
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChondrocyte-mimetic therapeutic microcarriers for synergistic chemo-mechanical signaling in cartilage regeneration†
Na-Hyun
Lee‡
 ab,
Zechu
Zhou‡
a,
Ji-Young
Yoon‡
ad,
Nandin
Mandakhbayar
a,
Cheng Ji
Li
abc,
Jung-Hwan
Lee
abcd,
Hae-Won
Kim
*abcd and
Hye Sung
Kim
*abc
ab,
Zechu
Zhou‡
a,
Ji-Young
Yoon‡
ad,
Nandin
Mandakhbayar
a,
Cheng Ji
Li
abc,
Jung-Hwan
Lee
abcd,
Hae-Won
Kim
*abcd and
Hye Sung
Kim
*abc
aInstitute of Tissue Regeneration Engineering (ITREN), Dankook University, Cheonan, 31116, Republic of Korea. E-mail: kimhw@dku.edu; hskim1213@dankook.ac.kr
bMechanobiology Dental Medicine Research Center, Dankook University, Cheonan, 31116, Republic of Korea
cNanobiomedical Science and BK21 NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan, 31116, Republic of Korea
dCell & Matter Institute, Dankook University, Cheonan, 31116, Republic of Korea
First published on 16th June 2025
Abstract
Traumatic and degenerative cartilage injuries pose a significant clinical challenge. Among emerging treatments, cell-free, tissue-mimicking approaches have shown promise for cartilage regeneration. Here we design chondrocyte-mimicking therapeutic microcarriers that stimulate host chondrocyte migration, proliferation, and maturation by recapitulating the native chondrogenic niche. Our microcarriers, featuring an interconnected microporous network, are functionalized with chondrocyte-derived cell membrane and chondrocyte-secreted exosome. The microcarrier framework provides structural support at the defect site while allowing cell anchorage and infiltration. The chondrocyte-derived cell membrane, replicating the native chondrocytes, retains cartilage-specific extracellular matrix (ECM), and serves as a reservoir for exosome delivery, facilitating anabolic responses through integrin-mediated mechanotransduction, particularly via the Rho/ROCK-actin-YAP axis. Furthermore, the chondrocyte-derived exosomes restore cartilage homeostasis by delivering chondrogenic miRNAs and regulating key metabolic processes, including lipid metabolism and ECM remodeling. Together, these therapeutic components play a synergistic role in driving chemo-mechanical signaling, promoting chondrocyte recruitment, proliferation, and maturation, ultimately coordinating cartilage repair. This biomimetic approach offers a promising strategy for cell-free cartilage regenerative therapies.
New conceptsWe introduce a transformative cell-free, tissue-mimicking platform that enables synergistic chemo-mechanical regulation for cartilage repair. Our concept leverages natural biomaterials – cell-secreted exosomes and cell membranes – to replicate the dynamic physicochemical environment of native cells. Unlike previous work that focused primarily on biochemical cues, our strategy integrates both biochemical signals such as cartilage-specific exosomal miRNA and biomechanical signals. This dual approach employs highly porous microcarriers that create a lacunae-like architecture to accelerate tissue integration, while a cell membrane coating acts as a drug reservoir and activates matrix-driven mechanotransduction signaling. For the first time, we reveal that this cell membrane-coated biointerface modulates integrin-mediated mechanotransduction by specifically activating the Rho/ROCK-actin-YAP pathway, an essential mechanism that significantly promotes host chondrocyte proliferation and maturation. This breakthrough deepens our understanding of cell membrane-associated designs and holds significant promise for the development of next-generation biomaterials. By synchronizing these multifaceted signals, our approach minimizes risks inherent to cell-based therapies and offers a scalable, broadly applicable framework for complex tissue regeneration. This work establishes a fresh conceptual paradigm in synergistic signaling for tissue repair, paving the way for innovative, multifunctional biomaterials. |
1. Introduction
Articular cartilage is a specialized connective tissue that facilitates smooth joint movement by distributing loads and minimizing friction between bones.1–3 Despite its vital functions, repairing damaged or degenerating articular cartilage remains a significant clinical challenge, primarily due to its avascular and aneural nature, severely impairing its regenerative capacity.4,5 With limited self-repair potential, articular cartilage injuries are common, affecting over 60% of patients undergoing arthroscopy.6,7 Even minor defects can progress to severe joint degeneration, often leading to osteoarthritis.8 Current clinical treatments, such as autologous chondrocyte transplantation, microfracture, and mosaicplasty, frequently fail to fully restore the structure and function of healthy cartilage. These methods face limitations, including tissue scarcity, donor site morbidity, mechanical weakness, and immune-related complications.9,10 Therefore, innovative clinical strategies are urgently needed to achieve effective cartilage repair.Recent advances in cartilage repair have increasingly highlighted the therapeutic potential of exosomes – nano-sized extracellular vesicles (40–160 nm) that deliver bioactive cargos (proteins, metabolites, and nucleic acids) with lower immunogenic and tumorigenic risks compared to cell therapies.11–15 Despite this promise, clinical application of exosomes faces significant challenges, particularly in maintaining their stability and retention in the body. Exosomes are often rapidly cleared by the immune system or accumulate in organs such as the liver, spleen, and lungs.16,17 Moreover, successful cartilage repair typically requires multiple doses of exosomes in large quantities, which increases treatment costs.18,19 To overcome these limitations, biomaterials such as hydrogels, microcarriers, and 3D-printed scaffolds are being explored to improve exosome retention and ensure controlled, localized release.20 Previous studies have demonstrated that scaffolds embedded or coated with exosomes can effectively promote cartilage regeneration.21,22 These scaffolds serve a dual role by acting as carriers for exosomes and providing a supportive matrix for cellular interactions, offering both biochemical and physical cues for cartilage repair. Such scaffold-mediated exosome delivery systems, therefore, hold significant promise for clinical translation.
In tissue engineering, various biomimetic scaffolds have been developed to enhance cartilage regeneration by replicating its physical and chemical characteristics. For rigid tissues, such as cartilage, the mechanical strength of the scaffolds is particularly crucial, as cartilage constantly withstands compressive stress during movement. Additionally, a porous structure is essential to facilitate tissue repair at the cellular level.23 Consequently, mechanically stable porous scaffolds, including 3D-printed scaffolds, microcarriers, and composite hydrogels, have proven more effective for cartilage repair than amorphous, soft hydrogel scaffolds.20,21,24,25 Among these materials, porous microcarriers have gained significant attention due to their multifaceted merits, enabled by open, interconnected pores.26,27 These microcarriers can deliver a wide range of bioactive molecules, including small molecules, growth factors, and cells.28,29 Additionally, the porous structure creates a favorable microenvironment for cell proliferation and infiltration while facilitating the exchange of oxygen and nutrients.30 Furthermore, microcarriers offer tunable physical properties, such as adjustable particle and pore sizes,31 allowing customization to meet specific tissue repair needs. For instance, the porous structure of microcarriers can mimic the lacunae found in cartilage – small spaces that house clusters of chondrocytes within the matrix.32 These lacunae provide a mechanical microenvironment that regulates chondrocyte function through biomechanical signaling. By replicating this structure, porous microcarriers support long-term chondrocyte viability and activity.33–35
While much research has focused on replicating the physical properties of cartilage, efforts to mimic its chemical features have primarily involved using cartilage extracellular matrix (ECM) components, such as hyaluronic acid, for scaffold preparation.36–38 However, the in vivo chemical composition of cartilage is likely far more intricate. We have previously demonstrated that scaffolds decorated with cell membranes can effectively replicate the physicochemical properties of the original cell sources, thereby facilitating specific cell–cell communications.39,40 For example, scaffolds decorated with mesenchymal stem cell membranes significantly enhanced the physical contact and adhesion of leukemia stem cells compared to non-decorated scaffolds.39 Similarly, T cells exhibited increased tethering on scaffolds coated with dendritic cell membranes.40 This cell membrane cloaking technique holds broad potential for biomimetic strategies in developing tissue-engineered scaffolds for cartilage repair.
In this study, we develop chondrocyte-mimicking scaffolds that replicate both the physical and chemical properties of cartilage microenvironments to enhance cartilage repair. The scaffold consists of porous microcarriers coated with chondrocyte membrane and loaded with chondrocyte-secreted exosomes. This design aims to promote host chondrocyte recruitment, proliferation, and maturation by replicating the physicochemical properties of the chondrocyte niche. We assess the effects of local exosome delivery through this scaffold on cartilage repair using a rat osteochondral defect model. Furthermore, we investigate the molecular mechanisms underlying the cellular processes mediated by chondrocyte-derived exosome and cell membrane, examining how the scaffold contributes to the restoration of matrix homeostasis during cartilage repair and regeneration.
2. Experimental section
2.1 Fabrication of microcarriers
Porous microcarriers were fabricated using a co-axial nozzle-based co-flow microfluidic device. The device consisted of an inner nozzle for the dispersed phase and an outer nozzle for the continuous phase (Coaxial Nozzle, NanoNC, South Korea). For the dispersed phase, polycaprolactone (PCL, MW 80 kDa, Sigma-Aldrich, USA) and camphene (Sigma-Aldrich, USA) were dissolved in chloroform (Daejung, South Korea) at concentrations of 2–5% (w/w) and 10–25% (w/w), respectively. The continuous phase was prepared by dissolving polyvinyl alcohol (PVA, MW 85–124 kDa, Sigma-Aldrich, USA) in distilled water (DW) at a concentration of 2% (w/v). The dispersed phase was injected through the inner nozzle using a flow rate of 1.0–2.5 ml h−1 using a syringe pump, while the continuous phase was simultaneously pumped through the outer nozzle at 10 –40 ml h−1. Micro-droplets formed at the nozzle junction due to shear forces between the two phases. Emulsified micro-droplets were collected in a water bath and solidified into microcarriers at room temperature for 4 h with orbital shaking at 60 rpm, allowing chloroform evaporation. The solidified microcarriers were purified using pluriStrainers® 300 μm (pluriSelect Life Science, Germany), washed with DW, and subsequently subjected to lyophilization for 12 h to sublimate the camphene, resulting in pore formation.Dense microcarriers were prepared using a dripping mode-based drop formation method, as previously reported.41 A 2% (w/w) PCL solution in chloroform was pumped at 1 ml h−1 through a vertically placed 27G nozzle into a 2% (w/v) PVA aqueous solution bath. Micro-droplets formed at the nozzle tip and detached when droplet weight overcame surface tension and viscous drag forces. These microdroplets were solidified and collected following the same procedure used for porous microcarriers.
2.2 Characterization of microcarriers
To measure the surface elastic modulus of the porous microcarriers before and after cell membrane coating, the microcarriers were placed in a well of a 6-well plate, submerged in phosphate-buffered saline (PBS), and analyzed using a nanoindenter (Pavone, Optics11 Life, Netherlands). A 27 μm diameter probe with a stiffness of 3.92 N m−1 was pressed 0.1 μm into the material to determine the surface elastic modulus.
2.3 Isolation and culture of rat mesenchymal stem cells and chondrocytes
Five-week-old male Sprague-Dawley rats were purchased from DBL Co. (South Korea) and housed in standard rat cages with unrestricted access to food and water. All experimental protocols and procedures involving the rats in this study were approved by the Institutional Animal Care and Use Committee of Dankook University (no. DKU-22-038).Mesenchymal stem cells (MSCs) were isolated from rat bone marrow, as previously reported.42 Briefly, the rats were euthanized via CO2 inhalation followed by cervical dislocation. Hind limbs were dissected, and the skin was removed to isolate femurs and tibias. Bone marrow was extracted by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 min. The extracted marrow was then resuspended in alpha minimum essential medium eagle, alpha modification (α-MEM, LM008-01, Welgene, South Korea) supplemented with 10% fetal bovine serum (FBS, 35-015-CV, Corning, USA) and 1% penicillin–streptomycin (P/S, 15140-163, Thermo Fisher Scientific, USA) and passed through a Falcon® 70 μm cell strainer (352350, Corning, USA) to remove bone debris and blood aggregates. After centrifugation at 783g for 5 min, the cell pellet was resuspended in growth medium, seeded in 100 mm culture dishes, and incubated at 37 °C in a humidified atmosphere of 5% CO2. The medium was changed every 2–3 days.
000g for 1 min. The extracted marrow was then resuspended in alpha minimum essential medium eagle, alpha modification (α-MEM, LM008-01, Welgene, South Korea) supplemented with 10% fetal bovine serum (FBS, 35-015-CV, Corning, USA) and 1% penicillin–streptomycin (P/S, 15140-163, Thermo Fisher Scientific, USA) and passed through a Falcon® 70 μm cell strainer (352350, Corning, USA) to remove bone debris and blood aggregates. After centrifugation at 783g for 5 min, the cell pellet was resuspended in growth medium, seeded in 100 mm culture dishes, and incubated at 37 °C in a humidified atmosphere of 5% CO2. The medium was changed every 2–3 days.
Rat chondrocytes were isolated using a previously described method.42 After euthanizing the rats via CO2 inhalation, an abdominal incision was made to extract costal cartilage from the anterior ribs. To minimize fibroblast contamination, the surrounding soft tissues were meticulously removed. The harvested cartilage was then minced into 1–2 mm3 pieces with micro scissors and incubated in 0.25% Trypsin/EDTA (T/E, 25200-056, Thermo Fisher Scientific, USA) at 37 °C for 1 h. Following incubation, the T/E solution was removed by centrifugation at 2000 rpm for 3 min. The cartilage pieces were then subjected to overnight digestion at 37 °C with a Collagenase II solution (2 mg ml−1, LS004176, Worthington Biochemical Corporation, USA). The next day, the tissue suspension was filtered through a 70 μm cell strainer to remove undigested tissue fragments. Filtered cells were collected by centrifugation at 1200 rpm for 5 min, and the supernatant was discarded. The cell pellet was resuspended in a growth medium consisting of High Glucose Dulbecco's Modified Eagle's medium (DMEM, LM001-26, Welgene, South Korea), 10% FBS, and 1% P/S. The resuspended cells were seeded in 100 mm culture dishes and incubated at 37 °C in a humidified atmosphere of 5% CO2. Subculturing was performed once the cells reached 70–80% confluence. Cells at passage 3 were used for all subsequent experiments to ensure consistency and reproducibility.
2.4 Preparation and characterization of chondrocyte-derived cell membranes
Cell membranes were isolated from rat chondrocytes following a previously reported protocol with minor modifications.40 Briefly, rat chondrocytes were lysed in a hypotonic lysis buffer (20 mM Tris–HCl (pH 7.5), 10 mM KCl, 2 mM MgCl2, and 0.2 mM EDTA) supplemented with a protease and phosphatase inhibitor cocktail (Halt™ Protease and Phosphatase Inhibitor Cocktail (100X), 78440, Thermo Fisher Scientific, USA). The cell lysate was then mechanically fragmented using an ultrasonic homogenizer (Scientz-IID, SCIENTZ, China) and then centrifuged at 3200g for 5 min. The supernatant was collected and centrifuged at 10 000g for 30 min at 4 °C. The resulting cell membrane pellet was resuspended in PBS containing 0.2 mM EDTA and the protease and phosphatase inhibitor cocktail to prevent protein degradation. The prepared cell membranes were stored at 4 °C and used within one month.To characterize the cell membranes, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis were performed. Cell membranes were lysed using RIPA lysis buffer (EBA-1149, ELPIS biotech, Korea) containing a protease and phosphatase inhibitor cocktail. Protein concentrations were determined using the Pierce™ BCA protein assay kit (23225, Thermo Scientific, USA). Proteins were then denatured by heating with 5× loading buffer (751-001, GeneAll, South Korea) at 100 °C for 10 min and subsequently electrophoresed on a polyacrylamide gel. The gel was washed in distilled water (DW) for 30 min, stained with Coomassie Blue (1610786, Bio-rad, USA) for 1 h at room temperature with gentle shaking, and washed again with DW for 30 min. The stained gel was imaged using an iBright imaging device (iBright™ FL1500, Invitrogen, USA).
For western blot analysis, equal amounts of total protein per lane were separated by electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membrane was blocked with 5% (w/v) bovine serum albumin (BSA) fraction V (SM-BOV-100, GeneAll) in 1× Tris-buffered saline containing 0.1% Tween (TBST, TR2007-105-80, Biosesang, South Korea) for 1 h, followed by incubation with primary antibodies in 5% (w/v) BSA in TBST solution overnight at 4 °C. After washing with TBST buffer, the membrane was incubated with an HRP-conjugated secondary antibody (1:10000, 7074, Cell Signaling, USA) at room temperature for 1 h. Protein signals were visualized using Supersignal west pico plus chemiluminescent substrate (34580, Thermo Fisher Scientific, USA) and captured by the iBright™ 1500 imaging system. Details of the antibodies used are described in Table S1 (ESI†).
2.5 Extraction and characterization of chondrocyte-derived exosomes
Rat chondrocytes were seeded at a density of 1.3 × 104 cells per cm2. After 24 h, the cells were washed with PBS, and the medium was replaced with DMEM supplemented with 2% exosome-depleted FBS (A2720803, Thermo Fisher Scientific, USA). Following 48 h of incubation, the conditioned medium was collected and centrifuged at 2000g for 20 min at 4 °C to remove cell debris. The supernatant was subsequently concentrated using an Amicon® ultra centrifugal filter 100 kDa MWCO (UFC9100, Millipore, USA) by centrifugation at 5000g for 30 min at 4 °C with a fixed-angle rotor. The concentrate was then filtered through a 0.2 μm syringe filter, mixed with ExoQuick-TC™ (EXOTC10A-1, System Bioscience, USA), and incubated overnight at 4 °C. The following day, the exosome fraction was collected by centrifugation at 1500g for 30 min at 4 °C. To preserve vesicle integrity for long-term storage, the exosomes were resuspended in a 25 mM trehalose solution and stored at −80 °C until use.The morphology of the isolated exosomes was observed using FE-SEM (Sigma 300, Carl Zeiss, Germany) and field-emission transmission electron microscopy (FE-TEM, JEM-F200, JEOL, Japan) after fixation in 2.5% glutaraldehyde at room temperature for 1 h. Before imaging, the fixed exosomes were further stained with 1.5% phosphotungstic acid (PTA, 79690, Sigma-Aldrich, USA) for TEM analysis.
The concentration, size, and distribution of the exosomes were evaluated using nanoparticle tracking analysis (NTA) on a Nanosight NS300 instrument (NS300, Malvern Panalytical Ltd, UK).43 The exosomes were diluted in PBS, and the measurements were conducted under the following conditions: detection threshold set to 5, temperature maintained at 25 °C, syringe pump speed set at 40 AU, syringe load at 100, and camera level set at 15. Data from four individual measurements were collected and analyzed using NanoSight NTA software.
