.0759.
Synthesis of Fe-1
Ligand-1 (0.2 g, 0.617 mmol) was dissolved in 100 mL of dehydrated tetrahydrofuran. After stirring and cooling with a liquid nitrogen/acetone slush bath, n-butyl lithium (nBuLi 3 mL, 4.8 mmol) was added, and the suspension was dissolved. When the mixture was turned dark, the slush bath was removed. After returning to room temperature, FeCl2 (0.1 g, 0.78 mmol) was added under a N2 atmosphere. After stirring overnight, the mixture was vacuum-filtered. The solvent was evaporated using a rotary evaporator until approximately 5–10 mL remained. The remaining solution was added to 75 mL of cold n-hexane, and a black solid was obtained by vacuum filtration. The yield was almost 100%. HRMS (ESI nano-spray negative mode) M− cacld for C16H8FeN4O4: 375.9895; found 375.9890.
Synthesis of 2,11-dinitro-5,8,13,16-tetrahydrodibenzo[b,h][1,4,7,10]tetraazacyclododecine-6,7,14,15-tetraone (ligand-2)
4-Nitrobenzene-1,2-diamine (0.1 g, 0.653 mmol) and oxalyl chloride (0.06 mL, 0.69 mmol) were dissolved in 100 and 10 mL of dehydrated tetrahydrofuran, respectively. The oxalyl chloride solution was added to the 4-nitrobenzene-1,2-diamine solution at a rate of 1 drop per s. After a 12 h reaction, the crude product was obtained by rotary evaporation, followed by column chromatography purification (dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 20:1). Yield: 44%. 1H NMR (300 MHz, d6-DMSO): δ 11.03 (s, 4 H), 8.46–8.47 (m, 2 H), 8.19–8.24 (m, 2 H), 7.90–7.96 (m, 2 H). HRMS (ESI negative mode) [M − H]− cacld for C16H9N6O8: 413.0487; found 413.0479.
:ethyl acetate = 20:1). Yield: 44%. 1H NMR (300 MHz, d6-DMSO): δ 11.03 (s, 4 H), 8.46–8.47 (m, 2 H), 8.19–8.24 (m, 2 H), 7.90–7.96 (m, 2 H). HRMS (ESI negative mode) [M − H]− cacld for C16H9N6O8: 413.0487; found 413.0479.
Synthesis of Fe-2
Ligand-2 (0.077 g, 0.18 mmol) was dissolved in 100 mL of dehydrated tetrahydrofuran. After stirring and cooling with a liquid nitrogen/acetone slush bath for 30 min, nBuLi (0.7 mL, 1.12 mmol) was added, and then the suspension was dissolved. When the mixture turned dark, the slush bath was removed. After returning to room temperature, FeCl2 (0.03 g, 0.24 mmol) was added under a N2 atmosphere. After being stirred overnight, the mixture was vacuum-filtered, and the solvent was removed using a rotary evaporator until a volume of approximately 5–10 mL remained. The remaining solution was added to 75 mL of cold n-hexane, and a black solid was obtained by vacuum filtration. The yield was almost 100%. MS (ESI negative mode) [M + H]− cacld for C16H7FeN6O8: 466.9675; found 467.
Synthesis of 2,11-dibromo-5,8,13,16-tetrahydrodibenzo[b,h][1,4,7,10]tetraazacyclododecine-6,7,14,15-tetraone (ligand-3)
4-Bromobenzene-1,2-diamine (0.1 g, 0.653 mmol) and oxalyl chloride (0.06 mL, 0.69 mmol) were dissolved in 100 and 10 mL of dehydrated tetrahydrofuran, respectively. The oxalyl chloride solution was added to the 4-bromobenzene-1,2-diamine solution at a rate of 1 drop per s. After a 12 h reaction, the crude product was obtained by vacuum filtration followed by rotary evaporation. After column chromatography purification (dichloromethane:methanol = 20:1), a white powder was obtained. Yield: 13%. 1H NMR (300 MHz, d6-DMSO): δ 10.72 (s, 4 H), 7.78–7.70 (m, 2 H), 7.53–7.53 (m, 4 H). HRMS (ESI positive mode) [2M + Na]+ cacld for C32H20N8O8NaBr4: 982.80298; found 982.8020.
