
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerating multicomponent phase-coexistence calculations with physics-informed neural networks†
Satyen
Dhamankar‡
,
Shengli
Jiang‡
and
Michael A.
Webb
 *
*
Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA. E-mail: mawebb@princeton.edu
First published on 24th December 2024
Abstract
Phase separation in multicomponent mixtures is of significant interest in both fundamental research and technology. Although the thermodynamic principles governing phase equilibria are straightforward, practical determination of equilibrium phases and constituent compositions for multicomponent systems is often laborious and computationally intensive. Here, we present a machine-learning workflow that simplifies and accelerates phase-coexistence calculations. We specifically analyze capabilities of neural networks to predict the number, composition, and relative abundance of equilibrium phases of systems described by Flory–Huggins theory. We find that incorporating physics-informed material constraints into the neural network architecture enhances the prediction of equilibrium compositions compared to standard neural networks with minor errors along the boundaries of the stable region. However, introducing additional physics-informed losses does not lead to significant further improvement. These errors can be virtually eliminated by using machine-learning predictions as a warm-start for a subsequent optimization routine. This work provides a promising pathway to efficiently characterize multicomponent phase coexistence.
Design, System, ApplicationAccurate phase coexistence characterization is critical for designing and optimizing systems and processes involving multiple components, yet traditional methods are often slow and computationally expensive. To overcome this, we developed a machine learning workflow grounded in physical principles to streamline and speed up these calculations. Using Flory–Huggins theory, we generated ternary phase diagrams and trained a theory-aware machine learning algorithm to predict equilibrium phases, compositions, and abundances. These predictions serve as an initial guess for numerical optimization, enabling fast and accurate determination of equilibrium states. This approach can be extended beyond ternary systems or applied to other free-energy models to describe a variety of chemical and biological processes. Ultimately, this method offers a promising way to accelerate chemical process simulations and drive innovations in multi-phase separations, as well as other system design workflows. |
1 Introduction
Phase coexistence in multicomponent systems is ubiquitous in nature and technology. Examples range from diverse purification processes in the chemical industry1–3 to the formation of membraneless organelles via liquid–liquid phase separation in biology.4–11 Thorough characterization of multicomponent phase equilibria involves not only identifying the phases present but also determining their composition and abundance, as the distribution and composition of species across phases significantly affect system properties and functions. This information guides processing methods and underlies calculations in process-simulation software. Critically, these calculations can constitute a substantial fraction of the overall computational time dedicated to simulation.12 Current multiphase flash calculation schemes require knowledge of the number of equilibrium phases,12,13 are sensitive to initial guesses,14 and can converge to spurious or trivial solutions if root-finding is not appropriately bounded and constrained.15,16 Therefore, efficient and accurate methods for predicting equilibrium states are valuable for both industrial applications and fundamental research.At equilibrium, species distribute across phases based on the extremization of an appropriate thermodynamic potential. For example, minimization of the Gibbs energy dictates equilibrium for a system at specified temperature T, pressure p, and global composition xi. Equilibrium phase-coexistence arises when species partition into distinct phases with equal chemical potentials driven by the extremization, rather than forming a homogeneous mixture. For a system at fixed T and p, this yields
| μαi(T, p, {xj}α) = μβi(T, p, {xj}β) ∀ i | (1) |
![[scr F, script letter F]](https://www.rsc.org/images/entities/char_e141.gif) = N −
= N − ![[scr P, script letter P]](https://www.rsc.org/images/entities/char_e52f.gif) + 2 where is the number of independent intensive relationships needed to specify a system of N species and phases). Provided a thermodynamic model for describing the mixture behavior as a function of intensive variables, eqn (1), or its equivalent at other conditions, functionally comprises N( − 1) equations to solve for the compositions of the various phases, with others fixed. The complexity of identifying equilibrium states can vary, even while the underlying thermodynamic framework is straightforward.
+ 2 where is the number of independent intensive relationships needed to specify a system of N species and phases). Provided a thermodynamic model for describing the mixture behavior as a function of intensive variables, eqn (1), or its equivalent at other conditions, functionally comprises N( − 1) equations to solve for the compositions of the various phases, with others fixed. The complexity of identifying equilibrium states can vary, even while the underlying thermodynamic framework is straightforward.
Determining the conditions, expected phases, and the chemical nature of species usually depends on appropriately parameterized equations-of-state or available free-energy models. For condensed phases and binary mixtures, there are several simple free-energy models like the Margules equations,17 the van Laar model,18 or the Guggenheim–Scatchard/Redlich–Kister equation.19 More complex models such as the Wilson models, non-random two-liquid (NRTL) models,20 universal quasi-chemical theory (UNIQUAC),21 UNIQUAC Functional-group Activity Coefficients (UNIFAC) models,13,22,23 Flory–Huggins theory24 can treat multicomponent systems. Although increasing complexity of the free-energy model or equation-of-state can facilitate more accurate representation of physical systems, the underlying calculations and theoretical principles for phase behavior remain the same for simple and complex models alike.
Given a thermodynamic model, calculating phase stability and equilibrium compositions can be approached in various ways. Simple models and binary mixtures may yield algebraic relationships that can be handled analytically or resolved using simple numerical schemes, such as self-consistent iteration or Newton's method. However, characterizing multicomponent phase coexistence typically requires dedicated software and more sophisticated numerical algorithms. Many algorithms are designed to work for only a specific set or number of phases.25 Direct solution methods based on Newton's root-finding algorithm can be effective but are computationally intensive and sensitive to the initial seed. Jindrová et al. refined Newton's algorithm and a successive substitution strategy to locate roots. Additionally, Nichita,26–28 Jindrová,14,16 and Castier29 independently performed volume stability analysis to obtain better initial guesses for the substitution strategy. There has been significant development in generating phase diagrams using constrained backmapping search algorithms.15,30–33
Indirect solution methods, based on thermodynamic principles and geometric criteria established via stability analysis, offer alternative approaches. Examples include Korteweg's tangent construction34 and Binous and Bellagi's arc extension method.35 Michelsen's multi-phase flash algorithm36 minimizes the distance between the tangent plane and the free energy surface to identify coexisting phases. Homotopy methods have also been used to calculate critical and saturation properties of mixtures.37,38 Additionally, Mao et al.39 generalized phase-diagram construction to multicomponent systems using a convex-hull construction40 applied to a discretized free-energy manifold, although accuracy and memory requirements depend on the mesh size. Overall, there is a need for simple, generalizable, and efficient methods for phase-coexistence calculations.
Machine learning (ML) techniques facilitate phase-coexistence calculations, offering prospective advantages relating to time- and memory-efficiency relative to more traditional optimization strategies.41–50 However, many efforts only address the issue of phase stability and neglect consideration of phase composition.42–46 Others have been restricted to binary systems with limited demonstration of more complex mixtures.47–50 Recently, Flory–Huggins (FH) theory has been combined with ML to improve the interpretability and accuracy of mixture behavior predictions, but limitations exist in their ability to handle complex interactions and multicomponent systems beyond binary mixtures.49,50 Nevertheless, such works highlight the potential of ML as part of a generalizable, accurate, efficient, and extensible framework for characterizing multicomponent phase behavior.
Here, we describe a data-driven workflow to characterize the phase behavior of multicomponent systems. Fig. 1 illustrates the overall approach in the context of ternary systems described by Flory–Huggins (FH) theory. Using FH theory as a representative free-energy model, we construct a series of phase diagrams across the model parameter space using labor-intensive methods. This data is then used to develop an ML surrogate model, based on neural network architectures, to predict the number, composition, and relative abundance of equilibrium phases from model parameters and total system composition. Surrogate models optimized with and without physics-informed architectures and loss functions are compared. Errors are assessed for classification (number of equilibrium phases) and regression (composition and abundance of phases). Predictions from the surrogate model, which are computationally efficient and improvable, are then used to warm-start a simple optimization to precisely and accurately characterize the system's phase behavior. This procedure exemplifies an efficient, accurate, and extensible approach to phase-coexistence calculations.
 | ||
| Fig. 1 Strategy for multi-component phase-coexistence prediction using machine learning. 1036 ternary phase diagrams are generated using the algorithm arc continuation algorithm (eqn (6)–(8)) and convex hull construction algorithm,39 and are used as training data for a physics-informed machine learning (ML) model to classify phase regions and predict equilibrium phase compositions. The ML predictions serve as initial guesses for the Newton-CG method to obtain equilibrium composition predictions. | ||
2 Methods
2.1 Thermodynamic framework
For demonstration, we consider the thermodynamics of ternary systems described by FH solution theory. Systems are comprised of species A, B, and C that occupy a lattice of n sites with volume V = nv0. The species can possess size disparities, reflected in their molar volumes vi. For a polymer comprised of Ni monomers that each occupy a single lattice site, vi = Niv0. Systems are incompressible such that where ni is the mole number for species i. System composition is specified by the volume fractions ϕi = (nivi)/V with
where ni is the mole number for species i. System composition is specified by the volume fractions ϕi = (nivi)/V with  .
.
The dimensionless, intensive (per lattice site) Helmholtz energy of mixing follows as
 | (2) |
Up to a constant, chemical potentials are obtained by partial differentiation of the Helmholtz energy of mixing:
 | (3) |
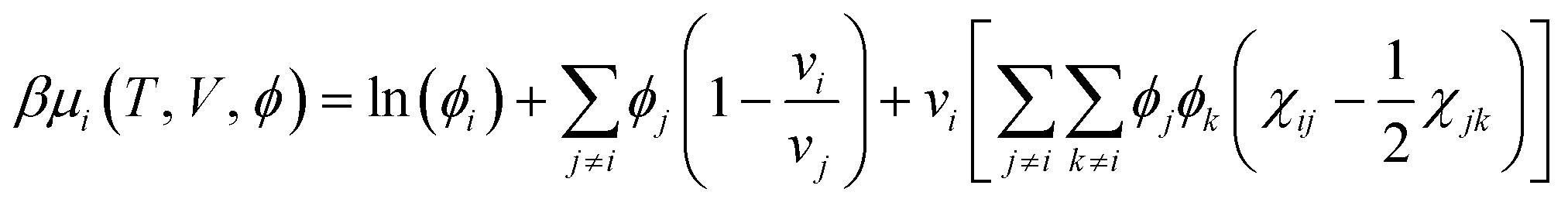 | (4) |
The thermodynamic stability of a mixture is assessed by considering the determinant of the Hessian matrix for the Helmholtz energy. For
 | (5) |
 | (6) |
 | (7) |
 | (8) |
For fixed total particle density (ρ = n/V) and constant temperature, Gibbs' phase rules indicate there can be at most three coexisting phases. To characterize three-phase coexistence, there are 12 variables. Nine correspond to the volume fractions in each phase: ϕα, ϕβ, and ϕγ, for which each ϕπ = (ϕAπ, ϕBπ, ϕCπ). Three correspond to the fractional abundances of each phase–wα, wβ, and wγ. Criteria for chemical equilibrium applied to each species across each phase
| μαi(T, p, ϕα) = μβi(T, p, ϕβ) = μγi(T, p, ϕγ) | (9) |
 | (10) |
 | (11) |
2.2 Phase-coexistence calculations
Two different algorithms are used to characterize phase-coexistence based on the principles outlined in section 2.1. For systems with at least one critical point and two-phase coexistence, an iterative and perturbative approach based on natural parameter continuation (NPC) is used to construct binodal curves originating from a critical point. Otherwise, the approach described by Mao et al.39 based on convex hull construction (CHC) is used. NPC is straightforward and computationally efficient but limited, while CHC is general but computationally intensive. Nevertheless, with this combination, the equilibrium composition of phases at coexistence can be reliably determined for models described by FH solution theory. The following algorithms are thus used to provide ground-truth results and requisite training data for the development of ML models (section 2.3).| Δμαβi(T, p, ϕα, ϕβ) ≡ μβi − μαi = 0 for i ∈ {A, B, C}. | (12) |
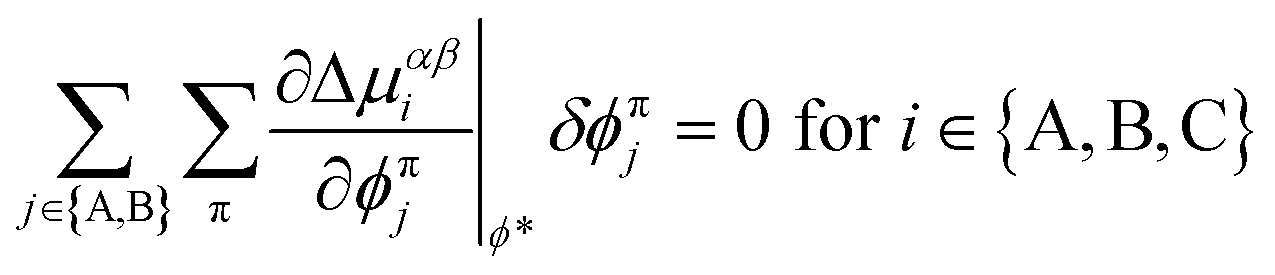 | (13) |
1. Define tolerance parameters δ∅ and δ0.
2. Identify and set the critical point to be ϕ*.
3. Generate a random, small perturbation on the composition δϕ, yielding two new compositions: ϕ′ = ϕ* + δϕ and ϕ′′ = ϕ* − δϕ.
4. Use the compositions ϕ′ and ϕ′′ as initial guesses to solve eqn (12), producing coexisting compositions ϕαnew and ϕβnew that are distinct from ϕ*.
5. Set ϕπold ← ϕπnew and use for eqn (13). Set one of the δϕπi (e.g., ϕβB) to a small perturbation and solve for the remaining δϕπi to produce δϕα′ and δϕβ′.
6. Set ϕ′ = ϕα(0) + δϕα′ and ϕ′′ = ϕβ(1) + δϕβ′ and use as initial guesses to solve eqn (12), producing new coexisting compositions ϕαnew and ϕβnew that are those distinct from those prior.
7. Repeat steps 5 and 6 until either ‖ϕαnew − ϕβnew‖ < δ∅, which indicates a closure of the coexistence curves, or when any ϕπi < δ0, which indicates termination at a composition boundary.
8. Verify validity of compositions by checking that all have |H![[f with combining tilde]](https://www.rsc.org/images/entities/i_char_0066_0303.gif) | > 0.
| > 0.
For the calculations described in this paper, δ∅ = δ0 = 10−9. Initial trials for random composition perturbations are set to have a magnitude of 10−6. Equations are solved numerically using fsolve from Python's SciPy module. Occasionally, the trial perturbations resulted in solutions that collapsed back to the critical point or other prior generated points, in which case new perturbations would be attempted with possibly different magnitudes.
The composition space (ϕA, ϕB) is discretized into a mesh of equilateral triangles, or two-dimensional simplices. Using a finer mesh results in more accurate calculations but also increases computational cost and memory requirements; this work uses a simplex edge-length of 0.0002. After generation of the mesh, the free energy surface (FES) is also discretized into points defined by the tuple (ϕA, ϕB, (ϕA, ϕB)). The convex hull (ϕCHA, ϕCHA, CH(ϕCHA, ϕCHB)) of the FES is calculated using the Quickhull algorithm.53 The convex hull of a non-convex FES will necessarily deform the original simplices and facilitate the identification of cotangent points on the FES. If one of the projected simplices has three unstretched sides (maximum edge length within five times initial mesh size39), the system is homogeneous (no phase-separation). If two sides are stretched (side length greater than five times the initial mesh size), the two farthest vertices are cotangent, indicating two coexisting phases. If all three sides are stretched, the three vertices of the simplex are cotangent, indicating three coexisting phases. With graph theoretic techniques, the number of equilibrium phases and their compositions can be determined.
2.3 Machine learning details
We explore machine learning algorithms as computationally expedient and generalizable alternatives to more traditional approaches for characterizing phase coexistence of multicomponent systems. Neural network architectures, with and without physics-informed loss functions, are optimized using data generated by the algorithms described in section 2.2. The performance of the ML models is evaluated based on predicting the number of coexisting phases, their compositions, and relative abundance for FH models not featured in training data.Parameters for the models are each selected from the range vi ∈ [1, 3] and χij ∈ [1, 3] where values for both ranges are discretized with a resolution of 0.1. Let s denote a parameter set and U denote the set of all possible parameter sets. With the given discretization, the total membership of U is then |U| = 216. Initially, 750 possible parameter sets are randomly selected from U with uniform probability to form S ⊂ U; care is taken to ensure that all parameter sets from this sampling are unique. From this initial sampling, only around 6.6% (≈50) of the selected parameter sets yielded three-phase coexistence. Using these parameter sets to define T ⊂ S, the representation of such rare systems is augmented by generating six additional parameter sets for each parameter set t ∈ T. Each new parameter set t′ is generated from t by adding a Gaussian random vector X. In particular, we use t′ = t + X with X ∼ ![[scr N, script letter N]](https://www.rsc.org/images/entities/char_e52d.gif) (0, σ2I) where σ = 0.005. All t′ that yielded three-phase coexistence are collected and added to S, resulting in a final membership of |S| = 1036 parameter sets.
(0, σ2I) where σ = 0.005. All t′ that yielded three-phase coexistence are collected and added to S, resulting in a final membership of |S| = 1036 parameter sets.
Input and output labels for the dataset are then generated as follows. First, the composition-space of the mixture is discretized into a uniform mesh with resolution 10−4. For each parameter set, if there are more than 1000 single-phase simplices, the centroid of 1000 randomly chosen simplices is added to the database; otherwise, the centroid of all single-phase simplices is added. For double-phase simplices, if there are more than 1000, a random point between the ends of 1000 randomly chosen double-phase separations is generated; otherwise, a random point between each double-phase separation is added. For multiple three-phase separations, a uniform number of points is generated in each region, ensuring a total of 1000 three-phase points in the database. Since the number of single and double-phase simplices are determined by the size of the discretization mesh, the number of data points per each parameter set can vary.
For each tuple (ϕA, ϕB) the number of equilibrium phases, their compositions, and their abundances are recorded. In this fashion, we define an input vector x = (χAB, χBC, χAC, vA, vB, vC, ϕA, ϕB) ∈ ![[Doublestruck R]](https://www.rsc.org/images/entities/char_e175.gif) 8 that is linked to two outputs. The first output is a one-hot encoded classification vector yc ∈ 3, for which a nonzero entry indicates the presence of one, two, or three phases at equilibrium. The second output is a vector yr ≡ (ϕαA, ϕαB, ϕβA, ϕβB, ϕγA, ϕγB, wα, wβ, wγ) ∈ 9, which describes the composition and abundances of the equilibrium phases. The phases are ordered such that ϕαA has the minimum value among all ϕA (ϕαA ≤ ϕβA ≤ ϕγA). If two phases have the same ϕA, they are further ordered according to ϕB. Such an ordering ensures a consistent representation of the equilibrium phases.
8 that is linked to two outputs. The first output is a one-hot encoded classification vector yc ∈ 3, for which a nonzero entry indicates the presence of one, two, or three phases at equilibrium. The second output is a vector yr ≡ (ϕαA, ϕαB, ϕβA, ϕβB, ϕγA, ϕγB, wα, wβ, wγ) ∈ 9, which describes the composition and abundances of the equilibrium phases. The phases are ordered such that ϕαA has the minimum value among all ϕA (ϕαA ≤ ϕβA ≤ ϕγA). If two phases have the same ϕA, they are further ordered according to ϕB. Such an ordering ensures a consistent representation of the equilibrium phases.
For systems with a single phase, ϕαA, ϕαB match the inputs ϕA, ϕB, and wα is set to unity; the abundance entries for phases β and γ are set to zero. However, the composition abundance entries for phases β and γ are assigned a value of 1/3. The value 1/3 is chosen to distribute errors uniformly across species. The absolute composition of these species in equilibrium will be determined by the abundance of the respective phases. For systems with two equilibrium phases, entries for the third phase compositions (i.e., ϕγA, ϕγB) are set to 1/3, and the abundance wγ is set to zero.
The basic model architecture consists of three, fully-connected hidden layers, each with m (tunable hyperparameter) hidden units; this yields a hidden vector h ∈ m. This hidden vector is then passed through a “classification layer” with softmax activation to yield ŷc. This vector is also fed into separate “regression layers” to predict the composition (ϕα, ϕβ, ϕγ) and abundance (w). Each regression layer consists of three hidden units, representing the composition of A, B, and C for each phase, and the abundance of α, β, and γ phases. Sigmoid activation is applied to limit predicted values on compositions and abundances to be between zero and unity, which avoids obviously unphysical values; however, overall composition and abundance constraints are not enforced. Since the composition of C depends on A and B, only the predictions for A and B compositions are kept and combined with the abundance predictions to form ŷr.
 | ||
| Fig. 2 Architectures of the machine learning models. In both the baseline and physics-informed (PI) models, the parameter vector x is fed into the model to produce an intermediate hidden vector h. The hidden vector h produces two outputs: (1) a phase classification probability vector ŷc, trained with cross-entropy (CE) loss, and (2) an equilibrium composition and abundance vector ŷr, trained with mean absolute error (MAE) loss. Softmax activation is applied in PI models to ensure that the equilibrium composition and abundance vectors sum to unity. Optional functionalities (indicated by dashed lines and boxes) include a “mask”, activated based on ŷc, which sets corresponding elements in ŷr to zero if an input is classified as one- or two-phase. For PI models, softmax renormalization is applied to the masked abundance to ensure the sum equals unity. Additionally, in PI models, a PI composite loss can be incorporated alongside the MAE loss during training for ŷr prediction. | ||
We also consider a variation on the basic model architecture that enforces consistency between ŷr and the majority class featured in ŷc. This is achieved using a mask-layer that sets abundance entries in ŷr to zero based on the plurality class indicated in ŷc (see dashed box in Fig. 2). For example, if one equilibrium phase is predicted, then abundance entries associated with the β and γ phase are set to zero. If two equilibrium phases are predicted, then entries associated with the γ phase are set to zero. If three equilibrium phases are predicted, then ŷr is preserved from the regression layer. Compositions of species for non-existent phases are set to 1/3, as described earlier. To enforce constraints on overall composition and abundance, the physics-informed (PI) model incorporates softmax activation functions, ensuring that predicted phase compositions and abundances sum to unity. As an alternative approach, following the masking, softmax normalization is applied to the abundances to ensure their sum equals unity.
![[script L]](https://www.rsc.org/images/entities/char_e144.gif) base = λCECE + MAE base = λCECE + MAE | (14) |
CE, and regression mean absolute error (MAE) loss, MAE. The weighting parameter (λCE = 0.1) is determined empirically to balance loss magnitudes throughout training. While a perfect and physically meaningful model would necessarily minimize base, with data limitations, simply minimizing the baseline loss function may not strictly satisfy all the criteria prescribed for thermodynamic systems at equilibrium. We therefore also consider augmented PI models (referred to as PI+) optimized with a composite loss function that includes additional regression targets| PI = base + λsplitsplit + λΔμΔμ + λff | (15) |
base while incorporating physical constraints. The specific functional forms for these PI losses are described next.
In eqn (15), the additional loss terms aim to satisfy different constraints on the thermodynamics of physical systems. In particular, split relates to constraints on the total composition of a given species distributed across phases:
 | (16) |
Δμ relates to the condition of equal chemical potentials for species across coexisting equilibrium phases. This loss is calculated as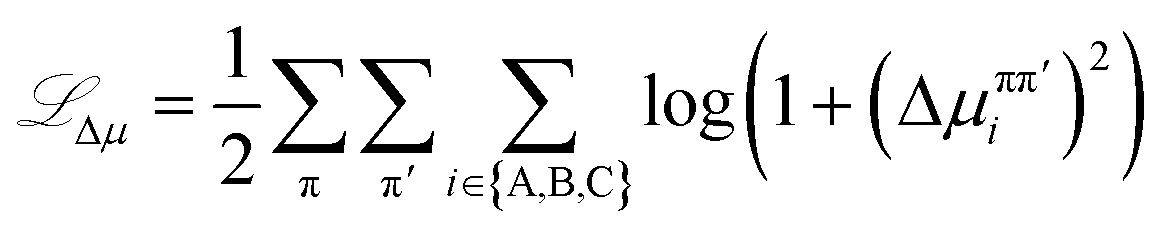 | (17) |
f promotes the minimization of the free energy of the equilibrium system: | (18) |
The overall training set is further divided into training and validation sets, using a similar five-fold CV approach (inner CV) to the outer CV process. Each fold of the inner CV is trained with 10% of the training data for efficient hyperparameter optimization. Tunable hyperparameters include batch sizes of {5000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 20000}, learning rates of {0.001, 0.005, 0.01}, the presence (or absence) of a mask, and the number of neurons selected from {64, 128, 256} for each hidden layer. The optimal hyperparameter setting for each fold is identified by the highest average validation composite score across five sub-folds, calculated as the sum of the F1 score for classification and the average R2 score.
000, 20000}, learning rates of {0.001, 0.005, 0.01}, the presence (or absence) of a mask, and the number of neurons selected from {64, 128, 256} for each hidden layer. The optimal hyperparameter setting for each fold is identified by the highest average validation composite score across five sub-folds, calculated as the sum of the F1 score for classification and the average R2 score.
Each fold of the outer CV uses the optimal hyperparameter settings identified from its corresponding five-fold inner CV and retrains the model for up to 500 epochs. The retraining involves selecting 80% of the overall training diagrams as the training set and 20% as the validation set, using the same stratified splitting. During the retraining process, the impact of training data sizes on model performance is assessed by using 1%, 5%, 10%, 20%, 30%, and up to 100% of the training data. Because each diagram contains a different number of data points, the number of training, validation, and test set data points ranges from 1447511 to 1458241, from 358987 to 366470, and from 452308 to 460074.
The nested CV strategy yields a mean and standard deviation of F1 and R2 scores as determined from the five-fold outer CV test sets. Given the imbalanced phase distribution in the dataset, the F1 score evaluates classification performance, while the R2 score assesses regression accuracy for the variables in yr.
2.4 Post-inference optimization
We implement a post-inference optimization procedure to correct some deficiencies in ML model predictions. This procedure uses the predictions from the ML model as a warm-start on initial values for more traditional optimization algorithms (e.g., truncated Newton method). The objective function for minimization is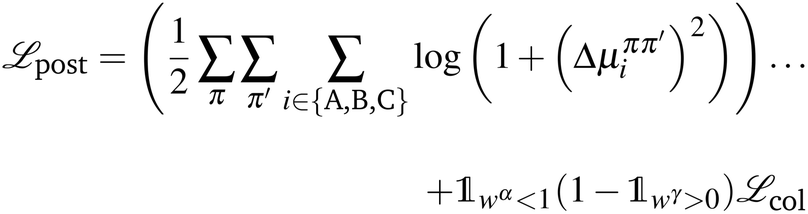 | (19) |
 is an indicator function equal to unity when the condition c is satisfied and zero otherwise and
is an indicator function equal to unity when the condition c is satisfied and zero otherwise and | (20) |
 excludes computation of col when there is only a single predicted phase, as this term would otherwise diverge. In fact, this procedure has no effect when only a single equilibrium phase is predicted. Relatedly, we note that this algorithm is asymmetrically robust against erroneous misclassification of the system phase behavior. If the predicted number of phases exceeds the true number of phases, then converged solutions will “collapse” compositions onto those of the true equilibrium phases. However, this procedure will not identify the true solution if the predicted number of phases is fewer than the ground-truth number.
excludes computation of col when there is only a single predicted phase, as this term would otherwise diverge. In fact, this procedure has no effect when only a single equilibrium phase is predicted. Relatedly, we note that this algorithm is asymmetrically robust against erroneous misclassification of the system phase behavior. If the predicted number of phases exceeds the true number of phases, then converged solutions will “collapse” compositions onto those of the true equilibrium phases. However, this procedure will not identify the true solution if the predicted number of phases is fewer than the ground-truth number.
The final optimization employs the Newton-CG optimizer in scipy module in Python. The Jacobian and Hessian matrix for the objective function are computed using the autograd package through automatic differentiation. The maximum number of iterations for optimization is limited to 10000. If newly optimized compositions are within a tolerance of 10−7 of the ideal value of the objective function, these values replace the predictions proffered by the ML model. Optimizations are only considered successful if they satisfy the stability criterion |H| > 0 (see eqn (6)).
3 Results
3.1 Performance with a basic architecture
With the standard loss functions (i.e.CE and MAE) and a basic architecture, the ML model predicts phase separation and equilibrium compositions reasonably well. Fig. 3a and b qualitatively depict performance in both classification and regression for some representative phase diagrams. Fig. 3c and d quantitatively summarize results across all phase diagrams.
 | ||
| Fig. 3 Performance summary of the baseline model. a) Classification of the number of coexisting phases. The background color in all phase diagrams denotes the ground truth phase: gray (one-phase), blue (two-phase), and red (three-phase). Scatter points represent the predicted phase splits for a given initial composition, and the legend colors indicate the types of predicted splits. The parameters of the phase diagrams are detailed in ESI† Tables S1 and S2. b) Predicted equilibrium compositions. Blue and orange scatter points represent two-phase equilibrium compositions. The yellow dashed line is a tie line for the two-phase split. Red scatter points depict composition that split into three phases. The red dashed triangle connects the three compositions at equilibrium. c) Confusion matrix for the predicted number of equilibrium phases. Diagonal entries represent correctly classified instances, while off-diagonal entries represent misclassifications. d) Parity plot for predicted equilibrium compositions. The diagonal dashed line represents perfect performance. | ||
The ML model capably predicts the number of phases at equilibrium with an overall accuracy rate of about 97% (Fig. 3c). The primary source of error (4.6%) stems from misclassifying three-phase points as two-phase, which is attributed to the relative paucity of three-phase splits (only 17% of the total data). Additionally, a portion of two-phase points (2.2%) are misclassified as one-phase. A closer inspection of predicted phase diagrams suggests that misclassifications mostly occur near binodals.
The model also performs well in predicting phase abundances and their compositions (Fig. 3d). By inspection, there are vertical error regions in the predicted abundance for all phases at true abundances of 0 and 1. These errors stem from inaccurate predictions of equilibrium abundance for non-existent phases, such as phases β and γ in a one-phase region and phase γ in a two-phase region. This misprediction also leads to similar error regions for equilibrium compositions around 1/3.
Table 1 provides the baseline expectations for a standard ML model. It highlights nuances in regression performance across different phase regions. The single-phase region has the lowest average MAE (0.006), followed by the two-phase (0.023) and three-phase (0.037) regions. This trend suggests increasing difficulty in predicting compositions as the number of coexisting phases increases. Notably, all R2 values remain high across all phases, with the three-phase region exhibiting a value above 0.88.
| MAE | R 2 | |||||
|---|---|---|---|---|---|---|
| Base | PI | PI+ | Base | PI | PI+ | |
| One-phase | 0.006 (0.001) | 0.005 (0.001) | 0.005 (0.001) | 0.982 (0.005) | 0.987 (0.004) | 0.988 (0.003) |
| Two-phase | 0.023 (0.003) | 0.022 (0.001) | 0.023 (0.003) | 0.912 (0.015) | 0.915 (0.009) | 0.913 (0.015) |
| Three-phase | 0.037 (0.006) | 0.038 (0.003) | 0.038 (0.008) | 0.884 (0.038) | 0.883 (0.023) | 0.889 (0.041) |
3.2 Performance with physics-informed losses and consistency constraints
To build on the prior model, we evaluate the potential of incorporating additional physical information on prediction accuracy (Fig. 4a). In particular, physical constraints on overall composition and abundance, along with several physics-informed losses (detailed in section 2.3) are implemented, and classification masks are used to zero the abundances of non-existent phases. While the baseline, PI, and PI+ models without classification masks achieve comparable F1 and R2 scores, models with masks significantly underperform in equilibrium composition prediction (Fig. 4b). | ||
| Fig. 4 Impact of physical constraints and data size on phase-coexistence prediction. a) Comparison of test set phase classification F1 and equilibrium composition prediction R2 across baseline (base), physics-informed (PI), and augmented PI (PI+) models, with and without classification masks, using five-fold cross-validation (CV). Bars represent mean values, and error bars indicate standard deviations. b) The impact of training data size on model performance. Each dot represents the average score calculated across the five-fold CV. | ||
The baseline model exhibits lower accuracy compared to the PI and PI+ models in one-phase and two-phase regions, while its performance is comparable to other models in three-phase scenarios (Table 1). The PI and PI+ models show similar performance across all scenarios under these metrics. The coexistence curve predictions of both the PI and PI+ models are similar (Fig. 5a and S6†), producing smooth and physically sensible two- and three-phase coexistence curves. In contrast, the baseline model generates erratic two-phase coexistence curves that significantly deviate from the true curves. This is also evident from the distribution of the MAE loss in Fig. 5b, where the baseline model (red) has a higher average MAE than the PI (blue) and PI+ (green) models, which perform similarly. The unphysical coexistence curves of the baseline model highlight the limitations of using broad performance metrics to assess improvements in predictive accuracy. Errors are better resolved by examining deviations in chemical equilibrium potential (Δμ) and split loss (split), where the baseline model shows significantly larger errors than both the PI and PI+ models.
 | ||
| Fig. 5 Comparison of performance metrics and physical constraints among baseline, PI, and PI+ models. a) Predicted phase coexistence curves for the baseline (left), PI (middle), and PI+ (right) models. Arrows indicate that both predictions are for the same phase diagram. The background color in all phase diagrams denotes the ground truth phase: gray (one-phase), blue (two-phase), and red (three-phase). Blue and orange scatter points represent two-phase equilibrium compositions. The yellow dashed line is a tie line for the two-phase split. Red scatter points depict composition that split into three phases. The red dashed triangle connects the three compositions at equilibrium. The parameters of the phase diagrams are detailed in ESI† Tables S1 and S2. b) Data distribution (shaded bars) and kernel density estimation fits (lines) for performance metrics and physical constraints. Vertical dashed lines indicate mean values. | ||
To further examine the impact on composition and abundance constraints, we analyze two additional metrics: unity, which relates to the volume fractions of each species within a given phase, and weight, which measures overall material conservation. These metrics are defined as:
 | (21) |
 | (22) |
unity and weight remain zero for these models, whereas the baseline model violates these constraints (Fig. 5b). The PI+ model, trained with additional constraints, demonstrates smaller deviations in chemical equilibrium potential (Δμ) and marginally improves composition prediction, split loss, and free energy minimization loss compared to the PI model. Overall, designing a physics-informed model architecture to enforce material constraints is essential; however, the addition of extra losses or masks complicates training without yielding significant improvements in phase classification or equilibrium composition prediction. Therefore, the PI architecture, without additional losses, emerges as the best practical choice for implementation.
3.3 Performance with limited data
Having achieved accurate phase-coexistence predictions with a dataset of over 1.4 million data points, we investigated whether PI models would be more data efficient and achieve comparable performance with less data. Fig. 4b demonstrates that even with only 10% of the data, the PI and PI+ models maintain high accuracy in phase classification (F1: 0.967 and 0.967, respectively) and equilibrium composition prediction (R2: 0.937 and 0.935, respectively). In contrast, the baseline model performs worse in equilibrium composition prediction (R2: 0.930) but achieves comparable accuracy in phase classification (F1: 0.967). The advantages of incorporating composition and abundance constraints are particularly evident in low-data scenarios (training with 1% and 5% of the data), where the PI and PI+ models significantly outperform the baseline. Although the PI+ model slightly outperforms the others with the full dataset (F1: 0.972, R2: 0.946), the improvement over using 10% of the data is marginal. The predicted phase diagrams with coexistence curves (Fig. S2, S4 and S5†) using 10% of the data are qualitatively accurate across the baseline, PI, and PI+ models. These findings underscore the critical role of physical constraints in enhancing model generalization under limited data conditions.3.4 Post-ML optimization
Seeded with ML predictions, a Newton-CG method can efficiently converge to arbitrarily accurate and precise equilibrium compositions (Fig. 6). This is demonstrated for the PI model, trained on the full dataset, where initial errors (Fig. 6a and b) can be virtually eliminated after the optimization procedure (Fig. 6c and S1†). The baseline and PI+ models also achieve comparable performance after post-ML optimization (Fig. S3–S6†), even when trained on only 10% of the data (Fig. S2, S4 and S5†). This combination of efficiency and accuracy could enable the handling of more complex systems and scaling to resource-intensive measurements, where data may be sparse or scarce.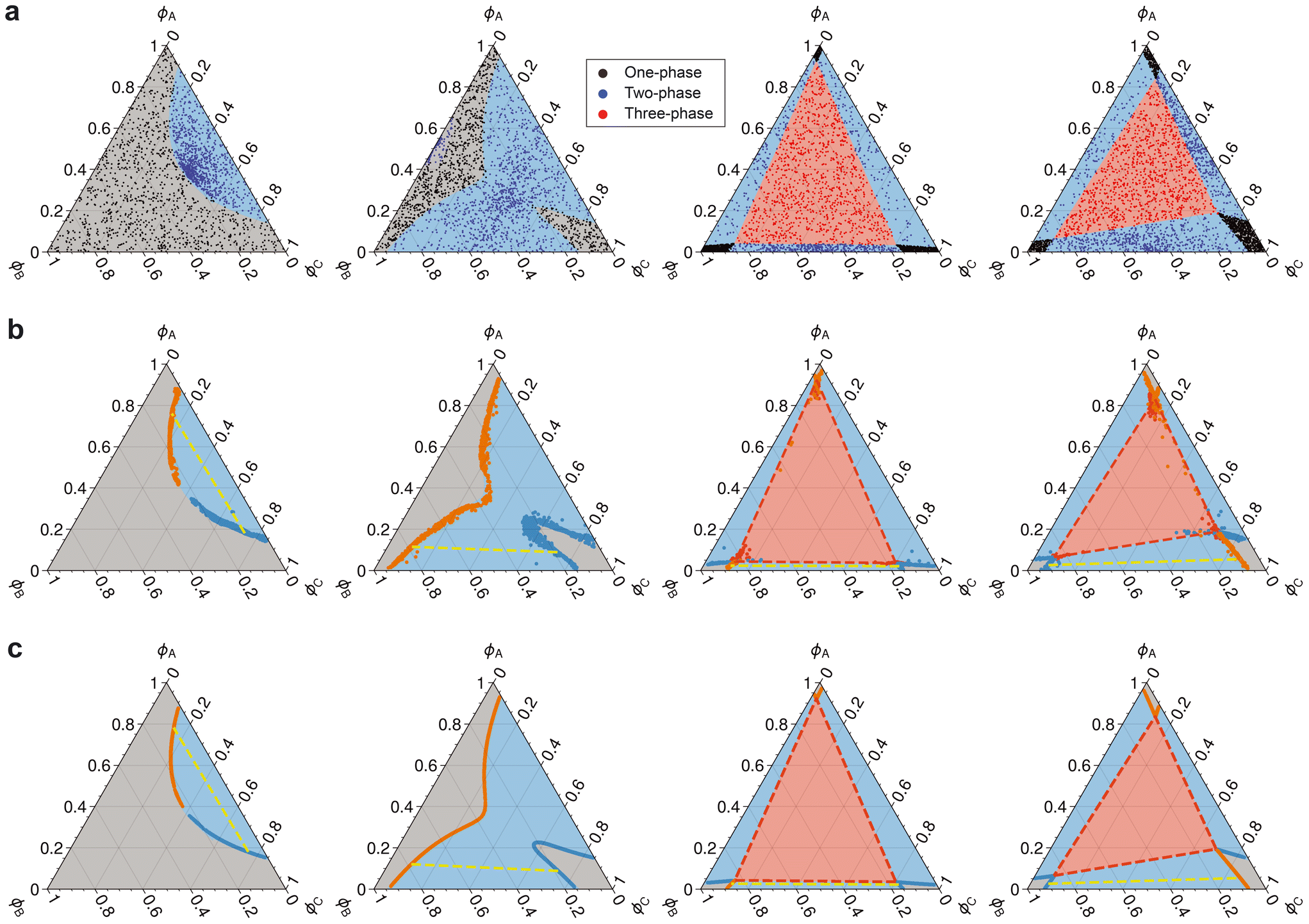 | ||
| Fig. 6 Refinement of PI model predictions. a) Classification of the number of coexisting phases. The background color in all phase diagrams denotes the true phase: gray (one-phase), blue (two-phase), and red (three-phase). The scatter points indicate the predicted phase splits for a given initial composition. Colors in the legend denote the types of predicted splits. The parameters of the phase diagrams are detailed in ESI† Tables S1 and S2. b) Predicted coexistence curves. Blue and orange scatter points indicate two-phase coexistence curves, with the yellow dashed line denoting an example tie line. The vertices of the red triangle indicate three-phase coexistence points. c) Coexistence curves produced with the post-ML optimization strategy. The results are obtained using ML inference to warm-start Newton-CG optimization. | ||
Errors for models trained on fold 1 data were analysed across a random sample of 187 two-phase and 76 three-phase equilibrium phase diagrams to better characterize post-ML optimization errors (Table 2 and S1†). The results indicate that Newton-CG optimization, initialized with predictions from ML models, achieves near-perfect success rates and significantly reduces deviations from true equilibrium compositions compared to individual ML model predictions. After post-ML optimization, the PI model trained on the full dataset outperforms both the baseline and PI+ models in predicting two-phase and three-phase coexistence. The relative ranking of performances post-optimization aligns well with the relative ranking of the ML predictions alone, underscoring the importance of initial ML prediction accuracy in determining the effectiveness of the post-ML optimization process. With limited training data, all models perform similarly, with PI+ showing slightly better overall performance. Errors remain comparable to those observed with the full dataset, although three-phase coexistence errors consistently exceed those for two-phase coexistence. This disparity likely stems from the relative scarcity of three-phase coexistence in the training set, which increases complexity and complicates precise prediction.
