
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating hydrogel mechanical properties with an electric field†
Hongyi
Cai
 a,
Max
Tepermeister
b,
Chenyun
Yuan
a and
Meredith N.
Silberstein
*bc
a,
Max
Tepermeister
b,
Chenyun
Yuan
a and
Meredith N.
Silberstein
*bc
aMaterials Science and Engineering, Cornell University, Ithaca, New York, USA
bSibley School of Mechanical and Aerospace Engineering, Cornell University, Ithaca, New York, USA. E-mail: meredith.silberstein@cornell.edu
cEngineered Living Materials Institute, Cornell University, Ithaca, New York, USA
First published on 5th May 2025
Abstract
Stimuli-responsive polymeric materials have attracted significant attention due to their ability to change properties in response to various external stimuli. Using an electric field as the stimulus is of particular interest as it possesses the potential for seamless integration of materials with electronic systems. While many materials with electric field responsive actuation have an associated mechanical property change, it is beneficial to develop materials that exhibit mechanical property changes without accompanying significant shape deformation. To address this challenge, here we designed a semi-interpenetrating polymer network (semi-IPN) hydrogel system containing both polyelectrolytes and salt ions, which enables electric field induced changes in mechanical properties while minimizing actuation. We first successfully verified the viability of our design by removing salt ions through a diffusion-only method where we witnessed the stiffness increased to 4.5 times the initial value while still being highly deformable. After this, we applied an electric field to transport the salt ions out of the hydrogel, as shown by both Raman spectroscopy and scanning electron microscopy. We were able to show a time-dependent stiffness increase, the maximum of which was 5 times the original stiffness. We quantified ion transport and water-splitting in the hydrogel by both experiments and simulations. Following this, we showed functional system reversibility by reversing the direction of the current to reinject salt ions into the semi-IPN hydrogel and reducing its stiffness to below the initial value. It's worth noting that our simulations enable us to understand the governing mechanisms behind ion generation and salt transport that leads to mechanical property changes. Finally, we were able to fabricate a spatially variable stiffness haptic interface with our hydrogel, with demonstrated reversibility and cyclability. This research can possibly find applications in soft robotics and haptics and also inspire the development of bio-compatible electronics related devices.
New conceptsWe present a novel concept for dynamically controlling mechanical properties of polymers via an electric field. Specifically, we use ionic current, stimulated by the electric field, to transport dissociated salt ions out of a polymer gel. The gel has bond strength diminished by ionic interactions, so salt removal results in a 5× stiffness increase with negligible dimensional change. Further, we show that this process is reversible; we restore the stiffness to its initial value by flowing the ions back into the gel. Finally, we demonstrated spatial controllability, reversibility, and cyclability by making devices with haptic interfaces. This work differs substantially from the now common electrically actuated polymers that primarily change shape and/or size, with property changes as a secondary effect. While we demonstrate this concept in an interpenetrating polymer network, it is quite general. We could vary the crosslinked gel, the polyelectrolyte type, and/or the salt type. Even more exciting, there are numerous interactions used to control polymer stiffness that are strongly influenced by ions, like metal–ligand coordination and cationic–aromatic interactions. This concept could influence materials science more broadly – all it requires is a matrix sufficient to maintain structural stability with an ion-sensitive stiffness regulating component. |
1 Introduction
In recent decades, stimuli-responsive polymeric materials1–3 have emerged as a compelling and rapidly evolving area of study within materials science. These materials respond to changes in the environment, including temperature,4,5 pH,6–8 light,9–11 humidity,4,12 solvent,13–15 magnetic fields,16,17 and electric fields,18,19 with property changes like mechanical stiffness,15,20 solubility,10,15 conductivity,21,22 shape,23,24 color,14,25 fluorescence,26,27 permeability,28,29 and so on. These features support various applications in controlled release,6,30,31 adaptive surfaces,13,32 soft robotics,33,34 and wearable electronics.35–37 Our lab previously reported a novel metal–ligand coordinated polydimethylsiloxane (PDMS) surface that undergoes a dramatic transformation from hydrophobic to hydrophilic upon contact with water droplets.13 Aubrecht et al. successfully synthesized biomimetic polyelectrolytes containing a geminal cation–aromatic group and demonstrated that such polymers exhibit diverse phase separation behaviors and tunable stiffness in response to specific ion interactions.15 Cheah et al. fabricated a conducting polymer hydrogel composite material with poly(3,4-ethylenedioxythiophene) (PEDOT) and gelatin methacryloyl (GelMA) and achieved controlled release of a model protein bovine serum albumin (BSA) over extended time periods up to 21 days, with the ability to modulate release rates using electrical stimulation.31 Among the different stimuli listed, an electric field is of particular interest, as it allows us to easily interface polymeric materials with the ever-developing electronic world, which can be easily combined with cutting-edge technologies. Moreover, we can manipulate the amplitude, phase, and frequency38 of an electric field, and all these can possibly lead to a change in the response of the material.One of the earliest electric-field-responsive polymer examples was piezoelectric polymers,39 most notably polyvinylidene fluoride (PVDF).40,41 Following that, there were breakthroughs made in the fields of liquid crystal elastomers (LCE),42,43 ionic polymer metal composites (IPMC)44–47 and dielectric elastomers.48–51 Hydrogels have also found applications as electric-field-responsive materials.18,52–55 From these previous works, it is apparent that having charged or partially charged polymers within the material is a promising route for realizing electric-field stimulated response. One thing worth noting is that many of these materials demonstrate mechanical property changes in association with actuation. Developing materials that undergo mechanical property changes independent of actuation would be beneficial.
One application combining the use of polyelectrolytes and an electric field without mechanical actuation, happens in the field of adhesion. For example semi-interpenetrating polymer network (semi-IPN),56 where densely charged polyelectrolytes are inserted into a covalently crosslinked polymer network enable polyelectrolytes to interact with electric fields through ion migration,57 while the covalent network holds the structure together. Asoh and coworkers successfully induced electrophoretic adhesion between two semi-IPN hydrogels by applying an electric field across them, one containing cationic polymers and the other having anionic polymers.58 Further studies were conducted with microbead adhesion59 and alternating current.60 However, these studies61 focus on adhesion while there is a lack of research on changing the bulk mechanical properties of the material. Layer-by-layer (LBL) assembly62–66 uses both cationic and anionic polymers to form a bulk material, and it is common to use an electric field to assist the assembly process.67–69 Following these two material concepts (polyelectrolyte semi-IPN and LBL), incorporating polyelectrolyte complexes (PEC),70,71 which are materials composed of both cationic polyelectrolytes and anionic ones, into semi-IPN seems to be an interesting possibility. PECs already have wide applications in drug/DNA deliveries72 and tough hydrogels.73–75 Highly-stretchable polymers inspired by PEC structure also show that ionic bonds among polymer chains can increase the stiffness and strength.76,77 Moreover, the morphologies and mechanical properties of PEC are responsive to the amount of salt doping, (i.e. saloplasticity).78,79 PECs show a continuous spectrum of states from solid to liquid as salt concentration increases. If we can use a covalent network to hold a PEC together, ensuring its integrity under high salt concentration, it is possible to use salts to modulate the mechanical properties of these semi-IPN hydrogels, while the concentration of ions is controlled by electric fields. The viability of fabricating semi-IPN containing PEC was confirmed by work from Li and coworkers80 as well as Pruthi et al.8 Furthermore, Sayed et al. showed that such PEC-based semi-IPN hydrogels can experience a stiffness drop when immersed in a water bath with higher KBr concentrations,81 though the effect of salt ions is still debatable as their swelling ratio increases.