2.6 Small RNA sequencing analysis of chondrocyte-derived exosomes
According to the manufacturer's instructions, total RNA was extracted from chondrocyte-derived exosomes using the miRNeasy Micro kit (217084, Qiagen, Germany). The RNA quality was assessed with the Agilent 2100 Bioanalyzer using the RNA 6000 Pico Chip (Agilent Technologies, Netherlands), and RNA quantification was performed using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). Library preparation was carried out for small RNA sequencing using the NEBNext® Multiplex Small RNA Library Prep kit (New England BioLabs, USA). The yield and size distribution of the constructed small RNA libraries were evaluated using the High-sensitivity DNA Assay on the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., USA). Sequencing was performed on the NextSeq500 system (Illumina, San Diego, CA., USA) to generate single-end 75 bp reads. Sequence reads were mapped to the reference genome for data analysis using the Bowtie2 software to generate alignment files (bam files). Mature miRNA sequences served as reference sequences for mapping. Read counts mapped to mature miRNA sequences were extracted from the alignment file using Bedtools (v2.25.0) and Bioconductor packages in the R statistical environment (version 3.2.2). These read counts were used to quantify miRNA expression levels. The top 100 miRNAs, based on average read counts across three individual batches of samples, were selected and visualized in a heatmap using MeV software (version 4.9.0). Gene ontology term enrichment analysis of the predicted target genes was performed using the DAVID Bioinformatics Resource, and KEGG pathway analysis was conducted using DIANA mirPath v.3. To compare the miRNA compositions between chExo and MSC exosomes, we used publicly available data on MSC exosomes from dataset #GSE 113868.2.7 Preparation and characterization of microcarriers coated with chondrocyte-derived cell membranes
To coat porous microcarriers with chondrocyte-derived cell membranes (chM), the microcarriers were mixed with chM at a weight ratio 1:0.1 and incubated with vigorous vortexing overnight at 4 °C. Following the coating process, the microcarriers were washed with PBS to remove unbound cell membranes.
The cell membrane layer on the chM-coated microcarriers was visualized by FE-TEM. Samples were fixed in 2.5% glutaraldehyde at room temperature for 1 h, embedded in O.C.T compound, cryosectioned to a thickness of 10 μm, and stained with 1.5% PTA before imaging. The average thickness of the cell membrane layers on the microcarriers was analyzed based on FE-TEM images using Image J software.
For additional visualization, the cell membranes were fluorescently labeled before extraction by incubating cells with Vybrant™ Dil Cell-Labeling solution (1 μl for 106 cells per ml, V22885, Invitrogen, USA). After coating the microcarriers with the fluorescently labeled cell membranes, the coating was observed by confocal laser scanning microscopy (CLSM, Eclipse Ti2, Nikon, Japan). Cross-sectional CLSM images were captured after cryosectioning the samples into 10 μm-thick sections.
Water contact angle measurements were conducted to assess the surface hydrophilicity of the coating. PCL films were prepared by casting 1 ml of a 3% (w/w) PCL solution in chloroform containing 20% (w/w) camphene onto a Teflon plate. The films were air-dried overnight and subsequently lyophilized. The cell membrane coating was then applied as described above. Water contact angles were measured using a contact angle goniometer (PHX300, SEO, South Korea).
2.8 chExo loading and release study
To load exosomes onto the microcarriers, 0.4 mg of chondrocyte cell membrane-coated microcarriers were mixed with 40 μg of chondrocyte-derived exosomes (chExo) in 5 μl of PBS. The mixture was incubated with gentle shaking for 1 h at 37 °C. For visualization and quantification of exosome loading, the exosomes were fluorescently labeled using VybrantTM DiO Cell-Labeling Solution (V22886, Invitrogen, USA). Briefly, 40 μg of chExo was incubated with 1 μl of DiO dye for 30 min at 37 °C. Excessive DiO dye was removed with Exosome Spin Columns MW 3000 (4484449, Invitrogen, USA). The DiO-labeled chExo was then mixed with chM-coated microcarriers at a weight ratio 1:10 and incubated for 1 h at 37 °C. After loading the exosomes, the fluorescence intensity of the supernatant containing the unloaded exosomes was measured using a spectrofluorophotometer (RF-6000, Shimadzu, Japan). The amount of exosomes loaded onto the microcarriers was calculated by subtracting the amount of unloaded exosomes from the initially added amount. To determine the release profiles of chExo from the microcarriers, 0.4 mg of chExo-loaded microcarriers were incubated in 20 μl of PBS with shaking at 250 rpm at 37 °C. At predetermined time points, 5 μl of the supernatant was collected, and the fluorescence intensity was quantified using a spectrophotometer. A standard curve with DiO-labeled chExo was used for quantification. After each collection, 5 μl of fresh PBS was replenished. Additionally, the surface charges of the chExo-loaded microcarriers dispersed in PBS were measured using a Zetasizer (Nano ZS90, Malvern Panalytical Ltd, UK).
2.9 Intracellular uptake of chExo
To evaluate the intracellular uptake of chExo released from the microcarriers, cells were treated with extracts from DiO-labeled chExo-loaded microcarriers. The extract was prepared by incubating 0.1 mg of microcarriers in 66.5 μl of DMEM for 24 h. Rat chondrocytes were seeded on glass coverslips at a density of 1 × 104 cells per cm2. The following day, the extract was added to the cells and incubated for 12 h. To assess whether the exosomes were internalized by cells, lysosomes were stained with Lysotracker Red DND-99 (L7528, Invitrogen, USA) and observed using CLSM. To investigate whether exosomes loaded onto microcarriers can be internalized by cells through direct contact, 104 rat chondrocytes were directly seeded onto 0.1 mg of DiO-labeled chExo-loaded microcarriers in a well of ultra-low attachment 96-well plates. After 12 h of incubation, the cells were stained with a Lysotracker and imaged by CLSM to visualize the internalization of the exosomes.2.10 Transwell migration assay
To evaluate rat chondrocytes or MSCs were seeded into the upper chamber of a transwell (353097, Corning, USA) at a density of 1.3 × 105 cells per cm2. In the lower chamber, 30 μg ml−1 of free exosomes, 0.5 mg of chM-coated microcarriers, or 0.5 mg of chExo/chM microcarriers (loaded with 10.5 μg of chExo) were added. After 12 h of incubation, the migrated cells were fixed with 4% formaldehyde and stained with 10% crystal violet solution (C0775-25G, Sigma-Aldrich, USA) in methanol. The stained cells were imaged using a light microscope (IX73, Olympus, Japan).2.11 Evaluation of cell adhesion, proliferation, and maturation on microcarriers
To assess cell adhesion and proliferation on the microcarriers, 1 × 105 chondrocytes or MSCs were seeded onto 1 mg of microcarriers (approximately 100 particles). On day 1 of culture, cells were fixed with 4% (v/v) paraformaldehyde and stained with Phalloidin (Alexa Fluor™ Plus 488, A32731, Invitrogen, USA) to visualize F-actin and with 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI, D9542, Sigma-Aldrich, USA) to stain nuclei. The number of adhered cells per microcarrier was quantified by counting nuclei from DAPI-stained images using Image J software. To image cell adhesion by SEM, fixed cells were dehydrated through a graded ethanol series (70%, 80%, 90%, 95%, and 100%), air-dried at room temperature, and observed using FE-SEM. The images were then pseudo-colored using Adobe Photoshop CS6 software. The proliferation profile was monitored using the Cell Counting Kit-8 (CK04, DOJINDO, Japan). At predetermined time points, 0.1 mg of microcarriers (10 particles) were transferred to a 96-well plate and incubated with medium containing 10% CCK-8 solution for 2 h at 37 °C. The absorbance of the resulting solution was measured using a Varioskan™ LUX multimode microplate reader (VLBL00D1, Thermo Scientific, USA).To evaluate rat chondrocyte maturation, chondrocyte-specific ECM deposition was confirmed by histological staining. After 20 days of culture on microcarriers, the cells were fixed, embedded in OCT compound, and cryosectioned to a thickness of 5 μm. The sections were stained using Masson's Trichrome (MT, HT15, Sigma-Aldrich, USA) and Safranin-O (Saf-O, HT90432, Sigma-Aldrich, USA). The percentage of positively stained areas was quantified and normalized to the relevant microcarrier cluster area (%). To evaluate microcarriers integration during cell culture, the number of microcarriers per cluster was counted using Fiji Image J software. A cluster was defined as a group where two or more microcarriers were connected, and a total of 100 microcarriers were counted per group.
2.12 Evaluation of cell infiltration to microcarriers
The cell infiltration from the surface into the inner space of the microcarriers was evaluated as previously reported.44 Rat chondrocytes were cultured on the microcarriers for 20 days. F-actin and nuclei were stained with Alexa FluorTM 546 Phalloidin (A22283, Invitrogen, USA) and DAPI, respectively. The stained microcarriers were embedded in O.C.T compound, cryosectioned into 20-μm-thick slices, and observed by CLSM. The microcarrier area was divided into three zones: core (C), middle (M), and outer (O), with a zone length ratio of C:M:O = 5:4:1. The fluorescence intensity of F-actin in each zone was measured using Fiji Image J software.
2.13 Real-time polymerase chain reaction (PCR)
Real-time PCR was conducted to assess gene expression levels in rat chondrocytes or MSCs cultured on microcarriers for 5 days. Total RNA was isolated using the Ribospin™II kit (341–150, GeneAll, South Korea) following the manufacturer's instructions. cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (4368813, Applied Biosystems, USA) with a HID Veriti® 96-Well Thermal Cycler (447907, Applied Biosystems, Singapore). Real-time PCR was carried out using the SensiMix™ SYBR Hi-ROX kit (QT605-05, Bioline, UK) and the StepOneTM Plus instrument (Applied Biosystems). Relative gene expression levels were quantified using the ΔΔCt method (2−ΔΔCt) and normalized to the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (Gapdh). Detailed primer sequences are described in Table S2 (ESI†).2.14 Bulk RNA sequencing analysis
To assess global changes in gene expression in rat chondrocytes cultured on microcarriers for 5 days, bulk RNA sequencing analysis was conducted. On day 5 of culture, total RNA was extracted using the Ribospin™ II kit. Quantitative sequencing was performed by E-biogen Inc. (South Korea). Libraries were constructed using the QuantSeq 3′ mRNA-Seq Library Prep Kit (Lexogen, Inc., Austria) according to the manufacturer's protocol. Briefly, 500 ng of total RNA per condition was hybridized with an oligo-dT primer containing an Illumina-compatible sequence at its 5′ terminus, followed by reverse transcription. The RNA template was degraded, and second-strand synthesis was initiated with a random primer that also contained an Illumina-compatible linker sequence at its 5′ end. Double-stranded libraries were purified using magnetic beads to remove reaction components. Amplification was then performed, and clusters were added to generate complete adapter sequences. The final libraries were purified from PCR components. High-throughput sequencing was performed using a single-end 75 sequencing approach on the NextSeq 500 platform (Illumina, Inc., USA). The resulting reads were aligned using Bowtie2 (Langmead and Salzberg, 2012). Differentially expressed genes were identified through coverage analysis using Bedtools (Quinlan AR, 2010). The read count data were normalized using the TMM + CPM method in R, implemented through EdgeR (R Development Core Team, 2020) within Bioconductor (Gentleman et al., 2004). Gene classification was conducted based on searches of the DAVID (https://david.abcc.ncifcrf.gov/) and Medline databases (https://www.ncbi.nlm.nih.gov/).2.15 Immunostaining
After 1 day of rat chondrocyte culture on microcarriers, the cells were fixed in 4% (v/v) paraformaldehyde for 30 min at room temperature. The fixed cells were then permeabilized using 0.1% (v/v) Triton X-100 in PBS for 15 min. To block non-specific binding, the cells were incubated with 5% (w/v) BSA for 1 h at room temperature with gentle shaking. Following blocking, the cells were incubated with primary antibodies overnight at 4 °C with gentle shaking, followed by treatment with secondary antibodies for 1 h at room temperature. Details of the antibodies used are described in Table S3 (ESI†). Subsequently, F-actin and nuclei were stained with Phalloidin (either Alexa Fluor™ Plus 488 or 546) and DAPI. Fluorescence images were obtained using CLSM. The cell spreading area and F-actin intensity were measured using Image J software. Additionally, the mean intensity of YAP and MRTF-A within the nucleus and cytosol was quantified using the freehand selection tool in Fiji.2.16 Inhibitor treatment
MK-0429 (MK, HY-15102, MedChemExpress, China), NSC 23766 (NSC, 2161, Tocris, UK), Y-27632 (Y, 1254, Tocris, UK), and verteporfin (VP, 5305, Tocris, UK) were used in this study. For dose–response screening, rat chondrocytes were seeded at a density of 5000 cells per well in a 96-well plate and treated with various concentrations of each inhibitor: MK (25, 50, 100, 200, 400, and 800 nM), NSC (12.5, 25, 50, 100, 200, and 400 μM), Y (12.5, 25, 50, 100, 200, and 400 μM), or VP (0.5, 1, 2, 4, 6, and 8 μM). After 24 h of treatment, cell viability was assessed using the CCK-8 assay. For experiments involving rat chondrocytes that were seeded onto microcarriers, cells were treated with 25 nM MK, 50 μM NSC, 100 μM Y, or 1 μM VP, which were added to the media at the time of cell seeding. Following 24 h of treatment, cells were stained for F-actin, MRTF-A, and YAP. The inhibitory effects were then observed using CLSM.2.17 In vivo cartilage regeneration using microcarriers in a rat osteochondral defect model
All animal experiments were conducted according to the ethical guidelines of the Animal Research Committee at Dankook University (no. DKU-22-038). Eight-week-old male Sprague-Dawley rats were used to establish an osteochondral defect model, as previously described.45 Rats were anesthetized with an intramuscular injection of ketamine (80 mg kg−1) and xylazine (10 mg kg−1) throughout the procedure. The leg hair on the knee was shaved, and the surgical site was disinfected with povidone-iodine followed by 70% ethanol. A linear incision was made through the skin, muscle, and synovial capsule of the knee joint using a surgical blade. The patella was luxated to either the right or left side to expose the trochlear groove, where a defect was created. Articular cartilage defects (1.6 mm in diameter and 1.6 mm in depth) were prepared on each side of the knee joint under cooling conditions with sterile saline, using a dental high-speed handpiece equipped with a 1.6 mm diameter dental bur (ISO No: 016, SBT No: 5). Eight defects from four rats were randomly assigned to six groups: intact, untreated, bare microcarriers, chM-coated microcarriers, chExo/chM-coated microcarriers, and chExo only treatment. For the microcarrier groups, 5 mg microcarriers were implanted into each defect. In the chExo/chM-coated microcarrier group, 100 μg of chExo was loaded onto 5 mg of microcarriers. For the chExo-only group, 100 μg of free chExo was applied for comparison and administered as a single dose on the day of surgery.After implantation, the muscle and skin were sutured with absorbable Vicryl® and non-absorbable 4-0 Prolene sutures (8603G, Ethicon, USA). Animals were housed individually to prevent wound manipulation. Eighteen weeks post-operation, the animals were euthanized by CO2 inhalation, and the knee joints were harvested and fixed in 10% neutral buffered formalin for subsequent histological analysis and micro-computed tomography (μCT) imaging.
2.18 μCT imaging
μCT images of the fixed tissues were obtained using X-ray settings of 65 kV and 385 μA, with an exposure time of 279 ms per section on a Skyscan 1176 system (Skyscan, Belgium). The reconstructed images were analyzed to assess hard tissue formation within the region of interest using CTAn Skyscan software. Three-dimensional images were generated and visualized using CTvol Skyscan software (ver. 2.3.2.0). Key parameters evaluated in the defect area included newly formed bone volume (mm3), percent bone volume (%), surface area (mm2), and tissue volume (mm3).2.19 Histological analysis
After tissue fixation, the samples were decalcified in RapidCal™ solution (6040, BBC Chemical Co., USA) for 2 weeks. Following decalcification, the tissues were dehydrated in a graded ethanol series and then embedded in paraffin. Sections were cut at a thickness of 5 μm using a microtome (Leica RM2245, Leica Biosystems, Germany) and mounted onto coated glass slides. The slides were deparaffinized and rehydrated through a series of xylene and graded ethanol solutions. The tissue sections were then stained with hematoxylin & eosin (H&E), Masson's trichrome (MT), Safranin O & Fast green (Saf O), and anti-collagen II (1:50 dilution, sc-52658, Santa Cruz, USA). Stained sections were imaged using a light microscope and further analyzed with Fiji Image J software. The percentage of positively stained areas for Saf-O and type II collagen was quantified and normalized to the relevant defect area (%). For histological scoring, a modified histological grading45 was employed to assess the quality of cartilage repair, as outlined in Table S4 (ESI†). Additionally, the regenerated cartilage was evaluated using the International Cartilage Repair Society (ICRS) macroscopic score method.462.20 Statistical analysis
All values were expressed as mean ± standard deviation. Statistical analyses were performed using either Student's t-test or one-way analysis of variance (ANOVA), followed by Tukey's multiple comparison test where applicable (GraphPad Prism). For the analysis of histological and ICRS scores, the Kruskal–Wallis test was used for global comparisons among groups. In all cases, p < 0.05 was considered statistically significant.3. Results and discussion
3.1 Preparation of porous microcarriers using a co-flow microfluidic system
We prepared porous microcarriers using a co-axial nozzle-based co-flow microfluidic device (Fig. 1(a)). In this setup, the dispersed phase channel was positioned within the continuous phase channel, allowing the simultaneous flow of both phases. A solution of PCL/camphene dissolved in chloroform served as the dispersed phase, while the continuous phase comprised an aqueous solution of PVA. The shear force exerted by the continuous phase fragmented the dispersed phase into emulsion droplets at the junction. These droplets were subsequently solidified through solvent extraction and lyophilization, where camphene sublimation resulted in the formation of pores. | ||
| Fig. 1 Fabrication of porous microcarriers using a co-flow microfluidic system and their characteristics. (a) Schematic illustration of the fabrication process for microcarriers using a co-axial nozzle-based co-flow microfluidic device (top). PCL/camphene dissolved in chloroform and a PVA aqueous solution serve as the dispersed and continuous phases, respectively. The shearing force exerted by the continuous phase causes the dispersed phase to break into emulsion droplets at the junction. These emulsified droplets are solidified through solvent extraction and lyophilization, during which camphene sublimates, forming pores. Bright-field images depict the emulsified droplets and the resulting porous microcarriers (bottom). Scale bars, 500 μm. (b) Optical image of the co-nozzle microfluidics device. (c) and (d) Particle and pore size distribution of microcarriers under various microfluidic operating conditions. Effect of PCL and camphene concentrations on microcarrier particle and pore sizes (c). Effect of flow rates of the continuous phase (Fc) and dispersed phase (Fd) on particle and pore sizes (d). The red arrow indicates the optimal condition for producing porous microcarriers: a mixture of 3% (w/w) PCL and 20% (w/w) camphene in the dispersed phase flowing at 1 ml h−1, with a PVA aqueous solution as the continuous phase co-flowing at 10 ml h−1. (e) Representative SEM images of the surface and cross-sections of porous or dense microcarriers. Scale bars: 100 μm (main images), 20 μm (porous microcarriers, inset, red box), and 5 μm (dense microcarriers, inset, red box). Cross-sectional areas are highlighted with red dotted lines. (f) Diameter and pore size measurements of microcarriers (n = 50 microcarriers per group). The coefficient of variation (CV) is defined by a ratio of the standard deviation to the average microcarrier size. (g) Elastic moduli of microcarriers (n = 9–10 microcarriers per group). (h) Degradation profiles of microcarriers under alkaline conditions over 9 days. Data are presented as means ± s.d. of n. ****p < 0.0001; Student's t-test for (f), (g); two-way ANOVA followed by Tukey's post hoc tests for (h). | ||
The emulsified droplets were transparent and uniformly sized (Fig. 1(b)). After a series of modifications, these droplets solidified into porous microcarriers. We could precisely control the size of PCL microcarriers by adjusting microfluidic parameters, including PCL concentration, camphene concentration, and flow rate (Fig. 1(c), (d) and Fig. S1, ESI†). Microcarriers produced using a coaxial nozzle of 27 G and 21 G were 300–500 μm in diameter.
