
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced thermal response of 3D-printed bilayer hydrogels via nanoclay incorporation†
Francis
Klincewicz
 a,
Subhash
Kalidindi
bc,
Siyuan
Liu
a,
Kritee
Sangroula
b and
LaShanda T. J.
Korley
*abc
a,
Subhash
Kalidindi
bc,
Siyuan
Liu
a,
Kritee
Sangroula
b and
LaShanda T. J.
Korley
*abc
aMaterials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: lkorley@udel.edu
bChemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
cCenter for Research in Soft matter & Polymers (CRiSP), University of Delaware, Newark, DE 19716, USA
First published on 11th June 2025
Abstract
There exist numerous opportunities to design stimuli-responsive bilayer hydrogels for enhanced actuation using simple and robust techniques. Specifically, digital light processing (DLP) 3D printing offers a robust technique for multi-layered hydrogel fabrication. However, nanocomposite hydrogels utilizing this technique have not yet been widely realized. Nanoclay incorporation has been shown to improve the actuation of poly(N-isopropyl acrylamide) (pNIPAAm) hydrogels; however, opportunities remain to study the relationship between clay morphology and thermal response, particularly in a 3D-printed bilayer system. In this work, we utilized an ethanol-water cosolvent, hydrogel precursor solution to incorporate montmorillonite (MMT) clay into 3D-printed pNIPAAm hydrogels. By varying the MMT loading, we demonstrated that a low loading of MMT (0.5 wt% relative to the mass of NIPAAm monomer) induced the greatest enhancement of the initial rate and final magnitude of actuation in the studied hydrogels. We utilized poly(2-hydroxyethyl acrylate) (pHEA) as a passive layer to form bilayers by sequentially printing pHEA before the pNIPAAm/MMT hydrogels, and used those hydrogels to demonstrate the accelerated actuation of 3D-printed pNIPAAm/MMT-pHEA bilayers compared to clay-free, pNIPAAm-pHEA bilayers. Through comparison to a mathematical framework and fabrication of an all-pNIPAAm bilayer, we suggested that the model has limitations for the prediction of bilayer curvature in these systems due to the inability of certain hydrogels to overcome the inertia of the passive layer. Overall, this work showcases the utility of MMT as a handle for tunability in 3D-printed pNIPAAm bilayer hydrogels.
Design, System, ApplicationNatural systems, such as the pinecone, utilize a bilayer structure to achieve curvature, in which an active layer responds to stimuli and a passive layer remains unresponsive. Design of synthetic systems inspired by the pinecone often encompasses tunability of the active component. In this work, we utilize nanoclay loading in a thermally-responsive hydrogel for optimization of the thermal response of the hydrogel. We targeted the initial rate and final magnitude of response as metrics and utilized the morphology of the clay and hydrogel porosity to further explore the thermoresponsive actuation. We revealed that mitigation of hydrophobic skin layer formation on the surface of the hydrogel led to increased exerted force during the expulsion of water, which enhanced bilayer actuation. This approach contributes to the molecular understanding of the design of soft actuators, particularly for soft robotics. |
1 Introduction
Natural structures, such as the bilayer scale of the pinecone, inspire the design of synthetic systems capable of changing their shape in response to external stimuli.1 A pinecone scale exhibits curvature upon hydration due to the exterior layer of the bilayer swelling more than the interior layer.2,3 Synthetic bilayer materials mimicking this behavior can be prepared by interfacing a responsive “active” layer and a non-responsive “passive” layer.4,5 The change in bilayer curvature, also known as actuation, is driven by the longitudinal contraction and modulus variation in the active layer.6 The quest for faster, complex, and tunable actuation in stimuli-responsive hydrogels has prompted numerous innovations in the development of soft sensors, drug delivery systems, and soft robotics.7,8 Although hydrogels having bilayer structures are a particularly exciting class of materials for these applications due to their intrinsic tunability and opportunity for biocompatibility, there remain opportunities to investigate the effects of design parameters such as nanoparticle inclusion on hydrogel properties, including actuation speed and magnitude of responsive hydrogels.As a model thermally-responsive hydrogel, poly(N-isopropyl acrylamide) (pNIPAAm) has shown significant promise for thermally-responsive bilayer hydrogels. The pendent moieties of pNIPAAm switch from favoring hydrophilic polymer-solvent interactions to preferring intra-chain hydrophobic interactions above a lower critical solution temperature (LCST) of ∼32 °C, causing a volume phase transition (VPT).9 Several studies have focused on modification of the VPT through techniques such as copolymerization with other monomers, including acrylic acid10 and hydroxyethyl acrylamide,11 induction of pore morphology changes through synthesis solvent, and inclusion of nanofillers.12,13 Despite the utility of these techniques, one limiting factor to fast actuation in pNIPAAm hydrogels is the formation of a “skin” layer, which is due to the rapid deswelling of the exterior of the hydrogel during the VPT, blocking further expulsion of water from the interior.14–17 Hydrophilic, non-covalently incorporated nanofillers have been shown to increase the extent to which pNIPAAm contracts during the VPT, which is attributed to either the inhibition of skin layer formation or increased hydrophilicity within the hydrogel.15,18 In work by Li and Bae, graphene oxide (GO) nanoparticles were shown to disrupt the formation of a skin layer, thus increasing actuation.15 Xia et al. showed that the introduction of hydrophilic montmorillionite (MMT) clay into a pNIPAAm hydrogel improved the magnitude of the thermal response, which was attributed to the introduction of hydrophilicity into the hydrogel; however, above a certain loading, contraction was hindered, potentially due to electrostatic repulsion between clay domains.18 MMT is a layered silicate in which oxygen is ionically bonded to multiple metal ions, yielding highly hydrophilic and overall negatively charged faces with slightly positively charged edges; unlike GO, the hydrophilicity in MMT is uniform throughout the face instead of being decorated by oxygenated moieties.19 The high hydrophilicity of MMT makes it an ideal candidate for the acceleration of actuation in pNIPAAm hydrogels, especially bilayer hydrogels in which pNIPAAm forms the active layer. Despite previous studies highlighting the use of pNIPAAm/clay nanocomposites for bilayer actuation, the relationship between clay morphology, single layer actuation, and bilayer actuation has not been fully studied.20,21 Although clay has previously been reported in hydrogels as a crosslinker, the focus of this paper is on non-covalently incorporated clay fillers to study the effects compared to structurally-similar, clay-free bilayer hydrogels.22,23
Although pNIPAAm hydrogels have traditionally been polymerized in molds via thermal- or photo-initiation, the molding process can be limited in the production of complex geometric materials and in the development of multi-step, subtractive manufacturing methods.24–26 Bilayer hydrogels are not necessarily well-suited for fabrication via molding, as they require a uniform surface on the first-polymerized layer and avoidance of overcuring. Both qualities can be difficult to achieve when the first-polymerized layer may polymerize out of plane and may be doubly cured when the second layer is added. Digital light processing (DLP) 3D printing is a fabrication method for stimuli-responsive polymers with excellent geometric control without expensive molds or harsh chemicals.27 In DLP 3D printing, a part is fabricated layer by layer via sequential polymerization of a photosensitive resin by projecting UV light onto a build plate.28 By fabricating individual sublayers sequentially, each part of the hydrogel receives an even amount of UV light; because the layers are sealed between the UV source and build plate, every layer is uniformly flat, so strong adhesion can be maintained for bilayer hydrogels.29 While an extensive library of polymers has been developed for DLP, including pNIPAAm,29,30 recent work has focused on varying processing parameters, such as solvent and temperature.16,31 Integration of clay nanoparticles (e.g., MMT) into DLP 3D-printed polymers remains a technical challenge due to inherent miscibility and dispersion issues between the polymer, solvent, and clay.32–35 Clay platelets contain many surface charges which are typically exchanged with positive cations, such as sodium or organic ligands, and, for this reason, are often dispersed in water.33 Despite the advantages of nanoclays in pNIPAAm hydrogel systems for accelerating actuation, these nanofillers have not yet been incorporated into DLP, 3D-printed pNIPAAm due to issues with the stability of the suspension during printing. Although MMT has been incorporated into DLP 3D-printed nanocomposites, it has previously been incorporated at very low (0.25%) loadings or in viscous printing solutions, which are rare for hydrogel printing resins.36,37 A recent review38 highlighted only two examples of clay nanocomposite fabrication via DLP 3D printing, neither of which addressed hydrogels.39,40 Therefore, in addition to accelerating the actuation of DLP 3D-printed bilayer hydrogels, there is also an opportunity to investigate the ability of MMT to maintain its dispersion during the DLP 3D printing process. In our previous work, a mixed solvent of ethanol and water in a pNIPAAm DLP 3D printing resin was utilized to induce morphological changes in the resulting hydrogels, which were correlated to the observed monolayer and bilayer actuation.16 The utilization of a mixed solvent system, which includes water, may facilitate the inclusion of clay in the printing resin. The clay may overcome the previously reported skin layer formation and the associated challenges with actuation speed and magnitude encountered in these pNIPAAm hydrogels.
In this work, we studied the addition of MMT to accelerate the actuation of 3D-printed, thermally-responsive bilayer hydrogels by improving the water transport within the bilayers and limiting the formation of a hydrophobic skin layer. Our methodology encompassed first studying the effect of MMT addition on single-layer active control hydrogels, and subsequently, on the bilayer hydrogels. For the active control hydrogels, we explored MMT morphology and the thermal response, focusing on the initial rate and final magnitude of actuation. To investigate the impact of the MMT on bilayer curvature, the active controls were interfaced with poly(2-hydroxyethyl acrylate) (pHEA) as the passive layer. The effect of clay loading on the curvature of the bilayer hydrogels was compared to the linear actuation of the single-layer pNIPAAm hydrogels, using a theoretical model as a guide. This study serves to elucidate the mechanics of thermally-responsive bilayer hydrogels and provide design principles for the incorporation of hydrophilic nanofillers for enhanced thermal response.
2 Experimental
2.1 Materials
2-Hydroxyethyl acrylate (HEA) and 2,4,6-trimethylbenzoyldiphenyl phosphine oxide (TPO) were purchased from Sigma Aldrich. N,N′-Methylenebis(acrylamide) (MBA) was purchased from Alfa Aesar. N-Isopropylacrylamide (NIPAAm) was purchased from Tokyo Chemical Industry Co. 200-proof anhydrous ethanol was purchased from Decon Laboratories, Inc. 2-isopropanol was purchased from Fisher Scientific. Sodium-exchanged montmorillonite (MMT) clay with a cation exchange capacity (CEC) of 145 mequiv/100 g was purchased from Nanocor Corporation.41 All chemicals were used as received without further purification. Deionized (DI) water was purified using a Milli-Q Academic (Millipore-Sigma).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50. For pNIPAAm-based resins, 18.4 g of NIPAAm monomer was added to 10 mL of an ethanol–water mixture. Then, MBA crosslinker was added at 1.5 mol% relative to the monomer (0.039 g mL−1 of solvent) to this NIPAAm monomer-mixed solvent solution. For the HEA-based resin for bilayer actuation, 19.6 mL of HEA monomer was added to 10 mL of the 50:50 ethanol–water mixture. Then, MBA crosslinker was added at 0.1 mol% relative to the monomer (0.003 g mL−1 of solvent) to this HEA monomer-mixed solvent solution. Finally, for both resins, TPO was added to the monomer solutions at a concentration of 0.06 g mL−1 of solvent.
:50. For pNIPAAm-based resins, 18.4 g of NIPAAm monomer was added to 10 mL of an ethanol–water mixture. Then, MBA crosslinker was added at 1.5 mol% relative to the monomer (0.039 g mL−1 of solvent) to this NIPAAm monomer-mixed solvent solution. For the HEA-based resin for bilayer actuation, 19.6 mL of HEA monomer was added to 10 mL of the 50:50 ethanol–water mixture. Then, MBA crosslinker was added at 0.1 mol% relative to the monomer (0.003 g mL−1 of solvent) to this HEA monomer-mixed solvent solution. Finally, for both resins, TPO was added to the monomer solutions at a concentration of 0.06 g mL−1 of solvent.
2.2 Hydrogel fabrication
Immediately before printing, resins were sonicated using a probe sonicator for 6 on–off cycles of 5 seconds. This sonication cycling was employed alongside a surrounding ice bath to minimize heating of the resin during sonication. Resins were visually homogenous for at least 15 min in preliminary experiments after sonication. When settling occurred after this time, manual shaking restored visual homogeneity. To minimize the effect of inhomogeneity from settling, all prints in this work were conducted within 5 min of printing time, except for the immersion dynamic mechanical analysis (DMA) experiments, which were 20 min. For these longer prints, the print was paused after 10 min, the resin stirred vigorously, and the print resumed.
To print the pNIPAAm monolayers independently, a NIPAAm monomer resin solution (∼3 mL) was poured into the resin vat of the printer and sequentially irradiated with UV light for 16 s per 0.1 mm printed sublayer. Printed monolayers for actuation were comprised of 10 printed sublayers, while the thicker immersion DMA samples were comprised of 40 printed sublayers. After printing, the monomers and bilayers were removed from the build plate using a metal scraper and inserted into a UV chamber for a 15 min, post-cure procedure to complete the reaction.
To manufacture the pNIPAAm-pHEA bilayer hydrogels, printing was paused upon the formation of ten pHEA printed sublayers, which was facilitated by the removal of the print head from the vat during this time. The HEA monomer resin was then poured out of the vat, followed by rinsing the vat with isopropanol and washing the print head with ethanol to remove excess unreacted resin. Then, NIPAAm monomer resin was poured into the cleaned vat, and upon continuation of printing, the layers of pNIPAAm were printed onto the existing pHEA layers to form pNIPAAm-pHEA bilayers. All prints were exposed in air to a 405 nm UV light (13 mW cm−2) in a UV chamber (SainSmart, Inc.) for 15 min to completely polymerize any unreacted initiator or monomer.
2.3 Characterization
A Thermo Scientific Talos L120C TEM equipped with a Thermo Scientific Ceta 16 M camera was utilized to observe the clay microstructure. Dried pNIPAAm gels were embedded in commercial two-part epoxy and sliced dry using a diamond knife in a Leica LC7 microtome at a thickness of ∼100 nm. Slices were placed on a copper grid and a gentle force applied to adhere the sample to the grid. Samples were imaged at a 120 kV accelerating voltage.
Thermal DMA. Dynamic mechanical analysis (DMA) was performed using a TA Instruments RSA-G2 operating in tension mode. To determine the plateau modulus, rectangle-shaped and dried (50 °C for 72 h) monolayers (1 mm thickness × 2 mm width × ∼5 mm gauge length) were heated at a rate of 3 °C min−1 from 25 °C to 250 °C at a frequency of 1 Hz and an amplitude of 0.05% strain.
Immersion DMA. To study the evolution of mechanical properties in water, cylinder-shaped hydrogels (∼6 mm height × 9 mm diameter) were used. During a single test, the cylinders were first conditioned in room-temperature water with an amplitude sweep (1 Hz, 0.1% to 1% strain) and frequency sweep (0.02 to 10 Hz, 0.1% strain). Next, the room-temperature water was evacuated, and pre-heated DI water was added to the immersion cup. Immediately, temperature control in the DMA chamber was enabled and set to 60 °C, and a sinusoidal compression (1 Hz, 0.1% strain) was applied.
Thermal actuation of monolayer hydrogels. To quantify the thermal contraction of the monolayers, cylindrical hydrogels (1 mm height × 7 mm diameter) were printed. Deionized water was heated in a 250 mL beaker equipped with a thermocouple-controlled heating loop. The monolayer pNIPAAm hydrogels were placed into the beaker. The temperature of the water bath was set to 60 °C as monitored via a thermocouple with an equilibration period of at least 30 min. Before submersion, and after 5, 10, 15, 20, 25, and 30 min, the hydrogels were removed from the water bath onto a paper towel, on which the diameters were measured with calipers, and subsequently the masses were measured on a balance. All actuation experiments were performed in triplicate to ensure reproducibility.
Thermal actuation of bilayer hydrogels. To quantify the thermal contraction of the pNIPAAm-pHEA bilayer hydrogels, rectangular prisms (7 mm × 20 mm × 2 mm) were printed and immersed into a glass petri dish filled with DI water and heated at 60 °C on a hot plate. For the first 5 min, images were captured using a phone camera with a resolution of 3472 × 4624 pixels every 2 min; for the next 25 min, images were captured every 5 min.
3 Results and discussion
3.1 Fabrication of nanocomposite hydrogels
Nanocomposite bilayer hydrogels were fabricated using a commercial DLP 3D printer (Fig. 1). Single layer hydrogels were printed as sequential sublayers (0.1 mm thickness, 10 sublayers). Bilayer hydrogels were printed by first printing ten sublayers with one resin, and then exchanging the solution in the resin vat with a second resin. After subsequent post-curing, the active layers and bilayers were swollen with DI water for >24 h. A representative N5MMT-pHEA bilayer is shown in Fig. 1 with the pHEA layer dyed red for improved contrast and visibility of the interface. | ||
| Fig. 1 Overall schematic for fabrication of pNIPAAm-pHEA bilayer hydrogels utilizing DLP 3D printing. | ||
Hydrogels prepared with X wt% of MMT relative to the solute, i.e., the combination of NIPAAm monomer, crosslinker, and photoinitiator, are noted NXMMT. For this study, 0.5 wt%, 1 wt%, and 5 wt% clay contents were selected to compare against the clay-free control to survey a wide range of clay contents. Although previous studies of nanocomposite hydrogels have investigated loadings of 0.5–1 wt%,18 5 wt% is the upper bound of MMT loading before a viscosity increase was observed. Resin compositions are summarized in Table S1.† The clay-free control (N0MMT) is similar to the mixed water-ethanol solvent sample from our previous work,16 except for the crosslinker content. We increased this amount to 1.5 mol% instead of 1 mol% to control bilayer flatness, i.e., to more closely match the equilibrium swelling ratio of the pHEA hydrogel layer. We note that the addition of extra crosslinker also accelerated both mass expulsion and length contraction in the pNIPAAm hydrogels, as shown in Fig. S1.† The effect of crosslinker content on pNIPAAm actuation has been investigated previously,17 and is not a focus of this current work.
To understand the effect of variation in loading of MMT on the swelling, LCST, and crosslink density of the active layers of the bilayer hydrogels, single-layer active controls were fabricated by only printing with pNIPAAm resin. TGA of dried gels shows that the clay that was incorporated into the nanocomposites is comparable to the nominally added weight percentage (Fig. S2†). Upon swelling with water for at least 24 hours, disc-shaped hydrogels swelled to ∼4 times their original mass and ∼1.6 times their initial diameter, as shown in Fig. S3.† Although the diameter swelling ratios were very similar, the N5MMT hydrogel swelled to a slightly lower extent, potentially due to the clay absorbing less water per unit mass than the pNIPAAm pores. The clay did not have a significant impact on the LCST of the hydrogels as observed in DSC (Fig. S4†) or on the storage modulus (Fig. S5†), suggesting that the clay impacts neither the onset of the VPT nor the crosslink density. Because a percolating clay network would potentially be expected to have a greater impact on these two properties, a dispersed, clay-rich domain morphology was predicted. However, microscopy techniques were required to directly assess the morphology (Fig. 2).
 | ||
| Fig. 2 Morphology of clay-containing hydrogels at multiple length scales. Top row: Photographs of printed hydrogels captured with a phone camera. Second row: Brightfield optical micrographs at 20× magnification, showing evenly dispersed clay-rich domains. Third row: TEM images of dried, as-printed, pNIPAAm gels with dark embedded clay particles at ∼17500× magnification. Bottom row: SEM images of lyophilized pNIPAAm hydrogels at 2000× (N0MMT) or ∼1000× (N0.5MMT, N1MMT, and N5MMT) magnification showing varying pore sizes. | ||
3.2 Effect of MMT clay on hydrogel morphology
Although MMT can form a variety of microstructures in polymer nanocomposites,32 the MMT concentrated into dispersed clay-rich domains, which increased in size as the MMT loading increased, in these hydrogels. The top row of images in Fig. 2 shows the pNIPAAm and pNIPAAm-MMT hydrogels as captured by a phone camera. The hydrogels were uniformly opaque, and that opacity and a characteristic brown color qualitatively increased as MMT loading increased (left to right). The opacity is likely due to heterogeneous hydrogel network formation due to rapid polymerization and the formation of MMT-rich domains upon clay inclusion. The clay-rich domains are visible as uniformly dispersed dots in the brightfield optical microscopy images in the second row of Fig. 2. To investigate the clay packing within the domains, unhydrated pNIPAAm nanocomposites were microtomed and imaged with TEM, as shown in the third row of Fig. 2. Crevasse artifacts (parallel lines) appear in all images, and are a product of the microtome procedure, but do not affect the visualization of the clay. The clay-rich domains increase in size with MMT loading (left to right), from ∼500 nm in the N0.5MMT gel to ∼5 μm in the N1MMT gel to ∼10 μm in the N5MMT gel, as shown in the zoomed-out TEM images in Fig. S6A.† Within the domains, the clay forms a mixture of exfoliated and intercalated structures, as shown in Fig. 2 and Fig. S6B.† The MMT platelets were expected to be primarily exfoliated, because the resins were sonicated immediately before printing, and resin that was deposited on a TEM grid yielded clay platelets that were predominantly exfoliated (Fig. S7†). Previously, clay-water–alcohol mixtures have been shown to form different microstructures at varying clay contents.33 The water is explained to preferentially solvate around the clay, leaving large alcohol-rich regions. Above a certain threshold alcohol content, the water surrounding the clay platelets collapses the clay platelets into clay-rich domains. We believe that a similar process occurs in this investigation; the water in the cosolvent system preferentially solvates the MMT, condensing the MMT into flocculates, which are incorporated into the hydrogel during polymerization as clay-rich domains. At higher MMT clay loadings, more clay is solvated by similar amounts of water, yielding larger clay-rich domains that minimize the surface area of the solvating water.33 A shift in the solvation of the NIPAAm monomer also can explain the dramatic increase in pore size observed in the clay-containing hydrogels compared to the N0MMT hydrogel, as shown in the bottom row of Fig. 2. Although the N0MMT hydrogel was printed from a resin in which NIPAAm monomer was solvated by both water and ethanol in a 50–50% ratio, the clay-containing hydrogels were printed from resins in which the water preferentially solvated the clay, leaving the NIPAAm monomer to be solvated primarily by ethanol. Consequently, the pore structure may differ between monomer solutions of different solvent ratios, because the solubility of the monomer in the solvent may change due to the cononsolvency effect, as has been explored in pNIPAAm hydrogels previously.16,42,433.3 Thermal response of active control hydrogels
The morphology of the clay-rich regions, explored in Fig. 2, led to the existence of an optimal loading of clay for enhanced thermal actuation. Generally, for thermally-responsive hydrogels, a rapid and large magnitude actuation response is desirable. The rate of actuation can be quantified utilizing two factors: the mass change of the hydrogel due to water being expelled, and the diameter contraction during the VPT. These values can be reported as the ratio of mass or diameter at a particular time point divided by the initial, hydrated mass or diameter. The initial rate of actuation after 5 min is the change in mass ratio or diameter ratio divided by the time (5 min) to yield an average rate (Fig. 3A). By both metrics (mass and diameter), the N0.5MMT active control hydrogel actuated the fastest, followed by the N1MMT, then the N5MMT, and finally the N0MMT hydrogels. Although clay has been shown to accelerate actuation in responsive polymers in a percolated network,41 less is known about the impact of clay filler clustered into clay-rich domains within hydrogels. An increase in response rate supports the hypothesis of clay improving water transport in the hydrogel above the LCST. We note that this trend correlates to the pore sizes shown in Fig. 2, and that it is difficult to separate the effect of clay increasing the water transfer compared to larger and more open pores enhancing the expulsion of free water. However, the increased rate of actuation is also evidence for mitigation of the skin layer effect that is common for pNIPAAm hydrogels.15–17 A skin layer is known to form when the exterior of a pNIPAAm hydrogel becomes hydrophobic and prevents further expulsion of water from the interior, yielding surface bubbling and decreasing the rate and magnitude of actuation. This skin layer can be observed in a lyophilized sample of the N0MMT that was flash-frozen after 5 min of actuation in 60 °C water, but not in the clay-containing hydrogels (Fig. S8†). The presence of an optimum loading for enhanced rate suggests that the clay does not itself assist in actuation; if it did, more clay would cause a faster actuation rate. Rather, the presence of a hydrophilic filler during the volume phase transition prevents skin layer formation. Therefore, a minimal amount of filler is desirable to avoid inter-domain electrostatic repulsion as suggested in previous studies, which would provide a counteracting force and slow the contraction of the hydrogel.18 | ||
| Fig. 3 Response of pNIPAAm and pNIPAAm/MMT hydrogels to 60 °C water. A. Average rate of mass expulsion (solid columns) and average rate of diameter contraction (striped columns) of pNIPAAm hydrogels over the first five min of submersion in 60 °C water. B. Ratio of mass (solid columns) and diameter (striped columns) of pNIPAAm hydrogels after 30 min of submersion in 60 °C water, relative to their as-printed mass. | ||
The final swelling ratio after actuation is an indicator of the presence of residual water within the hydrogel and a measure of the magnitude of actuation. Both the N0MMT and N0.5MMT active control hydrogels had final swelling ratios near 1, similar to the dry gel dimensions (Fig. 3B). Moreover, the N0.5MMT active control hydrogel consistently exhibited the highest degree of actuation of all pNIPAAm/MMT hydrogels across time points between 5 and 30 min (Fig. S9†), demonstrating the utility of a low clay loading for maximally-enhanced actuation. Although the N1MMT and N5MMT active control hydrogels had final diameter swelling ratios near 1, their final mass swelling ratios were between 1.5 and 2, suggesting residual water remaining in the hydrogel without expanding the hydrogel volume. We believe that this water is interstitial between the interlayers within the clay-rich domains. Similarly to these results, Xia et al. reported the presence of clay-rich domains leading to an optimal loading of clay for an enhanced magnitude of actuation.18 In that work, the greater clay loadings corresponded to decreased length contraction, which was attributed to electrostatic repulsion between clay domains. However, in our results, a decrease in length (diameter) contraction in the N1MMT and N5MMT hydrogels compared to the N0.5MMT hydrogel was observed only until ∼15 min, after which the diameter contractions were similar. Therefore, the electrostatic repulsion effect may be transient in these pNIPAAm/MMT hydrogels.
3.4 Thermal actuation of bilayers and modeling
In addition to accelerating the actuation of the single-layer active control hydrogels, MMT enhanced the rate and magnitude of the curvature of the bilayer hydrogels. The decreased skin layer effect and enhanced water transport induced by the clay led to increased curvature of the N0.5MMT-pHEA, N1MMT-pHEA, and N5MMT-pHEA bilayers compared to the N0MMT-pHEA bilayer, visually shown in Fig. 4. Despite the N0.5MMT active control hydrogel displaying the fastest response within the clay-control series, all three clay-containing bilayers had very similar curvatures over time. Additionally, the difference in bilayer curvature between the N0MMT-pHEA bilayer and the N0.5MMT-pHEA bilayer (Fig. 4A) is much greater than the difference between the initial actuation rates of the N0MMT and N0.5MMT active control hydrogels (Fig. 3A). The bilayer curvature and active control hydrogel diameter contraction are expected to be closely related as the contraction of the active layer effectively pulls the passive layer into the curved shape. One potential explanation for the mismatch between the bilayer curvature response and the active control contraction is the force that can be applied by the active hydrogel on the passive hydrogel. An increase in clay content has been previously demonstrated to increase the contractile force output in the volume phase transition of pNIPAAm.20 By this explanation, the active layer of the N0MMT-pHEA bilayer had residual internal stress as the active layer was thermodynamically driven to contract but was fixed in a stretched state by the passive layer. However, the clay-containing bilayers were all able to overcome the passive layer and achieve their maximum contraction due to the increased force compared to the N0MMT. To support this hypothesis, an all-pNIPAAm hydrogel bilayer was fabricated by interfacing an N0MMT hydrogel layer and an N1MMT hydrogel layer. The bilayer curved in favor of the N1MMT hydrogel (i.e., the N1MMT layer was on the concave side) (Fig. S10†), despite the two hydrogels not exhibiting dramatically different monolayer contraction behavior (Fig. S9B†), suggesting that the contraction of the N1MMT hydrogel layer dominated the N0MMT hydrogel layer. Because the main difference in the thermal response of the N0MMT and the N1MMT is the formation of the skin layer, we conclude that the skin layer prevents force transfer from pNIPAAm hydrogels during the volume phase transition. | ||
| Fig. 4 Bilayer actuation and modeling. A. Actuation of bilayers after immersion in 60 °C water. Scale bars 1 cm. B. Comparison of N0MMT and N0.5MMT bilayer curvature data from Fig. 4A (solid lines) and predicted curvature from eqn (1) (dashed lines). C. Comparison of N1MMT and N5MMT bilayer curvature data from Fig. 4A (solid lines) and predicted curvature from eqn (1) (dashed lines). D. Curvature over time of a printed butterfly after immersion in 60 °C water. Scale bars 1 cm. | ||
The bilayer curvature and active control curvature can be related via the Timoshenko model. This kinematic model for predicting bilayer curvature can be a useful tool for prediction of bilayer curvature.44 The change in curvature Δκ of the hydrogels can be related to the normalized length or diameter ratios  of the active and passive layers by the following equation:
of the active and passive layers by the following equation:
 | (1) |
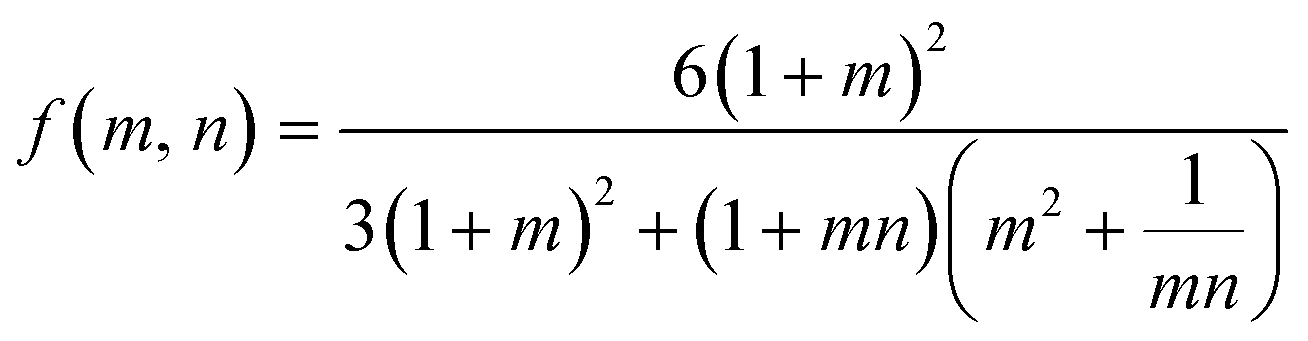 | (2) |
To assess the impact of the reduced skin layer on the predictive capability of the Timoshenko model, empirical parameters were utilized as inputs into the model and compared to the experimental bilayer curvature. Normalized ratios of the diameters of the pNIPAAm hydrogels over time at 60 °C are shown in Fig. S9B.† The normalized ratios of the diameter of the pHEA hydrogel from our previous work was used for the passive layer.16 Immersion DMA (Fig. S11†) was utilized to determine the moduli of the hydrogels over time upon exposure to water at 60 °C. We note that the signal shape of these storage moduli may be noisier than anticipated due to the complex acquisition mode and testing requirements; however, the smoothness of the thickness change of the hydrogel in Fig. S11D† indicates that these values are reasonable for use in the Timoshenko model.
A comparison of the model prediction (dots connected by dashed lines) and the experimentally-found curvature (dots connected by solid lines) is shown in Fig. 4B and C. Interestingly, the model predictions of all of the clay-containing hydrogels matched the experimental data substantially better than both the N0MMT control and the hydrogels shown in our previous work.16 The poor fit between the predictive model and the bilayer hydrogels was previously attributed to the skin layer.16 Inclusion of nanoclay as a method for decreasing the skin layer presents an exciting opportunity to validate the effect of skin layer on the predictive capabilities of the predictive model. The N0MMT hydrogel had the greatest skin layer effect, as well as the greatest deviation from the model's prediction of the four bilayers, which provides evidence that the Timoshenko model is better at curvature prediction when the skin layer is reduced. Interestingly, the N0MMT bilayer in this system achieves a lower curvature than predicted (Fig. 4B), rather than the underprediction of our previous work. The underperformance of the N0MMT hydrogel bilayer supports the hypothesis that the N0MMT active layer does not achieve the necessary force to pull the passive layer. Therefore, additional consideration of force transfer may be needed to accurately model the bilayer actuation of engineered hydrogel systems.
Finally, a butterfly-shaped bilayer was printed with N1MMT resin in the active layer and utilized to demonstrate the reversible actuation of the pNIPAAm/MMT-HEA bilayer system. A butterfly was chosen to highlight the utility of DLP 3D printing for fabrication of unique geometries for soft robotics applications due to its well-known motion of rapidly raising and lowering its wings during flight. The engineered butterfly fully raised its ‘wings’ within 5 min of heating in 60 °C water, and lowered its ‘wings’ to a position similar to its initial state after ∼60 min in ∼25 °C water (Fig. 4D). During the second and third cycles, shown in Fig. S12,† the butterfly raised its wings within 1 min after the first cycle, suggesting that the initial actuation process led to hysteresis and induced water channels that enhanced transport upon subsequent heating. Meanwhile, the butterfly wings did not return to the perfectly flat position, which can indicate the active hydrogel does not expand back to its original state, potentially maintaining some internal stress. Future studies are needed to explore the molecular origins of this hysteresis.
4 Conclusions
In this work, we demonstrated enhanced actuation in thermally-responsive, 3D-printed, clay-containing hydrogel bilayers and investigated the relationship between clay morphology, active layer actuation, and bilayer actuation. We fabricated pNIPAAm active control hydrogels to understand the impact of MMT inclusion on the morphology and actuation of the active layer of the bilayers before being interfaced with passive layers. Within the active control hydrogels, the MMT assembled into clay-rich domains, which increased in size as clay loading increased. The addition of clay also caused an increase in pore size, which was attributed to the water preferentially solvating the clay and increasing the solvation of the NIPAAm monomer by ethanol. The lowest loading of MMT within the single layer active control hydrogels corresponded to both the highest initial rate of actuation and the maximum final magnitude of actuation. However, as the MMT loading increased, both the initial rate and final magnitude of actuation decreased. All MMT-containing single layer hydrogels actuated faster than the clay-free control, which was attributed to the disruption of skin layer formation on the hydrogel during actuation. Residual water after hydrogel actuation at the two highest MMT loadings was attributed to water remaining within the clay-rich domains. The addition of clay enhanced the curvature of bilayer hydrogels fabricated by sequentially printing the pNIPAAm active hydrogels after pHEA hydrogels. Although the Timoshenko analytical model predicted the clay-containing bilayers well, the clay-free bilayer deviated greatly, potentially due to a hydrophobic skin layer preventing the active hydrogel from exerting the requisite force to overcome the passive hydrogel. Finally, a butterfly-shaped bilayer demonstrated the potential of these materials for soft robotics applications. Overall, this work highlights both the fabrication of clay nanocomposite hydrogels via DLP 3D printing as well as the impact that clay domain structure has on the thermal response properties of the hydrogels, especially the mechanics of bilayers fabricated using those hydrogels.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
F. K. and L. T. J. K. conceived the idea. F. K., S. K., S. L., and K. S. conducted the experiments. F. K. wrote the initial draft. L. T. J. K. supervised the work and obtained funding. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF) PIRE: Bio-inspired Materials and Systems under grant number OISE 1844463. F. K. also acknowledges financial support from the University of Delaware Graduate College through the Unidel Distinguished Graduate Scholar Award. The authors also thank the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under award DE-SC0021166 for the use of the TGA and RSA-G2 instrument. The authors acknowledge the use of facilities and instrumentation supported by NSF through the University of Delaware Materials Research Science and Engineering Center DMR 2011824. We thank Shannon Modla in the Bio-Imaging Center for assistance with microtoming and imaging the TEM samples. Microscopy access was supported by grants from the NIH-NIGMS (P20 GM103446), the NIGMS (P20 GM139760), and the State of Delaware. Microscopy equipment was acquired with the Unidel Foundation. Access to the SEM and Talos 200C TEM was provided by the Keck Microscopy Center at the University of Delaware. Fig. 1 was created in BioRender. Kalidindi, S. (2025) http://BioRender.com/d55e458.References
- Q. Zhao, Y. Wang, H. Cui and X. Du, J. Mater. Chem. C, 2019, 7, 6493–6511 RSC.
- C. Dawson, J. F. V. Vincent and A. M. Rocca, Nature, 1997, 390, 668–668 CrossRef CAS.
- K. Song, E. Yeom, S. J. Seo, K. Kim, H. Kim, J. H. Lim and S. Joon Lee, Sci. Rep., 2015, 5, 9963 CrossRef CAS PubMed.
- S. Basak and A. Bandyopadhyay, RSC Appl. Polym., 2024, 2(4), 583–605 RSC.
- E. Reyssat and L. Mahadevan, J. R. Soc. Interface, 2009, 6, 951–957 CrossRef CAS PubMed.
- S. L. M. Alexander, S. Ahmadmehrabi and L. T. J. Korley, Soft Matter, 2017, 13, 5589–5596 RSC.
- A. Sydney Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef CAS PubMed.
- Q. M. Zhang and M. J. Serpe, ChemPhysChem, 2017, 18, 1451–1465 CrossRef CAS PubMed.
- P. Chatterjee, A. Dai, H. Yu, H. Jiang and L. L. Dai, J. Appl. Polym. Sci., 2015, 132, 42776 CrossRef.
- J. Zhang, L.-Y. Chu, Y.-K. Li and Y. M. Lee, Polymer, 2007, 48, 1718–1728 CrossRef CAS.
- L. Tang, Y. Xu, F. Liu, S. Liu, Z. Chen, J. Tang and S. Wu, Macromolecules, 2023, 56, 6199–6207 CrossRef CAS.
- X.-Z. Zhang, X.-D. Xu, S.-X. Cheng and R.-X. Zhuo, Soft Matter, 2008, 4, 385 RSC.
- M. M. Rana and H. De La Hoz Siegler, Polymers, 2021, 13, 3154 CrossRef CAS PubMed.
- Y. Kaneko, R. Yoshida, K. Sakai, Y. Sakurai and T. Okano, J. Membr. Sci., 1995, 101, 13–22 CrossRef CAS.
- M. Li and J. Bae, Adv. Mater. Interfaces, 2023, 2300169 CrossRef CAS.
- F. Klincewicz, S. Kalidindi and L. T. J. Korley, RSC Appl. Polym., 2024, 2, 1062–1073 RSC.
- X. Zhang, D. Wu and C. Chu, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 582–593 CrossRef CAS.
- X. Xia, J. Yih, N. A. D'Souza and Z. Hu, Polymer, 2003, 44, 3389–3393 CrossRef CAS.
- F. Uddin, Metall. Mater. Trans. A, 2008, 39, 2804–2814 CrossRef.
- C. Yao, Z. Liu, C. Yang, W. Wang, X. Ju, R. Xie and L. Chu, Adv. Funct. Mater., 2015, 25, 2980–2991 CrossRef CAS.
- G. Gao, L. Wang, Y. Cong, Z. Wang, Y. Zhou, R. Wang, J. Chen and J. Fu, ACS Omega, 2018, 3, 17914–17921 CrossRef CAS.
- K. Haraguchi, T. Takehisa and S. Fan, Macromolecules, 2002, 35, 10162–10171 CrossRef CAS.
- K. Haraguchi and H.-J. Li, Angew. Chem., Int. Ed., 2005, 44, 6500–6504 CrossRef CAS PubMed.
- R. Foudazi, R. Zowada, I. Manas Zloczower and D. L. Feke, Langmuir, 2023, 39, 2092–2111 CrossRef CAS PubMed.
- J. Del Barrio and C. Sánchez Somolinos, Adv. Opt. Mater., 2019, 7, 1900598 CrossRef.
- I. Ertugrul, Micromachines, 2020, 11, 518 CrossRef PubMed.
- M. Champeau, D. A. Heinze, T. N. Viana, E. R. de Souza, A. C. Chinellato and S. Titotto, Adv. Funct. Mater., 2020, 30, 1910606 CrossRef CAS.
- H. Ding, M. Dong, Q. Zheng and Z. L. Wu, Mol. Syst. Des. Eng., 2022, 7, 1017–1029 RSC.
- J. Odent, S. Vanderstappen, A. Toncheva, E. Pichon, T. J. Wallin, K. Wang, R. F. Shepherd, P. Dubois and J. M. Raquez, J. Mater. Chem. A, 2019, 7, 15395–15403 RSC.
- D. Han, Z. Lu, S. A. Chester and H. Lee, Sci. Rep., 2018, 8, 1963 CrossRef PubMed.
- D. M. Solis and A. Czekanski, Soft Matter, 2022, 18, 3422–3429 RSC.
- F. Gao, Mater. Today, 2004, 7, 50–55 CrossRef CAS.
- Y. Kimura and K. Haraguchi, Langmuir, 2017, 33, 4758–4768 CrossRef CAS PubMed.
- G. Lagaly and S. Ziesmer, Adv. Colloid Interface Sci., 2003, 100–102, 105–128 CrossRef CAS.
- Suspensions: Fundamentals and Applications in the Petroleum Industry, ed. L. L. Schramm, American Chemical Society, Washington, DC, 1996, vol. 251 Search PubMed.
- V. B. Pereira, M. E. P. Goulart, L. V. R. Beltrami, D. Piazza, T. De Oliveira Gamba and A. J. Zattera, J. Mater. Eng. Perform., 2024, 1–11 Search PubMed.
- Z. Weng, Y. Zhou, W. Lin, T. Senthil and L. Wu, Composites, Part A, 2016, 88, 234–242 CrossRef CAS.
- H. A. Colorado, E. I. Gutierrez-Velasquez, L. D. Gil and I. L. De Camargo, Adv. Compos. Hybrid Mater., 2024, 7, 1 CrossRef CAS.
- H. Eng, S. Maleksaeedi, S. Yu, Y. Y. C. Choong, F. E. Wiria, C. L. C. Tan, P. C. Su and J. Wei, Procedia Eng., 2017, 216, 1–7 CrossRef.
- G. Hu, Z. Cao, M. Hopkins, J. G. Lyons, M. Brennan-Fournet and D. M. Devine, Procedia Manuf., 2019, 38, 1236–1243 CrossRef.
- N. D. Wanasekara, L. E. Matolyak and L. T. J. Korley, ACS Appl. Mater. Interfaces, 2015, 7, 22970–22979 CrossRef CAS PubMed.
- Y. Alsaid, S. Wu, D. Wu, Y. Du, L. Shi, R. Khodambashi, R. Rico, M. Hua, Y. Yan, Y. Zhao, D. Aukes and X. He, Adv. Mater., 2021, 33, 2008235 CrossRef CAS PubMed.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, A. K. Mishra and B. Ray, J. Appl. Polym. Sci., 2011, 121, 2422–2429 CrossRef CAS.
- S. Timoshenko, J. Opt. Soc. Am., 1925, 11, 233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5me00018a |
| This journal is © The Royal Society of Chemistry 2025 |