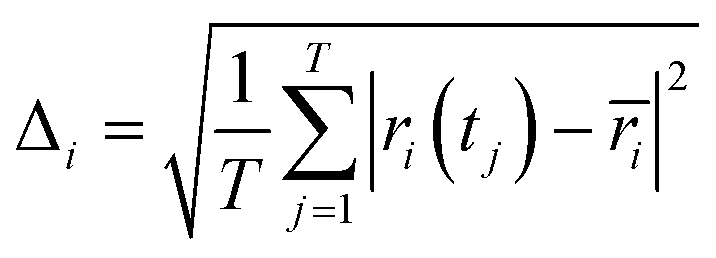Phenyl- versus cyclohexyl-terminated substituents: comparative study on aggregated structures and electron-transport properties in n-type organic semiconductors†
Received
6th July 2024
, Accepted 19th September 2024
First published on 15th October 2024
Abstract
Substituent engineering is a key route to high-performance functional molecular materials in the same way as the development of a π-electron core for organic (opto-)electronics. Here we demonstrate a comparative study between aromatic phenyl- and aliphatic cyclohexyl-terminated side-chain substituents on an electron-deficient π-electron core, 3,4,9,10-benzo[de]isoquinolino[1,8-gh]quinolinetetracarboxylic diimide (BQQDI), to get insights into the impact of intermolecular interactions between the substituents in the solid state on high-performance electron-transport properties. In the BQQDI system, both phenyl- and cyclohexyl-terminated ethyl substituents show similar packing structures, demonstrating the unobvious impact of terminal groups. However, solution-processed single-crystal transistor studies revealed a relatively low electron mobility of cyclohexyl-terminated BQQDI. Based on molecular dynamics simulations, we attribute this discrepancy to dynamic molecular motions coupled with electronic coupling in the solid state. While phenyl groups in the phenylethyl substituent show intermolecular C–H⋯π interactions which lead to less dynamic motions, the cyclohexyl counterpart does not show any specific intermolecular interactions. Hence, a low-dynamic feature thanks to inter-side-chain interactions is promising for excellent charge-transport properties. The present findings underline the crucial role of interactions between substituents in the development of organic materials via side-chain-engineered control of the solid-state dynamic motions.
Design, System, Application
Organic semiconductors (OSCs) offer a great opportunity of solution-processable devices, for which the molecular design involves a π-electron core and solubilizing side-chain substituents. Whereas the π-electron core plays a primary role in the packing motif via π–π and subsequent interactions such as C–H⋯π interactions, side-chain substituents can finely tune the packing structure and intermolecular π-orbital overlaps. In addition, the substituents may contribute to charge transport by modulating the thermal fluctuation of π-orbital overlaps. Hence, it is of great importance for high-performance OSCs to understand distinct roles of the π-electron core and side-chain substituents in packing structures and charge-transport properties. In this work, we comparatively studied two analogous n-type OSCs with phenyl- or cyclohexyl-terminated substituents. The solution-processed single-crystal thin-film transistor based on the cyclohexyl derivative exhibited the best electron mobility of 2.3 cm2 V−1 s−1, which is slightly lower than that based on the phenyl counterpart. While both derivatives form analogous packing structures, only the phenyl derivative shows weak C–H⋯π intermolecular interactions between neighboring phenyl groups. Hence, the higher mobility is attributed to suppressed molecular motions owing to the phenyl-to-phenyl interactions, which was successfully revealed by molecular dynamics simulations. Therefore, a potential impact of substituent-engineered molecular design is presented.
|
1. Introduction
An aggregated structure is a key factor of functional molecular materials. In charge-transporting organic semiconductors (OSCs), the solid-state packing structure of π-electron cores (π-cores) is particularly important since the charge transport is governed by the intermolecular overlap of π-orbitals.1,2 Whereas π-cores principally tend to form π-stacking structures, competitive intermolecular (e.g., C–H⋯π) interactions may induce the herringbone packing motif.3–5 On the other hand, incorporation of heteroatoms, such as O, N, S and halogens, into the π-core further complicates the resulting packing motif due to additional intermolecular interactions, such as hydrogen bonds,6,7 and the polarization of the π-core,8 resulting in the slipped/cofacial π-stacking motif, brickwork and so on. As such, the π-cores could play a basic role in the packing structure and charge-transport capability of OSCs.