By co-varying polymer/camphene ratios and emulsion flow rates, we simultaneously tuned microcarrier diameter and pore size. Increasing PCL content from 2% to 5% (w/w) raised solution viscosity, producing larger particles with smaller pores (due to inhibited camphene crystallization), whereas boosting camphene content at a fixed 3% PCL (w/w) expanded both pore volume and overall particle size. Under our optimized condition (3% (w/w) PCL, 20% (w/w) camphene; dispersed phase 1 ml h−1; continuous phase 10 ml h−1), microcarriers measured 454.5 ± 5.4 μm in diameter with uniform pores (Fig. 1(e) and (f)). Raising the continuous phase flow to 40 ml h−1 decreased particle size to ∼383 μm, while varying the dispersed phase flow rate (1.0–2.5 ml h−1) had only minor effect.
Compared to a conventional dripping-based method for PCL microcarrier fabrication,41,47 our co-flow microfluidic system yielded more uniform microcarriers, with a coefficient of variance (CV) of just 1.2% (Fig. 1(f)). These porous microcarriers, in contrast to non-porous “dense” PCL microcarriers of similar size, featured highly interconnected micropores (59.6 ± 11.1 μm) throughout the carriers (Fig. 1(e)).
We have carefully optimized fabrication conditions because both microcarrier particle and pore sizes are critical for clinical application. Particle sizes must be small enough to fill irregular cartilage defects effectively, yet large enough to allow easy handling in a clinical setting. Pore diameters of approximately 50–100 μm are essential to recreate lacunae-like niches that regulate chondrocyte behavior. Therefore, our optimized microcarriers – characterized by a particle size of ∼450 μm and a pore diameter of ∼60 μm – are expected to meet these design criteria and thereby support host cell ingrowth, provides physical support for cell proliferation, and facilitates nutrient and waste exchange, making them particularly promising for cartilage repair.44 Furthermore, the high porosity of the microcarriers reduced their elastic modulus while increasing their degradability (Fig. 1(g) and (h)). The bulk elastic modulus of the porous microcarriers was 0.4 ± 0.1 MPa, closely resembling that of native cartilage tissue.48–50 Due to their larger surface area and high porosity, the microcarriers exhibited a faster hydrolytic degradation rate in an alkaline solution compared to their dense counterparts (Fig. 1(h) and Fig. S2, ESI†). These characteristics suggest that porous microcarriers implanted in cartilage defects can support cartilage repair by providing a microenvironment that physically mimics native cartilage tissue while also being naturally degraded within the body.
3.2 Chondrocyte membrane coating of porous microcarriers alters the surface properties of microcarriers
To create a chondrocyte-mimicking scaffold for cartilage repair, we decorated porous PCL microcarriers with chondrocyte-derived cell membranes (chM) and exosomes (chExo) (Fig. 2(a)). The chM used for coating the microcarriers was extracted from primary rat chondrocytes through a series of hypotonic lysis and ultracentrifugation steps. SDS-PAGE analysis confirmed that the protein profile of the chM closely resembled that of chondrocyte lysates (Fig. 2(b)). Notably, the chondrocyte-specific surface marker, CD151, was highly preserved in chM, whereas the transcription factor, SOX9, was scarcely detected (Fig. 2(c)). Interestingly, despite the cell membrane extraction, significant amounts of chondrocyte-specific ECM proteins, such as aggrecan (ACAN) and type II collagen, remained associated with the chM. These findings suggest that microcarriers coated with chM can replicate the physicochemical characteristics of chondrocytes and their native microenvironment.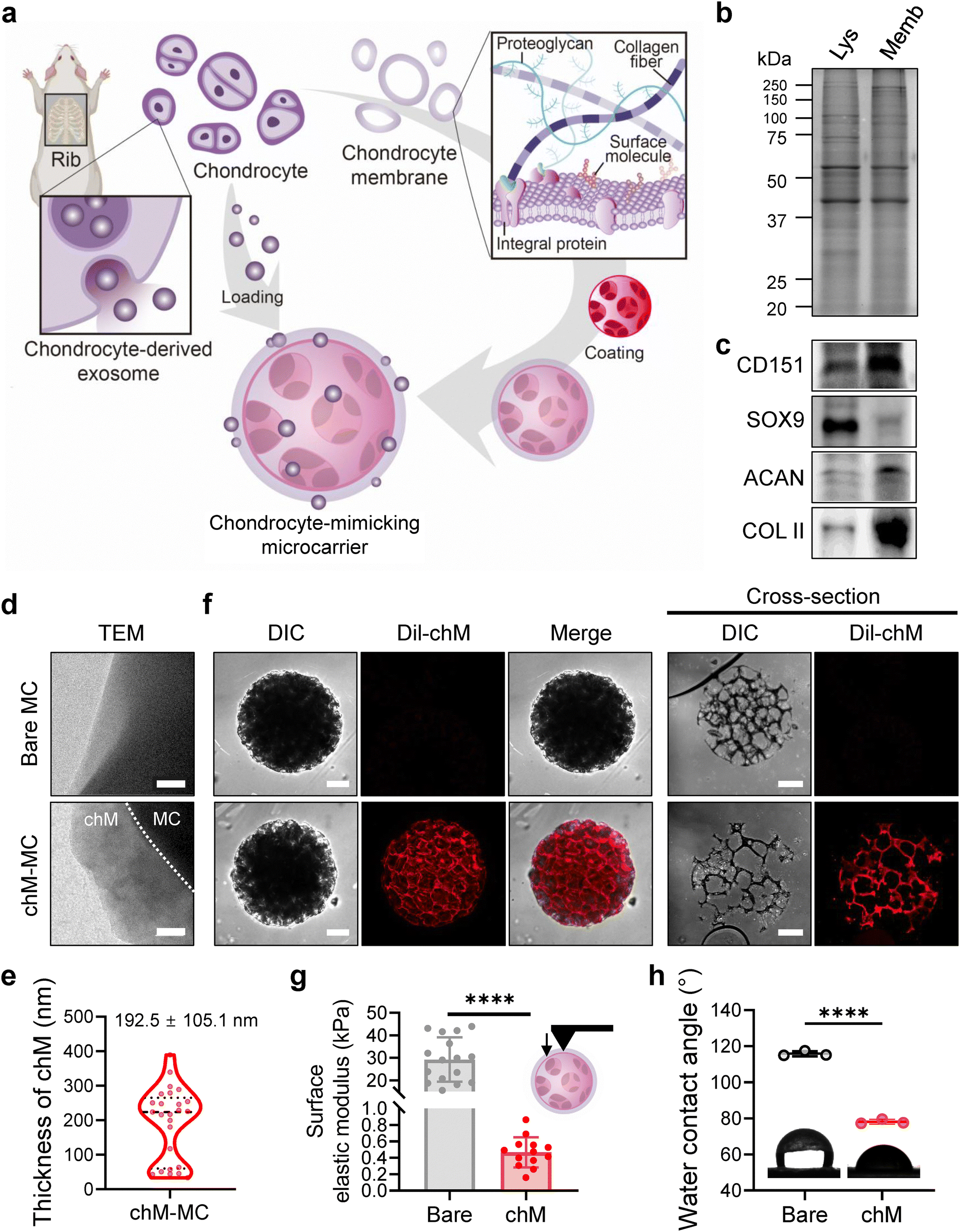 | ||
| Fig. 2 Preparation and characterization of chondrocyte-mimicking microcarriers. (a) Overview of the preparation process for chondrocyte-mimicking microcarriers. Porous microcarriers are decorated with the chondrocyte-derived cell membrane (chM) and exosome (chExo), referred to as chExo/chM microcarriers. (b) SDS-PAGE analysis and (c) western blots of lysate (Lys) and cell membranes (Memb) from chondrocytes. (d) Representative TEM images of bare microcarriers (MC) and chondrocyte membrane-coated microcarriers (chM-MC). Scale bar, 100 nm. (e) The thickness of the chondrocyte membrane layer coated on microcarriers (n = 25 fields from 4 microcarriers). (f) CLSM images of microcarriers, showing the cell membrane coating on the surface (left) and in cross-sections (right). Chondrocyte membranes are stained with Dil dye (red). Scale bar, 100 μm. (g) Surface elasticity of microcarriers (n = 13–16 microcarriers per group). (h) Water contact angle measurements of bare and chondrocyte membrane-coated substrates (n = 3 substrates per group). Data are presented as means ± s.d. of n. ****p < 0.0001; Student's t-test. | ||
We coated porous microcarriers with chM through membrane vesicle fusion. The chM vesicles, formed by ultrasonication, would readily fuse with the solid microcarrier substrate to minimize interfacial energy, plausibly through hydrophobic interactions, van der Waals forces, and electrostatic interactions.39,51 TEM revealed a thin cell membrane layer (192.5 ± 105.1 nm) on the surface of the chM-coated microcarriers (Fig. 2(d) and (e)), though membrane thickness varied, ranging from 50 nm to 200 nm (Fig. S3). Fluorescently labeled chM (Dil-chM) showed an evenly distributed fluorescence signal both on the surface and within the internal pores of the microcarriers (Fig. 2(f), Fig. S3a, and Movie S1, ESI†). Together, these results confirmed the successful and uniform coating of the porous microcarriers with chondrocyte-derived cell membranes.
The surface decoration of microcarriers with cell membranes altered the physical properties of the scaffold surface. While the surface elastic modulus of bare microcarriers was ∼30 kPa, that of the chM-coated microcarriers decreased to ∼0.5 kPa, which closely matches that of chondrocytes (0.5 to 1 kPa) (Fig. 2(g)).48–50 In addition to reduced surface elasticity, the surface hydrophobicity was significantly lowered, as confirmed by water contact angle measurement (Fig. 2(h)). These results imply that the modified surface properties, which mimic chondrocyte-specific chemical and physical characteristics, may act as bioactive cues to regulate chondrocyte behavior.
3.3 Chondrocyte-derived exosomes exhibit therapeutic potential for cartilage repair
We isolated chondrocyte-derived exosomes (chExo) from the same cells used for cell membrane extraction. To obtain chExo, we collected the supernatant of chondrocyte cultures, while the cell pellet was utilized for cell membrane extraction. SEM and TEM images revealed uniform spherical vesicles constructed with a lipid bilayer (Fig. 3(a) and (b)). Nanoparticle tracking analysis further indicated that the average size of chExo was 116.9 ± 33.6 nm (Fig. 3(c)), with no significant batch variations in size observed (Fig. S4a, ESI†). Importantly, our analysis confirmed the presence of exosomal surface marker proteins, including TSG101, ALIX, and CD63, while the endoplasmic reticulum marker Calnexin and Golgi apparatus marker GM130 were absent (Fig. 3(d)). This indicates successful isolation of chExo without contamination from other cellular organelles.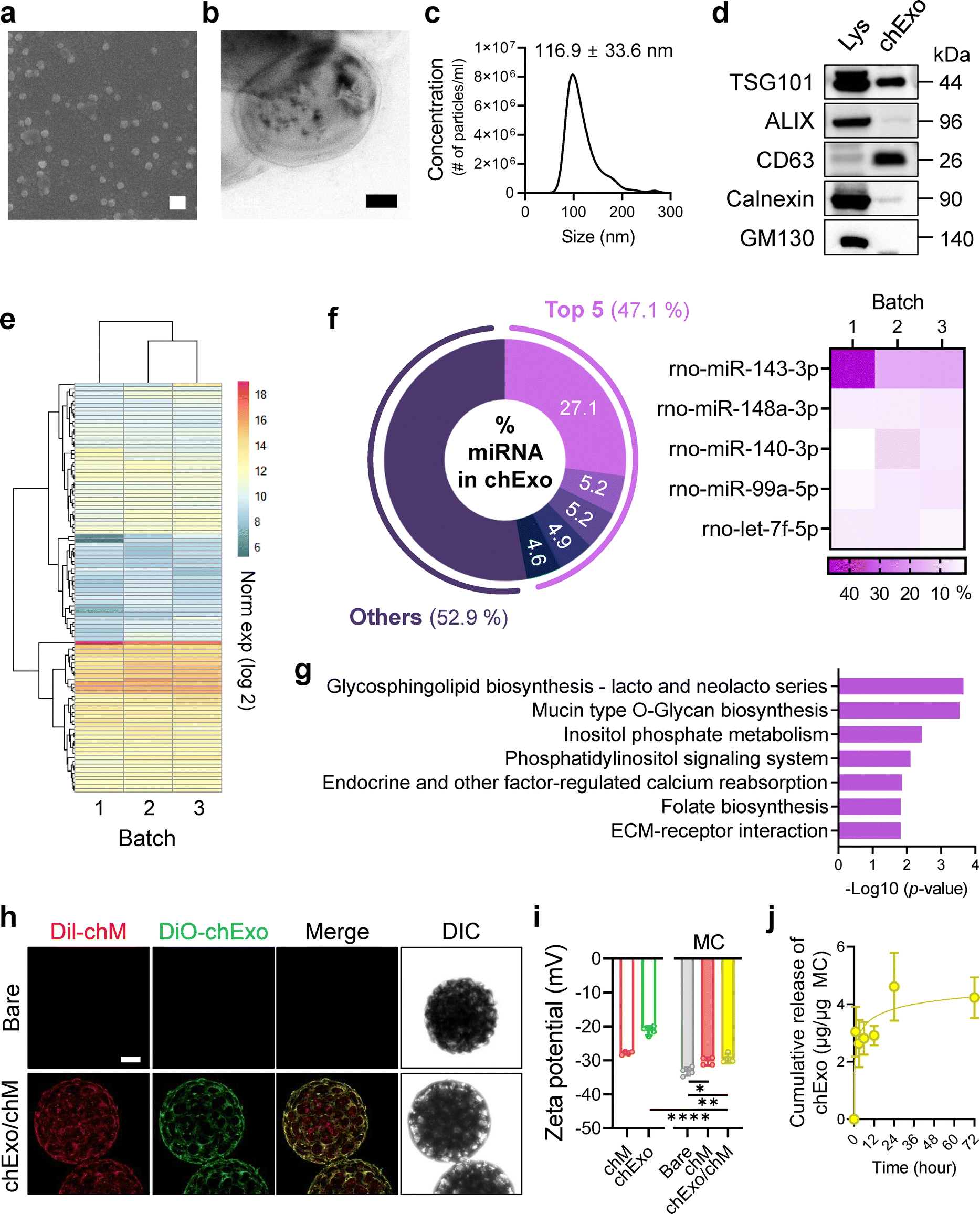 | ||
| Fig. 3 Characterization of chondrocyte-derived exosomes (chExo) and their integration onto the chondrocyte membrane-coated microcarriers. (a) Representative SEM showing the chExo morphology. Scale bar, 200 μm. (b) TEM images of chExo, illustrating their detailed structure. Scale bar, 20 nm. (c) Nanoparticle tracking analysis (NTA) showing the average size and concentration of chExo. (d) Western blot analysis of exosome-specific markers in lysates (Lys) and chExo. Exosomal surface marker proteins: TSG101, ALIX, and CD63; endoplasmic reticulum marker: Calnexin; Golgi apparatus marker: GM130. (e)–(g) Small RNA-sequencing analysis of chExo (n = 3 batches from independent experiments). (e) Heatmap and hierarchical clustering of the top 100 miRNAs enriched in chExo. (f) Pie chart (left) and heatmap (right) illustrating the most enriched top 5 miRNAs in chExo. (g) KEGG pathway analysis of the top 100 miRNAs in chExo. (h) CLSM images showing the integration of chExo onto chondrocyte membrane-coated microcarriers. Chondrocyte membranes are labelled with Dil (red) and chExo with DiO (green). Scale bar, 100 μm. (i) Surface charge measurements of free chM and chExo (left) and microcarriers decorated with none (bare), chM, or chExo/chM (right). (j) Cumulative release profiles of chExo from chExo/chM microcarriers over 72 h. Data represent mean ± s.d. of n and represent at least three independent experiments. *p < 0.05, **p < 0.01, and ****p < 0.0001; one-way ANOVA followed by Tukey's post hoc tests. | ||
Next, we analyzed the miRNA composition of chExo using small RNA sequencing across three independent batches of exosomes. Hierarchical clustering of the top 100 miRNAs demonstrated no significant differences in miRNA compositions among these batches (Fig. 3(e)). The most abundant miRNA identified in chExo was miR-143-3p (27.1%), followed by miR-148a-3p, miR-140-3p, miR-99a-5p, and let-7f-5p (Fig. 3(f) and Fig. S4b, c, ESI†). Furthermore, enriched KEGG pathway analysis revealed that the miRNAs in chExo were associated with the regulation of genes involved in lipid biosynthesis and metabolism, such as glycosphingolipid biosynthesis, inositol phosphate metabolism, phosphatidylinositol signaling system, folate biosynthesis, as well as extracellular signaling pathways such as mucin-type O-Glycan biosynthesis and ECM-receptor interaction (Fig. 3(g) and Table S5, ESI†).
To gain a better understanding, we compared the miRNA composition of chExo with that of mesenchymal stem cell (MSC)-derived exosomes, which are known to have therapeutic effects on cartilage repair.52 We found that half of the top 100 enriched miRNAs in chExo were distinct from those identified in MSC exosomes (Fig. S4d and e, ESI†). Furthermore, while the most abundant miRNA in MSC exosomes was miR-122-5p, chExo displayed miR-143-3p at the highest content (Fig. S4f, ESI†). These results suggest that chExo and MSC exosomes may regulate different pathways in host cells during the cartilage repair process.