Here, we present our novel idea of electric field induced modulation of the mechanical properties of a hydrogel without coupling to shape or size change. Specifically, the electric field drives electrodialysis of salt ions from a semi-IPN polyelectrolyte hydrogel. To start, we conducted a water-content-controlled study on how salt (NaCl) ion concentration would change the mechanical properties of the semi-IPN by simply letting salt ions diffuse into a water bath. The effect of molecular weights of the polyelectrolytes was also demonstrated. After that, we investigated how an electric field induced ionic current changed the NaCl content and distribution, the mechanical properties, and the ionic interactions of semi-IPN. We reinjected salt ions back into the semi-IPN to investigate whether this process is reversible. A newly developed equivalent ionic circuit model was used to support understanding of the time-evolving governing mechanisms. Finally, a spatially variable stiffness haptic interface was fabricated with demonstrated reversibility and cyclability. This research will offer insights into the development of novel stimuli-responsive materials with tunable characteristics for diverse applications.
2 Results and discussion
2.1 Hydrogel system design
The semi-IPN hydrogel was prepared using a one-pot procedure as described in the ESI.† A large amount of sodium chloride was added to the mixture prior to the addition of polydiallyldimethylammonium chloride (PDADMAC) and poly(sodium 4-styrenesulfonate) (PSS) to prevent the formation of PEC precipitation. We designed two different pathways to transport the salt ions (sodium cations and chloride anions) out of the semi-IPN hydrogel (Fig. 1). In the first path, termed diffusion, the semi-IPN specimens were submerged in deionized (DI) water so that the salt ions diffused into the water surrounding the hydrogel, thus transporting salt ions out of the hydrogel. Due to water absorption during this process, the water content in the hydrogel specimens dramatically increased compared to their as-prepared state. To return the water content to the same as the as-prepared semi-IPN hydrogel, the swollen hydrogel specimens were exposed to air to let the water evaporate until the mass reached its pre-soaked value. After this, the hydrogel specimens were placed in a humidity chamber (humidity set at 100%) so that they could equilibrate to avoid serious surface-bulk inhomogeneity. The second path is electrodialysis, the core of this paper. In this path, the semi-IPN was sandwiched between ion-exchange membranes and polyacrylamide (PAam) hydrogels. On the one side of the semi-IPN hydrogel, a Cl-form anion exchange membrane FAA-3-PK-75 was placed, followed by PAam hydrogel and a platinum electrode, which was connected to the positive side of the power supply. A Na-form cation exchange membrane Nafion 117 was put on top of the other side of the semi-IPN hydrogel, then a PAam hydrogel, Pt electrode, and the negative side of the power supply. The idea behind the design is that, when the electric field is activated, Cl ions can travel through the anion exchange membrane into the PAam hydrogel and are stored there, while the hydronium ions generated from water splitting are blocked by the membrane from transporting back into the semi-IPN hydrogel. Similarly, the Na ions can be transport across the Nafion membrane and then stay in the PAam hydrogel while hydroxyl ions are prevented from injecting into the semi-IPN. We propose that during the gel preparation process and in the initial semi-IPN gels, interactions among oppositely charged polyelectrolytes, PDADMAC and PSS, are limited because of the ion screening from the abundant presence of the salt ions. After salt ions are removed through either diffusion or electrodialysis, ionic bonds are formed among PDADMAC and PSS chains thus changing the mechanical properties of the semi-IPN hydrogel.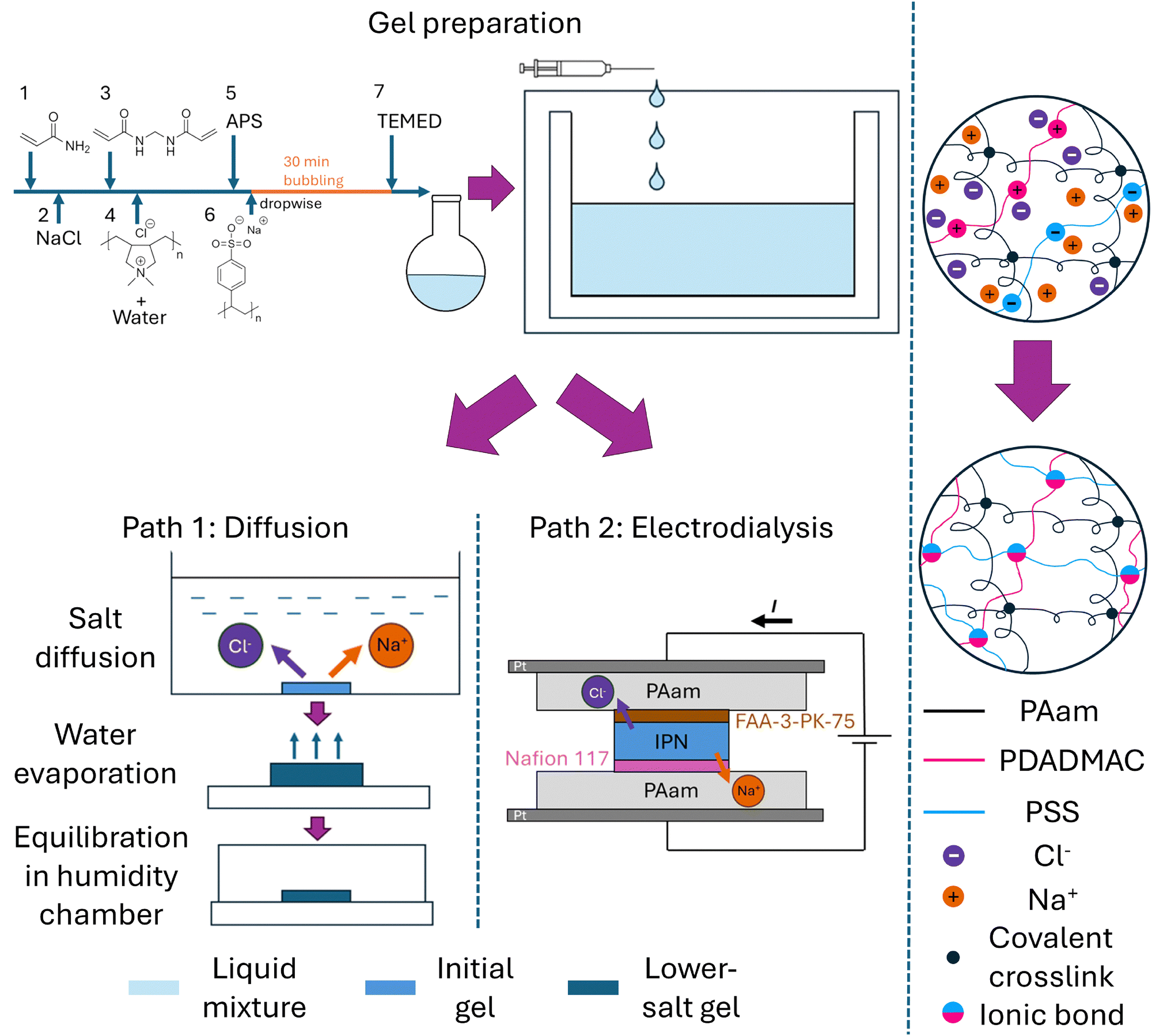 | ||
| Fig. 1 Material preparation. Top: In the gel preparation step, acrylamide monomers, crosslinkers, NaCl, polyelectrolytes, and initiators were mixed and injected into molds with two different thicknesses. The molds were kept in an oven at 50 °C for polymerization. Bottom left: After the gel was solidified, the thinner gel underwent a diffusion process (path 1), where the gel was submerged in DI water and the counterions within the gel diffused into water. After this step, the gel was exposed to air to removed excess water so that the gel weight remained the same as before diffusion. In the final step, the gel was kept in a humidity chamber to be equilibrated. Bottom middle: The thicker gel underwent an electrodialysis process (path 2), where it was sandwiched between polyacrylamide gels and ion-exchange membranes to have the counterions removed with currents induced by an electric field. Right: We propose that during gel preparation, limited interchain ionic interaction exist due to salt ion screening; after salt ions are removed, ionic bonds are formed among oppositely charged polyelectrolytes. | ||
2.2 Verifying salt-based stiffness modulation via the diffusion path
We first investigated how NaCl content in the semi-IPN hydrogel would affect its mechanical properties when the water content is constant, and how the molecular weight (MW) of polyelectrolytes would play a role in this process with two available polyelectrolyte pairs, 70 k MW PSS and 100 k MW PDADMAC (noted as IPN*), and 200 k MW PSS and 200 k MW PDADMAC (noted as IPN). Please note that when we talk about semi-IPNs, we are referring to the material microstructure design, which is a covalently crosslinked structure blended with PECs. When we use “IPN”, we are specifically referring to this material, PAam with 200 k MW PSS and 200 k MW PDADMAC. The diffusion path was used to control the NaCl content. To demonstrate the effect of NaCl leaving the semi-IPN hydrogel, IPN specimens were prepared and soaked in DI water for different time, ranging from 10 s to 300 s (Fig. 2a). We discover that when the time of IPN specimens in the DI water increases, the turbidity of the specimens increases, and the size also increases due to swelling. This trend matches the prototypical transition pattern of PECs in a salt solution with different salt concentrations, where at higher concentration the PEC acts as a homogeneous solution while at lower concentration the PEC behaves like a solid.78 Following this discovery, we set three different levels of NaCl concentration in the semi-IPN hydrogel