| Data size | Two-phase (×10−2) | Three-phase (×10−2) | |||
|---|---|---|---|---|---|
| ML prediction | Post-optimization | ML prediction | Post-optimization | ||
| Base | 100% | 2.40 (0.01) | 0.88 (0.02) | 3.66 (0.03) | 2.76 (0.03) |
| PI | 100% | 2.39 (0.01) | 0.87 (0.02) | 3.19 (0.02) | 2.45 (0.03) |
| PI+ | 100% | 2.94 (0.01) | 0.95 (0.02) | 4.26 (0.03) | 3.32 (0.03) |
| Base | 10% | 2.92 (0.01) | 0.96 (0.02) | 3.93 (0.02) | 2.94 (0.06) |
| PI | 10% | 2.95 (0.01) | 1.03 (0.02) | 4.58 (0.03) | 3.56 (0.03) |
| PI+ | 10% | 2.36 (0.01) | 0.83 (0.02) | 3.98 (0.03) | 3.00 (0.03) |
The post-ML optimization process is also efficient and parallelizable – taking less than 1 second to converge to the optimal solution (Table S1†). The ML model training requires less than 1 MB for 140000 parameters, a substantial reduction in memory usage compared to arc continuation or convex hull methods, which demand approximately 1 GB of storage and 50 GB of memory per run. Overall, the accurate ML predictions of equilibrium compositions enable rapid convergence to highly accurate solutions, offering significant advantages in both memory- and time-efficiency.
4 Conclusions
In this work, we presented an efficient and extensible machine learning-based approach for calculating phase coexistence in ternary systems. A neural network trained on phase coexistence data was able to predict the number and compositions of equilibrium phases for a solution prepared at a given composition under a specific mixing potential. Incorporating physical constraints into the neural network architecture enhanced prediction accuracy, while additional physics-informed losses offered no significant improvement. The physics-constrained architecture produced higher-quality models with less data, offering advantages in scenarios where data acquisition is labor- or resource-intensive. However, the resulting models still exhibit errors that may be unacceptable for certain applications, such as process simulation software. To achieve precise results, a Newton conjugate gradient method was used, with machine-learning predictions serving as a warm start for optimization to determine final equilibrium phase compositions. This integration of neural networks with numerical refinement enabled rapid and accurate predictions of coexisting phases, their compositions, and abundances.This work motivates several areas of future inquiry. Extensions to systems with more components would increase utility for complex industrial and biological processes. Expanding beyond the Flory–Huggins theory by incorporating other free energy models or data from molecular simulation, perhaps in a single framework, would further enhance its generalizability across diverse chemical systems. Additionally, exploring more advanced physics-informed learning strategies, incorporating uncertainty quantification, and refining neural network architectures could boost prediction efficiency and reliability. Collectively, these directions could enhance both the theoretical and practical impact of leveraging ML for phase coexistence calculations.
Data availability
The data associated with this study are publicly accessible at https://doi.org/10.5281/zenodo.13776946. The code associated with this study is publicly accessible at https://github.com/webbtheosim/ml-ternary-phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. D. and M. A. W. acknowledge support from the National Science Foundation. The development of certain machine learning approaches and analyses is based on work supported by the National Science Foundation under Grant No. 2118861. The investigation of phase behavior in polymer solutions is supported by the National Science Foundation under Grant No. 2237470. S. J. acknowledges support from the Princeton Catalysis Initiative. Calculations were performed using resources from Princeton Research Computing at Princeton University, which is a consortium led by the Princeton Institute for Computational Science and Engineering (PICSciE) and Office of Information Technology's Research Computing. These resources include a GPU-based computing cluster purchased with support from the National Science foundation under Grant No. 2320649. The authors also acknowledge Andrej Košmrlj and Qiwei Yu for helpful discussions regarding the convex hull algorithm.Notes and references
- G. R. Guillen, Y. Pan, M. Li and E. M. V. Hoek, Ind. Eng. Chem. Res., 2011, 50, 3798–3817 CrossRef CAS.
- D. R. Lloyd, S. S. Kim and K. E. Kinzer, J. Membr. Sci., 1991, 64, 1–11 CrossRef CAS.
- Y. Fily and M. C. Marchetti, Phys. Rev. Lett., 2012, 108, 235702 CrossRef.
- J. Runnström, Dev. Biol., 1963, 7, 38–50 CrossRef.
- H. Walter and D. E. Brooks, FEBS Lett., 1995, 361, 135–139 CrossRef CAS PubMed.
- M. Muschol and F. Rosenberger, J. Chem. Phys., 1997, 107, 1953–1962 CrossRef CAS.
- F. J. Iborra, Theor. Biol. Med. Modell., 2007, 4, 15 CrossRef.
- S. Weber and C. Brangwynne, Curr. Biol., 2015, 25, 641–646 CrossRef CAS PubMed.
- A. Molliex, J. Temirov, J. Lee, M. Coughlin, A. Kanagaraj, H. Kim, T. Mittag and J. Taylor, Cell, 2015, 163, 123–133 CrossRef CAS PubMed.
- C. P. Brangwynne, T. J. Mitchison and A. A. Hyman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4334–4339 CrossRef CAS.
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Jülicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS.
- J. M. Michael Michelsen, Thermodynamic Modelling: Fundamentals and Computational Aspects, Tie-Line Publications, 2004 Search PubMed.
- D.-Y. Peng and D. B. Robinson, Ind. Eng. Chem. Fundam., 1976, 15, 59–64 CrossRef CAS.
- T. Jindrová and J. Mikyška, Fluid Phase Equilib., 2013, 353, 101–114 CrossRef.
- Y. Zhan, Z. Hu, J. Kou and Q. Su, Phys. Fluids, 2024, 36, 083351 CrossRef.
- T. Jindrová and J. Mikyška, Fluid Phase Equilib., 2015, 393, 7–25 CrossRef.
- N. A. Gokcen, J. Phase Equilib., 1996, 17, 50–51 CrossRef CAS.
- J. J. v. Laar, Z. Phys. Chem., 1910, 72, 723–751 CrossRef.
- M. L. McGlashan, J. Chem. Educ., 1963, 40, 516 CrossRef CAS.
- H. Renon and J. M. Prausnitz, AIChE J., 1968, 14, 135–144 CrossRef CAS.
- G. Maurer and J. Prausnitz, Fluid Phase Equilib., 1978, 2, 91–99 CrossRef CAS.
- A. Fredenslund, R. L. Jones and J. M. Prausnitz, AIChE J., 1975, 21, 1086–1099 CrossRef CAS.
- X. Feng, M.-H. Chen, Y. Wu and S. Sun, J. Comput. Phys., 2022, 463, 111275 CrossRef.
- P. J. Flory, J. Chem. Phys., 1941, 9, 660 CrossRef CAS.
- J. Kou, S. Sun and X. Wang, Comput. Geosci., 2016, 20, 283–295 CrossRef.
- D. V. Nichita, Fluid Phase Equilib., 2018, 466, 31–47 CrossRef CAS.
- D. V. Nichita, Fluid Phase Equilib., 2017, 447, 107–124 CrossRef CAS.
- D. V. Nichita, J.-C. de Hemptinne and S. Gomez, Pet. Sci. Technol., 2009, 27, 2177–2191 CrossRef CAS.
- M. Castier, Fluid Phase Equilib., 2014, 379, 104–111 CrossRef CAS.
- T. Zhang, Y. Li, Y. Li, S. Sun and X. Gao, Comput. Methods Appl. Mech. Eng., 2020, 369, 113207 CrossRef.
- P. Civicioglu, Appl. Math. Comput., 2013, 219, 8121–8144 CrossRef.
- H. Wang, Z. Hu, Y. Sun, Q. Su and X. Xia, Comput. Intell. Neurosci., 2018, 2018, 1–27 Search PubMed.
- S. Wang, X. Da, M. Li and T. Han, J. Syst. Eng. Electron., 2016, 27, 395–406 Search PubMed.
- J. L. Sengers, How Fluids Unmix: Discoveries by the School of Van der Waals and Kamerlingh Onnes, Edita-the Publishing House of the Royal, 1st edn, 2002 Search PubMed.
- H. Binous and A. Bellagi, Eng. Rep., 2020, 3(3), e12296 Search PubMed.
- M. L. Michelsen, Fluid Phase Equilib., 1982, 9, 1–19 CrossRef CAS.
- H. Sidky, J. K. Whitmer and D. Mehta, AIChE J., 2016, 62, 4497–4507 CrossRef CAS.
- H. Sidky, A. C. Liddell, D. Mehta, J. D. Hauenstein and J. K. Whitmer, Ind. Eng. Chem. Res., 2016, 55, 11363–11370 CrossRef CAS.
- S. Mao, D. Kuldinow, M. P. Haataja and A. Košmrlj, Soft Matter, 2019, 15, 1297–1311 RSC.
- C. B. Barber, D. P. Dobkin and H. Huhdanpaa, ACM Trans. Math. Softw., 1996, 22, 469–483 CrossRef.
- Y. An, M. A. Webb and W. M. Jacobs, Sci. Adv., 2024, 10, eadj2448 CrossRef PubMed.
- K. Terayama, K. Han, R. Katsube, I. Ohnuma, T. Abe, Y. Nose and R. Tamura, Scr. Mater., 2022, 208, 114335 CrossRef CAS.
- G. Deffrennes, K. Terayama, T. Abe and R. Tamura, Mater. Des., 2022, 215, 110497 CrossRef CAS.
- J. C. R. Thacker, D. J. Bray, P. B. Warren and R. L. Anderson, J. Phys. Chem. B, 2023, 127, 3711–3727 CrossRef CAS.
- R. Tamura, G. Deffrennes, K. Han, T. Abe, H. Morito, Y. Nakamura, M. Naito, R. Katsube, Y. Nose and K. Terayama, Sci. Technol. Adv. Mater.:Methods, 2022, 2, 153–161 Search PubMed.
- J. G. Ethier, R. K. Casukhela, J. J. Latimer, M. D. Jacobsen, B. Rasin, M. K. Gupta, L. A. Baldwin and R. A. Vaia, Macromolecules, 2022, 55, 2691–2702 CrossRef CAS.
- J. Lund, H. Wang, R. D. Braatz and R. E. García, Mater. Adv., 2022, 3, 8485–8497 RSC.
- Y. Li, T. Zhang and S. Sun, Ind. Eng. Chem. Res., 2019, 58, 12312–12322 CrossRef CAS.
- Y. Aoki, S. Wu, T. Tsurimoto, Y. Hayashi, S. Minami, O. Tadamichi, K. Shiratori and R. Yoshida, Macromolecules, 2023, 56, 5446–5456 CrossRef CAS.
- J. G. Ethier, D. J. Audus, D. C. Ryan and R. A. Vaia, Giant, 2023, 15, 100171 CrossRef CAS.
- H. Tompa, Trans. Faraday Soc., 1949, 45, 1142 RSC.
- R. L. Scott, J. Chem. Phys., 1949, 17, 268–279 CrossRef CAS.
- C. B. Barber, D. P. Dobkin and H. Huhdanpaa, ACM Trans. Math. Softw., 1996, 22, 469–483 CrossRef.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein and L. Antiga, et al. , Adv. Neural Inf. Process. Syst., 2019, 32, 8026–8037 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional optimized phase diagrams; phase classification confusion matrices; equilibrium composition prediction parity plots; post-ML optimization performance. See DOI: https://doi.org/10.1039/d4me00168k |
| ‡ Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2025 |