To assess cellular responses to chExo, we treated chondrocytes and bone marrow-derived MSCs with chExo. Chondrocytes treated with chExo exhibited increased cell migration in a dose-dependent manner, with no toxicity at concentrations of up to 30 μg ml−1 (Fig. S5, ESI†). Moreover, chExo treatment significantly accelerated the proliferation of chondrocytes compared to the untreated control (Fig. S5c, ESI†). Intriguingly, the gene expression of both chondrogenic and osteogenic markers, such as Sox9, Col2a1, Runx2, and Alp, was significantly up-regulated in the chExo-treated MSCs, while the expression of adipogenic and neurogenic genes remained unchanged (Fig. S6, ESI†). These results suggest that chondrocyte-derived exosomes may play a beneficial role in stimulating host cells in osteochondral defects toward the repair process.
To mimic chondrocyte-specific functions, we loaded chExo onto chM-coated microcarriers (Fig. 3(h)). DiO-labeled chExo (green) was successfully loaded onto the Dil-labeled chM-coated microcarriers (red), as indicated by the green fluorescence overlapping with the red signals.
This overlap confirmed the loading of chExo onto the cell membrane layers of the microcarriers. We speculate that exosomes associate with the cell membrane coat via lipid–lipid fusion, receptor–ligand binding, electrostatic interactions, and non-specific adsorption.53 Although the cell membrane coating slightly increased the surface charge of the microcarriers from −33.3 ± 1.3 mV to −30.3 ± 1.2 mV, loading chExo did not further alter the surface charge (Fig. 3(i)). On average, the chM-coated microcarriers contained 13.2 ± 1.3 μg chExo per μg microcarrier. The cumulative release profile of chExo from the chExo-loaded and chM-coated (chExo/chM) microcarriers exhibited an initial burst release of 35%, followed by a plateau within 24 h (Fig. 3(j)). These results suggest that the cell membrane layers of the microcarriers can act as a reservoir for chExo, allowing for a more sustained delivery compared to bolus exosome delivery via local injection.
3.4 Chondrocyte-mimicking microcarriers induce chondrocyte-specific recruitment and improve chondrocyte proliferation while maintaining the anabolic phenotype
The chExo/chM microcarriers demonstrated that chExo was effectively taken up by chondrocytes (Fig. 4(a)–(c)). Notably, chExo released from the microcarriers were internalized by cells in a manner comparable to soluble chExo (Fig. 4(b)). Furthermore, cells adhering to the chExo/chM microcarriers were able to internalize the chExo directly from the microcarriers (Fig. 4(c)). Importantly, the released chExo stimulated chondrocyte migration to a degree similar to that of free chExo (Fig. 4(d)), suggesting that the exosomes remained intact and functional upon release. Interestingly, chExo did not induce any migration in rat mesenchymal stem cells, highlighting their specific ability to recruit chondrocytes (Fig. S7, ESI†).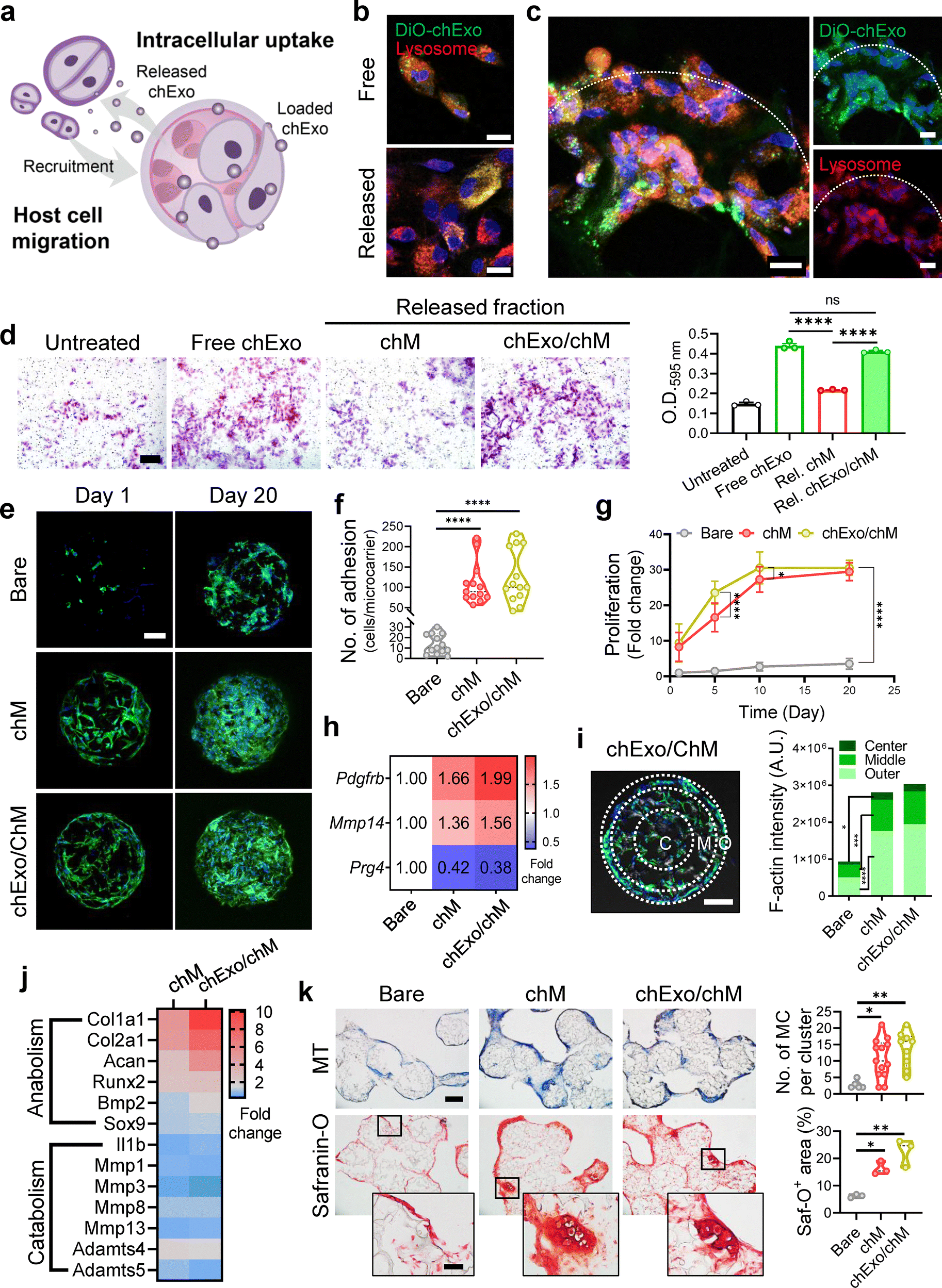 | ||
| Fig. 4 Chondrocyte-mimicking microcarriers enhance chondrocyte recruitment and proliferation while maintaining the anabolic phenotype. (a) Schematic illustrating the migration of host cells induced by chExo released from chExo/chM microcarriers and the subsequent intracellular uptake of chExo by recruited and adhered cells. (b) and (c) CLSM images demonstrating intracellular uptake of chExo by chondrocytes. chExo are labeled with DiO dye (green), and lysosomes are stained with LysoTracker™ Red DND-99 (red). Image taken at 12 h after (b) treating chondrocytes with either free chExo or chExo released from chExo/chM microcarriers (scale bar, 20 μm), or (c) culturing chondrocytes on chExo/chM microcarriers for 12 h (scale bar, 20 μm). The boundaries of the microcarriers were indicated with a dotted line. (d) Transwell migration assay results following treatment with released fractions from chM microcarriers or chExo/chM microcarriers for 12 h (left) and corresponding quantification (n = 3 replicates per condition). (e) CLSM images showing chondrocyte adhesion and proliferation on microcarriers at day 1 and 20. F-actin (green) and nuclei (blue). Scale bar, 100 μm. (f) The number of chondrocytes adhered to microcarriers on day 1 (n = 13 microcarriers per group). (g) Proliferation profiles of chondrocytes on microcarriers over 20 days. (h) Heatmap depicting the fold changes in gene expressions related to chondrocyte migrations in chondrocytes cultured on microcarriers for 5 days (n = 3 replicates per group). (i) Cell infiltration analysis based on cross-sectional CLSM images of microcarriers after 20 days of chondrocyte culture. F-actin (green) and nuclei (blue). Scale bar, 100 μm. The cross-sectional areas of microcarriers are divided into center, middle, and outer regions at a ratio of 5:3.5:1.5, and F-actin intensity in each region is quantified (n = 18–20 fields from 10 microcarriers). (j) Bulk RNA-sequencing analysis showing expression levels of genes associated with anabolic and catabolic phenotypes of chondrocytes in the chM microcarrier and chExo/chM microcarrier group versus bare microcarrier group. (k) Representative optical images of Masson's trichrome (MT) and safranin-O (Saf-O) staining after 20 days of chondrocyte culture on microcarriers (left), with quantification of the number of microcarriers per cluster (n = 5–13 fields per group) and Saf-O-stained area (n = 3 fields per group) (right). Scale bar: 200 μm (main) and 50 μm (inset). Data represent mean ± s.d. of n. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; one-way ANOVA followed by Tukey's post hoc tests for (d), (f), (k); two-way ANOVA followed by Tukey's post hoc tests for (g). | ||
On the first day of culture, chondrocyte adhesion to chM or chExo/chM microcarriers was approximately 8- to 9-fold greater than to bare microcarriers (Fig. 4(e) and (f)). Both chM and chExo/chM microcarriers further supported rapid cell proliferation compared to bare ones (Fig. 4(g)). Furthermore, chondrocytes adhered to the microcarriers continued to migrate toward the center of the microcarriers, accompanied by the up-regulation of migration-related genes (Fig. 4(h) and (i)). Considerable cell infiltration was observed in chExo/chM microcarriers compared to chM or bare microcarriers on the first day of culture, and this infiltration persisted through day 20 (Fig. S8, ESI†).
Bulk RNA sequencing analysis revealed that chondrocytes maintained their anabolic phenotype while actively proliferating on the chondrocyte-mimicking microcarriers (Fig. 4(j)). Compared to the bare microcarrier group, both chM and chExo/chM microcarriers significantly up-regulated the expression of genes associated with anabolic phenotypes (Col2a1, Sox9, Comp, Acan, and Col1a1) while down-regulating catabolic phenotype-related genes (Mmps and Adamts) in chondrocytes. Additionally, anti-inflammatory genes (Tgfbi, Tgfbl, Tgfb2, and Tgfb3) were up-regulated, while pro-inflammatory genes (Il1b, Nos2, and Tnf) were down-regulated in chondrocytes cultured on both chM and chExo/chM microcarriers (Fig. S9, ESI†). Although the overall trends in gene expression profiles were similar between chM and chExo/chM microcarriers, the fold change was more pronounced in the chExo/chM microcarrier group. This suggests that the loading of chExo onto chM microcarriers had an additional enhancing effect on chondrocyte function and maturation. The chondrocyte-mimicking microcarriers supported chondrocytes in maintaining their anabolic phenotype while rapidly proliferating, enabling significant deposition of chondrocyte-specific ECM proteins, including collagens and aggrecans (Fig. 4(k) and Fig. S10, ESI†).
3.5 Chondrocyte-mimicking microcarriers not only increase cell adhesion and ECM synthesis but also modulate metabolic activity in chondrocytes
To examine how chM and chExo influence chondrocyte behavior, we analyzed changes in the transcriptomic levels of chondrocytes using bulk RNA sequencing (Fig. 5 and Fig. S11, ESI†). Hierarchical clustering of global gene expression profiles revealed that chondrocytes cultured on chExo/chM microcarriers clustered closely with those on chM microcarriers, distinct from the gene profiles of chondrocytes on bare microcarriers (Fig. 5(a)). In chExo/chM microcarriers, 687 differentially expressed genes (DEGs, 1.5-fold change) were identified compared to bare microcarriers, with 413 up-regulated and 274 down-regulated. When normalized to chM microcarriers, 139 DEGs were detected, consisting of 63 up-regulated and 76 down-regulated genes (Fig. 5(b) and (c)).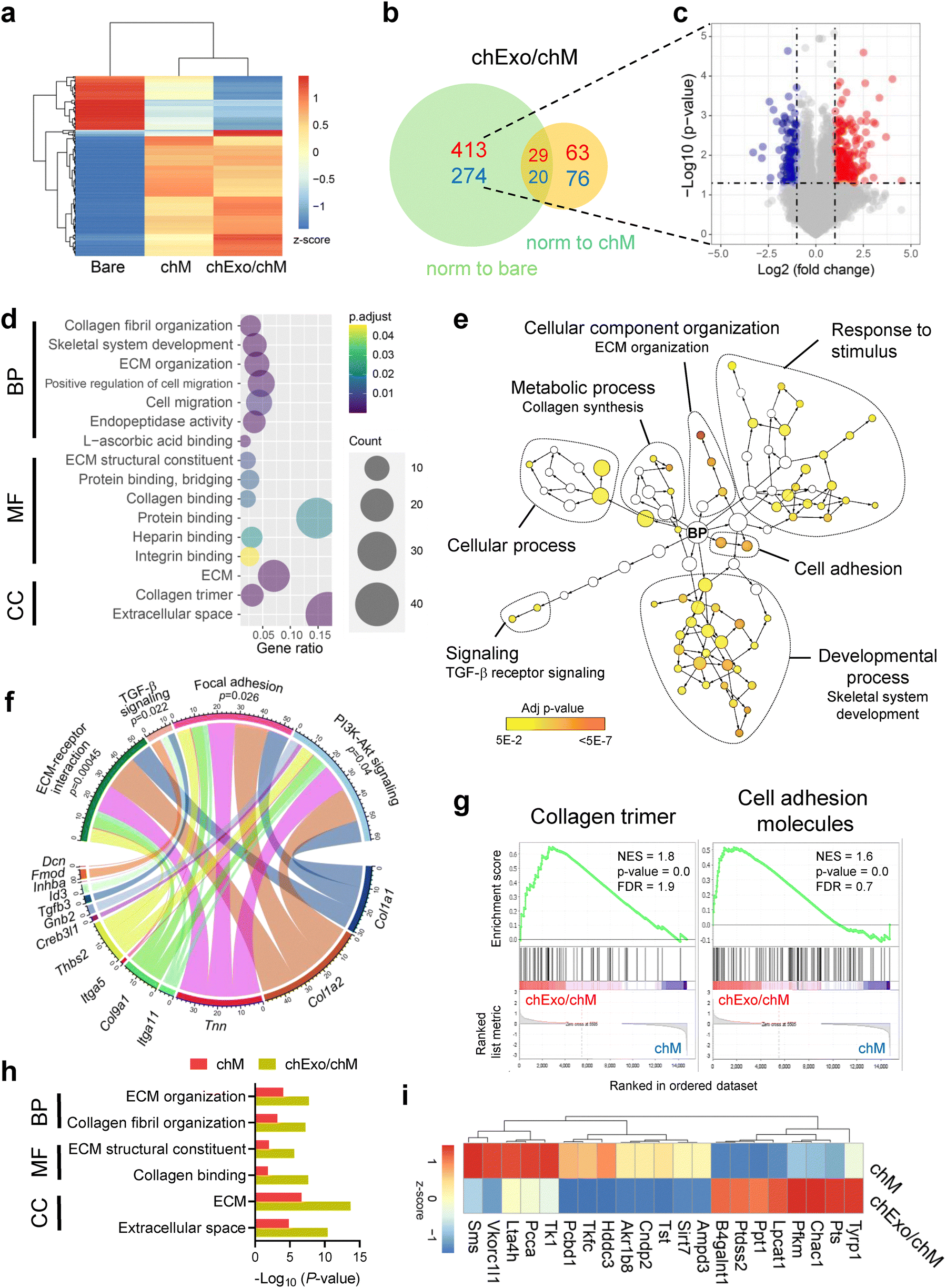 | ||
| Fig. 5 Chondrocyte-mimicking microcarriers enhance cell adhesion and ECM synthesis and modulate metabolic activity in chondrocytes. Bulk RNA sequencing analysis was performed to assess the impact of chondrocyte-mimicking microcarriers on transcriptomic changes in chondrocytes cultured on bare, chM, or chExo/chM microcarriers for 5 days (n = 3 replicates per group). (a) Heatmap and hierarchical clustering of differentially expressed genes (DEGs, 2-fold change, p < 0.05) across the three microcarrier groups. (b) Venn diagram showing the number of up-regulated (red) or down-regulated (blue) genes in the chExo/chM microcarrier group compared to the bare (green circle) or chM microcarrier (yellow circle) groups. (c) Volcano plot depicting DEGs up-regulated (red) or down-regulated (blue) in the chExo/chM microcarrier group relative to the bare microcarrier group (cut off: 2-fold change, p = 0.05). (d) Bubble plot showing enriched gene ontology terms for DEGs in the chExo/chM microcarrier group compared to the bare microcarrier group. BP, biological process; CC, cellular component; MF, molecular function. (e) Network visualization of BP terms associated with up-regulated DEGs in the chExo/chM microcarrier group compared to the bare microcarrier group. Node size represents the number of markers associated with each term, and node color indicates the p-value. (f) Chord diagram illustrating enriched KEGG pathways and their corresponding core genes in the chExo/chM microcarrier group compared to the bare microcarrier group. (g) Gene set enrichment analysis of gene sets enriched in the chExo/chM microcarrier group relative to the chM microcarrier group. FDR, false discovery rates; NES, normalized enrichment score. (h) Gene ontology analysis of DEGs enriched in both chM and chExo/chM microcarrier groups. (i) KEGG pathway analysis highlighting DEGs related to lipid metabolism uniquely enriched in the chExo/chM microcarrier group compared to the chM microcarrier group. | ||
Gene ontology (GO) analysis confirmed that the DEGs in chondrocytes cultured on chExo/chM microcarriers, compared to bare microcarriers, were significantly enriched in processes related to ECM remodeling (collagen fibril organization, ECM organization, and ECM structural constituent), cell migration, and protein binding (heparin, integrin, and collagen binding) (Fig. 5(d)). A network visualization of enriched GO terms, particularly those involving biological processes, revealed that up-regulated genes were linked to cellular component organization (including ECM organization), response to stimulus, developmental process (including skeletal system development), metabolic process (including collagen synthesis), cell adhesion, and TGF-β receptor signaling (Fig. 5(e)). The chord plot demonstrated that specific genes, such as Col1a1, Col1a2, and Tnn, were mainly involved in ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathways (Fig. 5(f)). Additionally, genes, such as Tgfb3, Id3, Inhba, Fmod, and Dcn, were linked to TGF-β signaling pathways. These results indicate that the chondrocyte-mimicking microcarriers enhance cell adhesion and migration and stimulate ECM synthesis in chondrocytes, potentially through the activation of TGF-β and PI3K-Akt signaling pathways.