Synthesis of Fe-3
Ligand-3 (0.103 g, 0.213 mmol) was dissolved in 100 mL of dehydrated tetrahydrofuran. After stirring and cooling in a liquid nitrogen/acetone slush bath for 30 min, nBuLi (0.7 mL, 1.12 mmol) was added, and the suspension was dissolved. When the mixture turned dark, the slush bath was removed. After returning to room temperature, FeCl2 (0.032 g, 0.252 mmol) was added under a N2 atmosphere. After being stirred overnight, the mixture was vacuum-filtered, and the solvent was removed using a rotary evaporator until a volume of approximately 5–10 mL remained. The remaining solution was added to 75 mL of cold n-hexane, and a black solid was obtained by vacuum filtration. The yield was almost 100%. HRMS (ESI negative mode) M− cacld for C16H6N4O4FeBr2: 531.8111; found 531.8109.
Cation exchange of Fe-TAMLs
The ion-exchange polymer was activated with 1 M hydrochloric acid. The remaining hydrochloric acid was removed by a DI-water rinse, followed by a tetramethylammonium aqueous solution rinse until a pH of 7 was reached. The Fe-TAML complex was dissolved in 10 mL of methanol and a pre-treated ion-exchange resin was added. After 30 min of stirring, the resin was removed by vacuum filtration, and the methanol in the filtrate was removed by rotary evaporation to obtain the product.
XAS measurements
Fe L3-edge XAS were obtained in total-electron yield mode at the BL20A beamline. Measurements were performed at the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). The powder samples were subjected to ultrahigh (∼1 × 10−9 torr) vacuum treatment. The absorption energies were calibrated with reference to the maximum peak of Fe2O3 at 709.6 eV. Fe K-edge (7112 eV) XAS spectra were obtained in total-fluorescence mode, using a Lytle detector at the BL01C1 beamline with a double-crystal Si(111) monochromator under an ambient atmosphere. Measurements were performed at the NSRRC. The operando measurements were obtained using a homemade PTFE cell with a Kapton-tape-sealed shutter. Before each measurement, the energy was calibrated using a standard Fe foil. XAS data normalization, Fourier transform, phase correction, and L3-edge peak fitting were processed by the Athena software.86 Pre-edge fitting in K-edge XAS was processed by the Larch software.87 Wavelet transforms were processed using the MATLAB code from Muñoz and co-workers.88
XES measurements
Fe Kβ XES data were collected at the 12XU beamline at Spring-8, Japan. The incident energy was 7142 eV, and the emission energy for collection ranged from 7025 to 7080 eV with an energy step of 0.25 eV. All XES data were normalized to unity based on the area of each XES spectrum.
Electrochemical setup
All the electrochemical measurements were carried out on a BioLogic VSP potentiostat (Seyssinet-Pariset, France) with a standard three-electrode setup. An O2-saturated 0.1 M KOH aqueous solution was selected as the electrolyte. The catalyst inks were prepared by adding 15 μmol of the sample and 100 μL of a 5% Nafion solution into 900 μL of ethanol. Nafion solution (10 μL, 0.1%) was dropped onto the glassy carbon (GC) disk electrode (OD: 5.61 mm) of a rotating ring-disk electrode (RRDE, E7R9, Pine Research Instrumentation, Durham, NC, USA), after 30-min vacuum drying, followed by 10 μL catalyst ink to achieve a catalyst loading of 6.07 × 10−4 mmol cm−2. A graphite rod and an Ag/AgCl (3 M KCl) electrode were chosen as the counter and reference electrodes, respectively. The RRDE was rotated at 1600 rpm, the scan rate was set at 10 mV s−1, and the Pt ring of the RRDE was kept at 1.20 VRHE. The potential was corrected using eqn (3). The peroxide selectivity and the average electron transfer number (n) were calculated using eqn (4) and (5), respectively.| | | ERHE = Emeasure + 0.210 + 0.05916 × pH − Idisk × Ru | (3) |
| | 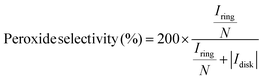 | (4) |
| | 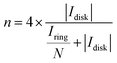 | (5) |
here, Idisk and Iring are the measured currents of the disk and ring electrodes of the RRDE, respectively; Ru is the uncompensated resistance between the working and counter electrodes, measured by potential electrochemical impedance spectroscopy (PEIS). N is 0.37 for the collection efficiency of the Pt ring.