as one variable, crossed with two pairs of polyelectrolytes with different MW (see ESI† for procedure). Critically, we maintained water content across the specimens, such that salt content is the controlling factor in mechanical properties. Salt removed from the semi-IPN hydrogels was calculated by measuring ionic conductivity of the removed salts dissolved them in DI water (Fig. 2b). High-NaCl is not plotted as the removal ratio is merely 0 according to the mentioned definition. As can be seen from the plot, there is a clear distinction in the amount of removed NaCl across three different NaCl levels. At medium-NaCl level, 35% of the initial NaCl are removed from IPN and 24% from IPN*, while at low-NaCl level almost all the initial NaCl are diffused out of the hydrogel. This result evidently demonstrates the viability of using the diffusion path to remove NaCl from the initial semi-IPN hydrogel in a controlled manner. Following this, monotonic tensile tests were performed to investigate how the mechanical properties change when NaCl leaves the hydrogel (Fig. 2c, modulus in Table S5, ESI†). Of the six sample preparations with varied MW and NaCl content, four are highly stretchable and were still intact when the travel limit of our universal tester was reached. The other two exceeded strains of 500% before breaking. At high NaCl content, both the IPN* and the IPN samples exhibit a more compliant behavior compared to the reference PAam hydrogel (Fig. S7, ESI†). This finding matches the result shown by Li et al.80 Interestingly, the salt PAam hydrogel, which has the same NaCl content as the high NaCl content IPN gel, shows a slightly higher stiffness and lower maximum strain than the PAam hydrogel (Fig. S7, ESI†). Therefore, if there is a stiffening effect when NaCl is removed from the IPN, its origin is the polyelectrolytes rather than NaCl interactions with PAam. Indeed, as the NaCl content decreases to medium and then low level, the IPN hydrogel becomes stiffer and less stretchable, showing that the removal of salt ions and the formation of ionic crosslinks among polyelectrolyte chains can indeed stiffen and strengthen the hydrogel. This is similar to our previous finding about ionic crosslinks in elastomers.76,77 The response of the IPN* sample to NaCl content shows the same trend, but is muted in magnitude. The stress–strain curve of the medium NaCl level overlaps that of the high NaCl level. A larger change in stiffness and strength can be seen at low NaCl level. Given the relative salt dependence of IPN and IPN*, we have chosen the 200 k–200 k MW polyelectrolyte pair (IPN) for our electric field response study. Based on our data for this system, we expect that a stiffness change ratio of 4.5 is achievable with an electric field. | ||
| Fig. 2 Dependence of semi-IPN hydrogels on NaCl content, obtained via path 1: diffusion. Three different levels of NaCl content were investigated, noted as high, medium and low. Two pairs of polyelectrolytes with different molecular weights (MW) were used. IPN contains 200 k MW PSS and 200 k MW PDADMAC while IPN* has 70 k MW PSS and 100 k MW PDADMAC. (a) A photo showing the increase in turbidity of IPN hydrogels as they were stirred in DI water for a longer period of time, ranging from no soaking at all to 300 s. (b) NaCl removal ratio from IPN and IPN* at two different NaCl content levels, assigned as medium and low. Note that high level NaCl content is not plotted here, because it is the initial level, meaning there is no NaCl removed. (c) Stress–strain curves from monotonic tensile tests of IPN and IPN* hydrogels with three different levels of NaCl content. | ||
2.3 Stiffening by an electric field
After our investigation utilizing diffusion (path 1), we moved onto proving that electrodialysis (path 2) could realize a similar effect. For forward operation, a device was assembled in the fashion shown in Fig. 1, and the current across the gels was manually kept at 100 mA by adjusting the output voltage of the power supply. This current level was determined based on consideration of time and heat generation. In short, a higher current can save time, which prevents excessive water evaporation, but it also generates more heat, which enhances water loss. The voltage required for our device was around 5 V when the current was stable, which corresponds to a power of 0.5 W. The total thickness of our device is around 9 mm, which gives us an electric field of 5.6 V cm−1. The voltage we used is in the relatively low regime of hydrogel-based actuators, which typically operate in ranges from 2 V to 20 kV. As a comparison, a reverse operation was also employed where the direction of the current was reversed relative to the normal operation, simply by swapping the connections between the Pt electrodes and the power supply (Fig. S3, ESI†). We have discovered that after a forward operation, the two ion exchange membranes were strongly adhered to the IPN and peeling them off seriously deformed the gel. Therefore, all compression tests were done with both the membranes on the hydrogels. We can calculate the small strain elastic modulus of the hydrogel from the stiffness of the stack since the membrane thickness and stiffness values are known (see ESI† Calculations section). Comparison of the compression stress–strain curves of the as-prepared initial IPN, the hydrogel after 30 min of forward operation, and the one after 30 min of reverse operation is shown in Fig. 3a. Evidently, the IPN becomes stiffer after 30 min of forward operation, which supposedly transports salt ions out of the IPN across the ion-exchange membranes, resulting in increased ionic bond formation in the PEC structure. The IPN after reverse operation becomes more compliant than the initial IPN. This also makes sense as the salt ions are blocked by ion-exchange membranes from traveling into the PAam buffer hydrogels on the sides, while hydronium and hydroxyl ions generated from water splitting are allowed to migrate into the IPN across the membranes, after which they recombine and become water, thus swelling the IPN. To support our mechanistic assumptions, we measured the weight change of the IPN involved in the process before and after drying, and calculated the salt ion and the water content change (Fig. 3b). A 62% drop in NaCl content can indeed be seen after 30 min of forward operation. Water content also decreases by 26%. To eliminate the possible effect of water content drop on stiffening of the hydrogel, IPN with matching lower water content were prepared by evaporating water from the as-prepared initial IPN and tested (Fig. S8, ESI†). No significant change in mechanical properties were detected, with only a 7.6% increase in modulus detected. As for the IPN after 30 min reverse operation, the water content has increased by 33%, which should be the probable cause of the softening. To investigate the effect of water content increase on the stiffness, IPN with 33% more water were prepared by placing the original IPN in a humidity chamber set at 25 °C and 95% for 4.5 h and then tested under compression. This time we witnessed a significant drop in modulus to 8.6 kPa, similar to the modulus of IPN after reverse operation. Interestingly, NaCl content also drops by 11%, showing that there is still some ion leakage across the ion-exchange membranes, yet the leakage is very limited relative to the forward operation. | ||
| Fig. 3 Comparison of IPN hydrogels under path 2 electrodialysis in three different conditions: forward operation, reverse operation, and initial state. (a) Stress–strain curves from compression tests of IPN hydrogels, including initial IPN, IPN after a 30 min forward operation electrodialysis, and IPN after a 30 min reverse operation. Note that these compression tests are performed with the thin and relatively rigid ion exchange membranes still attached. (b) Amount of NaCl and water remaining in the IPN after a 30 min forward or reverse operation, shown as fractions of the initial composition. Note that the water content in the IPN after a 30 min reverse operation increases from the initial state. (c) SEM images of initial IPN and IPN after a 60 min forward operation. EDS mapping of Na and Cl shows that the latter contains a much less NaCl. (d) Raman spectra of IPN with high and low NaCl content used in the diffusion path, and as-prepared initial IPN (high-NaCl) and IPN after a 60 min forward operation (low-NaCl) used in the electrodialysis path. The arrows indicate the peak of interest around 983 cm−1, which corresponds to PDADMAC ring deformation and C–N bond stretching. All spectra are normalized to the respective highest peak. | ||
Scanning electron microscopy – energy-dispersive X-ray spectroscopy (SEM-EDS) was used to find additional evidence of salt ion removal from the hydrogel (Fig. 3c). In the as-prepared initial IPN, there is an abundant distribution of small crystal-like objects across the whole scanned area of the dried hydrogel, which have both sodium ions and chloride ions confirmed by EDS. In the IPN after 60 min forward operation, only a few of these objects can be seen. This is clear evidence that salt ions are indeed removed by electrodialysis while there is limited change in the microstructure of the IPN.
To gather more evidence of salt removal and to check the consistency between the diffusion path and the electrodialysis path, Raman spectra of the IPN were collected (Fig. 3d). A peak at around 983 cm−1 can be identified in the as-prepared (high salt) IPN, for specimens used in both electrodialysis and diffusion. This peak corresponds to PDADMAC ring deformation and C-N bond stretching.82 Meanwhile, this peak is much smaller in the IPN after a 60 min forward operation. The same finding applies to the low salt NaCl IPN obtained via the diffusion path. This similarity also confirms that both the diffusion path and the electrodialysis path reach the same goal, which is removing salt ions in the IPN to drive polyelectrolyte ionic crosslinking.
Next we investigated how the duration of the forward operation will progressively change the properties of the IPN. We have observed that as the forward operation time increases, the turbidity of the IPN increases (Fig. 4a), which matches our findings from the diffusion path, showing an increasing degree of salt ion removal. The current–voltage–time plot of the device is shown in Fig. 4b. As we can see, the current is very low at the beginning due to the lack of mobile charge carriers in the PAam buffer gels. As more charge carriers are generated by water splitting and salt ions moving into the PAam buffer gels, the current gradually increases. To keep the current at around 100 mA, output voltage from the power supply also drops and therefore, the voltage across the device gets lower. After this stage, the resistance is stable and both the current and voltage are kept at a relatively constant level. In the final stage, as salt ions are gradually depleted from the IPN, the resistance goes up and the voltage has to be tuned up again. The operation comes to a stop when we can no longer keep the current at 100 mA level (due to the absence of ion conductors in the IPN) as the power supply reaches its output limit. We would like to point out that the output voltage of the power source was manually controlled by turning the adjustment knob, which caused sharp changes in both current and voltage during adjustment. Based on this plot, we chose three time points to compare: 15 min, which is around the beginning of the stable stage; 30 min, which is in the middle of the stable stage; and 60 min, which is the end of the operation. Compression test results (Fig. 4c) show that the semi-IPN hydrogel is the most stiff after 60 min operation, followed by 30 min, 15 min, and then the initial IPN. The modulus of the semi-IPN hydrogel is approximately 5× that of the initial gel, analogous to the long duration diffusion result. This result again confirms our design of removing salt ions from the IPN to enhance its stiffness. NaCl content change is confirmed by calculation based on weight measurements, and the salt ion content drops as the operation time goes longer (Fig. 4d). We also measured the amount of acid generated in the FAA-3-PK-75 membrane side PAam buffer gel and base in the Nafion 117 membrane side PAam buffer gel (Fig. 4e). In forward operation, hydrochloric acid should be generated in the FAA-3-PK-75 side PAam gel while sodium hydroxide should be formed in the Nafion side. As expected, more acid and base are generated as operation time increases. The amount of acid is also slightly higher than the amount of base. pH can affect the behavior of polyelectrolytes. The IPN after a 60 min forward operation was immersed in 20 mL water overnight and the pH was measured to be 5 by pH strips. Since the acid form of PSS is a strong acid83 and charges on PDADMAC are independent of pH values,84 the PEC formed by PSS and PDADMAC should show minimal response to pH values,85 meaning the stiffening effect should primarily come from salt removal.