Gene set enrichment analysis (GSEA) identified that gene sets related to collagen trimer formation and cell adhesion molecules were significantly enriched in chondrocytes cultured on chExo/chM microcarriers compared to those on chM microcarriers (Fig. 5(g)). GO analysis further indicated that many of the enriched gene sets in chExo/chM microcarriers overlapped with those in chM microcarriers. However, chExo/chM microcarriers exhibited more pronounced changes in fold change and p-values, indicating a more substantial alteration in gene expressions attributed to the additional exosomes delivered through the chM microcarriers (Fig. 5(h)). Notably, gene sets related to metabolic pathways were uniquely enriched in chExo/chM microcarriers compared to chM microcarriers (Fig. 5(i)), including genes involved in lipid metabolism (such as B4galnt1, Ptdss2, Lpcat1, Lta4h, Pcca, and Tkfc) and fructose and mannose metabolism (including Tkfc, Pfkm, and Akr1b8). These results suggest that incorporating chExo into chM-coated microcarriers modulates the metabolic activity of chondrocytes, potentially contributing to the restoration of chondrocyte homeostasis at defect sites.
3.6 Chondrocyte membrane-coated microcarriers induce robust ECM synthesis via integrin-mediated mechanotransduction
Interestingly, chM microcarriers were found to impact the global gene expression profiles of chondrocytes, even in the absence of soluble factors like chExo. When chondrocytes were cultured on chM microcarriers, they exhibited increased expression of key anabolic genes while reducing the expression of catabolic genes (Fig. 4(j) and Fig. S9 and S11, ESI†). This suggests that chM microcarriers maintained a more anabolic phenotype in chondrocytes than bare microcarriers. Additionally, GSEA revealed that gene sets related to ECM structural constituents and proteoglycans were significantly enriched in chondrocytes cultured on chM microcarriers versus bare microcarriers (Fig. S11c, ESI†), resulting in substantial ECM accumulation on chM microcarriers after 20 days of culture (Fig. 4(k) and Fig. S10, ESI†). GO term analysis also demonstrated that up-regulated genes were enriched in molecular functions such as actin filament binding, GTPase activator activity, and integrin binding (Fig. 6(a)). Additionally, genes associated with biological processes like cell proliferation, migration, and response to mechanical stimulus were highly up-regulated in chM microcarriers (Fig. 6(b)). KEGG pathway analysis indicated that up-regulated genes in chM microcarriers were involved in cytoskeleton remodeling, including adherent junction, Hippo signaling pathway, focal adhesion, Rap1 signaling, and actin cytoskeleton regulation (Fig. 6(c)). These findings suggest that mechanotransduction signaling in chondrocytes is activated upon chM treatment.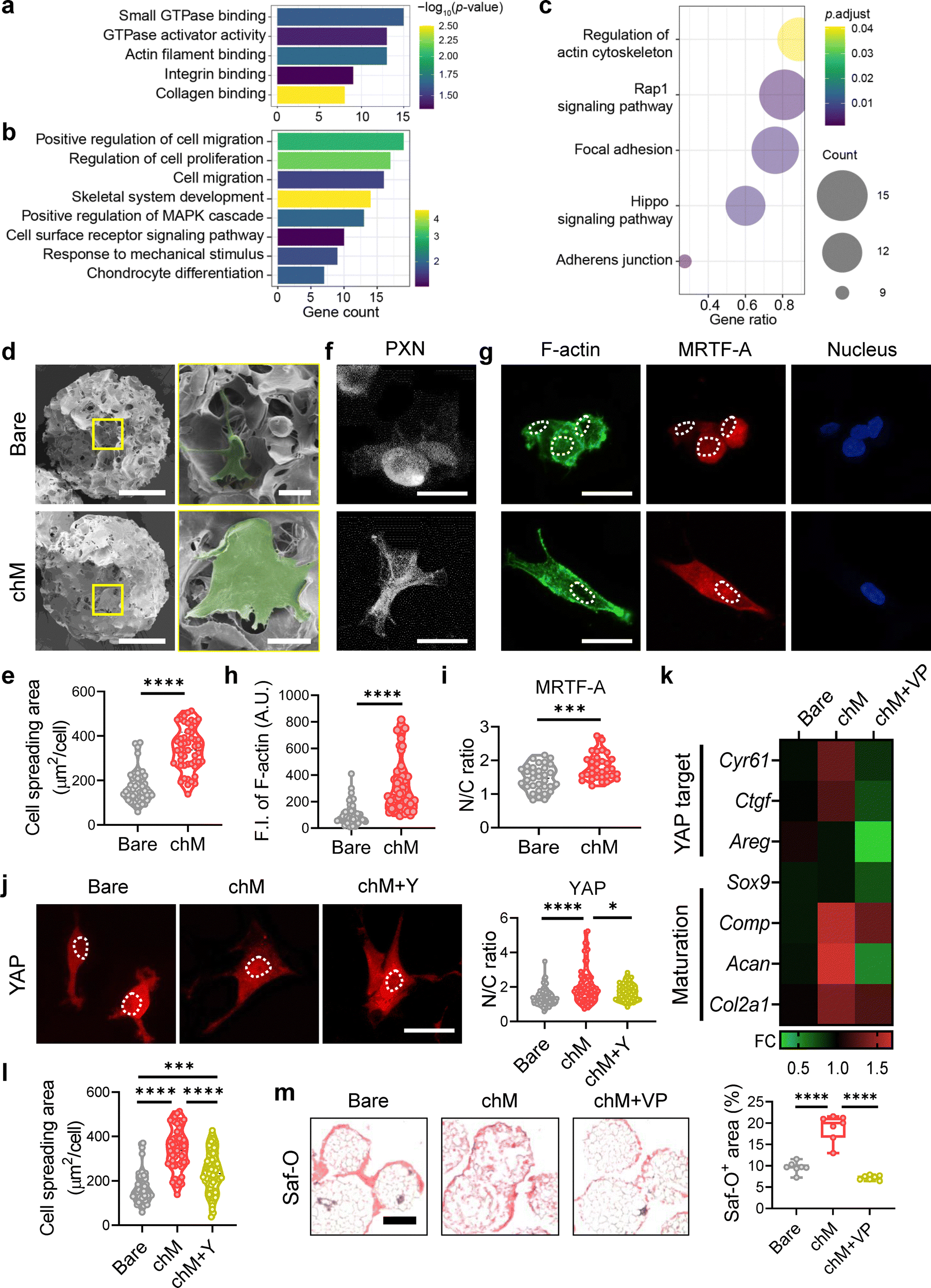 | ||
| Fig. 6 Chondrocyte membrane-coated microcarriers induce robust ECM synthesis via integrin-mediated mechanotransduction, potentially through the Rho/ROCK-actin-YAP axis. To elucidate the impact of chM microcarriers on the global gene expression profiles in chondrocytes, independent of soluble factors like chExo, we analyzed up-regulated DEGs (≥1.5-fold change, p < 0.05) in the chM microcarrier group relative to the bare microcarrier group. (a) and (b) Gene ontology analysis for molecular function (a) or biological process (b), and (c) KEGG pathway analysis of up-regulated DEGs in the chM microcarrier group compared to the bare microcarrier group. (d)–(j) Morphological and molecular analysis of chondrocytes attached to microcarriers after 1 day of culture. (d) Representative SEM images of chondrocytes attached on microcarriers. Cells are pseudo-colored with green. Scale bars: 100 μm (main images) and 20 μm (insets, yellow boxes). (e) Cell spreading area (μm2 per cell) of chondrocytes on microcarriers (n = 50 cells per group). (f) Inverted fluorescence images of chondrocytes stained for paxillin (PXN, white). Scale bar, 20 μm. (g) Representative co-staining images of chondrocytes for F-actin (left, green), MRTF-A (middle, red), and nuclei (right, blue). Scale bar, 20 μm. The nuclear area was outlined with dotted lines. (h) Fluorescent intensity (F.I.) of F-actin per cell (n = 60 cells per group). A.U.: arbitrary unit. (i) Nuclear-to-cytoplasmic (N/C) ratio of MRTF-A measured based on fluorescence intensity (n = 32–33 cells per group). (j) Immunostaining for YAP (red) in chondrocytes cultured on microcarriers with or without ROCK inhibitor Y-27632 (Y) treatment (left; scar bar, 20 μm), and corresponding quantification of the YAP intensity ratio in the nucleus and cytoplasm (right) (n = 57–74 cells per group). The nuclear area was outlined with dotted lines. (k) Heatmap showing the expression levels of YAP target genes (upper panel) and chondrocyte maturation-specific genes (lower panel) after 5 days of culture on microcarriers, with or without YAP inhibitor verteporfin (VP) treatment (n = 9 replicates per group). (l) Quantification of cell spreading areas after 1 day of culture with or without Y-27632 treatment (n = 29–50 cells per group). (m) Safranin-O (Saf-O, red) staining after 20 days of culture following VP treatment (left; scale bar, 200 μm), and corresponding quantification of Saf-O-positive stained areas normalized to total microcarrier areas per field (n = 7 fields per group). Data represent mean ± s.d. of n. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; Student's t-test for (e), (h), and (i); one-way ANOVA followed by Tukey's post hoc tests for (j), (i), and (m). | ||
Indeed, chM microcarriers facilitated greater cell adhesion and spreading than bare microcarriers (Fig. 6(d) and (e)). Chondrocytes cultured on chM microcarriers formed more pronounced focal adhesions, subsequently promoting actin development (Fig. 6(f)–(h) and Fig. S12, ESI†). This can be attributed to cartilage-specific ECM proteins associated with the chondrocyte membranes, such as aggrecan and type II collagen (Fig. 2(c)), which likely improved the adhesion of chondrocytes to chM microcarriers through integrins. Consequently, the nuclear localization of MRTF-A increased in chondrocytes cultured on chM microcarriers, driven by F-actin polymerization that dissociates G-actin from MRTF-A (Fig. 6(i)).54,55 Given that nuclear shuttling of mechanotransducers like MRTF-A and YAP is known to be mediated by actin cytoskeletons during the integrin-mediated mechanotransduction,56,57 we also assessed the nuclear localization of YAP. We observed that a more prominent nuclear localization of YAP in chondrocytes cultured on chM microcarriers compared to bare microcarriers (Fig. 6(j)). Furthermore, the expression of YAP target genes, including Cyr61, Ctgf, and Areg, was significantly up-regulated in chM microcarriers compared to bare ones, again indicating enhanced YAP nuclear activity (Fig. 6(k)). Given the crucial role of YAP in chondrocyte maturation,58,59 inhibiting YAP activity with verteporfin (VP) treatment led to a significant reduction in the expression of both YAP target genes and chondrocyte maturation markers (Fig. 6(k) and Fig. S13, ESI†).
To verify whether chondrocyte maturation depends on actin formation, we treated cells on chM microcarriers with specific inhibitors: the integrin activation inhibitor MK-0429, the Rho/ROCK inhibitor Y-27632, and the Rac1 inhibitor NSC-23766. Inhibitor concentrations were carefully selected to avoid cytotoxicity in subsequent experiments (Fig. S14, ESI†). While all inhibitors significantly decreased the cell spreading area, Rho/ROCK inhibition by Y-27632 notably disrupted nuclear YAP localization compared to integrin or Rac1 inhibition, suggesting that Rho/ROCK might regulate actin-mediated mechanotransduction via YAP in chondrocytes cultured on chM microcarriers (Fig. 6(l) and Fig. S15, ESI†). Consequently, this YAP inhibition substantially decreased the production of cartilage-specific ECM (Fig. 6(m) and Fig. S13c, ESI†). Together, these findings suggest that chM microcarriers preserve the anabolic phenotype of chondrocytes through integrin-mediated mechanotransduction, resulting in robust cartilage-specific ECM synthesis.
When compared to bare microcarriers, chondrocytes cultured on chM microcarriers exhibited significantly increased expressions of genes related to cell migration (Fig. 6(b)). Notably, the cellular infiltration of adhered cells in chM microcarriers was comparable to that in chExo/chM microcarriers (Fig. 4(i)). These findings indicate that chondrocyte-derived membranes on microcarriers enhance cellular contractility and mobility by promoting the formation of more focal adhesions and actin structures. While chondrocyte-derived exosomes released from the microcarriers induce robust chondrocyte recruitment over long distances, the chondrocyte membranes on the microcarriers subsequently expedite cell adhesion, infiltration, proliferation, and maturation in synergy with the exosomes.
3.7 chExo/chM improves cartilage repair in a rat osteochondral defect model
We assessed the efficacy of cartilage repair using chondrocyte-mimicking microcarriers in a rat osteochondral defect model. Critical defects, 1.6 mm in diameter, were created in the trochlear groove of rats and filled with microcarriers (approximately 50 particles per defect) (Fig. 7(a)). After 18 weeks of implantation, visual examination revealed that defects treated with chExo/chM microcarriers were fully filled with neo-tissue, seamlessly integrated with surrounding tissues (Fig. 7(b)). In contrast, defects treated with a single administration of free chExo or left untreated displayed rough surfaces, indicating incomplete recovery. Safranin-O staining showed a larger positively stained area in the chExo/chM microcarrier group compared to the other groups (Fig. 7(c) and (d)). Furthermore, the chExo/chM microcarrier group exhibited a more extensive area of positive type II collagen staining compared to the other groups (Fig. S16, ESI†). Masson's trichrome staining demonstrated the most well-reconstructed cartilage ECM in the chExo/chM group (Fig. S16, ESI†). Notably, host cells not only covered the defects but also infiltrated the implanted microcarriers (Fig. 7(e)). In the chExo/chM microcarrier group, these infiltrated cells displayed the characteristic spherical morphology of chondrocytes within lacunae, resembling hyaline cartilage. | ||
| Fig. 7 chExo/chM microcarriers enhance cartilage repair in a rat osteochondral defect model. (a) Schematic overview of the in vivo treatment with chondrocyte-mimicking microcarriers in a rat osteochondral defect model. In vivo evaluations were conducted 18 weeks post-implantation. The regenerative capacity of microcarriers (Bare, chM, or chExo/chM microcarriers) was compared to healthy cartilage (Intact), untreated cartilage defects (Untreated), and a free chExo treatment group (single administration on day 1). (b) Gross appearance of defects post-treatment. Scale bar, 1 cm. (c) Representative Saf-O-staining images, with black arrowheads indicating the margin of the original defects. Scale bar, 500 μm. (d) Quantitative analysis of positively stained areas for Saf-O (upper) and type II collagen (Col II, bottom) (n = 4–5 defects per group). UT: untreated, MC: bare microcarriers, chM-MC; chM-coated microcarriers, chExo/chM-MC; chExo and chM-decorated microcarriers, chExo; free chExo. (e) High-magnification images of Saf-O, MT, and COL II staining in defects treated with chExo/chM microcarriers. Scale bar, 100 μm (Saf-O and MT) and 50 μm (COL II). Superficial: superficial regions of regenerated tissues, Inside; interior of defects where microcarriers are located. The regions where microcarriers are located were marked with dashed lines. (f) International Cartilage Repair Society (ICRS) microscopic (upper) and histological (lower) scores. Data represent mean ± s.d. of n. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; one-way ANOVA followed by Tukey's post hoc tests for (d); Kruskal–Wallis test followed by Dunn's multiple comparisons test for (f). | ||
We conducted histological grading to assess the quality of cartilage repair, following the criteria outlined in Table S4 (ESI†). The International Cartilage Repair Society (ICRS) macroscopic scoring evaluated various aspects, including the degree of defect repair, edge integration, surface roughness, defect filling, and macroscopic appearance.46 The chExo/chM microcarrier group showed significantly higher ICRS macroscopic scores than the free chExo and untreated groups (Fig. 7(f)). Moreover, the histological grading scores of the chExo/chM microcarrier group were 1.3- and 2-fold higher than those of the free chExo and untreated groups, respectively. In comparison to the free chExo group, the chExo/chM microcarrier group exhibited statistically significant differences in surface regularity (p = 0.0388), integration (p = 0.0278), macroscopic appearance (p = 0.0082), and overall repair assessment (p = 0.0069). These findings suggest that the current scaffold-mediated exosome delivery system can achieve more effective cartilage repair by prolonging the therapeutic effects of exosomes beyond those with traditional bolus exosome treatment.
Regarding average histological scores, all microcarrier groups exhibited accelerated repair compared to the non-microcarrier-treated groups. Also, the chExo/chM microcarrier group showed statistically significant improvement in cell morphology compared to the bare microcarrier group (p = 0.0084). This result suggests that highly porous microcarriers are suitable as a scaffold material for cartilage repair. Moreover, the microcarriers appear to increase host cell recruitment through chExo release, which, coupled with their physical bridging effect, facilitates the engagement of cells from adjacent intact cartilage to the defect site, as demonstrated by the increased microcarrier clustering observed in vitro (Fig. 4(k)).
While cartilage regeneration was largely influenced by the composition of the microcarriers, all three types, bare, chM, and chExo/chM microcarriers, significantly enhanced bone regeneration compared to the untreated group and the free chExo treatment (Fig. S17, ESI†). Additionally, the free chExo treatment group showed no significant improvement in bone regeneration compared to the untreated group. This suggests that the greater bone regeneration observed in the microcarrier groups is due to the intrinsic bone regeneration capacity of the PCL material and the porous structure of the microcarriers.28,60,61 These findings indicate that the chondrocyte-mimicking microcarriers are particularly effective for cartilage repair. Although articular cartilage was not fully restored by week 18, the chondrocyte-mimicking microcarriers accelerated cartilage repair in osteochondral defects more effectively than either the chM microcarrier or exosome-only treatment, as evidenced by the regeneration of hyaline cartilage-like tissue and its successful integration with surrounding normal cartilage.
4. Discussion
The treatment of cartilage defects remains a significant clinical challenge worldwide, driven by the increasing prevalence of trauma, degenerative diseases, and aging populations.62–64 While autologous cartilage transplantation is a common approach for restoring functionality and appearance, it is hindered by limitations such as restricted availability and complications at the donor site.65–68 Although stem cell therapies have been explored as an alternative, these too carry risks such as cartilage fibrosis and potential tumorigenicity. Therefore, here we propose a cell-free approach that employs biomimetic scaffolds incorporating natural chondrocyte-derived bioactive components – specifically exosomes and cell membranes. Compared to cell-based methods, our approach is not only less immunogenic and tumorigenic but also offers off-the-shelf platforms that are cost-effective, stable, and reproducible – features that directly benefit clinical practice. Unlike cell-based products, these ready-to-use materials can be handled easily by surgeons without specialized cell-processing expertise, making them highly practical for routine clinical application.In addition, our streamlined production process – where exosomes and membranes are harvested from the same chondrocyte culture – achieves high yield and reproducibility, making large-scale production feasible. Thus, our biomimetic cartilage scaffolds represent a promising alternative, poised to overcome the limitations of autografts and meet the growing demand for effective cartilage repair.69–71
Chondrocytes, the primary cells responsible for cartilage regeneration, are essential for synthesizing extracellular matrix (ECM). In this study, we propose chondrocyte-mimicking scaffolds designed to stimulate host chondrocyte migration, proliferation, and maturation by replicating the chondrogenic niche. The scaffold comprises three key components: porous microcarriers, chondrocyte-derived exosomes, and chondrocyte-derived cell membranes (Fig. 8). The porous microcarriers provide structural support at the injury site and promote cell adhesion. The chondrocyte-derived exosomes help restore cartilage homeostasis by delivering chondrogenic microRNAs (miRNAs) and regulating metabolic processes. Meanwhile, the chondrocyte-derived membranes mimic the physicochemical properties of chondrocytes, promoting anabolic responses through mechanotransduction. Together, these components synergistically enhance cartilage repair.
