
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-step functionalization of hydrogels through mechano- and photo-responsive linkages†
Zihao
Li
 ac,
Chavinya D.
Ranaweera
ac,
Kang
Lin
bc,
Yuwan
Huang
d,
Thomas G.
Molley
bc,
Lei
Qin
a,
Jamie J.
Kruzic
d and
Kristopher A.
Kilian
*abc
ac,
Chavinya D.
Ranaweera
ac,
Kang
Lin
bc,
Yuwan
Huang
d,
Thomas G.
Molley
bc,
Lei
Qin
a,
Jamie J.
Kruzic
d and
Kristopher A.
Kilian
*abc
aSchool of Chemistry, UNSW Sydney, Sydney, NSW 2052, Australia. E-mail: k.kilian@unsw.edu.au
bSchool of Materials Science and Engineering, UNSW Sydney, Sydney, NSW 2052, Australia
cAustralian Centre for NanoMedicine, UNSW Sydney, Sydney, NSW 2052, Australia
dSchool of Mechanical and Manufacturing Engineering, UNSW Sydney, Sydney, NSW 2052, Australia
First published on 27th January 2025
Abstract
Patterning soft materials with cell adhesion motifs can be used to emulate the structures found in natural tissues. While patterning in tissue is driven by cellular assembly, patterning soft materials in the laboratory most often involves light-mediated chemical reactions to spatially control the presentation of cell binding sites. Here we present hydrogels that are formed with two responsive crosslinkers—an anthracene-maleimide adduct and a disulfide linkage—thereby allowing simultaneous or sequential patterning using force and UV light. Hydrogels were formed using poly(ethylene glycol)-based crosslinkers, yielding homogeneous single networks where the mechanical properties can be controlled with crosslinker content. Compression with a PDMS stamp inked with a cysteine-terminated peptide leads to (1) force-mediated retro-Diels Alder revealing a pendant maleimide and (2) subsequent Michael-type addition of the peptide. Successful functionalization was verified through monitoring anthracene fluorescence and via cell adhesion to the immobilized peptides. The material was further functionalized using UV light to open the disulfide bond in the presence of a maleimide-terminated peptide, thereby allowing a second immobilization step. Sequential derivatization was demonstrated by adding a second cell type, yielding patterns of multiple cell populations. In this way, force and light serve as complementary triggers to create geometrically structured heterotypic cell cultures for next-generation bioassays and materials for tissue engineering.
New conceptsWhile stimuli responsive linkages are widely used in polymer science and engineering, the integration of multiple responsive elements has received less attention. In this paper, we report the inclusion of two separate crosslinkers with dynamic covalent chemistry that are responsive to applied force and light. One linkage presents an anthracene–maleimide adduct that undergoes a retro Diels–Alder reaction in response to compression. In contrast to previous reports of this activity in polymers, the hydrogel environment promotes bond opening at an order of magnitude lower force. We harness this facile conversion for immobilization of thiol containing molecules through Michael-type addition in response to strain within the hydrogel. A disulfide linkage makes up the second crosslinker, which we later activate with UV light to facilitate an additional immobilization of maleimide containing molecules. Using a structured stamp inked with functionalized cell adhesion peptides, we show how force and light can be used to trigger two distinct immobilization events, for stepwise pairing of two cell types. This approach of integrating multiple stimuli responsive crosslinkers is broadly applicable to virtually all hydrogel systems, thereby providing a general means for spatiotemporal control of chemistry and functionality using combinations of force and light. |
Introduction
Human tissues are home to hundreds of unique cell types which are assembled in complex patterns to facilitate tissue form and function. Recreating the spatiotemporal positioning of cells in the laboratory has been a primary goal of tissue engineers.1–3 To understand the communication between different cell types, model systems that allow spatial arrangement of cells across planar substrates have proven useful for fundamental studies.4–6 Mrksich, Whitesides and colleagues pioneered the use of stimuli responsive surface chemistry for dynamic control of cell adhesion.7–10 In the ensuing years, approaches to pattern multiple cell types on a surface have been demonstrated using light triggers,11–15 electroactive surfaces,9,16 temperature responsive materials17,18 and exploiting the natural self-segregation programs of dissimilar cells (e.g. cancer cells and fibroblasts).19A versatile approach to functionalizing soft materials involves the use of applied force, i.e. mechanochemistry, with a rich history for instilling dynamic activities into polymer networks.20–22 Force-responsive molecular linkages have been integrated with a range of polymer systems with examples of changes in optical properties,23–25 mechanics,26–28 conductivity,29,30 the immobilization and release of molecules,31–34 and polymer degradation.35,36 However, mechanochemistry in hydrogels has not received much attention, primarily because (1) the hydrophobicity of the mechanophores is not compatible with aqueous solutions, (2) mechanochemical activity is different in water compared to bulk polymer environments, and (3) hydrogels are generally brittle, disallowing the magnitude of force required for activation. Approaches to translate mechanochemistry concepts to hydrogel materials are needed to exploit the value of this underutilized chemistry in biotechnology and biomedical applications.