 | ||
| Fig. 4 Comparison of IPN hydrogels with different forward operation time. (a) A photo showing the increase in turbidity of IPN hydrogels as the forward operation time increased. (b) A current–voltage–time plot of both the experimental data and the simulation result of the stacked-gel device throughout a 60 min forward operation. Note that during the experiment, the current was manually controlled at around 100 mA by adjusting the voltage output from the power supply, while in the simulation the current was set at 100 mA. (c) Stress–strain curves from compression tests of IPN hydrogels with different forward operation time, ranging from no operation to 60 min. (d) A plot showing both experimental data and simulation results of how sodium ion content and chloride ion content in the IPN hydrogel change during a 60 min forward operation. Note that experimental data was calculated based on the assumption that the number of sodium ions in IPN is the same as the number of chloride ions. (e) A plot showing both experimental data and simulation results of how much acid and base were generated in PAam buffer hydrogels during a 60 min forward operation. Acid is in anode side PAam buffer gel and base is in cathode side PAam buffer gel. | ||
To further understand the ion transport in the stack-hydrogel structure, we adapted a recently developed equivalent ionic circuit model86 to simulate ion concentration change during the operation (see ESI† Simulation section). Using the model, we are able to have continuous time varying predictions of voltage and current of the device, as well as concentrations of ion species present in the hydrogel, including sodium ions, chloride ions, hydroxyl ions, and hydronium ions. In Fig. 4b, the simulation result is plotted along with the experimental result, showing a good match, meaning we are able to capture the effective resistance change in the device with our model. In Fig. 4d, the salt ion concentration change in the IPN from the model is also in good agreement with the experimental measurements. The model also predicts the acid and base generation reasonably well (Fig. 4e), though it does slightly over-predict quantities at long times, possibly due to the acid or base starting to react with PAam at very high concentration, or the PAam gel not being in equilibrium with the water in the vial by the end of the acid/base extraction process.
2.4 Reversibility
Finally, we investigated whether we could reinject salt ions into the IPN by reversing current after a forward operation to return the gel to its initial mechanical properties (Fig. 5a same-buffer). The current was again kept at 100 mA level by manually adjusting the output voltage of the power supply. Interestingly, the voltage required to maintain this current starts lower than the steady-state forward voltage, increases gradually to above this value over approximately 10 min, and then gradually decreases over the next 20 min until we end the process (Fig. 5b). The simulation also shows this increase and decrease, although the predicted changes are reduced in magnitude and slower (peak at 15.5 min). The simulation indicates that the PAam buffer gel on the FAA-3-PK-75 side switched from acidic to basic, and vice versa for the buffer gel on the Nafion 117 side at this peak position (Fig. S11, ESI†). Experimentally, we see an increase in the salt fraction in the IPN from 0.38 up to 0.76 of the original value over the 30 minutes (Fig. 5c). Interestingly, the simulation predicted salt fraction change over time exhibits a peak at the same time as the voltage peak. To gain insight into this peak we plotted the flux of ions across the membranes (Fig. S12 and S13, ESI†) and found out that after the turning point, there is an increasing flux of hydroxyl ions across FAA-3-PK-75 into the IPN and the flux of chloride ions decreases quickly. There is still a leaking flux of chloride ions across Nafion 117. These factors in combination lead to a drop of chloride ion concentration in the IPN. The deviation of simulation results from experimental measurements could be due to higher salt ion leakage flux across membranes in simulations, meaning the exclusion effect of membranes are under-estimated in our simulations. We measure the mechanical behavior of the gel at this time and find that the stiffness has decreased below the original stiffness (Fig. 5d), even though the salt has not fully been restored. This decrease in stiffness beyond the starting point is possibly due to an increased water content in the IPN, which was 117% of the initial water content (Fig. S10, ESI†). Our model predicts the pH in the IPN to be 13.4 consistent with a simple titration measurement result of 13.6. Since under such basic conditions PSS is fully ionized while PDADMAC is not pH sensitive, the over-softening should be due to higher water content. | ||
| Fig. 5 Reinjection of sodium and chloride ions into IPN hydrogels after a 30 min forward operation on IPN. (a) Scheme showing two different paths to reinject sodium and chloride ions into IPN after the forward operation. In the “same-buffer” path, the direction of the current was simply reversed. In the “new-buffer” path, in addition to reversing the direction of the current, the PAam buffer gels on both sides were swapped with PAam gels having high NaCl content. (b) A current–voltage–time plot of both the experimental data and the simulation result of the stacked-gel device during a 30 min same-buffer reinjection operation. (c) A plot showing both experimental data and simulation results for how sodium ion content and chloride ion content in IPN hydrogel change during a 30 min same-buffer reinjection operation. (d) Stress–strain curves from compression tests of the initial IPN, IPN undergoing the “same-buffer” path for 30 min, and IPN undergoing the “new-buffer” path for 30 min. (e) A current–voltage–time plot of both the experimental data and the simulation result of the stacked-gel device during a 30 min new-buffer reinjection operation. (f) A plot showing both experimental data and simulation results for how sodium ion content and chloride ion content in IPN hydrogel change during a 30 min new-buffer reinjection operation. Note that experimental data was calculated based on the assumption that the number of sodium ions in the IPN is the same as the number of chloride ions. | ||
To examine what would happen if we fully restore the salt, we replace the original PAam buffers with high-NaCl-content PAam hydrogel of the same dimensions prior to applying the reverse direction current (Fig. 5a new-buffer). Again, we kept the current at 100 mA by manually adjusting the output voltage of the power supply. We see both from experiments and simulation that we are able to maintain this current with a voltage that starts at the steady state value of the forward process and only slightly decreases throughout the 30 min (Fig. 5e). For this approach, we see that the salt content in the IPN increases monotonically over the full time period (Fig. 5f). Experimentally, at the end of this process we restored 98% of the salt ions in the IPN and the water content was 0.96 of the original amount, meaning this approach nearly fully restored both the salt and the water content. Similarly to reversing current with the original buffer, this new-buffer approach yields a stress–strain response less than the initial IPN. Our model predicts the pH in the IPN to be 0.32, while a simple titration measurement gave us 0.08. Under such strong acidic conditions PSS should be in its acid form, losing the charges and thus weakening the interactions with PDADMAC, making the IPN less stiff, which explains why the IPN here is less stiff than the original even with full water and salt restoration.
2.5 Spatially variable stiffness haptic interface
To demonstrate potential application for our electrically modulated IPN hydrogel, we have designed a customized 3D-printed device which resembles the previous experimental setup but also allows direct pressing or indentation onto the hydrogel, which in principle works as a haptic interface with spatially variable stiffness. For each 3D-printed device (Fig. S15, ESI†), a piece of Pt electrode was placed in the back that was covered with copper tape. The inside of the device was filled with 0.1 M NaCl aqueous solution while PAam hydrogels with low salt content were fabricated in the windows of the device, blocking the solution from leaking out. These two parts together work as the buffer gels from the previous setup. A piece of ion-exchange membrane (Nafion 117 or FAA-3-PK-75) was placed on top of the PAam hydrogel at the window on the side with an electrode while a piece of polytetrafluoroethylene (PTFE) film was put at the window without an electrode, acting as insulation. The IPN was placed on top of the membranes, and during an operation with current going through, it was sandwiched between two identical devices (the ion-exchange membranes are different). We first conducted cyclic indentation tests with a spherical indenter before any current operation to obtain a baseline of the IPN's mechanical response (Fig. 6b and Fig. S16, ESI†). The area on top of the left window and the right window exhibit the same mechanical response. Although the IPN shows residual strain at the unloading process, the maximum force of the second loading cycle is the same as the first one, showing that the residual strain at unloading is likely due to viscoelasticity rather than plastic deformation. We then assembled the setup and applied a 25 V voltage for 5 min. Visually the IPN had a spatial specific look – the area on top of the left window had higher turbidity, matching our findings earlier. A subsequent indentation showed that this area with current applied across it gave a maximum force response approximately 2× the original, while the one without current applied across it remained the same as before. To investigate if this design can also demonstrate reversibility, we applied a reverse voltage of 25 V until the current–time integral was approximately the same as the forward bias (Fig. S17, ESI†). Similar to previous results, the IPN showed a softened response compared to the original state. We further investigated the cyclability of our design by applying the forward–reverse bias cycle multiple times (Fig. 6c). Within the 5 cycles we tested, the IPN successfully demonstrated a strengthened-softened behavior corresponding to forward–reverse bias, with the maximum force of the strengthened status being 2 to 3 times higher than that of the softened status.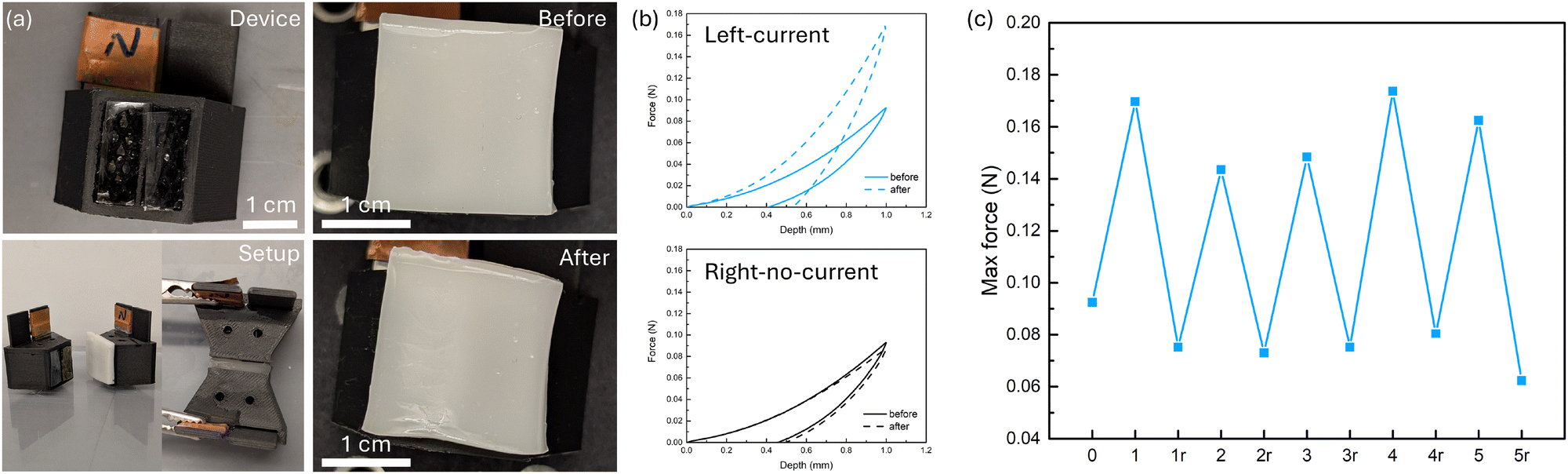 | ||
| Fig. 6 IPN with spatially varying current coverage shows area-specific stiffness change. (a) Pictures showing the 3D-printed device, the setup, and IPN appearance before and after forward potential bias was applied. (b) Force–depth curves obtained from cyclic indentation tests of IPN hydrogel before and after applying 25 V for 5 min. The current only went through the left side. (c) Maximum force recorded from cyclic indentation tests of IPN during five 5-min forward–reverse current cycles. Number represents the number of current cycles and “r” represents reverse current. | ||
3 Conclusions
In this work, we successfully developed a multi-layer stacking structure with highly deformable semi-IPN hydrogels containing both PECs and salt ions that exhibits electric field regulated mechanical properties, achieving a five-fold increase in stiffness of the hydrogel. We confirmed that the cause of this effect is removal of salt ions via (1) mechanical tests of semi-IPN hydrogels with salt ions removed by a diffusion process where we witnessed a 4.5 times increase in stiffness, and (2) SEM-EDS and Raman spectroscopy offering evidence of salt ion removal and ionic crosslink formation. We have also shown that this stiffening effect is reversible by simply reinjecting salt ions back into the hydrogel. In addition to experiments, we also developed a model to simulate ion concentrations in the stacking hydrogels, which enables us to investigate the continuous change of ion concentration in the gels, helping us develop a deeper understanding of ion generation and transport mechanism during the electrodialysis process. With this hydrogel, we were able to create a haptic interface with reversible, spatially variable stiffness. This device also highlights tunability of system timescale through geometry, showing changes on a 5 minute time scale similar to those of the 15 minute timescale in the original geometry. More work can be done in investigating how to achieve a more drastic stiffness change in a shorter period of time, for example by using a more compliant host matrix and by using thinner gels. Using lower initial salt concentration and choosing a material system that can dissipate heat faster will also help with realizing swifter response, as we limited the current magnitude in this study to prevent overheating. Other mechanical properties like strength and toughness are also tunable, though we would need to design distinct experiments to characterize these changes. What's more, future researchers may use the simulation tool in this work to first screen potential material and device structure candidates before actual fabrication, thus accelerating new material development. The findings of this work can potentially expand beyond polyelectrolytes, as there are many other polymer interactions that are strongly influenced by ions, for example metal–ligand coordination and cationic–aromatic interactions. This work can possibly lead to applications in soft robotics and inspire materials design for bio-compatible electronic related devices.Author contributions
H. C. contributed to conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review and editing, visualization. M. T. contributed to conceptualization, methodology, software, formal analysis, investigation, writing – review and editing. C. Y. contributed to investigation, formal analysis, writing – review and editing. M. N. S. contributed to conceptualization, writing – review and editing, supervision, project administration, funding acquisition.Data availability
Data for this communication are available at Zenodo at https://doi.org/10.5281/zenodo.13917209.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award # DE-SC0019141. This work made use of the Cornell Center for Materials Research shared instrumentation facilities which are supported through the NSF MRSEC program (DMR-1719875).Notes and references
- Z. M. Png, C. G. Wang, J. C. C. Yeo, J. J. C. Lee, N. E. Surat'man, Y. L. Tan, H. Liu, P. Wang, B. H. Tan, J. W. Xu, X. J. Loh and Q. Zhu, Mol. Syst. Des. Eng., 2023, 8, 1097–1129 RSC
.
- A. Tariq, Z. U. Arif, M. Y. Khalid, M. Hossain, P. I. Rasool, R. Umer and S. Ramakrishna, Adv. Eng. Mater., 2023, 25, 2301074 CrossRef CAS
- S. Amirthalingam, A. K. Rajendran, Y. G. Moon and N. S. Hwang, Mater. Horiz., 2023, 10, 3325–3350 RSC
- X. Yang, L. Lan, X. Pan, Q. Di, X. Liu, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2023, 14, 2287 CrossRef CAS PubMed
- Y. Wang, J. Zhang, M. Li, M. Lei, Y. Wang and Q. Wei, J. Polym. Res., 2022, 29, 243 CrossRef CAS
- J. Singh and P. Nayak, J. Polym. Sci., 2023, 61, 2828–2850 CrossRef CAS
- H. Ceylan, N. Olcay Dogan, I. Ceren Yasa, M. Nur Musaoglu, Z. Umut Kulali and M. Sitti, Sci. Adv., 2021, 7, eabh0273 CrossRef CAS PubMed
- V. Pruthi, V. Hirschberg and P. Théato, Adv. Mater. Technol., 2024, 9, 2400416 CrossRef CAS
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed
- T. Ueki, Y. Osaka, K. Homma, S. Yamamoto, A. Saruwatari, H. Wang, M. Kamimura and J. Nakanishi, Macromol. Rapid Commun., 2024, 2400419 CrossRef CAS PubMed
- Y. Wang, R. Yin, L. Jin, M. Liu, Y. Gao, J. Raney and S. Yang, Adv. Funct. Mater., 2023, 33, 2210614 CrossRef CAS
- D. Druvari, F. Kyriakopoulou, G. C. Lainioti, A. Vlamis-Gardikas and J. K. Kallitsis, ACS Appl. Mater. Interfaces, 2023, 15, 11193–11207 CrossRef CAS PubMed
- X. Zhang, R. Crisci, J. A. Finlay, H. Cai, A. S. Clare, Z. Chen and M. N. Silberstein, Adv. Mater. Interfaces, 2022, 9, 2200430 CrossRef CAS
- T. Watabe and H. Otsuka, Angew. Chem., Int. Ed., 2023, 62, e202216469 CrossRef CAS PubMed
- F. J. Aubrecht, K. Orme, A. Saul, H. Cai, T. A. Ranathunge, M. N. Silberstein and B. R. McDonald, Angew. Chem., Int. Ed., 2024, 63, e202408673 CrossRef CAS PubMed
- X. Cao, S. Xuan, S. Sun, Z. Xu, J. Li and X. Gong, ACS Appl. Mater. Interfaces, 2021, 13, 30127–30136 CrossRef CAS PubMed
- Y. He, J. Tang, Y. Hu, S. Yang, F. Xu, M. Zrínyi and Y. Mei Chen, Chem. Eng. J., 2023, 462, 142193 CrossRef CAS
- J. Yang, W. Huang, K. Peng, Z. Cheng, L. Lin, J. Yuan, Y. Sun, N. J. Cho and Y. Chen, Adv. Funct. Mater., 2024, 34, 2313725 CrossRef CAS
- Z. Ren, S. Kim, X. Ji, W. Zhu, F. Niroui, J. Kong and Y. Chen, Adv. Mater., 2022, 34, 2106757 CrossRef CAS PubMed
- L. Chen, C. Zhao, J. Huang, J. Zhou and M. Liu, Nat. Commun., 2022, 13, 6821 CrossRef CAS PubMed
- H. Liu, M. Li, C. Ouyang, T. J. Lu, F. Li and F. Xu, Small, 2018, 14, 1801711 CrossRef PubMed
- X. Liu, X. Chen, X. Chi, Z. Feng, C. Yang, R. Gao, S. Li, C. Zhang, X. Chen, P. Huang, A. Dong, D. Kong and W. Wang, Nano Energy, 2022, 92, 106735 CrossRef CAS
- Y. Xiang, C. Liu, S. Ma, X. Wang, L. Zhu and C. Bao, Adv. Funct. Mater., 2023, 33, 2300416 CrossRef CAS
- P. Zhang, M. G. Debije, L. T. De Haan and A. P. Schenning, ACS Appl. Mater. Interfaces, 2022, 14, 20093–20100 CrossRef CAS PubMed
- Y. Yang, A. Li, Y. Yang, J. Wang, Y. Chen, K. Yang, B. Z. Tang and Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202308848 CrossRef CAS PubMed
- M. Lei, Q. Wang, R. Gu and D. Qu, Responsive Mater., 2024, 2, e20230027 CrossRef
- G. Zhu, T. Yu, J. Chen, R. Hu, G. Yang, Y. Zeng and Y. Li, ACS Appl. Mater. Interfaces, 2023, 15, 11033–11041 CrossRef CAS PubMed
- D. Yu, X. Xiao, C. Shokoohi, Y. Wang, L. Sun, Z. Juan, M. J. Kipper, J. Tang, L. Huang, G. S. Han, H. S. Jung and J. Chen, Adv. Funct. Mater., 2023, 33, 2211983 CrossRef CAS
- S. Uredat, A. Gujare, J. Runge, D. Truzzolillo, J. Oberdisse and T. Hellweg, Phys. Chem. Chem. Phys., 2023, 26, 2732–2744 RSC
- M. E. Alkahtani, M. Elbadawi, C. A. Chapman, R. A. Green, S. Gaisford, M. Orlu and A. W. Basit, Adv. Healthcare Mater., 2024, 13, 2301759 CrossRef CAS PubMed
- E. Cheah, M. Bansal, L. Nguyen, A. Chalard, J. Malmström, S. J. O'Carroll, B. Connor, Z. Wu and D. Svirskis, Acta Biomater., 2023, 158, 87–100 CrossRef CAS PubMed
- H. Zhou, S. Ye, M. Xu, L. Hao, J. Chen, Z. Fang, K. Guo, Y. Chen and L. Wang, Biomaterials, 2023, 301, 122200 CrossRef CAS PubMed
- S. Panda, S. Hajra, P. M. Rajaitha and H. J. Kim, Micro Nano Syst. Lett., 2023, 11, 2 CrossRef
- Z. Shen, F. Chen, X. Zhu, K. T. Yong and G. Gu, J. Mater. Chem. B, 2020, 8, 8972–8991 RSC
- C. Lim, Y. Joseph Hong, J. Jung, Y. Shin, S.-H. Sunwoo, S. Baik, O. Kyu Park, S. Hong Choi, T. Hyeon, J. Hoon Kim, S. Lee and D.-H. Kim, Sci. Adv., 2021, 7, eabd3716 CrossRef CAS PubMed
- Z. Xiong, S. Achavananthadith, S. Lian, L. Edward Madden, Z. Xin Ong, W. Chua, V. Kalidasan, Z. Li, Z. Liu, P. Singh, H. Yang, S. P. Heussler, S. M. P. Kalaiselvi, M. B. H. Breese, H. Yao, Y. Gao, K. Sanmugam, B. C. K. Tee, P.-Y. Chen, W. Loke, C. Teck Lim, G. Shu Hui Chiang, B. Yeow Tan, H. Li, D. Laurence Becker and J. S. Ho, Sci. Adv., 2021, 7, eabj1617 CrossRef CAS PubMed
- A. Barhoum, O. Sadak, I. A. Ramirez and N. Iverson, Adv. Colloid Interface Sci., 2023, 317, 102920 CrossRef CAS PubMed
- S. Gratz-Kelly, G. Rizzello, M. Fontana, S. Seelecke and G. Moretti, Adv. Funct. Mater., 2022, 32, 2201889 CrossRef CAS
- M. Smith and S. Kar-Narayan, Int. Mater. Rev., 2022, 67, 65–88 CrossRef CAS
- Y. Abe, M. Kakizaki and T. Hideshima, Jpn. J. Appl. Phys., 1969, 8, 975–976 CrossRef
- Z. Feng, Z. Zhao, Y. Liu, Y. Liu, X. Cao, D. G. Yu and K. Wang, Adv. Mater. Technol., 2023, 8, 2300021 CrossRef CAS
- H. Finkelmann, H. Kock and G. Rehage, Macromol. Rapid Commun., 1981, 2, 317–322 CrossRef CAS
- R. Annapooranan, Y. Wang and S. Cai, Adv. Mater. Technol., 2023, 8, 2201969 CrossRef CAS
- K. Oguro, Y. Kawami and H. Takenaka, J. Micromach. Soc., 1992, 5, 27–30 Search PubMed
- M. Shahinpoor, Smart Mater. Struct., 1992, 1, 91 CrossRef
- K. Sadeghipour, R. Salomon and S. Neogi, Smart Mater. Struct., 1992, 1, 172 CrossRef CAS
- H. Zhang, Z. Lin, Y. Hu, S. Ma, Y. Liang, L. Ren and L. Ren, Adv. Sci., 2023, 10, 2206135 CrossRef CAS PubMed
- R. Pelrine, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836–839 CrossRef CAS PubMed
- E. Hajiesmaili and D. R. Clarke, J. Appl. Phys., 2021, 129, 151102 CrossRef CAS
- C. Tang, B. Du, S. Jiang, Z. Wang, X. J. Liu and H. Zhao, Adv. Intell. Syst., 2024, 6, 2300047 CrossRef
- Y. Guo, L. Liu, Y. Liu and J. Leng, Adv. Intell. Syst., 2021, 3, 2000282 CrossRef
- D. Morales, E. Palleau, M. D. Dickey and O. D. Velev, Soft Matter, 2014, 10, 1337–1348 RSC
- Y. Yang, C. Li, L. C. Palmer and S. I. Stupp, Sci. Adv., 2023, 9, eadi4566 CrossRef CAS PubMed
- W. Jiang, L. Wang, B. Chen and H. Liu, J. Micromech. Microeng., 2023, 33, 015003 CrossRef CAS
- W. Li, Q. Guan, M. Li, E. Saiz and X. Hou, Prog. Polym. Sci., 2023, 140, 101665 CrossRef CAS
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS
- Y. Ren, Z. Liu, G. Jin, M. Yang, Y. Shao, W. Li, Y. Wu, L. Liu and F. Yan, Adv. Mater., 2021, 33, 2008486 CrossRef CAS PubMed
- T. A. Asoh and A. Kikuchi, Chem. Commun., 2010, 46, 7793–7795 RSC
- T.-a Asoh and A. Kikuchi, Chem. Commun., 2012, 48, 10019–10021 RSC
- T. A. Asoh, W. Kawai and A. Kikuchi, Colloids Surf., B, 2014, 123, 742–746 CrossRef CAS PubMed
- T. A. Asoh, Polym. J., 2016, 48, 1095–1101 CrossRef CAS
- J. J. Kirkland, Anal. Chem., 1965, 37, 1458–1461 CrossRef CAS
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed
- W. Yuan, G. M. Weng, J. Lipton, C. M. Li, P. R. Van Tassel and A. D. Taylor, Adv. Colloid Interface Sci., 2020, 282, 102200 CrossRef CAS PubMed
- J. Lipton, G. M. Weng, J. A. Röhr, H. Wang and A. D. Taylor, Matter, 2020, 2, 1148–1165 CrossRef
- V. Kozlovskaya, M. Dolmat and E. Kharlampieva, Langmuir, 2022, 38, 7867–7888 CrossRef CAS PubMed
- G. Zhang, X. Gao, S. Ji and Z. Liu, J. Membr. Sci., 2008, 307, 151–155 CrossRef CAS
- Y. H. Ko, Y. H. Kim, J. Park, K. T. Nam, J. H. Park and P. J. Yoo, Macromolecules, 2011, 44, 2866–2872 CrossRef CAS
- T. Wang, J. Lu, L. Mao and Z. Wang, J. Membr. Sci., 2016, 515, 125–133 CrossRef CAS
- A. S. Michaels, Ind. Eng. Chem., 1965, 57, 32–40 CrossRef CAS
- V. S. Meka, M. K. Sing, M. R. Pichika, S. R. Nali, V. R. Kolapalli and P. Kesharwani, Drug Discovery Today, 2017, 22, 1697–1706 CrossRef CAS PubMed
- D. Wu, L. Zhu, Y. Li, X. Zhang, S. Xu, G. Yang and T. Delair, Carbohydr. Polym., 2020, 238, 116126 CrossRef CAS PubMed
- F. Zhu, L. Cheng, J. Yin, Z. L. Wu, J. Qian, J. Fu and Q. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 31304–31310 CrossRef CAS PubMed
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, X. Li, H. Guo, Y. Huang, H. Zhang and J. P. Gong, Polymer, 2017, 116, 487–497 CrossRef CAS
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed
- H. Cai, Z. Wang, N. W. Utomo, Y. Vidavsky and M. N. Silberstein, Soft Matter, 2022, 18, 7679–7688 RSC
- Z. Wang, H. Cai and M. N. Silberstein, Mech. Mater., 2023, 179, 104604 CrossRef
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS
- P. Schaaf and J. B. Schlenoff, Adv. Mater., 2015, 27, 2420–2432 CrossRef CAS PubMed
- D. Li, T. Göckler, U. Schepers and S. Srivastava, Macromolecules, 2022, 55, 4481–4491 CrossRef CAS
- J. Es Sayed, A. Mukherjee, S. El Aani, N. Vengallur, M. Koch, A. Giuntoli and M. Kamperman, Macromolecules, 2024, 57, 3190–3201 CrossRef CAS PubMed
- G. Francius, A. Razafitianamaharavo, M. Moussa, M. Dossot, E. André, J. Bacharouche, B. Senger, V. Ball and J. F. Duval, J. Phys. Chem. C, 2016, 120, 5599–5612 CrossRef CAS
- B. N. Dickhaus and R. Priefer, Colloids Surf., A, 2016, 488, 15–19 CrossRef CAS
- Z. Sui, J. A. Jaber and J. B. Schlenoff, Macromolecules, 2006, 39, 8145–8152 CrossRef CAS
- M. G. Elshof, W. M. de Vos, J. de Grooth and N. E. Benes, J. Membr. Sci., 2020, 615, 118532 CrossRef CAS
- M. Tepermeister and M. N. Silberstein, arXiv, 2024, preprint, arXiv:2410.04310 DOI:10.48550/arXiv.2410.04310.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5mh00308c |
| This journal is © The Royal Society of Chemistry 2025 |