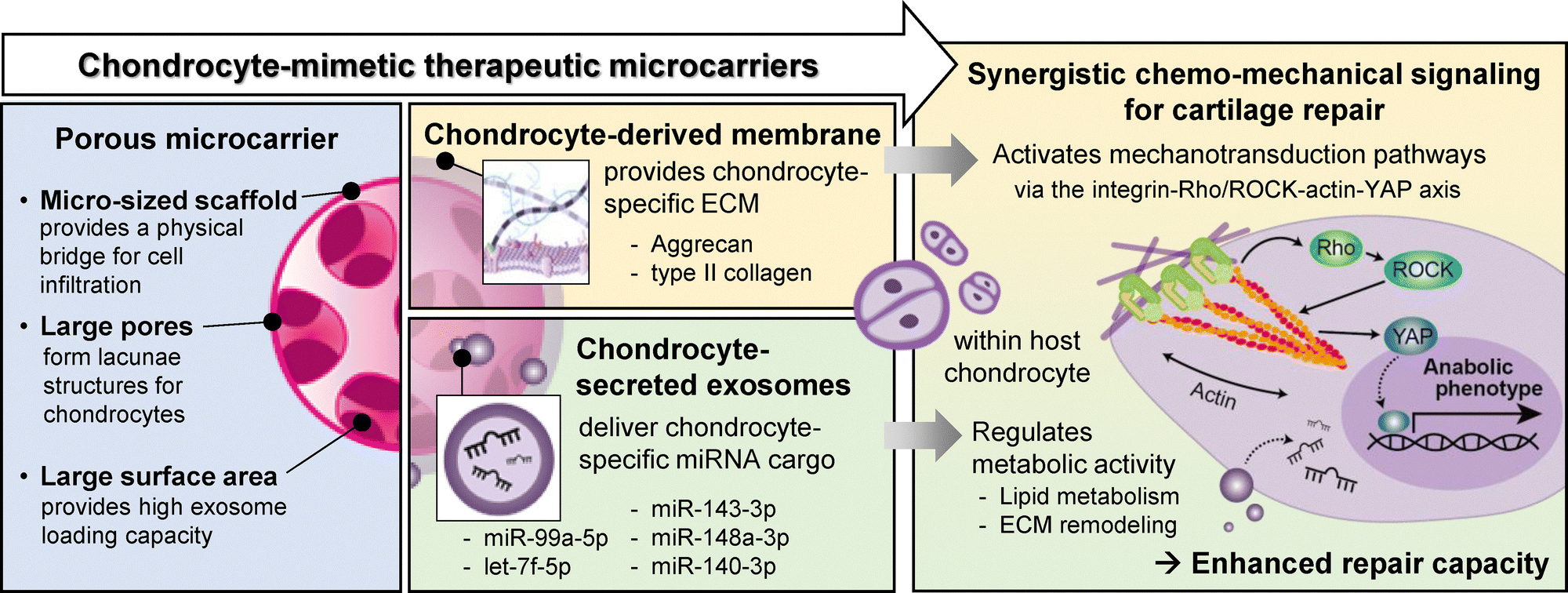 | ||
| Fig. 8 Mechanistic depiction of chondrocyte-mimetic therapeutic microcarriers orchestrating synergistic chemo-mechanical signaling in cartilage repair. These microcarriers consist of an interconnected microporous PCL scaffold that combines large, lacunae-forming pores with a high surface area to support host chondrocyte anchorage, infiltration, and exosome loading. A cloaking layer of chondrocyte-derived membrane preserves cartilage-specific ECM components such as aggrecan and type II collagen, recapitulating the native cell–matrix interface. The exosomes themselves are enriched in chondrogenic miRNAs (miR-143-3p, miR-148a-3p, miR-140-3p, miR-99a-5p, and let-7f-5p) that restore cartilage homeostasis by fine-tuning lipid metabolism and ECM remodeling. Engagement of integrins with this membrane-coated scaffold triggers the Rho/ROCK-actin-YAP mechanotransduction cascade in host chondrocytes to upregulate anabolic gene expression, while the delivered miRNAs further modulate metabolic pathways. Together, these complementary mechanical and biochemical cues recruit, proliferate, and mature chondrocytes at the defect site, ultimately enhancing the capacity for cartilage regeneration. | ||
Porous microcarriers were selected as the backbone scaffold material due to their numerous advantages in cartilage repair. These modular scaffolds can effectively fill defects of varying sizes and shapes.26,30,72 Previous studies have shown that the porous structure not only accelerates nutrient and oxygen transport but also serves as an excellent reservoir for both cells and drugs.73,74 For instance, Li et al. demonstrated that increasing the pore size of microcarriers significantly improved the thermal stability, storage stability, and reusability of drugs.75 In another study, Wei et al. reported that highly open porous microcarriers, with pore sizes ranging from 10 to 60 μm and interconnected passages averaging 8.8 μm, led to significantly higher cell adhesion and proliferation compared to less porous microcarriers.44 In our study, we also demonstrated that the large surface area and interconnected porous networks of the microcarriers support long-term cell proliferation while enhancing control over exosome loading and sustained release. Additionally, the cell membrane coating on the microcarriers acts as a drug reservoir,39,40 improving exosome delivery efficacy further compared to traditional bolus delivery systems. This approach addresses current limitations in exosome delivery, such as low stability and retention within the body.16,17 The cell membrane layer also allows localized exosome loading on the scaffold surface, reducing the dosage and administration frequency needed for effective cartilage repair. Together, our scaffold-mediated exosome delivery system significantly enhances exosome delivery efficiency, thereby promoting cartilage regeneration.
Notably, the microcarriers serve as a physical bridge in cartilage defects, accelerating host cell migration by filling voids.76 Normally, chondrocytes do not migrate readily due to the surrounding dense, proteoglycan-rich pericellular matrix.77 Recent studies have demonstrated that enhancing host chondrocyte migration from intact regions to the remote injury site can accelerate cartilage repair.78 For instance, clinical use of collagenase at poorly integrated boundaries between cartilage graft and host cartilage has been shown to facilitate chondrocyte migration and cartilage fusion.77,79,80 Additionally, overexpression of matrix remodeling gene Mmp14 has been found to accelerate articular chondrocyte migration and enhance cartilage repair.81 In our approach, we enhanced host chondrocyte migration and infiltration by coating the microcarriers with homologous chondrocyte cell membranes, which improved cell affinity to the scaffold surface. This resulted in significantly enhanced host cell infiltration, with up-regulated migration-related genes, such as Pdgfrb, Mmp14, and Prg4 (Fig. 4(h)), which are also markers of cartilage stem/progenitor cells.82–84 Our findings suggest that the physical bridging provided by the scaffolds stimulates chondrocytes to adopt a highly mobile, proliferative phenotype resembling cartilage stem/progenitor cells, thereby facilitating cartilage repair.
In addition, the pores in the microcarriers mimic the lacunae structure of native cartilage, which is beneficial for cartilage repair.34,35 This lacunar structure has been shown to facilitate chondrocyte-driven remodeling of the ECM, promoting the regeneration of hyaline cartilage microtissue. Also, it prevents a metabolic shift from anaerobic to aerobic conditions due to geometric constraints.35 In our study, we observed that chondrocytes initially attached to the surface and pores of the microcarriers, gradually infiltrating and filling the interconnected pores over time (Fig. 4(i)). Actively proliferating chondrocytes on the microcarriers formed large clusters that expanded between the microcarriers, recreating a lacunae-like structure along with substantial deposition of cartilage ECM (Fig. 4(k)). Notably, in vivo results showed that chondrocytes migrated and infiltrated into microcarriers within the defects, forming a lacunae structure (Fig. 7(e)), indicating successful regeneration of hyaline cartilage supported by the porous microcarriers.
Our most notable finding was that coating the porous microcarriers with chondrocyte-derived cell membranes significantly enhanced chondrocyte adhesion and proliferation compared to non-coated microcarriers. Cell membrane cloaking is one of the most effective biomimetic strategies as it retains the unique characteristics of parental cells, enabling specific cell–cell communication.85,86 For instance, cancer cell membrane-coated nanoparticles have demonstrated enhanced targeting efficiency toward homologous cancer cells due to their inherent affinity.87 Similarly, nanofibers coated with pancreatic beta-cell membranes have been shown to promote beta-cell proliferation and function.88 In our previous studies, we also observed that even suspension cells, such as leukemia stem cells (LSCs) and T cells, exhibited high reactivity to cell membrane-coated scaffolds.39,40 For example, LSCs displayed a strong affinity for bone marrow-derived mesenchymal stem cell (BMSC) membrane-coated scaffolds, likely due to VLA-4/VCAM-1 interaction.39 Likewise, T cells formed clusters on dendritic cell membrane-coated scaffolds, potentially initiated by MHC I-antigen interactions that triggered T cell receptor clustering, leading to successful T cell activation and expansion.40
While our previous studies focused on the interactions between cell membrane surface molecules and target cells, the current study further demonstrated that ECM components associated with the cell membranes also play a crucial role in enhancing cell adhesion. We confirmed that chondrocyte-derived cell membranes contain significant amounts of cartilage-specific ECM components, such as aggrecan and type II collagen (Fig. 2(c)). Coating the microcarriers with these membranes reduced surface stiffness and increased hydrophilicity, leading to significantly enhanced chondrocyte and MSC attachment compared to bare microcarriers (Fig. 4(e), (f) and Fig. S7a, ESI†). Notably, there was no difference in initial cell adhesion between chondrocyte membrane-coated microcarriers (chM microcarriers) and those loaded with exosomes (chExo/chM microcarriers), indicating that the cell membrane coating, rather than exosome delivery, played the primary role in promoting cell adhesion.
Interestingly, even without exosomes, chondrocytes cultured on cell membrane-coated microcarriers exhibited a significant up-regulation of genes related to anabolic chondrogenic phenotype and cartilage-specific ECM synthesis. Genes associated with cytoskeleton rearrangement and ECM remodeling were also enriched in this group, suggesting that this effect is driven by the activation of mechanotransduction signaling. This activation is likely triggered by cartilage-derived ECM molecules present on the cell membrane-coated scaffolds. The cell membranes provide cartilage-specific physical cues, such as stiffness and the type and density of adhesion moieties, which lead to pronounced morphological changes that enhance chondrocyte functions and maturations through integrin-mediated mechanotransduction.
Mechanotransduction is the process by which cells convert mechanical stimuli, such as substrate stiffness, topography, and mechanical loading, into biochemical responses. Cells sense these mechanical cues and respond by altering cytoskeletal architecture, forming focal adhesions, and modulating transcription factor activity.89–91 As a mechanoadaptive tissue, articular cartilage relies on chondrocytes, its predominant cell type, to adapt to mechanical forces.92 It is well established that chondrocytes transduce dynamic physiological forces to regulate the expression of key matrix molecules, increasing anabolic activity in response to physiological loading and shifting to catabolic activity under pathological or injury conditions.3,93 While the precise molecular mechanisms by which chondrocytes adapt to their physical microenvironment are still under investigation, recent evidence highlights the critical role of actin dynamics in controlling the chondrocyte phenotype.94 The Rho/ROCK pathway, which promotes F-actin fiber formation, has been shown to activate YAP, a key mechanotransducer that responds to mechanical signals, thereby regulating chondrogenesis in vivo.95 YAP is pivotal in chondrocyte mechanotransduction, as its expression levels and nuclear localization directly influence the expression of canonical chondrogenic genes, including Sox9, Col2a1, and Acan,95 as well as matrix remodeling enzymes.96 Furthermore, substrate stiffness is a critical mechanical cue that governs the chondrocyte phenotype. Chondrocytes cultured on supraphysiological substrates, such as tissue culture plastic (∼10 GPa), tend to remodel their actin cytoskeleton into a stress fiber-dominant cytoarchitecture.97,98 High substrate stiffness (∼55 kPa) has been shown to reduce the chondrocyte phenotype by increasing stress fiber formation, whereas softer substrates (∼5 kPa)95 or 3D culture systems97,98 can rescue this phenotype by disrupting stress fibers.
Interestingly, in this study, we observed that chondrocytes with a more spread morphology on the chondrocyte membrane-coated microcarriers exhibited more significant chondrogenic maturation compared to those on non-coated microcarriers, where cells remained more spherical with limited spreading. This finding contrasts with previous reports. On the cell membrane-coated microcarriers, chondrocytes formed significantly more focal adhesions and actin structures, likely due to the presence of ECM components associated with the cell membranes. This increased adhesion promoted chondrocyte spreading, leading to YAP nuclear localization and activation. Consequently, chondrogenic gene expression was significantly elevated on the cell membrane-coated microcarriers compared to the non-coated microcarriers, where cells showed reduced spreading, along with decreased focal adhesion formation and stress fiber development.
Importantly, when chondrocytes were cultured on the cell membrane-coated microcarriers with Rho/ROCK pathway inhibition, YAP nuclear localization was significantly suppressed. Furthermore, direct inhibition of YAP nuclear localization led to a marked reduction in chondrogenic gene expression. Although these findings differ from previous reports, they align with the consensus that the Rho/ROCK-actin-YAP axis is a critical signaling pathway in chondrocyte mechanotransduction. We propose that the exceptionally soft surface of the cell membrane-coated microcarriers (∼0.5 kPa) serves as a reversible mechanical cue. Interestingly, while YAP gene expression was not significantly up-regulated, gene sets associated with the Hippo signaling pathway were enriched in the cell membrane-coated microcarrier group compared to the non-coated group. This indicates that YAP activity, regulated by its nuclear localization rather than its expression level, is modulated by integrin-mediated mechanoactivation in our scaffold system. These results further suggest that actin-dependent YAP nuclear localization is a crucial mechanism driving chondrocyte proliferation and maturation in our chondrocyte-mimicking scaffolds.
While YAP is undoubtedly a key mechanotransducer in chondrogenesis,58,93 its role is highly context-dependent. Although YAP positively regulates the proliferation of primary chondrocytes in vitro, its primary function in cartilage development appears to control tissue morphogenesis through the regulation of ECM rather than directly influencing cell proliferation or fate in vivo.96 Our future research will focus on understanding how substrate stiffness and adhesion affinity specifically regulate chondrogenesis and how these factors can be harnessed to modulate the chondrocyte phenotype therapeutically. Taken together, our study offers new insights into the mechanical regulation of the chondrocyte phenotype induced by biomaterials, which can be leveraged for therapeutic approaches in cartilage repair. Future studies will aim to identify the optimal combination of physical cues such as pore size, curvature, and stiffness of porous microcarriers that can specifically guide cells toward a chondrogenic phenotype.
Our final key component is chondrocyte-derived exosomes. Recently, exosomes have been recognized as principal agents mediating the therapeutic efficacy of cell-based regenerative medicine approaches for cartilage repair.19,99–101 Exosomes are attractive candidates due to their pivotal roles in cellular communication and tissue repair, where they regulate vital processes such as proliferation, differentiation, and recruitment.12,14,102 However, identifying the optimal cell sources for isolating exosomes with high chondrogenic potential remains an active area of research. While exosomes derived from MSCs have been extensively studied, the impact of chondrocyte-derived exosomes on cartilage regeneration has received comparatively little attention. Given that chondrocytes, along with MSCs, are critical cell types in cartilage tissue, and their proximity to the cartilaginous niche suggests a strong potential for promoting tissue repair. Moreover, since the primary goal of cartilage repair therapy is to restore the ECM to its native state, using compositionally healthy, physiologically relevant exosomes derived from chondrocytes is a logical approach. Despite this, limited research has been conducted on chondrocyte-derived exosomes.
Emerging evidence suggests that chondrocytes can steer the chondrogenesis of stem cells through paracrine effects, both in vitro and in vivo.103,104 For instance, chondrocyte-derived exosomes have been shown to induce chondrogenic differentiation in MSCs by transferring miRNAs, thereby promoting cartilage regeneration.105 Additionally, these exosomes positively regulate chondrocyte proliferation, migration, and differentiation and efficiently induce the polarization of M1 to M2 macrophages, alleviating osteoarthritis (OA) symptoms.106 Interestingly, recent studies have demonstrated that chondrocyte-derived exosomes reduce angiogenesis during cartilage regeneration compared to exosomes derived from bone marrow MSC (BMSC).104 While BMSC-derived exosomes up-regulate angiogenic factors such as SDF-1 and VEGF, potentially leading to ectopic cartilage hypertrophy, chondrocyte-derived exosomes offer the advantage of limiting vascular invasion – one of the critical drivers of hypertrophic cartilage differentiation – thus making them more favorable for successful cartilage regeneration.
Exosome composition varies depending on the cell source. Among the various cargoes, exosomal miRNAs have been linked to cartilage development and homeostasis, with dysregulation of these miRNAs associated with aging and OA.107–109 Recently, the roles of specific miRNAs in maintaining cartilage differentiation and homeostasis have gained increasing recognition. Our results confirmed that, compositionally, 30% of the miRNAs in chondrocyte-derived exosomes overlap with those in MSC-derived exosomes, suggesting that these miRNAs are evolutionarily conserved.99,110 However, the most abundant miRNAs in chondrocyte-derived exosomes, distinct from those in MSC-derived exosomes, were miR-143-3p, followed by miR-148a-3p, miR-140-3p, miR-99a-5p, and let-7f-5p.