Besides, recent progress in OSCs demonstrates molecular structures composed of not only a π-core but also side-chain substituents,9,10 the latter of which primarily solubilize π-conjugated molecules for solution-processable electronics.11–13 In this context, aliphatic alkyl chains are frequently adopted for OSCs due to their flexibility and affinity for organic solvents conventionally used for aromatic compounds, making both synthesis and device fabrication convenient. In addition, the side-chain substituents are crucial for the packing structure, ranging from fine-tuning to drastic rearrangements, and thus for charge transport capability.12,14–17 Furthermore, the side-chain substituents could impact suppressing thermally excited molecular motions, which is attributed to a fluctuation of intermolecular electronic coupling, namely, dynamic disorder and non-local electron–phonon couplings, leading to higher charge-carrier mobilities.18–20
Although alkyl substituents play an appropriate role in high-mobility hole-transporting (p-type) OSCs with the herringbone motif,18,19,21 the role is not versatile for an enormous range of π-cores. For example, some of electron-transporting (n-type) OSCs demonstrated high electron mobilities with the brickwork packing motif, which is organized by fluoroalkyl or cyclohexyl substituents rather than alkyl ones.22–24 Hence, it is necessary to clarify distinct roles of the π-core and side-chain substituents in the packing structure and charge-transport properties, which leads to an opportunity of side-chain substituent engineering to achieve the optimal packing structure for high OSC performances with each designed π-core.
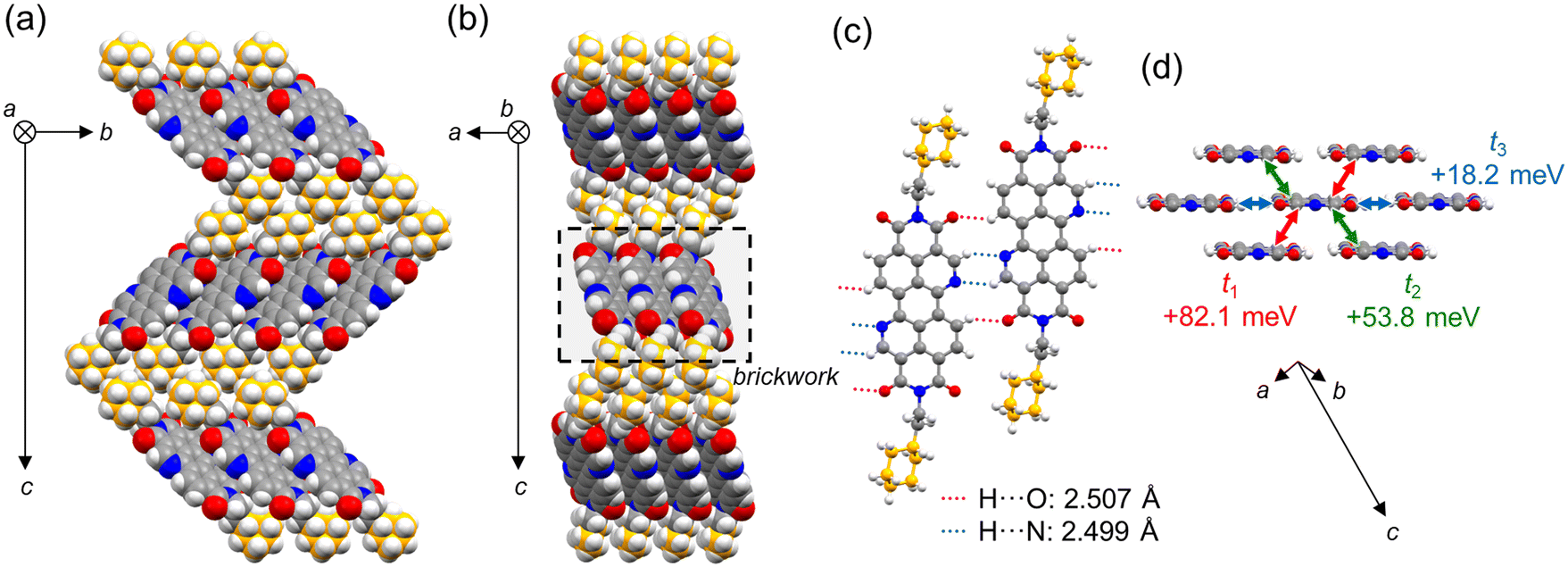
Our group has recently developed n-type OSCs based on the 3,4,9,10-benzo[de]isoquinolino[1,8-gh]quinoline diimide (BQQDI) π-core.25,26 A typical characteristic of BQQDI derivatives is the layered brickwork structure triggered by intermolecular C–H⋯O and C–H⋯N attractive interactions in the lateral direction of the π-core. In the BQQDI family reported, the 2-phenylethyl-substituted derivative (PhC2–BQQDI) exhibits high electron mobility up to 3.0 cm2 V−1 s−1 under ambient atmosphere.25 In the layered brickwork structure of PhC2–BQQDI, the terminal phenyl groups further contribute to C–H⋯π interactions (Fig. 1a). This C–H⋯π interaction between substituents could suppress the thermal motion of molecules in the aggregated structure in cooperation with the C–H⋯O and C–H⋯N interactions between π-cores, resulting in a suppressed dynamic disorder, which is a detrimental factor decreasing charge-carrier mobility.27,28 Although the ethyl linker has been modified with different alkyl chain lengths,29 it is also interesting to study the impact of the terminal phenyl group for future solution-processable OSC developments.