Computational details
All calculations including geometry optimization, vibrational frequency, and single-point energy were based on broken-symmetry DFT performed at the B3LYP/6-311G level with GD3 dispersion force correction using the Gaussian 16 software.89 The solvation model was taken into account and performed with the SMD method in water.90 The results were visualized and analyzed using the GaussView 6. Stationary points of geometry optimizations have been characterized finding that all harmonic vibrational frequencies of local minima are positive. For each species, we calculated structures corresponding to all possible spin multiplicities and selected the structure with the lowest free energy. Charge decomposition analysis (CDA) was performed using Multiwfn software.91,92
The Bader charge analysis was performed using Henkelman's code.93 The absorption energy (ΔGabs) between the catalyst (Fe-TAML) and the OOH adduct was calculated following eqn (6):
| | | ΔGabs(OOH) = G(Fe–OOH) − G(Fe–TAML) − G(OOH) | (6) |
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data supporting this article have been included as part of the SI.
1H NMR, ESI-MS spectra, Fe K-edge XAS pre-edge fittings, cyclic voltammetry, 2-hour electrochemical stability test, operando Fe Kβ XES, Fe L3-edge XAS and fittings, overall Fe Kβ XES and fittings (including Fe standards), operando Fe K-edge XANES, EXAFS, DFT-optimized structures of Fe-TAMLs, Marcus curves, detailed thermodynamic information from DFT calculations, Fe L3-edge XAS fitting parameters, and atomic coordinates of Fe-TAMLs and Fe–OOH intermediates. See DOI: https://doi.org/10.1039/d5mh00615e
Acknowledgements
This work was supported by the National Science and Technology Council, Taiwan (contracts no. NSTC 113-2123-M-002-005, 113-2639-M-002-009-ASP, 112-2926-I-002-513-G, and 113-2113-M-032-002). The authors thank the computational resources provided by the National Center for High-Performance Computing of Taiwan. We gratefully acknowledge support from MCUT Formosa Center, and National Taiwan University (NTU-111L880513). We thank the interdisciplinary project of NSRRC for providing assistance with the synchrotron-based X-ray experiments.
References
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6(6), 1593–1610 RSC.
- A. Kumar, S. Ibraheem, T. Anh Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef.
- J. Li, J. Ma, Z. Ma, E. Zhao, K. Du, J. Guo and T. Ling, Adv. Energy Sustainable Res., 2021, 2(8), 2100034 CrossRef.
- Z. Li, R. Ma, Q. Ju, Q. Liu, L. Liu, Y. Zhu, M. Yang and J. Wang, Innovation, 2022, 3(4), 100268 Search PubMed.
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H. J. Gao and H. Yang,
et al.
, Nat. Commun., 2021, 12(1), 2608 CrossRef.
- Z. Chen, H. Niu, J. Ding, H. Liu, P. H. Chen, Y. H. Lu, Y. R. Lu, W. Zuo, L. Han and Y. Guo,
et al.
, Angew. Chem., Int. Ed., 2021, 60(48), 25404–25410 CrossRef.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B. A. Lu, X. Xue, H. Yin, W. Cheng and J. Cheng,
et al.
, Nat. Commun., 2021, 12(1), 1734 CrossRef PubMed.
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 61(21), e202117617 Search PubMed.
- X. T. Wang, T. Ouyang, L. Wang, J. H. Zhong, T. Ma and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58(38), 13291–13296 Search PubMed.
- S. M. Fukuzumi and T. Tanaka, Chem. Lett., 1989,(1), 27–30 Search PubMed.
- S. M. Fukuzumi and T. Tanaka, Inorg. Chem., 1989, 28(12), 2459–2465 CrossRef.
- C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134(12), 5444–5447 CrossRef.
- B. D. Matson, C. T. Carver, A. Von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48(90), 11100–11102 RSC.
- X. Chen, Y. Dai, H. Zhang and X. Zhao, Colloids Surf., A, 2023, 663, 131091 CrossRef.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55(6), 878–892 CrossRef PubMed.
- Z. Tian, Y. Wang, Y. Li, G. Yao, Q. Zhang and L. Chen, iScience, 2022, 25(7), 104557 Search PubMed.
- S. Amanullah, P. Saha and A. Dey, Faraday Discuss., 2022, 234(0), 143–158 RSC.
- X. Y. Zhou, C. Xu, P. P. Guo, W. L. Sun, P. J. Wei and J. G. Liu, Chem. – Eur. J., 2021, 27(38), 9898–9904 CrossRef PubMed.
- A. C. Brezny, S. I. Johnson, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2020, 142(9), 4108–4113 CrossRef.
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2019, 141(20), 8315–8326 Search PubMed.
- C. C. Leyla Gidi, M. J. Aguirre, F. Armijo and G. Ramírez, Int. J. Electrochem. Sci., 2018, 13(2), 1666–1682 Search PubMed.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137(13), 4296–4299 Search PubMed.
- Z. Ou, A. Lu, D. Meng, S. Huang, Y. Fang, G. Lu and K. M. Kadish, Inorg. Chem., 2012, 51(16), 8890–8896 CrossRef PubMed.
- N. Levy, A. Mahammed, A. Friedman, B. Gavriel, Z. Gross and L. Elbaz, ChemCatChem, 2016, 8(17), 2832–2837 Search PubMed.
- W. Fan, Z. Duan, W. Liu, R. Mehmood, J. Qu, Y. Cao, X. Guo, J. Zhong and F. Zhang, Nat. Commun., 2023, 14(1), 1426 Search PubMed.
- S. Yuan, J. Peng, Y. Zhang, D. J. Zheng, S. Bagi, T. Wang, Y. Román-Leshkov and Y. Shao-Horn, ACS Catal., 2022, 12(12), 7278–7287 CrossRef.
- Y. Wang, T. Zhou, S. Ruan, H. Feng, W. Bi, J. Hu, T. Chen, H. Liu, B. Yuan and N. Zhang,
et al.
, Nano Lett., 2022, 22(16), 6622–6630 CrossRef PubMed.
- M. Wang, Y. Wu, X. Li, Y. Wang, X. Wu, G. Li, L. Yang and Y. Lin, Nanoscale, 2022, 14(23), 8255–8259 RSC.
- Y.-Y. Li, Y.-C. Wang, Z.-Y. Zhou and S.-G. Sun, Electrochem. Commun., 2022, 141, 107357 CrossRef.
- H. Yabu, K. Nakamura, Y. Matsuo, Y. Umejima, H. Matsuyama, J. Nakamura and K. Ito, ACS Appl. Energy Mater., 2021, 4(12), 14380–14389 CrossRef.
- C. F. Olguin, N. Agurto, C. P. Silva, C. P. Candia, M. Santander-Nelli, J. Oyarzo, A. Gomez, J. F. Silva and J. Pavez, Molecules, 2021, 26(6), 1631 CrossRef.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li and H. Liu,
et al.
, Nat. Commun., 2020, 11(1), 4173 Search PubMed.
- E. T. Acar, T. A. Tabakoglu, D. Atilla, F. Yuksel and G. Atun, Polyhedron, 2018, 152, 114–124 CrossRef.
- A. Alsudairi, J. Li, N. Ramaswamy, S. Mukerjee, K. M. Abraham and Q. Jia, J. Phys. Chem. Lett., 2017, 8(13), 2881–2886 CrossRef.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105(6), 2253–2278 Search PubMed.
- M. E. Albertolle and F. Peter Guengerich, J. Inorg. Biochem., 2018, 186, 228–234 CrossRef.
- M. Sankaralingam, Y. M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2017, 56(9), 5096–5104 CrossRef PubMed.
- B. Chandra, P. De and S. Sen Gupta, Chem. Commun., 2020, 56(60), 8484–8487 RSC.
- M. Salavati-Niasari, J. Mol. Catal. A: Chem., 2007, 278(1–2), 22–28 CrossRef.
- E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136(15), 5603–5606 CrossRef.
- C. Panda, J. Debgupta, D. Diaz Diaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136(35), 12273–12282 CrossRef CAS PubMed.
- S. Pattanayak, D. R. Chowdhury, B. Garai, K. K. Singh, A. Paul, B. B. Dhar and S. S. Gupta, Chem. – Eur. J., 2017, 23(14), 3414–3424 Search PubMed.
- J. Xiong, Q. Liu, B. Lavina, M. Y. Hu, J. Zhao, E. E. Alp, L. Deng, S. Ye and Y. Guo, Chem. Sci., 2023, 14(11), 2808–2820 RSC.
-
P. Vanysek, Electrochemical Series. In CRC Handbook of Chemistry and Physics: A ready-reference book of Chemical and physical data, ed. D. R. Lide, CRC Press, 2003 Search PubMed.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2009, 1(6), 466–472 CrossRef CAS PubMed.
- J. Suntivich, E. E. Perry, H. A. Gasteiger and Y. Shao-Horn, Electrocatalysis, 2013, 4(1), 49–55 CrossRef CAS.
- T. J. Collins, R. D. Powell, C. Slebodnick and E. S. Uffelman, J. Am. Chem. Soc., 1991, 113(22), 8419–8425 CrossRef CAS.
- L. L. Diaddario, W. R. Robinson and D. W. Margerum, Inorg. Chem., 1983, 22(7), 1021–1025 Search PubMed.
- W. T. Chen, C. W. Hsu, J. F. Lee, C. W. Pao and I. J. Hsu, ACS Omega, 2020, 5(10), 4991–5000 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119(27), 6297–6314 CrossRef CAS.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55(13), 4327–4331 CrossRef CAS.
- S. S. Xue, X. X. Li, Y. M. Lee, M. S. Seo, Y. Kim, S. Yanagisawa, M. Kubo, Y. K. Jeon, W. S. Kim and R. Sarangi,
et al.
, J. Am. Chem. Soc., 2020, 142(36), 15305–15319 CrossRef PubMed.
- B.-K. Teo, J. Am. Chem. Soc., 1981, 103(14), 3990–4001 CrossRef.
- J. Wang, C. S. Hsu, T. S. Wu, T. S. Chan, N. T. Suen, J. F. Lee and H. M. Chen, Nat. Commun., 2023, 14(1), 6576 CrossRef.
- C.-W. Tung, W. Zhang, T. Y. Lai, J. Wang, Y.-C. Chu, G.-B. Wang, C.-S. Hsu, Y.-F. Liao, N. Hiraoka, H. Ishii, X. C. Zeng and H. M. Chen, Nat. Sustainability, 2025, 8, 793–805 CrossRef.
- G. Vankó, T. Neisius, G. Molnár, F. Renz, S. Kárpáti, A. Shukla and F. M. de Groot, J. Phys. Chem. B, 2006, 110(24), 11647–11653 CrossRef PubMed.
- R. K. Hocking, S. D. George, K. N. Raymond, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2010, 132(11), 4006–4015 CrossRef PubMed.
- M. Agote-Arán, I. Lezcano-González, A. G. Greenaway, S. Hayama, S. Díaz-Moreno, A. B. Kroner and A. M. Beale, Appl. Catal., A, 2019, 570, 283–291 CrossRef.
- R. K. Hocking, E. Wasinger, Y.-L. Yan, F. M. F. deGroot, F. Ann Walker, K. O. Hodgson, B. Hedman and E. I. Solomon., J. Am. Chem. Soc., 2007, 129(1), 113–125 CrossRef PubMed.
- L. Bondì, A. L. Garden, F. Totti, P. Jerabek and S. Brooker, Chem. – Eur. J., 2022, 28(22), e202104314 CrossRef PubMed.
-
R. K. Hocking and E. I. Solomon, Ligand Field and Molecular Orbital Theories of Transition Metal X-ray Absorption Edge Transitions. In Molecular Electronic Structures of Transition Metal Complexes I, Structure Bonding, 2011, pp. 155–184 Search PubMed.
- J. Zhou, P. N. Duchesne, Y. Hu, J. Wang, P. Zhang, Y. Li, T. Regier and H. Dai, Phys. Chem. Chem. Phys., 2014, 16(30), 15787–15791 RSC.
- Y.-C. H. Lin, I.-J. Hsu, H.-H. Hsieh, J.-F. Lee, J.-M. Chen, J.-M. Lee and Y. Wang, J. Chin. Chem. Soc., 2006, 53(6), 1571–1576 CrossRef CAS.
- V. A. Saveleva, K. Ebner, L. Ni, G. Smolentsev, D. Klose, A. Zitolo, E. Marelli, J. Li, M. Medarde and O. V. Safonova,
et al.
, Angew. Chem., Int. Ed., 2021, 60(21), 11707–11712 CrossRef CAS PubMed.
- H.-Y. Tan, S.-C. Lin, J. Wang, J.-H. Chen, C.-J. Chang, C.-H. Hou, J.-J. Shyue, T.-R. Kuo and H. M. Chen, J. Am. Chem. Soc., 2023, 145(49), 27054–27066 CrossRef CAS PubMed.
- Y.-C. Chu, K.-H. Chen, C.-W. Tung, H.-C. Chen, J. Wang, T.-R. Kuo, C.-S. Hsu, K.-H. Lin, L. D. Tsai and H. M. Chen, Adv. Mater., 2024, 36(26), 2400640 CrossRef CAS PubMed.
- C.-S. Hsu, J. Wang, Y.-C. Chu, J.-H. Chen, C.-Y. Chien, K.-H. Lin, L. D. Tsai, H.-C. Chen, Y.-F. Liao, N. Hiraoka and H. M. Chen,
et al.
, Nat. Commun., 2023, 14(1), 5245 CrossRef CAS PubMed.
- J. Wang, H.-Y. Tan, M.-Y. Qi, J.-Y. Li, Z.-R. Tang, N.-T. Suen, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2023, 52(15), 5013–5050 RSC.
- C.-J. Chang, Y.-A. Lai, Y.-C. Chu, C.-K. Peng, H.-Y. Tan, C.-W. Pao, Y.-G. Lin, S.-F. Hung, H.-C. Chen and H. M. Chen, J. Am. Chem. Soc., 2023, 145(12), 6953–6965 CrossRef CAS.
- Y. Zhu, G. Chen, Y.-C. Chu, C.-S. Hsu, J. Wang, C.-W. Tung and H. M. Chen, Angew. Chem., Int. Ed., 2022, 61(48), e202211142 CrossRef CAS.
- C.-W. Tung, T.-R. Kuo, Y.-P. Huang, Y.-C. Chu, C.-H. Hou, Y. Li, N.-T. Suen, J. Han and H. M. Chen, Adv. Energy Mater., 2022, 12(28), 2200079 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364(6445), 1091–1094 CrossRef CAS PubMed.
- J.-W. Wang, P. Sahoo and T.-B. Lu, ACS Catal., 2016, 6(8), 5062–5068 CrossRef CAS.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5(4), 1281–1291 CrossRef CAS.
- Y. Zhu, H.-C. Chen, C.-S. Hsu, T.-S. Lin, C.-J. Chang, S.-C. Chang, L.-D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4(4), 987–994 CrossRef CAS.
- X. Han, T. Zhang, W. Chen, B. Dong, G. Meng, L. Zheng, C. Yang, X. Sun, Z. Zhuang and D. Wang,
et al.
, Adv. Energy Mater., 2020, 11(6), 2002753 CrossRef.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W. C. Cheong, R. Shen, X. Wen, L. Zheng and A. I. Rykov,
et al.
, Nat. Commun., 2018, 9(1), 5422 CrossRef CAS.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S. M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(40), e2110036118 CrossRef CAS.
- K. Y. Vinogradov, A. V. Bulanova, R. V. Shafigulin, E. O. Tokranova, A. M. Mebel and H. Zhu, ACS Omega, 2022, 7(8), 7066–7073 CrossRef CAS PubMed.
- J. Zhang, F. Li, W. Liu, Q. Wang, X. Li, S. F. Hung, H. Yang and B. Liu, Angew. Chem., Int. Ed., 2024, 63(51), e202412245 CrossRef CAS.
- C.-Y. Huang, H.-M. Lin, C.-H. Chiang, H.-A. Chen, T.-R. Liu, S. K. Vishnu, J.-W. Chiou, R. Sankar, H.-M. Tsai, W.-F. Pong and C.-W. Chen,
et al.
, Adv. Funct. Mater., 2023, 33(43), 2305792 CrossRef CAS.
- T. Wu, X. Ren, Y. Sun, S. Sun, G. Xian, G. G. Scherer, A. C. Fisher, D. Mandler, J. W. Ager and A. Grimaud,
et al.
, Nat. Commun., 2021, 12(1), 3634 CrossRef CAS.
- X. Zhou, Y. Min, C. Zhao, C. Chen, M. K. Ke, S. L. Xu, J. J. Chen, Y. Wu and H. Q. Yu, Nat. Commun., 2024, 15(1), 193 CrossRef.
- J. M. Mayer, J. Phys. Chem. Lett., 2011, 2(12), 1481–1489 CrossRef CAS.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. T. Kim, K. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef PubMed.
- B. Ravel and M. Newville, Phys. Scr., 2005, 2005, 1007–1010 CrossRef.
- M. Newville, J. Phys. Conf. Ser., 2013, 430, 012007 CrossRef.
- M. Muñoz, P. Argoul and F. Farges, Am. Mineral., 2003, 88(4), 694–700 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Gaussian Inc., 2016 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105(8), 2999–3094 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef.
- T. Lu, J. Chem. Phys., 2024, 161(8), 082503 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *d,
Chih-Hsin
Chen
*d,
Chih-Hsin
Chen