Chondrocytes in the superficial zone of articular cartilage, which possess stem or progenitor capacity,110,111 actively respond during repair and secrete exosomes enriched with specific miRNAs, including miR-221, miR-222, miR-143, and miR-145.112 The expression of miR-140 and miR-143 positively correlates with a chondrogenic phenotype,112 while miR-221 and miR-145 negatively regulate chondrogenesis.113,114 Previous reports have shown that miR-140 knock-out mice exhibit a mild skeletal phenotype with age-related OA-like changes, such as ECM loss and cartilage fibrillation. In contrast, transgenic mice overexpressing miR-140 in cartilage are resistant to antigen-induced arthritis.115 Additionally, ADAMTS-5 metalloproteinase, a key enzyme in cartilage matrix remodeling, has been identified as a target gene for miR-140.115 Expression of miR-140 is reduced in cartilage progenitor/stem cells derived from OA cartilage, while treatment with miR-140 has been shown to alleviate OA-like changes in these cells.116 Similarly, treatment of chondrocytes with exosomes overexpressing miR-140 increases secretion of ECM components, such as collagen and aggrecan, and intra-articular injection of these exosomes enhances cartilage regeneration in an OA rat model.117
In the current study, KEGG pathway analysis revealed that miRNAs in chondrocyte-derived exosomes are likely to induce chondrocytes into an active state of lipid synthesis and matrix production, mainly through the glycan and lipid biosynthesis pathways (Fig. 3(g) and Table S5, ESI†). This finding underscores the importance of maintaining cell membrane integrity and extracellular interactions, both critical for cartilage function. Glycosphingolipids, essential for cell-to-cell communication, may help preserve cartilage matrix integrity and respond to environmental stresses.118 Mucins, heavily glycosylated proteins, contribute to cellular protection and lubrication, indicating a potential increase in the production of protective ECM components by chondrocytes.119–121
Subsequently, the chondrocyte-derived exosome treatment altered the expression of genes related to lipid metabolism and ECM production in chondrocytes (Fig. 5(i)). For example, the up-regulation of Lpcat1, involved in phospholipid remodeling, may reflect an adaptive response to maintain membrane homeostasis or address altered metabolic conditions in chondrocytes. Similarly, Ppt1 up-regulation, associated with the degradation of lipid-modified proteins, could indicate increased protein turnover and degradation.122Ptdss2 plays a role in synthesizing phosphatidylserine, a crucial phospholipid in membrane composition and signaling, while B4galnt1, involved in glycosphingolipid biosynthesis, enhances glycosphingolipid production, which is important for membrane structure and cell signaling. Moreover, genes associated with ECM remodeling were more enriched in the chondrocyte-derived exosome-loaded microcarrier group compared to the non-loaded group (Fig. 5(f)–(h)), suggesting that chondrocytes actively modify their ECM and lipid composition in response to exosome treatment.
Notably, treatment with chondrocyte-derived exosomes increased chondrocyte migration, while no similar effect was observed on MSC migration (Fig. S7, ESI†). Although the mechanism behind this chondrocyte-specific migration is not yet fully understood, we believe that exosomes derived from healthy cartilage chondrocytes may stimulate these cells to become more active and functional. These exosomes likely carry signals that help maintain functional zonal differences and cartilage homeostasis by regulating mechanotransduction pathways, as the miRNAs in chondrocyte-derived exosomes are also enriched in weight-bearing regions of cartilage.112,123 In conjunction with exosomal miRNAs, the surface physical properties of the chondrocyte membrane-coated microcarriers and the exosomes can synergistically activate integrin-mediated mechanotransduction, promoting chondrocyte migration, proliferation, and maturation, ultimately leading to successful cartilage regeneration.
Although our chondrocyte-mimetic system effectively recruits and supports host chondrocytes, it may be insufficient for large or severely degenerative defects such as osteoarthritis or aged cartilage. To enhance functional integration and long-term durability, future work could combine this cell-free approach with additional potent biochemical cues such as TGF-β3 and engineer zonal or gradient architecture by layering scaffolds with different stiffnesses or pore structures.
5. Conclusions
We successfully developed chondrocyte-mimicking microcarriers by coating porous microcarriers with chondrocyte-derived membranes and loading them with chondrocyte-secreted exosomes. The cell membrane coating replicates the physicochemical properties of native chondrocytes, containing chondrocyte-specific surface molecules and ECM. This coating enhanced chondrocyte adhesion and activated anabolic responses through integrin-mediated mechanotransduction. Additionally, the exosome restored cartilage homeostasis by delivering chondrogenic miRNAs, regulating lipid metabolism, and extracellular remodeling. Using scaffold-mediated delivery, our system achieves a highly localized and sustained release of exosomes at the implanted sites, in contrast to conventional bolus delivery. Collectively, our scaffold system synergizes chemo-mechanical signaling, significantly promoting cartilage repair in a rat osteochondral defect model. This study presents a promising approach to advancing cartilage regenerative therapies.Author contributions
Conceptualization, H. S. K.; methodology, J. H. L., Z. Z., J. Y. Y., and N. H. L.; investigation, N. M., C. J. L., Z. Z., J. Y. Y., and N. H. L.; writing – original draft, H. S. K., N. H. L., Z. Z. and J. Y. Y.; writing – review & editing, H. W. K. and H. S. K.; funding acquisition, H. W. K. and H. S. K.; resources, J. H. L. and H. S. K.; supervision, H. W. K. and H. S. K.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The authors thank Dr. Hyung-Sik Kim (Pusan National University) for his kind support for the experiments. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2021-NR060095, RS-2024-00353064, 2022R1I1A1A01070131, RS-2024-00463860, RS-2024-00348908, and RS-2023-00220408).References
- W. Zhang, C. Ling, A. Zhang, H. Liu, Y. Jiang and X. Li, et al., An all-silk-derived functional nanosphere matrix for sequential biomolecule delivery and in situ osteochondral regeneration, Bioact. Mater., 2020, 5(4), 832–843 Search PubMed.
- Y. Li, L. Li, M. Wang, B. Yang, B. Huang and S. Bai, et al., O-alg-THAM/gel hydrogels functionalized with engineered microspheres based on mesenchymal stem cell secretion recruit endogenous stem cells for cartilage repair, Bioact. Mater., 2023, 28, 255–272 CAS.
- A. J. Sophia Fox, A. Bedi and S. A. Rodeo, The basic science of articular cartilage: structure, composition, and function, Sports Health, 2009, 1(6), 461–468 CrossRef PubMed.
- Z. Yang, H. Li, Z. Yuan, L. Fu, S. Jiang and C. Gao, et al., Endogenous cell recruitment strategy for articular cartilage regeneration, Acta Biomater., 2020, 114, 31–52 CrossRef CAS PubMed.
- T. Zhao, X. Li, H. Li, H. Deng, J. Li and Z. Yang, et al., Advancing drug delivery to articular cartilage: From single to multiple strategies, Acta Pharm. Sin. B, 2023, 13(10), 4127–4148 CrossRef CAS PubMed.
- D. C. Flanigan, J. D. Harris, P. M. Brockmeier and R. A. Siston, The effects of lesion size and location on subchondral bone contact in experimental knee articular cartilage defects in a bovine model, Arthroscopy, 2010, 26(12), 1655–1661 CrossRef PubMed.
- W. Widuchowski, J. Widuchowski and T. Trzaska, Articular cartilage defects: Study of 25124 knee arthroscopies, Knee, 2007, 14(3), 177–182 CrossRef CAS PubMed.
- L. Zhou, J. Xu, A. Schwab, W. Tong, J. Xu and L. Zheng, et al., Engineered biochemical cues of regenerative biomaterials to enhance endogenous stem/progenitor cells (ESPCs)-mediated articular cartilage repair, Bioact. Mater., 2023, 26, 490–512 CAS.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanasiou, Repair and tissue engineering techniques for articular cartilage, Nat. Rev. Rheumatol., 2015, 11(1), 21–34 CrossRef CAS PubMed.
- A. R. Armiento, M. J. Stoddart, M. Alini and D. Eglin, Biomaterials for articular cartilage tissue engineering: Learning from biology, Acta Biomater., 2018, 65, 1–20 CrossRef CAS PubMed.
- N. T. Chan, M.-S. Lee, Y. Wang, J. Galipeau, W.-J. Li and W. Xu, CTR9 drives osteochondral lineage differentiation of human mesenchymal stem cells via epigenetic regulation of BMP-2 signaling, Sci. Adv., 2022, 8(46), eadc9222 CrossRef CAS PubMed.
- F. Tan, X. Li, Z. Wang, J. Li, K. Shahzad and J. Zheng, Clinical applications of stem cell-derived exosomes, Signal Transduction Targeted Ther., 2024, 9(1), 17 CrossRef PubMed.
- S. Sadeghi, F. R. Tehrani, S. Tahmasebi, A. Shafiee and S. M. Hashemi, Exosome engineering in cell therapy and drug delivery, Inflammopharmacology, 2023, 31(1), 145–169 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu, H. Liu and W. H. Tang, Exosomes: biogenesis, biologic function and clinical potential, Cell Biosci., 2019, 9(1), 19 CrossRef PubMed.
- M. Dimik, P. Abeysinghe, J. Logan and M. Mitchell, The exosome: a review of current therapeutic roles and capabilities in human reproduction, Drug Delivery Transl. Res., 2023, 13(2), 473–502 CrossRef PubMed.
- A. K. Riau, H. S. Ong, G. H. F. Yam and J. S. Mehta, Sustained Delivery System for Stem Cell-Derived Exosomes, Front. Pharmacol., 2019, 10, 1368 CrossRef CAS PubMed.
- T. Imai, Y. Takahashi, M. Nishikawa, K. Kato, M. Morishita and T. Yamashita, et al., Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice, J. Extracell. Vesicles, 2015, 4(1), 26238 CrossRef PubMed.
- S. Zhang, W. C. Chu, R. C. Lai, S. K. Lim, J. H. Hui and W. S. Toh, Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration, Osteoarthritis Cartilage, 2016, 24(12), 2135–2140 CrossRef CAS PubMed.
- S. Zhang, S. J. Chuah, R. C. Lai, J. H. P. Hui, S. K. Lim and W. S. Toh, MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity, Biomaterials, 2018, 156, 16–27 CrossRef CAS PubMed.
- P. Chen, L. Zheng, Y. Wang, M. Tao, Z. Xie and C. Xia, et al., Desktop-stereolithography 3D printing of a radially oriented extracellular matrix/mesenchymal stem cell exosome bioink for osteochondral defect regeneration, Theranostics, 2019, 9(9), 2439–2459 CrossRef CAS PubMed.
- H. Hu, L. Dong, Z. Bu, Y. Shen, J. Luo and H. Zhang, et al., miR-23a-3p-abundant small extracellular vesicles released from Gelma/nanoclay hydrogel for cartilage regeneration, J. Extracell. Vesicles, 2020, 9(1), 1778883 CrossRef CAS PubMed.
- M. Zhai, Y. Zhu, M. Yang and C. Mao, Human Mesenchymal Stem Cell Derived Exosomes Enhance Cell-Free Bone Regeneration by Altering Their miRNAs Profiles. Advanced, Science, 2020, 7(19), 2001334 CAS.
- T.-M. De Witte, L. E. Fratila-Apachitei, A. A. Zadpoor and N. A. Peppas, Bone tissue engineering via growth factor delivery: from scaffolds to complex matrices, Regener. Biomater., 2018, 5(4), 197–211 CrossRef CAS PubMed.
- Y. Xie, Q. Guan, J. Guo, Y. Chen, Y. Yin and X. Han, Hydrogels for Exosome Delivery in Biomedical Applications, Gels, 2022, 8(6), 328 CrossRef CAS PubMed.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy and A. Khademhosseini, et al., Controlling the Porosity and Microarchitecture of Hydrogels for Tissue Engineering, Tissue Eng., Part B, 2010, 16(4), 371–383 CrossRef CAS PubMed.
- V. Gupta, Y. Khan, C. J. Berkland, C. T. Laurencin and M. S. Detamore, Microsphere-Based Scaffolds in Regenerative Engineering, Annu. Rev. Biomed. Eng., 2017, 19, 135–161 CrossRef CAS PubMed.
- L. Bai, X. Zhang, Z. Han, X. Yang and Y. Hao, Injectable porous microspheres for articular cartilage regeneration through in situ stem cell recruitment and macrophage polarization, Acta Biomater., 2024, 185, 429–440 CrossRef CAS PubMed.
- Q. Li, B. Chang, H. Dong and X. Liu, Functional microspheres for tissue regeneration, Bioact. Mater., 2023, 25, 485–499 CAS.
- R. J. Kulchar, B. R. Denzer, B. M. Chavre, M. Takegami and J. Patterson, A Review of the Use of Microparticles for Cartilage Tissue Engineering, Int. J. Mol. Sci., 2021, 22(19), 10292 CrossRef CAS PubMed.
- X. Li, X. Li, J. Yang, J. Lin, Y. Zhu and X. Xu, et al., Living and Injectable Porous Hydrogel Microsphere with Paracrine Activity for Cartilage Regeneration, Small, 2023, 19(17), 2207211 CrossRef CAS PubMed.
- C. Martins, F. Sousa, F. Araújo and B. Sarmento, Functionalizing PLGA and PLGA Derivatives for Drug Delivery and Tissue Regeneration Applications, Adv. Healthcare Mater., 2018, 7(1), 1701035 CrossRef CAS PubMed.
- Y. Y. Gong, J. X. Xue, W. J. Zhang, G. D. Zhou, W. Liu and Y. Cao, A sandwich model for engineering cartilage with acellular cartilage sheets and chondrocytes, Biomaterials, 2011, 32(9), 2265–2273 CrossRef CAS PubMed.
- R. E. Wilusz, J. Sanchez-Adams and F. Guilak, The structure and function of the pericellular matrix of articular cartilage, Matrix Biol., 2014, 39, 25–32 CrossRef CAS PubMed.
- H. Li, T. Zhao, Z. Yuan, T. Gao, Y. Yang and R. Li, et al., Cartilage lacuna-biomimetic hydrogel microspheres endowed with integrated biological signal boost endogenous articular cartilage regeneration, Bioact. Mater., 2024, 41, 61–82 CAS.
- S.-L. Ding, X.-Y. Zhao, W. Xiong, L.-F. Ji, M.-X. Jia and Y.-Y. Liu, et al., Cartilage Lacuna-Inspired Microcarriers Drive Hyaline Neocartilage Regeneration, Adv. Mater., 2023, 35(30), 2212114 CrossRef CAS PubMed.
- M. Wang, Z. Deng, Y. Guo and P. Xu, Designing functional hyaluronic acid-based hydrogels for cartilage tissue engineering, Mater. Today Bio, 2022, 17, 100495 CrossRef CAS PubMed.
- A. Karimizade, E. Hasanzadeh, M. Abasi, S. E. Enderami, E. Mirzaei and N. Annabi, et al., Collagen short nanofiber-embedded chondroitin sulfate–hyaluronic acid nanocomposite: A cartilage-mimicking in situ-forming hydrogel with fine-tuned properties, Int. J. Biol. Macromol., 2024, 266, 131051 CrossRef CAS PubMed.
- P. C. O. Ching, F.-H. Chen, I. H. Lin, D.-T. Tran, L. L. Tayo and M.-L. Yeh, Evaluation of Articular Cartilage Regeneration Properties of Decellularized Cartilage Powder/Modified Hyaluronic Acid Hydrogel Scaffolds, ACS Omega, 2024, 9(31), 33629–33642 CrossRef CAS PubMed.
- T.-C. Ho, H. S. Kim, Y. Chen, Y. Li, M. W. LaMere and C. Chen, et al., Scaffold-mediated CRISPR-Cas9 delivery system for acute myeloid leukemia therapy, Sci. Adv., 2021, 7(21), eabg3217 CrossRef CAS PubMed.
- H. S. Kim, T.-C. Ho, M. J. Willner, M. W. Becker, H.-W. Kim and K. W. Leong, Dendritic cell-mimicking scaffolds for ex vivo T cell expansion, Bioact. Mater., 2023, 21, 241–252 CAS.
- B. Chang, G. Nave and S. Jung, Drop formation from a wettable nozzle, Commun. Nonlinear Sci. Numer. Simul., 2012, 17(5), 2045–2051 CrossRef.
- C. J. Li, J.-H. Park, G. S. Jin, N. Mandakhbayar, D. Yeo and J. H. Lee, et al., Strontium/Silicon/Calcium-Releasing Hierarchically Structured 3D-Printed Scaffolds Accelerate Osteochondral Defect Repair, Adv. Healthcare Mater., 2024, 13(20), 2400154 CrossRef CAS PubMed.
- L. Simon, J. Constanzo, C. Terraza-Aguirre, Z. Ibn Elfekih, J. Berthelot and B. T. Benkhaled, et al., Surface modification of extracellular vesicles with polyoxazolines to enhance their plasma stability and tumor accumulation, Biomaterials, 2025, 313, 122748 CrossRef CAS PubMed.
- D.-X. Wei, J.-W. Dao and G.-Q. Chen, A Micro-Ark for Cells: Highly Open Porous Polyhydroxyalkanoate Microspheres as Injectable Scaffolds for Tissue Regeneration, Adv. Mater., 2018, 30(31), 1802273 CrossRef PubMed.
- H. S. Kim, N. Mandakhbayar, H.-W. Kim, K. W. Leong and H. S. Yoo, Protein-reactive nanofibrils decorated with cartilage-derived decellularized extracellular matrix for osteochondral defects, Biomaterials, 2021, 269, 120214 CrossRef CAS PubMed.
- M. P. J. van den Borne, N. J. H. Raijmakers, J. Vanlauwe, J. Victor, S. N. de Jong and J. Bellemans, et al., International Cartilage Repair Society (ICRS) and Oswestry macroscopic cartilage evaluation scores validated for use in Autologous Chondrocyte Implantation (ACI) and microfracture, Osteoarthritis Cartilage, 2007, 15(12), 1397–1402 CrossRef CAS PubMed.
- S.-M. Hong, J.-Y. Yoon, J.-R. Cha, J. Ahn, N. Mandakhbayar and J. H. Park, et al., Hyperelastic, shape-memorable, and ultra-cell-adhesive degradable polycaprolactone-polyurethane copolymer for tissue regeneration, Bioeng. Transl. Med., 2022, 7(3), e10332 CrossRef CAS PubMed.
- J. Song, X. Zeng, C. Li, H. Yin, S. Mao and D. Ren, Alteration in cartilage matrix stiffness as an indicator and modulator of osteoarthritis, Biosci. Rep., 2024, 44(1), BSR20231730 CrossRef CAS PubMed.
- W. R. Jones, H. Ping Ting-Beall, G. M. Lee, S. S. Kelley, R. M. Hochmuth and F. Guilak, Alterations in the Young's modulus and volumetric properties of chondrocytes isolated from normal and osteoarthritic human cartilage, J. Biomech., 1999, 32(2), 119–127 CrossRef CAS PubMed.
- E. M. Darling, R. E. Wilusz, M. P. Bolognesi, S. Zauscher and F. Guilak, Spatial Mapping of the Biomechanical Properties of the Pericellular Matrix of Articular Cartilage Measured In Situ via Atomic Force Microscopy, Biophys. J., 2010, 98(12), 2848–2856 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana and D. Dehaini, et al., Nanoparticle biointerfacing by platelet membrane cloaking, Nature, 2015, 526(7571), 118–121 CrossRef CAS PubMed.