 |
| | Fig. 1 Research target in this work. (a) Chemical and crystal structures of phenyl-terminated PhC2–BQQDI (CCDC: 1938483), and schematic drawing of the interlayer C–H⋯π interaction between phenyl groups. C, gray and green (for phenyl group); H, white; N, blue; O, red. (b) Chemical structure of cyclohexyl-terminated ChxC2–BQQDI featured in this work. | |
In this work, we have studied the crucial role of terminal phenyl groups in PhC2–BQQDI. For this, 2-cyclohexylethyl-substituted BQQDI (ChxC2–BQQDI) was synthesized because the aliphatic cyclohexyl ring is the representative opposite of the phenyl group24,30–34 and promises a lack of the aforementioned C–H⋯π interactions (Fig. 1b). Through comparative studies on crystallography and charge-transport properties, the cyclohexyl replacement was not so influential for the crystal packing; however, molecular dynamics simulations revealed that the absence of C–H⋯π interaction in ChxC2–BQQDI could cause relatively large thermal motions and dynamic disorder in the crystalline states, which leads to a lower electron mobility than PhC2–BQQDI, while it is still as high as 2.3 cm2 V−1 s−1. Hence, this paper shows a significant role of intermolecular interactions between side-chain substituents on the π-core in enhancing charge-transport properties through a suppression of dynamic molecular motions in crystalline molecular solids.
2. Experimental
2.1. Materials
Benzo[de]isoquinolino[1,8-gh]quinolinetetracarboxylic dianhydride (BQQ–TCDA) and PhC2–BQQDI were prepared according to the literature.25 The other reagents and solvents were purchased and used without further purification, whereas o-dichlorobenzene was purified by using a solvent purification system (GlassContour, Nikko Hansen Co., Ltd.) prior to use.
2.2. Instruments
1H nuclear magnetic resonance (NMR) spectra were acquired with a JEOL ECS400 spectrometer (400 MHz) in deuterated sulfuric acid (D2SO4) at 25 °C. Chemical shifts are reported in δ ppm. 1H NMR spectra are referenced to residual protons of sulfuric acid (δ 10.90 ppm) as an internal standard. The data are presented in the following format: chemical shift, multiplicity (s = singlet, d = doublet, br = broad), coupling constant in Hertz (Hz), signal area integration in natural numbers. Elemental analysis was performed on a J-Science Lab JM10 CHN analyzer.
2.3. X-ray crystallography
Single-crystal X-ray diffraction data were collected on a Rigaku R-AXIS RAPID II imaging plate diffractometer with CuKα radiation (λ = 1.54187 Å). The structures were solved by direct methods (SHELXT35 (ver. 2014/4)) and refined by full-matrix least-squares procedures on F2 for all reflections (SHELXL36 (ver. 2014/7)) by using the CrystalStructure interface (Rigaku (ver. 4.2.2)). While all hydrogen atoms were placed at calculated positions, all other atoms were refined anisotropically. Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication, and from the Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/data_request/cif (CCDC 2232687).
2.4. Quantum chemical calculations
The intermolecular interaction energies between pairs of adjacent molecules were obtained at the M06-2X/6–31++G(d,p) level of density functional theory (DFT) with counterpoise correction37,38 for the basis set superposition error. Transfer integrals between the LUMO of adjacent molecules were calculated at the PBEPBE/6-31G(d) level of DFT.
2.5. Band calculation
Band calculation was conducted by the tight-binding approximation. Effective masses of electrons were calculated from the curvature of the LUMO-band bottom according to the following equation:| |  | (1) |
where m* is the effective mass, ħ is the Dirac's constant, E is the energy, and k is the wave vector. In band theory, a charge-carrier mobility (μ) is expressed by the following equation:39where q and τ are the elementary charge and relaxation time of charge carriers, respectively.
2.6. Molecular dynamics simulation
All-atom molecular dynamics (MD) simulations were performed using GROMACS 2016.3. The generalized Amber force field40 parameters were used for the force field parameters of both ChxC2–BQQDI and PhC2–BQQDI and their atomic charges were calculated using the restrained electrostatic potential (RESP)41 methodology based on DFT calculations (B3LYP/6-31G(d)) using the GAUSSIAN09 program.42 The time step was set to 1 fs. The long-range Coulomb interactions were calculated with the smooth particle-mesh Ewald (PME)43 method with a grid spacing of 0.30 nm. The real space cutoff for both Coulomb and van der Waals interactions was 1.2 nm. The B-factors were calculated by the following equation:| |  | (3) |
where Δi is the root mean square fluctuation (RMSF) of atom i. The RMSF values were estimated by the following equation:| |  | (4) |
where T is the time step, ri(tj) is the position coordinate of atom i, and ri is the average of ri(tj). A 10 × 10 × 3 super cell including 600 molecules was prepared based on the single crystal structure, and 5 and 50 ns MD runs were sequentially performed by the number–temperature–volume (NTV) and the number–temperature–pressure (NTP) ensembles, respectively, at the temperatures the same as that in the crystallographic data (295 and 296 K for ChxC2–BQQDI and PhC2–BQQDI, respectively). The pre-equilibrium NTV run was carried out by using a Berendsen44 thermostat, whereas the NTP process was conducted by using a Nosé–Hoover45–47 thermostat and Parrinello–Rahman barostat.48
2.7. Synthesis of N,N′-di(2-cyclohexylethyl)-3,4,9,10-benzo[de]isoquinolino[1,8-gh]quinolinetetracarboxylic diimide (ChxC2–BQQDI)
A Schlenk flask was charged with BQQ–TCDA (200 mg; 0.51 mmol), 2-cyclohexylethylamine (0.22 mL; 1.52 mmol), propionic acid (3.80 mL; 50.8 mmol) and o-dichlorobenzene (17 mL) and sealed. The mixture was stirred at 130 °C for 5 h under an argon atmosphere to yield a red-brown suspension. The precipitates were collected by suction filtration, washed with excess methanol, and dried in vacuo. After recrystallization from o-dichlorobenzene (0.143 g L−1 dissolved at 150 °C), 271 mg of red-brown solids were collected (yield: 87% based on BQQ–TCDA). Further purification was performed by sublimation under vacuum for compound characterization and device studies. 1H NMR (400 MHz, D2SO4, 25 °C): δ 9.56 (s, 2H, ArH), 9.37 (d, J = 8.4 Hz, 2H, ArH), 9.31 (d, J = 8.0 Hz, 2H, ArH), 4.18 (br, 4H, NCH<), 1.72 (br, 4H, –CH2–), 1.65–1.56 (br, 10H, –CH2–), 1.37 (br, 2H, –CH2–), 1.24–1.08 (br, 6H, –CH2–), 1.00–0.92 (br, 4H, –CH2–). 13C NMR could not be obtained due to poor solubility. Anal. calcd. for C38H36N4O4: C, 74.49; H, 5.92; N, 9.14. Found: C, 74.51; H, 5.99; N, 9.11.
2.8. Device fabrication and characterization
Single-crystal organic thin-film transistors (OTFTs) were fabricated in the bottom-gate/top-contact geometry. An n++-Si/SiO2 (200 nm) wafer encapsulated with a thermally cross-linked insulating polymer AL-X601 (AGC Inc.; 40 nm) was used as the substrate, where n++-Si and the SiO2/AL-X601 bilayer act as the gate electrode and gate dielectric layer, respectively. Single-crystalline thin films of ChxC2–BQQDI were solution-crystallized by the edge casting method49 from its 1-methylnaphthalene solution. Au layers (40 nm) were vacuum-deposited onto the thin films through a metal shadow mask as the top-contact source and drain electrodes, followed by laser etching to afford the single-crystal OTFTs. A post-annealing process at 100 °C was conducted for 2 h under vacuum prior to electrical evaluations.
Electrical evaluations were carried out with a Keithley 4200-SCS semiconductor parameter analyzer under ambient atmosphere. Electron mobilities in the linear and saturation regimes were estimated using the conventional equations:
| |  | (5) |
and
| |  | (6) |
respectively, where
W is the channel width,
L is the channel length,
Ci is the gate capacitance per unit area (
Ci = 14.2 nF cm
−2 measured by the Keithley 4200-SCS),
ID is the drain current,
VD is the drain voltage,
Vth is the threshold voltage, and
μlin and
μsat are the electron mobilities in the linear and saturation regimes, respectively.
3. Results and discussion
3.1. Synthesis, X-ray crystallography and transport calculations
ChxC2–BQQDI was prepared in a similar way as PhC2–BQQDI (Scheme S1, ESI†).25 A red plate-like single crystal of ChxC2–BQQDI enough for X-ray crystallography was obtained by recrystallization from nitrobenzene. The X-ray single-crystal structure of ChxC2–BQQDI is shown in Fig. 2, where the crystallographic system of monoclinic P21/n resembles that of PhC2–BQQDI,25 whereas the a- and b-axes are exchanged (Table S1†). Thus, ChxC2–BQQDI exhibits the brickwork aggregated structure supported by the attractive C–H⋯N and C–H⋯O interactions between the BQQDI π-cores with close H⋯O and H⋯N contacts (2.507 and 2.499 Å, respectively) (Fig. 2c), despite a lack of specific weak intermolecular C–H⋯π interactions between phenyl groups observed in PhC2–BQQDI (Fig. 1a). This fact can reveal that the attractive C–H⋯N and C–H⋯O interactions and the volumetric filling arising from the similar molecular structures are significant for their packing structures. An apparent interdigitation of 2-phenylethyl substituents is loosened in ChxC2–BQQDI, leading to a ∼10% increase in lattice length in the c-axis while the comparable dimensions in the ab-plane. The force constant for the laterally aligned ChxC2–BQQDI dimer is calculated to be −8.45 kcal mol−1, which is comparable to that for PhC2–BQQDI (−8.48 kcal mol−1)25 regardless of their different substituents. Similar aggregated structures are also revealed by the short- and long-axis misalignments in π-stacked BQQDI skeletons (Fig. S3a and S3b, ESI†). The long-axis misalignments (dy1 and dy2) are +1.00 and +5.47 Å for ChxC2–BQQDI, whereas they are +1.01 and +5.51 Å for PhC2–BQQDI, respectively, showing comparable values. Also, the π–π distances dπ1 and dπ2 are also comparable between ChxC2–BQQDI (3.31 and 3.24 Å, respectively) and PhC2–BQQDI (3.30 and 3.25 Å, respectively). On the other hand, the short-axis misalignments (dx1 and dx2) show a slight mismatch. In ChxC2–BQQDI, dx1 and dx2 are −3.57 and +4.39 Å, respectively, which are smaller than the corresponding values in PhC2–BQQDI (−3.68 and +5.14 Å).29 That is, ChxC2–BQQDI demonstrates larger geometrical overlaps of the BQQDI π-cores than PhC2–BQQDI. However, transfer integral calculations suggested a slight reduction of the LUMO overlaps in ChxC2–BQQDI than that in PhC2–BQQDI. Despite their comparable transfer integrals for the lateral direction (t3) being +18 meV, those for the π-stacked dimers (t1 and t2) are +82.1 and +53.8 meV, respectively, for ChxC2–BQQDI, which are slightly smaller than those for PhC2–BQQDI (+90.7 and +58.5 meV).25 This difference is attributed to the difference in the short-axis misalignments, which the direction crosses the major nodes of the LUMOs of the BQQDI π-core (Fig. S3c, ESI†).
 |
| | Fig. 2 Single crystal structure of ChxC2–BQQDI. (a and b) Packing structures viewed along the a- and b-axes. (c) Intermolecular C–H⋯O and C–H⋯N interactions in the lateral direction. van der Waals radius:50 H, 1.20 Å; N, 1.55 Å; O, 1.52 Å. Close contacts are estimated relative to a sum of the van der Waals radii. (d) Geometry of the brickwork structure and transfer integrals corresponding to the layer highlighted by a black-lined square in (b). C, orange and gray (for cyclohexyl moiety and the others, respectively); H, white; N, blue; O, red. | |
3.2. Computational insights into the molecular conformation in the single crystal
Here we would like to address the relationship of molecular conformation between the crystal structure and geometry optimization by quantum chemical calculations to consider the engineering ability of the aggregated structure by adopting the 2-cyclohexylethyl substituent. In Fig. 3, single-molecule geometries are compared between the X-ray diffraction (XRD) analysis at room temperature and a geometry optimization by the B3LYP/6-31+G(d) level of DFT, where both ChxC2–BQQDI and PhC2–BQQDI show good similarities at a glance. As summarized in Table 1, for more details, the differences of some specific geometrical parameters between XRD and DFT results are smaller for ChxC2–BQQDI than those for PhC2–BQQDI. This might be attributed to the general steric effects in aliphatic alkyl chains, which subjects the 2-cyclohexylethyl substituent to an almost fixed conformation, in contrast to the phenethyl substituent having less intramolecular steric repulsions. Yet, in reality, the argument for the 2-cyclohexylethyl group depends on the π-core. As the representative, a crystal structure of 2-cyclohexylethyl substituted naphtho[2,3-b:6,7-b']dithiophene diimide (NDTI) has been reported by Takimiya and co-workers.51 In this case, the deviation between XRD and DFT results is far remarkable (Fig. S4 and Table S2, ESI†). Therefore, the 2-cyclohexylethyl-substituted ChxC2–BQQDI improves the chemistry of aggregated structures of BQQDI derivatives and provides an interesting insight into the 2-cyclohexyl substituent for organic semiconductors.
 |
| | Fig. 3 Comparison of single-molecule geometries between XRD and DFT. C, gray; H, white; N, blue; O, red. | |
Table 1 Summary of bond and torsion angles for ChxC2–BQQDI and PhC2–BQQDI
| Measured angle |
ChxC2–BQQDI |
PhC2–BQQDI |
| XRD |
DFT |
XRD |
DFT |
| Bond angle ∠(N1-C1-C2) (°) |
113.2 |
112.2 |
113.0 |
112.4 |
| Bond angle ∠(C1-C2-C3) (°) |
112.4 |
113.7 |
109.1 |
111.4 |
| Torsion angle ∠(N1-C1-C2-C3) (°) |
11.3 |
4.1 |
14.9 |
0.0 |
| Torsion angle ∠(C1-C2-C3-C4) (°) |
11.3 |
11.0 |
80.1 |
89.3 |
| Torsion angle ∠(C1-C2-C3-C5) (°) |
67.7 |
67.1 |
97.2 |
89.2 |
3.3. Solution-crystallized single-crystal transistors
Charge-transport properties of ChxC2–BQQDI were investigated by solution-crystallized single-crystal OTFTs. The OTFTs were fabricated in a bottom-gate/top-contact geometry with the gold source and drain electrodes (Fig. 4a). Single-crystal channels were edged by means of laser ablation to assess the charge-carrier mobility properly (Fig. 4b). As shown in Fig. 4c–f, ChxC2–BQQDI exhibits typical n-channel OTFT characteristics under ambient atmosphere. The best device exhibited high electron mobilities of 1.92 and 2.35 cm2 V−1 s−1 in the linear (μlin) and saturation regimes (μsat), respectively, where the μsat value is slightly lower than that of PhC2–BQQDI (3.0 cm2 V−1 s−1).25 XRD measurement on the OTFT assigned the channel direction to [0 1 0] (i.e., the b-axis direction) (Fig. S5, ESI†). To consider theoretical electron-transport capabilities with anisotropy, band calculations were further carried out. The inverse of the effective mass of electrons (m*)−1 within the ab plane is azimuthally plotted in Fig. S6, ESI,† where horizontal and vertical directions correspond to the [0 1 0] and [1 0 0] directions in the crystal structures of both derivatives. Although the overall size of a peanut-like (m*)−1 distribution is larger for PhC2–BQQDI than that for ChxC2–BQQDI due to their relative transfer integrals, the (m*)−1 value along the [0 1 0] direction, which corresponds to the channel direction of solution-crystallized single-crystal OTFTs based on both derivatives, is larger in ChxC2–BQQDI. Since a charge-carrier mobility is proportional to (m*)−1 in band theory (eqn 2), these results imply that ChxC2–BQQDI could exhibit a higher electron mobility than PhC2–BQQDI.
 |
| | Fig. 4 Solution-crystallized single-crystal OTFT based on ChxC2–BQQDI. (a) Schematic device structure. (b) Cross-polarized microscopy image. Channel length L is 200 μm. (c) Output and (d and e) transfer characteristics. (f) VG-dependent electron mobility. | |
3.4. Molecular dynamics simulations and dynamic disorder
To study the inconsistency between the calculational and experimental results, MD simulations were performed on ChxC2–BQQDI based on the crystal structure. Fig. 5a and b show B-factor distributions after the MD runs, which depict the isotropic thermal fluctuations resolved into the atoms. The MD simulation of ChxC2–BQQDI revealed an effectively suppressed thermal motion of the BQQDI π-core and ethyl moieties (colored exclusively by blue) similarly to PhC2–BQQDI (Fig. S8a, S8b and Table S3, ESI†), owing to the lateral C–H⋯N and C–H⋯O attractive interactions.25 On the other hand, cyclohexyl rings are regularly colored by light blue, suggesting their potential thermal disordering due to the absence of specific intermolecular interactions. In particular, C atoms at the 2-, 3-, 5- and 6-positions of the cyclohexyl ring in a few ChxC2–BQQDI molecules within the supercell are highlighted by red color (Fig. 5b), whose B-factors exceed 30 Å2. This irregular cyclohexyl rings are associated with a rotation of the cyclohexyl ring around an axis linking C atoms at the 1- and 4-positions (Fig. 5c). Although it is curious if such rotations occur in a real crystal, this result would possibly suggest local structural disorders and defects in the single crystals. Besides, in comparison with the single crystal data, the unit cell volume after the MD simulations was more expanded in ChxC2–BQQDI (∼1.4%) than in PhC2–BQQDI (~0.6%) (Fig. S7†), suggesting the effect of the C–H⋯π interactions in the robust aggregated structure of PhC2–BQQDI.
 |
| | Fig. 5 MD simulation for ChxC2–BQQDI. (a and b) Color-coded B-factor distribution (unit: Å2) obtained from the trajectory of the crystal structure during the last 10 ns of 50 ns MD runs at 295 K. (c) Representative molecules extracted from (a) with the regular and irregular B-factor distributions at the cyclohexyl moiety. (d) Schematic depiction of the rotation of the cyclohexyl moiety corresponding to the irregular molecule in (c). (e and f) Statistics of transfer integrals t1 and t2 based on the MD simulations of ChxC2–BQQDI, and (g and h) of PhC2–BQQDI. Red and blue solid lines show the corresponding normal distributions. The thick black line indicates the transfer integrals calculated from the crystal structure. | |
In addition, the thermal molecular motions are coupled with a dynamic variation of transfer integrals, i.e., dynamic disorder.27,28 The degree of dynamic disorder was estimated by calculating a population of transfer integrals after MD runs; in ChxC2–BQQDI, the average transfer integral values (tavg) corresponding to t1, t2 and t3 are 71.6 (27.2), 37.8 (15.3) and 11.6 (2.6) meV, respectively, where the value in the parentheses indicates a standard deviation (σ) (Fig. 5e, f, S7c, and Table S4, ESI†). The resulting coefficient of variation (σ/tavg) is calculated as a brief measure of dynamic disorder to be 0.38, 0.41 and 0.22. On the other hand, for PhC2–BQQDI, the corresponding σ/tavg values are 0.32, 0.36 and 0.23 (Fig. 5g, h, S7d, and Table S4, ESI†) (note that these tavg and σ values are slightly different from those in ref. 25 because of the sophisticated parameters for simulation). Hence, ChxC2–BQQDI could show a larger dynamic disorder that leads to lower electron mobilities than PhC2–BQQDI, which is consistent with our experimental results.
4. Conclusions
Here we reported a comparative study on a 2-phenylethyl-substituted high-performance n-type OSC PhC2–BQQDI and its 2-cyclohexylethyl-substituted analogue, ChxC2–BQQDI. ChxC2–BQQDI bearing only aliphatic hydrocarbons has a very similar aggregated structure to PhC2–BQQDI, which crystallizes in a layered brickwork structure supported by C–H⋯π interactions between phenyl groups. Solution-crystallized single-crystal transistors based on ChxC2–BQQDI demonstrated a high electron mobility of 2.3 cm2 V−1 s−1 in air. This mobility is lower than that reported for PhC2–BQQDI, whereas theoretical calculations suggest higher electron mobility of ChxC2–BQQDI. Further analysis by means of MD simulations revealed the more serious dynamic disorder of ChxC2–BQQDI, demonstrating a crucial role of interactions between substituents in charge-carrier transport in molecular semiconductors. The present results will be helpful for the materials design of crystalline, high-performance OSCs by side-chain-engineered suppression of molecular motions.
Data availability
Crystallographic data has been deposited in the Cambridge Crystallographic Data Centre (CCDC) under 2232687 and can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/data_request/cif.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge AGC Inc. for supplying AL-X601. This project was partially supported by the JST-PRESTO and JST-CREST programs “Scientific Innovation for Energy Harvesting Technology” (no. JPMJPR17R2 and JPMJCR21Q1) and “Exploring Innovative Materials in Unknown Search Space” (no. JPMJCR21O3) by JSPS KAKENHI (grant no. JP18H01856, JP19H05716, JP19H05718, JP22K14743, and JP24H00472).
Notes and references
- J. L. Brédas, J. P. Calbert, D. A. Da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- E. F. Valeev, V. Coropceanu, D. A. Da Silva Filho, S. Salman and J. L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882–9886 Search PubMed.
- G. R. Desiraju and A. Gavezzotti, J. Chem. Soc., Chem. Commun., 1989, 621–623 RSC.
- B. Schatschneider, J. Phelps and S. Jezowski, CrystEngComm, 2011, 13, 7216–7223 Search PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 Search PubMed.
- E. D. Głowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 Search PubMed.
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CAS.
- K. E. Maly, Cryst. Growth Des., 2011, 11, 5628–5633 Search PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 Search PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS.
- M. C. R. Delgado, E. Kim, S. Filho and J. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef.
- J. Vura-Weis, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 1738–1739 CrossRef CAS PubMed.
- Y. Tsutsui, G. Schweicher, B. Chattopadhyay, T. Sakurai, J. B. Arlin, C. Ruzié, A. Aliev, A. Ciesielski, S. Colella, A. R. Kennedy, V. Lemaur, Y. Olivier, R. Hadji, L. Sanguinet, F. Castet, S. Osella, D. Dudenko, D. Beljonne, J. Cornil, P. Samorì, S. Seki and Y. H. Geerts, Adv. Mater., 2016, 28, 7106–7114 CrossRef CAS.
- C. P. Yu, S. Kumagai, T. Kushida, M. Mitani, C. Mitsui, H. Ishii, J. Takeya and T. Okamoto, J. Am. Chem. Soc., 2022, 144, 11159–11167 CrossRef CAS PubMed.
- S. Illig, A. S. Eggeman, A. Troisi, L. Jiang, C. Warwick, M. Nikolka, G. Schweicher, S. G. Yeates, Y. H. Geerts, J. E. Anthony and H. Sirringhaus, Nat. Commun., 2016, 7, 10736 CrossRef CAS PubMed.
- G. Schweicher, G. D'Avino, M. T. Ruggiero, D. J. Harkin, K. Broch, D. Venkateshvaran, G. Liu, A. Richard, C. Ruzié, J. Armstrong, A. R. Kennedy, K. Shankland, K. Takimiya, Y. H. Geerts, J. A. Zeitler, S. Fratini and H. Sirringhaus, Adv. Mater., 2019, 31, 1902407 CrossRef CAS.
- M. A. Dettmann, L. S. R. Cavalcante, C. A. Magdaleno and A. J. Moulé, Adv. Funct. Mater., 2023, 33, 2213370 CrossRef CAS.
- T. Okamoto, C. P. Yu, C. Mitsui, M. Yamagishi, H. Ishii and J. Takeya, J. Am. Chem. Soc., 2020, 142, 9083–9096 CrossRef CAS PubMed.
- T. He, Y. Wu, G. D'Avino, E. Schmidt, M. Stolte, J. Cornil, D. Beljonne, P. P. Ruden, F. Würthner and C. D. Frisbie, Nat. Commun., 2018, 9, 2141 CrossRef PubMed.
- M. A. Stoeckel, Y. Olivier, M. Gobbi, D. Dudenko, V. Lemaur, M. Zbiri, A. A. Y. Guilbert, G. D'Avino, F. Liscio, A. Migliori, L. Ortolani, N. Demitri, X. Jin, Y. G. Jeong, A. Liscio, M. V. Nardi, L. Pasquali, L. Razzari, D. Beljonne, P. Samorì and E. Orgiu, Adv. Mater., 2021, 33, 2007870 Search PubMed.
- C. P. Yu, N. Kojima, S. Kumagai, T. Kurosawa, H. Ishii, G. Watanabe, J. Takeya and T. Okamoto, Commun. Chem., 2021, 4, 155 CAS.
- T. Okamoto, S. Kumagai, E. Fukuzaki, H. Ishii, G. Watanabe, N. Niitsu, T. Annaka, M. Yamagishi, Y. Tani, H. Sugiura, T. Watanabe, S. Watanabe and J. Takeya, Sci. Adv., 2020, 6, eaaz0632 CrossRef CAS.
- S. Kumagai, H. Ishii, G. Watanabe, C. P. Yu, S. Watanabe, J. Takeya and T. Okamoto, Acc. Chem. Res., 2022, 55, 660–672 CrossRef CAS.
- A. Troisi and G. Orlandi, Phys. Rev. Lett., 2006, 96, 086601 CrossRef.
- S. Fratini, S. Ciuchi, D. Mayou, G. T. De Laissardière and A. Troisi, Nat. Mater., 2017, 16, 998–1002 CrossRef CAS PubMed.
- S. Kumagai, H. Ishii, G. Watanabe, T. Annaka, E. Fukuzaki, Y. Tani, H. Sugiura, T. Watanabe, T. Kurosawa, J. Takeya and T. Okamoto, Chem. Mater., 2020, 32, 9115–9125 CrossRef CAS.
- K. Kunal, C. G. Robertson, S. Pawlus, S. F. Hahn and A. P. Sokolov, Macromolecules, 2008, 41, 7232–7238 CrossRef CAS.
- J. M. Torres, C. Wang, E. B. Coughlin, J. P. Bishop, R. A. Register, R. A. Riggleman, C. M. Stafford and B. D. Vogt, Macromolecules, 2011, 44, 9040–9045 CrossRef CAS.
- M. Hopkins Hatzopoulos, J. Eastoe, P. J. Dowding, I. Grillo, B. Demé, S. E. Rogers, R. Heenan and R. Dyer, Langmuir, 2012, 28, 9332–9340 CrossRef CAS PubMed.
- O. Munkhbat, M. Garzoni, K. R. Raghupathi, G. M. Pavan and S. Thayumanavan, Langmuir, 2016, 32, 2874–2881 CrossRef CAS.
- T. Yu, M. D. Marquez and T. R. Lee, Langmuir, 2022, 38, 13488–13496 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
-
S. M. Sze, Semiconductor Devices: Physics and Technology, Wiley, Hoboken, New Jersey, 2nd edn, 2001 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian Inc., Pittsburgh, PA, 2009 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 Search PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 Search PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CAS.
- T. Uemura, Y. Hirose, M. Uno, K. Takimiya and J. Takeya, Appl. Phys. Express, 2009, 2, 111501 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CAS.
- M. Nakano, D. Hashizume and K. Takimiya, Molecules, 2016, 21, 981 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Takeru
Koguma
b,
Yutaro
Arai
c,
Go
Watanabe
*ab,
Takeru
Koguma
b,
Yutaro
Arai
c,
Go
Watanabe