Recently, we developed mechanochemistry in hydrogels through the use of double network systems, where a covalent hydrogel is strengthened by virtue of a secondary network.37 Jayathilaka and colleagues integrated a “flex-activated”, oxanorbornadiene-based mechanophore into a double network of polyacrylamide and alginate, showing release of pendant cargo at relatively low forces, e.g. < 100 kPa.37 Similarly, anthracene–maleimide mechanochemistry, also based on a retro-Diels–Alder reaction, has been explored for its ability to produce fluorescence signal in polymer network upon activation rather than release of cargo.24 However, this chemistry has so far only been demonstrated in organic polymer networks, and its application in hydrogels remains an unexplored opportunity. In addition to release, force-mediated immobilization was demonstrated using disulfide linked hydrogels, where the presence of a thiol reactive group would lead to in situ derivatization under compression or tension.34,38 Disulfide linked hydrogels are also amenable to modification through UV light activation of thiyl radicals,39 making this molecular linkage flexible for modification using both force and light. Considering the diverse “toolbox” of responsive linkages available, the use of multi-responsive linkages in a single material holds great promise for revealing novel functionalities and for new approaches in biofabrication.
In this communication, we demonstrate the first example of a mechano- and photo-responsive hydrogel system, where two stimuli-responsive motifs can be activated alone or in combination. Poly(ethylene glycol) (PEG) hydrogels were synthesized with an anthracene–maleimide (A–M) mechanophore or with internal disulfide bonds (S–S). Co-polymerization led to a homogenous hydrogel (e.g. single network) with two responsive moieties (A–M and S–S). Mechanochemical activation of the A–M linked hydrogels decreased the fracture strain compared to pure PEG networks, with a continual decrease during cyclic loading. Compression in the presence of thiol compounds led to Michael-type addition with the maleimide groups from A–M activation, which we demonstrated using a thiol containing fluoresceine derivative and a cysteine terminated peptide. Compression with the cell adhesion peptide GRGDSC was used to promote cell attachment, which we demonstrated using soft lithography for patterned cell adhesive regions. The dual network could be further activated by UV light, breaking the S–S linkages for further reaction, which we demonstrated using a maleimide-terminated GRGDS peptide to facilitate adhesion of a second cell type. This work provides a new platform for geometric patterning of multiple cell types using force and light, with broad flexibility for integration with virtually any soft material.
Results
Anthracene–maleimide linked poly(ethylene glycol) hydrogels
Due to the conjugated molecular structures, the anthracene–maleimide mechanophore has low solubility in water and previous studies have primarily been investigated in organic systems.24,33,40,41 We aimed to address this issue by leveraging the excellent water solubility and versatile end-functionality of polyethylene glycol (PEG) polymers. Thus, we designed the anthracene–maleimide linked poly(ethylene glycol) dimethacrylate (PEG-A-M-DM, Fig. 1a) macromonomer. The synthetic scheme for PEG-A-M-DM is depicted in Fig. S3 (ESI†). Briefly, a Diels–Alder reaction was first carried out between 9-anthracenemethanol and maleic anhydride. Aminolysis between the anthracene-maleic anhydride precursor (1) and diamino-terminated PEG (10 kDa) led to the formation of anthracene–maleimide mechanophore at both ends of the PEG molecule. Dimethacrylate groups were introduced by methacryloyl chloride to produce the desired PEG-A-M-DM macromonomer (3). Detailed information about the synthesis can be found in the ESI.† | ||
| Fig. 1 (a) Chemical structures of anthracene–maleimide linked poly(ethylene glycol) dimethacrylate (PEG-A-M-DM), disulfide linked poly(ethylene glycol) dimethacrylate (PEG-S-S-DM) and poly(ethylene glycol) dimethacrylate (PEG-DM). (b) Schematic overview of sequential cell patterning with dual-functional hydrogel. Scale bar = 500 μm. | ||
Rheological analysis comparing PEG-A-M-DM hydrogel and the non-mechanophore containing PEG-DM (same length as the PEG-A-M-DM, 10 kDa) hydrogel revealed the force-activated breakage of the anthracene–maleimide moiety within the hydrogels. The incorporation of anthracene–maleimide into the PEG-A-M-DM hydrogel was evident by its significantly higher storage modulus, 25 kPa after complete gelation, compared to PEG-DM, which was around 7 kPa as observed in Fig. 2a. Importantly, during oscillatory shear the PEG-A-M-DM hydrogel exhibited an earlier breakage point at 15% shear strain, in contrast to PEG-DM hydrogel requiring 40% shear strain for failure (Fig. 2b). This result suggests that inclusion of the anthracene–maleimide linage reduces the strain required for network rupture in PEG-based hydrogels, presumably due to the lower energy required for a force-mediated retro-Diels Alder reaction compared to breaking a C–C bond. To evaluate iterative mechanophore opening, we performed thixotropy testing, where the hydrogels were exposed to alternating high and low shear regimes. The hydrogels were cycled between 20 min at 0.2% shear strain and 3 min at 400% shear strain, which revealed dynamic changes in the rheological properties over cycles. PEG-A-M-DM hydrogel demonstrated a continuous decrease in storage modulus due to ongoing breakage of anthracene–maleimide mechanophores, while PEG-DM hydrogel stabilized after two cycles (Fig. 2c and d). Notably, after six cycles, the storage modulus of PEG-A-M-DM hydrogel diminished to only 10% of its initial value, whereas PEG-DM hydrogel retained 50% of its original storage modulus, as illustrated in Fig. 2e.
 | ||
| Fig. 2 Rheological characterization of 10 wt% hydrogels by (a) oscillatory time sweeps, (b) strain sweeps and (c) thixotropic test (alternating between 20 minutes of 0.2% shear strain and 3 minutes of 400% shear strain). (d) Maximum storage modulus and (e) normalized maximum storage modulus after each thixotropic test cycle in (c). | ||
Force-mediated functionalization and cell adhesion
Next, to support the mechanochemical mechanism we sought to demonstrate the presence of free maleimide and anthracene groups from mechanophore breakage using microscopy techniques. To verify the formation of maleimide groups after activation, a hydrogel composition of 10 wt% 1/1 PEG-A-M-DM/PEG-DM was prepared on a glass coverslip. This hydrogel was compressed using a 500 g weight (47 kPa) on a PDMS stamp (500 μm2 circular-patterned) coated with a commercially available thiol-containing dye, FITC-PEG-SH (1 kDa, Broadpharm), for 5 minutes to strain the network. Upon opening of the mechanophore, free maleimide groups were expected to react with the thiol groups in FITC-PEG-SH via spontaneous thiol–ene reaction. After washing with detergent to remove physically bound dye, the compressed hydrogels exhibited circular patterned fluorescence under confocal microscopy, with a higher fluorescence signal around the edge, possibly due to higher pressure at the interface of patterned/non-patterned areas (Fig. 3a). Notably, the interior of compressed areas still displayed significantly higher fluorescence signals than non-compressed areas (Fig. S7a, ESI†). In addition to the maleimide group, mechanophore activation results in release of a tethered anthracene moiety. To support the evidence of exposed maleimide functionalities, we exploited the natural fluorescence of anthracene (397 nm); using the same process with a blank PDMS stamp which demonstrated markedly higher anthracene signals at the compressed area when compared to the non-compressed area (Fig. 3b and Fig. S7b, ESI†). Notably, in a more extreme scenario, where a disc-shaped 3D hydrogel was cut by a razor blade, the fluorescence of the activated anthracene moieties was clearly observable and could be captured under a 365 nm UV lamp using a digital camera (Fig. S8a, ESI†). To estimate the minimum pressure required for anthracene–maleimide mechanophore activation within the hydrogel, disc-shaped 3D hydrogels were prepared, compressed at indicated pressures (0 to 150 kPa) for 5 minutes, and stained with FITC-PEG-SH. Confocal imaging revealed observable mechanophore activation in hydrogels at 100 and 150 kPa (Fig. 3c), significantly less than previously reported study where MPa pressures were required for anthracene–maleimide activation in organic polymer networks,24 highlighting the importance of solvent effects for mechanophore activation. Furthermore, increased anthracene fluorescence signals were observed in compressed 3D hydrogels (Fig. S8b and c, ESI†), further validating the force-activation mechanism. This result is consistent with previous work demonstrating how hydrogel environments provide faster mechanochemical activation at lower applied force.37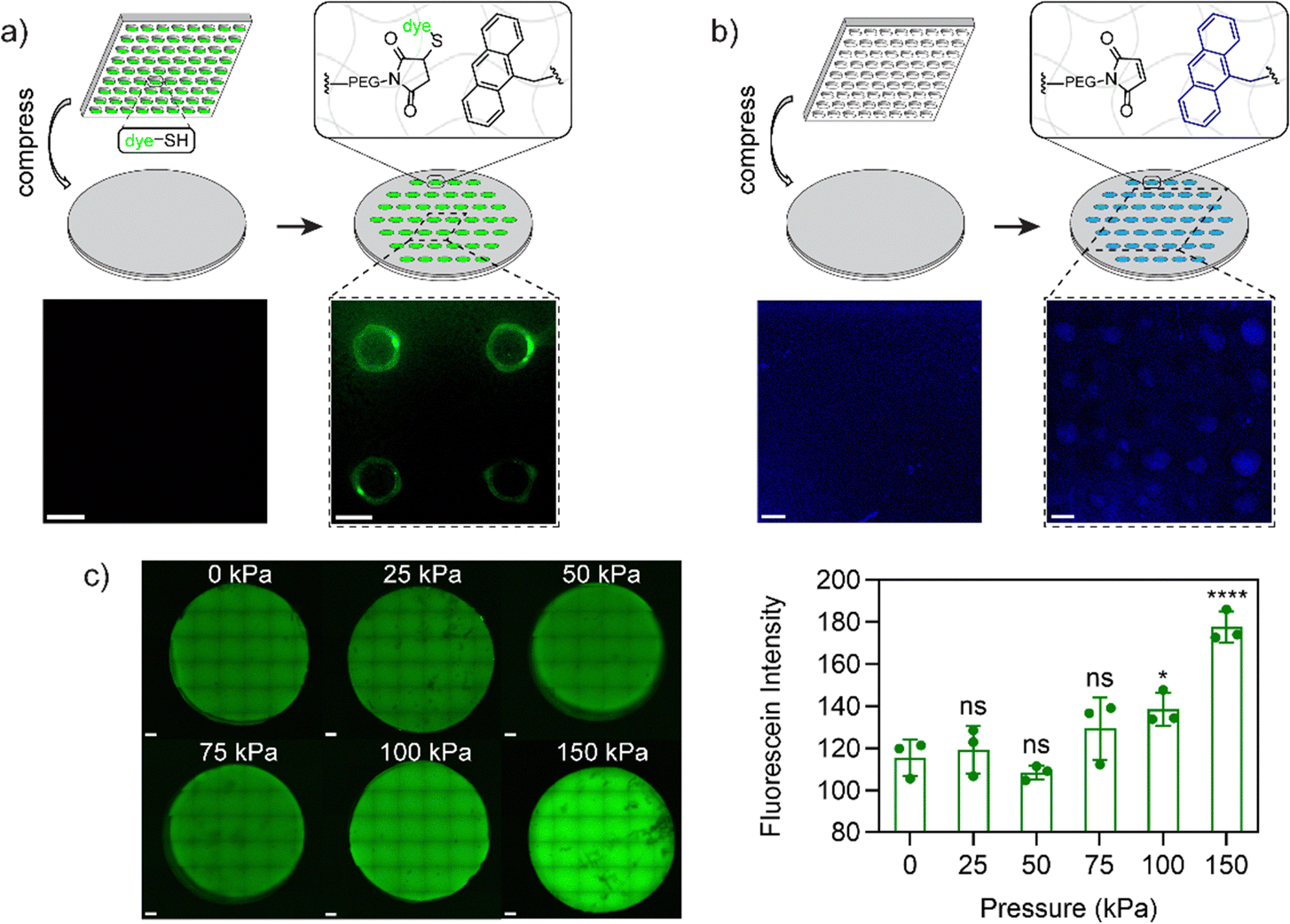 | ||
| Fig. 3 (a) Confocal image for hydrogels before (left) and after (right) compression with FITC-PEG-SH inked PDMS stamp. (b) Fluorescence image for hydrogels (anthracene fluorescence, 397 nm) before (left) and after (right) compression with PDMS stamp. (c) Representative z-stacked confocal images (left) and total fluorescence intensity (right) for FITC-PEG-SH stained 3D hydrogels (3 repeats) after compression at indicated pressures for 5 min. All hydrogels had a composition of 10 wt% 1/1 PEG-A-M-DM/PEG-DM. p values were calculated via one-way ANOVA in compression with 0 kPa (ns: not significant, p > 0.05; *: p < 0.05; ****: p < 0.0001). Scale bars = 500 μm. | ||
The lower force activation property and induced presentation of maleimide handles render this system a suitable candidate for biomedical applications, particularly for force-patterned functionalization and subsequent cell attachment. To validate this concept, hydrogels composed of 10 wt% 1/1 PEG-A-M-DM/PEG-DM were patterned with a thiol-containing cell-binding peptide, GRGDSC, using inked PDMS stamps. Immediately after patterning and sterilization, human foreskin fibroblast cells (HFF-1) were added to the hydrogel samples. Following 4 days of incubation, the fibroblast cells exclusively attached and proliferated only in the patterned area, displaying a healthy spindle-like morphology (Fig. 4a and b). There were very few cells adherent to the negative control samples, and they showed a rounded morphology (Fig. 4c–g). This demonstrates how force can be used to immobilize cell adhesion ligands for patterning cell populations.
 | ||
| Fig. 4 (a) Schematic representation and brightfield microscopy image of HFF-1 cells on GRGDSC patterned 10 wt% 1/1 PEG-A-M-DM/PEG-DM hydrogels 96 hours after cell seeding. (b) Zoomed in picture from the selected area in (a). (c)–(g) Negative controls with the scrambled peptide GRDGSC; not compressed; 10 wt% PEG-DM; 10 wt% PEG-DM compressed the scrambled peptide GRDGSC; and not compressed 10 wt% PEG-DM respectively. Scale bars = 200 μm. | ||
Dual responsive networks for stepwise patterning
One area that has received limited attention is the integration of multiple responsive linkages to increase functionality. The versatility of the A–M mechanophore extends to compatibility with other responsive moieties. Inspired by a photo-responsive disulfide hydrogel system,39 we incorporated disulfide groups to enable stepwise patterning by force and light. To maintain a homogeneous system, we incorporated disulfide groups into the hydrogels in the form of disulfide-linked poly(ethylene glycol) dimethacrylate (PEG-S-S-DM, see Fig. 1a), similar to the PEG-A-M-DM structure. The full synthetic scheme and details for PEG-S-S-DM are provided in Fig. S5 and S6 (ESI†). Briefly, the two amino groups in cystamine dihydrochloride were initially hetero-functionalized with BOC anhydride and methacryloyl chloride. Subsequently, the BOC-protected amine was deprotected using trifluoroacetic acid and functionalized using N,N′-disuccinimidyl carbonate. Aminolysis with diamino-terminated PEG (10 kDa) resulted in the formation of the desired PEG-S-S-DM macromonomer.The concept of stepwise patterning was initially demonstrated on hydrogels composed of 10 wt% 1/1 PEG-A-M-DM/PEG-S-S-DM using two fluorophores: FITC-PEG-SH for maleimide from anthracene–maleimide activation and thiol fluorescence probe IV (Merck) for thiyl radicals from disulfide activation. After compression with a FITC-PEG-SH inked PDMS stamp with circular features, a solution containing 65 μM thiol fluorescence probe IV and 1 mM photoinitiator LAP was applied onto the hydrogels. Subsequently, the hydrogels were irradiated with 395 nm UV light (2 W) for 1 minute to open disulfide bonds and promote the subsequent reaction with the fluorescence probe IV (Fig. 5a). Confocal images of the patterned samples clearly demonstrated the controlled presentation of green FITC signals only at the compressed area, while the blue signals were evenly distributed across the surface area, confirming the success of stepwise patterning (Fig. 5b).
 | ||
| Fig. 5 (a) Schematic representation and (b) confocal images of stepwise patterning for 10 wt% 1/1 PEG-A-M-DM/PEG-S-S-DM hydrogels with two fluorophores, FITC-PEG-SH and thiol fluorescence probe IV. Scale bars = 500 μm. | ||
The presence of two responsive groups offers opportunities for selective patterning of multiple cell types on the same culture scaffold. To demonstrate this concept, the fluorophores were substituted with two cell-adhesive peptides: the thiol-containing RGD peptide GRGDSC to attach with maleimide groups and a maleimide-containing RGD peptide Mal-GRGDS to react with thiols/thiyl radicals. To facilitate clear distinguishability between different cell types, we employed two fluorescently labeled cell lines: green fluorescent protein (GFP) expressing metastatic human melanoma cells (WM266-4) and mCherry expressing metastatic human melanoma cells (A374-MA2).42
The experimental procedure was initiated by the compression of the mixed 10 wt% 1/1 PEG-A-M-DM/PEG-S-S-DM hydrogel using a GRGDSC inked PDMS stamp (circular-patterned). Subsequently, WM266-4 (GFP) cells were introduced and allowed to incubate for 24 hours to ensure firm attachment. Next, unattached cells were removed and media containing 2 mM Mal-GRGDS and 1 mM LAP was evenly applied to the hydrogel surface. The surface was then irradiated briefly with UV light to initiate reaction of opened disulfides with the maleimide terminated peptides. Finally, A374-MA2 (mCherry) cells were introduced and allowed to incubate for an additional 18 hours to ensure complete cell attachment (Fig. 1b). In compression-only hydrogels, both WM266-4 (GFP) and A374-MA2 (mCherry) cells are preferentially attached to the compressed areas, as they served as the sole sites for cell attachment, due to the force-mediated conjugation of the GRGDSC peptide. Conversely, in UV-only treated hydrogels, no WM266-4 (GFP) cells were observed, and A374-MA2 (mCherry) cells were evenly distributed across the surface, attaching to the UV-mediated conjugation of the Mal-GRGDS peptide. Notably, in dual-patterned hydrogels, WM266-4 (GFP) cells displayed selective attachment at the compressed areas, while A374-MA2 (mCherry) cells were evenly distributed across the UV-treated surface (Fig. 6a and b). These results demonstrate how multiple responsive triggers can be integrated for selective patterning of multiple cell types. This approach provides the first method where force and light can be used independently and together to functionalize hydrogels, with scope for immobilization of virtually any appropriately tagged molecule and flexibility in the order and timing of activation.
 | ||
| Fig. 6 (a) Confocal images of WM266-4 (GFP) and A374-MA2 (mCherry) co-cultured cells on stepwise patterned (compressed with GRGDSC inked PDMS stamp, UV in the presence of LAP and Mal-GRGDS) 10 wt% 1/1 PEG-A-M-DM/PEG-S-S-DM hydrogels. The schematic representation of stepwise cell patterning can be found in Fig. 1b. (b) Fluorescence intensities (analyzed in ImageJ by selecting 8 independent areas for the indicated regions) of the compressed and non-compressed areas for the combined confocal images in (a). p values were calculated via one-way ANOVA (ns: not significant, p > 0.05; *: p < 0.05; **: p < 0.005; ****: p < 0.0001;). Scale bars = 500 μm. | ||
Conclusions
In conclusion, we have demonstrated a multi-responsive hydrogel system capable of dynamic functionalization using both force and UV light, thereby enabling the activation of different responsive motifs independently or in combination. Our synthesis of poly(ethylene glycol) hydrogels with anthracene–maleimide mechanophores and internal disulfide bonds resulted in a homogeneous hydrogel with distinct responsive regions. This system showed decreased fracture strain under mechanochemical activation and enabled controlled cell attachment through compression and UV irradiation. Our approach, utilizing soft lithography for patterning cell adhesive motifs, provides a unique system for spatial control of cell and tissue composition and geometry. The flexibility afforded with two distinct stimuli responsive linkages, that are amenable to inclusion in virtually any soft material with any combination of immobilized molecular species, provides broad versatility spanning applications in designer culture surfaces for cell biology, coatings for implants, materials for scaffolds, and functionalizable interfaces for soft devices and robotics.Data availability
This study generated data including shear rheology and spectroscopy files which are stored in instrument specific software formats and in ASCiI file format, and microscopy files and images. The datasets generated during the current study are not publicly available due to further analysis related to future publications but are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Australian Research Council for funding this work through the Discovery Project Grant DP210103654. We also thank Associate Prof. Kate Poole from the School of Biomedical Sciences, UNSW Sydney for providing fluorescently labeled cell lines. We gratefully acknowledge the assistance of Alaa Ajam who contributed supplementary data in support of the revision process. The authors acknowledge the help and support of staff at the Katharina Gaus Light Imaging Facility (KGLMF) of the UNSW Mark Wainwright Analytical Centre.References
- W. F. Liu and C. S. Chen, Mater. Today, 2005, 8, 28–35 CrossRef.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS.
- J. Deng, C. Zhao, J. P. Spatz and Q. Wei, ACS Nano, 2017, 11, 8282–8291 CrossRef CAS.
- A. Joshi, T. Kaur and N. Singh, Langmuir, 2021, 37, 4933–4942 CrossRef CAS.
- A. Joshi and N. Singh, ACS Omega, 2023, 8, 34249–34261 CrossRef CAS.
- M. Mrksich, C. S. Chen, Y. Xia, L. E. Dike, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10775–10778 CrossRef CAS PubMed.
- M. Mrksich, Chem. Soc. Rev., 2000, 29, 267–273 Search PubMed.
- M. N. Yousaf, B. T. Houseman and M. Mrksich, Proc. Natl. Acad. Sci., 2001, 98, 5992–5996 CrossRef CAS PubMed.
- B. T. Houseman, E. S. Gawalt and M. Mrksich, Langmuir, 2003, 19, 1522–1531 CrossRef CAS.
- K. Jang, K. Sato, Y. Tanaka, Y. Xu, M. Sato, T. Nakajima, K. Mawatari, T. Konno, K. Ishihara and T. Kitamori, Lab Chip, 2010, 10, 1937 RSC.
- H. Ogawa, Y. Yamazawa, T. Nakaji-Hirabayashi, H. Kitano, Y. Saruwatari and K. Matsuoka, Mater. Adv., 2022, 3, 5753–5759 RSC.
- L. S. Prahl, C. M. Porter, J. Liu, J. M. Viola and A. J. Hughes, iScience, 2023, 26, 106657 CrossRef CAS PubMed.
- C. Wang, M. Fadeev, J. Zhang, M. Vázquez-González, G. Davidson-Rozenfeld, H. Tian and I. Willner, Chem. Sci., 2018, 9, 7145–7152 RSC.
- F. Huang, M. Chen, Z. Zhou, R. Duan, F. Xia and I. Willner, Nat. Commun., 2021, 12, 1–14 Search PubMed.
- O. Guillaume-Gentil, M. Gabi, M. Zenobi-Wong and J. Vörös, Biomed. Microdevices, 2011, 13, 221–230 CrossRef PubMed.
- X. Cheng, Y. Wang, Y. Hanein, K. F. Böhringer and B. D. Ratner, J. Biomed. Mater. Res. A, 2004, 70A, 159–168 CrossRef CAS PubMed.
- M. Yamato, C. Konno, M. Utsumi, A. Kikuchi and T. Okano, Biomaterials, 2002, 23, 561–567 CrossRef CAS.
- S. Nemec, J. Lam, J. Zhong, C. Heu, P. Timpson, Q. Li, J. Youkhana, G. Sharbeen, P. A. Phillips and K. A. Kilian, Adv. Biol., 2021, 5, 2000525 CrossRef CAS.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- B. A. Versaw, T. Zeng, X. Hu and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 21461–21473 CrossRef CAS PubMed.
- E. M. Lloyd, J. R. Vakil, Y. Yao, N. R. Sottos and S. L. Craig, J. Am. Chem. Soc., 2023, 145, 751–768 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- C. P. Kabb, C. S. O’Bryan, C. D. Morley, T. E. Angelini and B. S. Sumerlin, Chem. Sci., 2019, 10, 7702–7708 RSC.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 1979, 2019(363), 504–508 Search PubMed.
- Z. Wang, X. Zheng, T. Ouchi, T. B. Kouznetsova, H. K. Beech, S. Av-Ron, T. Matsuda, B. H. Bowser, S. Wang, J. A. Johnson, J. A. Kalow, B. D. Olsen, J. P. Gong, M. Rubinstein and S. L. Craig, Science, 1979, 2021(374), 193 Search PubMed.
- S. Wang, Y. Hu, T. B. Kouznetsova, L. Sapir, D. Chen, A. Herzog-Arbeitman, J. A. Johnson, M. Rubinstein and S. L. Craig, Science, 1979, 2023(380), 1248–1252 Search PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 1979, 2017(357), 475–479 Search PubMed.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2014, 136, 1276–1279 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- J. Li, T. Shiraki, B. Hu, R. A. E. Wright, B. Zhao and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 15925–15928 CrossRef CAS.
- J. Lee, M. N. Silberstein, A. A. Abdeen, S. Y. Kim and K. A. Kilian, Mater. Horiz., 2016, 3, 447–451 RSC.
- T.-G. Hsu, J. Zhou, H.-W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, J. Am. Chem. Soc., 2020, 142, 2100–2104 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS PubMed.
- P. B. Jayathilaka, T. G. Molley, Y. Huang, M. S. Islam, M. R. Buche, M. N. Silberstein, J. J. Kruzic and K. A. Kilian, Chem. Commun., 2021, 57, 8484–8487 RSC.
- Y. H. Tran, M. J. Rasmuson, T. Emrick, J. Klier and S. R. Peyton, Soft Matter, 2017, 13, 9007–9014 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- D. C. Church, G. I. Peterson and A. J. Boydston, ACS Macro Lett., 2014, 3, 648–651 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- A. Patkunarajah, J. H. Stear, M. Moroni, L. Schroeter, J. Blaszkiewicz, J. L. E. Tearle, C. D. Cox, C. Fürst, O. Sánchez-Carranza, M. D. Á. Ocaña Fernández, R. Fleischer, M. Eravci, C. Weise, B. Martinac, M. Biro, G. R. Lewin and K. Poole, eLife, 2020, 9, 1–25 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00761a |
| This journal is © The Royal Society of Chemistry 2025 |