- E. Mianehsaz, H. R. Mirzaei, M. Mahjoubin-Tehran, A. Rezaee, R. Sahebnasagh and M. H. Pourhanifeh, et al., Mesenchymal stem cell-derived exosomes: a new therapeutic approach to osteoarthritis?, Stem Cell Res. Ther., 2019, 10(1), 340 CrossRef PubMed.
- L. A. Mulcahy, R. C. Pink and D. R. F. Carter, Routes and mechanisms of extracellular vesicle uptake, J. Extracell. Vesicles, 2014, 3(1), 24641 CrossRef PubMed.
- K. V. Iyer, S. Pulford, A. Mogilner and G. V. Shivashankar, Mechanical activation of cells induces chromatin remodeling preceding MKL nuclear transport, Biophys. J., 2012, 103(7), 1416–1428 CrossRef CAS PubMed.
- M. M. Schofield, A. Rzepski, J. Hammerstedt, S. Shah, C. Mirack and J. Parreno, Targeting F-actin stress fibers to suppress the dedifferentiated phenotype in chondrocytes, Eur. J. Cell Biol., 2024, 103(2), 151424 CrossRef CAS PubMed.
- Z. Sun, S. S. Guo and R. Fässler, Integrin-mediated mechanotransduction, J. Cell Biol., 2016, 215(4), 445–456 CrossRef CAS PubMed.
- R. F. Loeser, Integrins and chondrocyte–matrix interactions in articular cartilage, Matrix Biol., 2014, 39, 11–16 CrossRef CAS PubMed.
- M. Zarka, E. Haÿ and M. Cohen-Solal, YAP/TAZ in Bone and Cartilage Biology, Front. Cell Dev. Biol., 2022, 9, 788773 CrossRef PubMed.
- Y. Deng, A. Wu, P. Li, G. Li, L. Qin and H. Song, et al., Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair, Cell Rep., 2016, 14(9), 2224–2237 CrossRef CAS PubMed.
- A. Luciani, V. Coccoli, S. Orsi, L. Ambrosio and P. A. Netti, PCL microspheres based functional scaffolds by bottom-up approach with predefined microstructural properties and release profiles, Biomaterials, 2008, 29(36), 4800–4807 CrossRef CAS PubMed.
- Z. Cai, H. Jiang, T. Lin, C. Wang, J. Ma and R. Gao, et al., Microspheres in bone regeneration: Fabrication, properties and applications, Mater. Today Adv., 2022, 16, 100315 CrossRef CAS.
- M. Wang, Y. Wu, G. Li, Q. Lin, W. Zhang and H. Liu, et al., Articular cartilage repair biomaterials: strategies and applications, Mater. Today Bio, 2024, 24, 100948 CrossRef CAS PubMed.
- J. Yang, Y. S. Zhang, K. Yue and A. Khademhosseini, Cell-laden hydrogels for osteochondral and cartilage tissue engineering, Acta Biomater., 2017, 57, 1–25 CrossRef CAS PubMed.
- Y. Du, H. Liu, Q. Yang, S. Wang, J. Wang and J. Ma, et al., Selective laser sintering scaffold with hierarchical architecture and gradient composition for osteochondral repair in rabbits, Biomaterials, 2017, 137, 37–48 CrossRef CAS PubMed.
- L. Zhang, J. Hu and K. A. Athanasiou, The role of tissue engineering in articular cartilage repair and regeneration, CRC Crit. Rev. Bioeng., 2009, 37(1–2), 1–57 Search PubMed.
- D. J. Huey, J. C. Hu and K. A. Athanasiou, Unlike Bone, Cartilage Regeneration Remains Elusive, Science, 2012, 338(6109), 917–921 CrossRef CAS PubMed.
- B. Del Frari and A. H. Schwabegger, Diced Autologous Rib Cartilage for Primary Treatment or Refinement of Minor Chest Wall Deformities, Plast. Reconstr. Surg., 2011, 128(1), 154–162 CrossRef CAS PubMed.
- A. Haisch, S. Kläring, A. Gröger, C. Gebert and M. Sittinger, A tissue-engineering model for the manufacture of auricular-shaped cartilage implants, Eur. Arch. Otorhinolaryngol., 2002, 259(6), 316–321 CrossRef PubMed.
- B. J. Huang, J. C. Hu and K. A. Athanasiou, Cell-based tissue engineering strategies used in the clinical repair of articular cartilage, Biomaterials, 2016, 98, 1–22 CrossRef CAS PubMed.
- A. R. Brunelle, C. B. Horner, K. Low, G. Ico and J. Nam, Electrospun thermosensitive hydrogel scaffold for enhanced chondrogenesis of human mesenchymal stem cells, Acta Biomater., 2018, 66, 166–176 CrossRef CAS PubMed.
- J. M. Patel, S. A. Ghodbane, A. Brzezinski, C. J. Gatt and M. G. Dunn, Tissue-Engineered Total Meniscus Replacement With a Fiber-Reinforced Scaffold in a 2-Year Ovine Model, Am. J. Sports Med., 2018, 46(8), 1844–1856 CrossRef PubMed.
- Y. Zeng, X. Li, X. Liu, Y. Yang, Z. Zhou and J. Fan, et al., PLLA Porous Microsphere-Reinforced Silk-Based Scaffolds for Auricular Cartilage Regeneration, ACS Omega, 2021, 6(4), 3372–3383 CrossRef CAS PubMed.
- T. K. Kim, J. J. Yoon, D. S. Lee and T. G. Park, Gas foamed open porous biodegradable polymeric microspheres, Biomaterials, 2006, 27(2), 152–159 CrossRef CAS PubMed.
- D. Ghosh Dastidar, S. Saha and M. Chowdhury, Porous microspheres: Synthesis, characterisation and applications in pharmaceutical & medical fields, Int. J. Pharm., 2018, 548(1), 34–48 CrossRef CAS PubMed.
- Y. Li, F. Gao, W. Wei, J.-B. Qu, G.-H. Ma and W.-Q. Zhou, Pore size of macroporous polystyrene microspheres affects lipase immobilization, J. Mol. Catal. B: Enzym., 2010, 66(1), 182–189 CrossRef CAS.
- J. E. Won, Y. S. Lee, J. H. Park, J. H. Lee, Y. S. Shin and C. H. Kim, et al., Hierarchical microchanneled scaffolds modulate multiple tissue-regenerative processes of immune-responses, angiogenesis, and stem cell homing, Biomaterials, 2020, 227, 119548 CrossRef CAS PubMed.
- Y. Shiromoto, Y. Niki, T. Kikuchi, Y. Yoshihara, T. Oguma and K. Nemoto, et al., Increased migratory activity and cartilage regeneration by superficial-zone chondrocytes in enzymatically treated cartilage explants, BMC Musculoskeletal Disord., 2022, 23(1), 256 CrossRef CAS PubMed.
- T. I. Morales, Chondrocyte moves: clever strategies?, Osteoarthritis Cartilage, 2007, 15(8), 861–871 CrossRef CAS PubMed.
- L. M. Janssen, C. D. I. der Maur, P. K. Bos, J. A. Hardillo and G. J. V. M. van Osch, Short-Duration Enzymatic Treatment Promotes Integration of a Cartilage Graft in a Defect, Ann. Otol., Rhinol., Laryngol., 2006, 115(6), 461–468 Search PubMed.
- A. J. McGregor, B. G. Amsden and S. D. Waldman, Chondrocyte repopulation of the zone of death induced by osteochondral harvest, Osteoarthritis Cartilage, 2011, 19(2), 242–248 CrossRef CAS PubMed.
- Y. Lei, J. Peng, Z. Dai, Y. Liao, Q. Liu and J. Li, et al., Articular Cartilage Fragmentation Improves Chondrocyte Migration by Upregulating Membrane Type 1 Matrix Metalloprotease, Cartilage, 2021, 13(2_suppl), 1054S–1063S CrossRef CAS PubMed.
- M. K. Kosinska, T. E. Ludwig, G. Liebisch, R. Zhang, H.-C. Siebert and J. Wilhelm, et al., Articular Joint Lubricants during Osteoarthritis and Rheumatoid Arthritis Display Altered Levels and Molecular Species, PLoS One, 2015, 10(5), e0125192 CrossRef PubMed.
- S. Ikegawa, M. Sano, Y. Koshizuka and Y. Nakamura, Isolation, characterization and mapping of the mouse and human PRG4 (proteoglycan 4) genes, Cytogenet. Cell Genet., 2000, 90(3–4), 291–297 CrossRef CAS PubMed.
- G. D. Jay, D. E. Britt and C. J. Cha, Lubricin is a product of megakaryocyte stimulating factor gene expression by human synovial fibroblasts, J. Rheumatol., 2000, 27(3), 594–600 CAS.
- N.-H. Lee, S. You, A. Taghizadeh, M. Taghizadeh and H. S. Kim, Cell Membrane-Cloaked Nanotherapeutics for Targeted Drug Delivery, Int. J. Mol. Sci., 2022, 23(4), 2223 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Cell Membrane Coating Nanotechnology, Adv. Mater., 2018, 30(23), e1706759 CrossRef PubMed.
- D. Shao, M. Li, Z. Wang, X. Zheng, Y.-H. Lao and Z. Chang, et al., Bioinspired Diselenide-Bridged Mesoporous Silica Nanoparticles for Dual-Responsive Protein Delivery, Adv. Mater., 2018, 30(29), 1801198 CrossRef PubMed.
- W. Chen, Q. Zhang, B. T. Luk, R. H. Fang, Y. Liu and W. Gao, et al., Coating nanofiber scaffolds with beta cell membrane to promote cell proliferation and function, Nanoscale, 2016, 8(19), 10364–10370 RSC.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti and M. Cordenonsi, et al., Role of YAP/TAZ in mechanotransduction, Nature, 2011, 474(7350), 179–183 CrossRef CAS PubMed.
- T. P. Driscoll, B. D. Cosgrove, S. J. Heo, Z. E. Shurden and R. L. Mauck, Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells, Biophys. J., 2015, 108(12), 2783–2793 CrossRef CAS PubMed.
- A. Elosegui-Artola, I. Andreu, A. E. M. Beedle, A. Lezamiz, M. Uroz and A. J. Kosmalska, et al., Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores, Cell, 2017, 171(6), 1397–1410 CrossRef CAS PubMed.
- J. C. Mansfield, J. S. Bell and C. P. Winlove, The micromechanics of the superficial zone of articular cartilage, Osteoarthritis Cartilage, 2015, 23(10), 1806–1816 CrossRef CAS PubMed.
- J. Sanchez-Adams, H. A. Leddy, A. L. McNulty, C. J. O’Conor and F. Guilak, The Mechanobiology of Articular Cartilage: Bearing the Burden of Osteoarthritis, Curr. Rheumatol. Rep., 2014, 16(10), 451 CrossRef PubMed.
- J. C. Lauer, M. Selig, M. L. Hart, B. Kurz and B. Rolauffs, Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function, Int. J. Mol. Sci., 2021, 22(6), 3279 CrossRef CAS PubMed.
- G. F. Hallström, D. L. Jones, R. C. Locke, E. D. Bonnevie, S. Y. Kim and L. Laforest, et al., Microenvironmental mechanoactivation through Yap/Taz suppresses chondrogenic gene expression, Mol. Biol. Cell, 2023, 34(7), ar73 CrossRef PubMed.
- H. K. Vanyai, F. Prin, O. Guillermin, B. Marzook, S. Boeing and A. Howson, et al., Control of skeletal morphogenesis by the Hippo-YAP/TAZ pathway, Development, 2020, 147(21), dev187187 CrossRef PubMed.
- A. Woods, G. Wang and F. Beier, RhoA/ROCK Signaling Regulates Sox9 Expression and Actin Organization during Chondrogenesis, J. Biol. Chem., 2005, 280(12), 11626–11634 CrossRef CAS PubMed.
- D. Kumar and A. B. Lassar, The transcriptional activity of Sox9 in chondrocytes is regulated by RhoA signaling and actin polymerization, Mol. Cell. Biol., 2009, 29(15), 4262–4273 CrossRef CAS PubMed.
- R. C. Lai, R. W. Y. Yeo and S. K. Lim, Mesenchymal stem cell exosomes, Semin. Cell Dev. Biol., 2015, 40, 82–88 CrossRef CAS PubMed.
- A. J. Poe and A. A. Knowlton, Exosomes as agents of change in the cardiovascular system, J. Mol. Cell. Cardiol., 2017, 111, 40–50 CrossRef CAS PubMed.
- T. M. Ribeiro-Rodrigues, T. L. Laundos, R. Pereira-Carvalho, D. Batista-Almeida, R. Pereira and V. Coelho-Santos, et al., Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis, Cardiovasc. Res., 2017, 113(11), 1338–1350 CrossRef CAS PubMed.
- R. Kalluri and V. S. LeBleu, The biology, function, and biomedical applications of exosomes, Science, 2020, 367, 6478 CrossRef PubMed.
- Y. S. Kim, A. J. Chien, J. L. Guo, B. T. Smith, E. Watson and H. A. Pearce, et al., Chondrogenesis of cocultures of mesenchymal stem cells and articular chondrocytes in poly(L-lysine)-loaded hydrogels, J. Controlled Release, 2020, 328, 710–721 CrossRef CAS PubMed.
- Y. Chen, K. Xue, X. Zhang, Z. Zheng and K. Liu, Exosomes derived from mature chondrocytes facilitate subcutaneous stable ectopic chondrogenesis of cartilage progenitor cells, Stem Cell Res. Ther., 2018, 9(1), 318 CrossRef CAS PubMed.
- K. Ma, B. Zhu, Z. Wang, P. Cai, M. He and D. Ye, et al., Articular chondrocyte-derived extracellular vesicles promote cartilage differentiation of human umbilical cord mesenchymal stem cells by activation of autophagy, J. Nanobiotechnol., 2020, 18(1), 163 CrossRef CAS PubMed.
- X. Sang, X. Zhao, L. Yan, X. Jin, X. Wang and J. Wang, et al., Thermosensitive Hydrogel Loaded with Primary Chondrocyte-Derived Exosomes Promotes Cartilage Repair by Regulating Macrophage Polarization in Osteoarthritis, Tissue Eng. Regener. Med., 2022, 19(3), 629–642 CrossRef CAS PubMed.
- P. Balaskas, K. Goljanek-Whysall, P. D. Clegg, Y. Fang, A. Cremers and A. Smagul, et al., MicroRNA Signatures in Cartilage Ageing and Osteoarthritis, Biomedicines, 2023, 11(4), 1189 CrossRef CAS PubMed.
- R. Coutinho de Almeida, Y. F. M. Ramos, A. Mahfouz, W. den Hollander, N. Lakenberg and E. Houtman, et al., RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage, Ann. Rheum. Dis., 2019, 78(2), 270 CrossRef PubMed.
- Y. F. M. Ramos, R. Coutinho de Almeida, N. Lakenberg, E. Suchiman, H. Mei and M. Kloppenburg, et al., Circulating MicroRNAs Highly Correlate to Expression of Cartilage Genes Potentially Reflecting OA Susceptibility—Towards Identification of Applicable Early OA Biomarkers, Biomolecules, 2021, 11(9), 1356 CrossRef CAS PubMed.
- R. W. Y. Yeo, R. C. Lai, B. Zhang, S. S. Tan, Y. Yin and B. J. Teh, et al., Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery, Adv. Drug Delivery Rev., 2013, 65(3), 336–341 CrossRef CAS PubMed.
- S. Hattori, C. Oxford and A. H. Reddi, Identification of superficial zone articular chondrocyte stem/progenitor cells, Biochem. Biophys. Res. Commun., 2007, 358(1), 99–103 CrossRef CAS PubMed.
- E. Hong and A. H. Reddi, Dedifferentiation and Redifferentiation of Articular Chondrocytes from Surface and Middle Zones: Changes in MicroRNAs-221/-222, -140, and -143/145 Expression, Tissue Eng., Part A, 2013, 19(7–8), 1015–1022 CrossRef CAS PubMed.
- D. Kim, J. Song and E.-J. Jin, MicroRNA-221 Regulates Chondrogenic Differentiation through Promoting Proteosomal Degradation of Slug by Targeting Mdm2, J. Biol. Chem., 2010, 285(35), 26900–26907 CrossRef CAS PubMed.
- B. Yang, H. Guo, Y. Zhang, S. Dong and D. Ying, The microRNA expression profiles of mouse mesenchymal stem cell during chondrogenic differentiation, BMB Rep., 2011, 44(1), 28–33 CrossRef CAS PubMed.
- S. Miyaki, T. Sato, A. Inoue, S. Otsuki, Y. Ito and S. Yokoyama, et al., MicroRNA-140 plays dual roles in both cartilage development and homeostasis, Genes Dev., 2010, 24(11), 1173–1185 CrossRef CAS PubMed.
- Y. Chen, H. Huang, W. Zhong, L. Li, Y. Lu and H.-b Si, miR-140-5p protects cartilage progenitor/stem cells from fate changes in knee osteoarthritis, Int. Immunopharmacol., 2023, 114, 109576 CrossRef CAS PubMed.
- Y. Liu, Y. Zeng, H.-B. Si, L. Tang, H.-Q. Xie and B. Shen, Exosomes Derived From Human Urine–Derived Stem Cells Overexpressing miR-140-5p Alleviate Knee Osteoarthritis Through Downregulation of VEGFA in a Rat Model, Am. J. Sports Med., 2022, 50(4), 1088–1105 CrossRef PubMed.
- K. Homan, T. Onodera, M. Matsuoka and N. Iwasaki, Glycosphingolipids in Osteoarthritis and Cartilage-Regeneration Therapy: Mechanisms and Therapeutic Prospects Based on a Narrative Review of the Literature, Int. J. Mol. Sci., 2024, 25(9), 4890 CrossRef CAS PubMed.
- G. Petrou and T. Crouzier, Mucins as multifunctional building blocks of biomaterials, Biomater. Sci., 2018, 6(9), 2282–2297 RSC.
- Y. Lee, J. Choi and N. S. Hwang, Regulation of lubricin for functional cartilage tissue regeneration: a review, Biomater. Res., 2018, 22(1), 9 CrossRef PubMed.
- L. A. Navarro, D. L. French and S. Zauscher, Advances in mucin mimic synthesis and applications in surface science, Curr. Opin. Colloid Interface Sci., 2018, 38, 122–134 CrossRef CAS.
- Y. Yuan, P. Li, J. Li, Q. Zhao, Y. Chang and X. He, Protein lipidation in health and disease: molecular basis, physiological function and pathological implication, Signal Transduction Targeted Ther., 2024, 9(1), 60 CrossRef PubMed.
- J. Gu, W. Rao, S. Huo, T. Fan, M. Qiu and H. Zhu, et al., MicroRNAs and long non-coding RNAs in cartilage homeostasis and osteoarthritis, Front. Cell Dev. Biol., 2022, 10, 1092776 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5mh00593k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |