
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small molecule drug discovery for Ebola virus disease
Destiny
Durante
 a,
Venkatesh
Murugesh
a,
Tyler
Kalanquin
a,
Irina N.
Gaisina
ab,
Lijun
Rong
c and
Terry W.
Moore
*ad
a,
Venkatesh
Murugesh
a,
Tyler
Kalanquin
a,
Irina N.
Gaisina
ab,
Lijun
Rong
c and
Terry W.
Moore
*ad
aDepartment of Pharmaceutical Sciences, Retzky College of Pharmacy, University of Illinois Chicago, IL 60612, USA. E-mail: twmoore@uic.edu
bUICENTRE for Drug Discovery, University of Illinois Chicago, IL 60612, USA
cDepartment of Microbiology and Immunology, College of Medicine, University of Illinois Chicago, IL 60612, USA
dUniversity of Illinois Cancer Center, University of Illinois Chicago, IL 60612, USA
First published on 6th August 2025
Abstract
Known for its widespread outbreaks, including the 2013–2016 epidemic that infected almost 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 individuals and resulted in approximately 11300 deaths, Ebola virus (EBOV) and related filoviruses remain a current threat as consecutive filoviral outbreaks have occurred between 2021 through 2025. Due to high fatality rates of 40–90% among infected individuals, researchers have invested significant efforts to discover effective treatments for Ebola virus disease. Small molecules hold great potential for treating Ebola virus disease because they can target various stages of the filoviral life cycle, such as entry, transcription, replication, and egress; however, the FDA has not yet approved any small molecule treatments for EBOV. In this review, we report both historic and recent progress in the discovery of small molecule drugs for EBOV.
000 individuals and resulted in approximately 11300 deaths, Ebola virus (EBOV) and related filoviruses remain a current threat as consecutive filoviral outbreaks have occurred between 2021 through 2025. Due to high fatality rates of 40–90% among infected individuals, researchers have invested significant efforts to discover effective treatments for Ebola virus disease. Small molecules hold great potential for treating Ebola virus disease because they can target various stages of the filoviral life cycle, such as entry, transcription, replication, and egress; however, the FDA has not yet approved any small molecule treatments for EBOV. In this review, we report both historic and recent progress in the discovery of small molecule drugs for EBOV.
EBOV background
EBOV taxonomy, disease, and recent filoviral outbreaks
Since its 1976 discovery, Ebola virus (EBOV) has commanded worldwide attention as a pathogen of significant concern. As a member of the Filoviridae family and Orthoebolavirus genus, EBOV is a zoonotic RNA virus.1 The likely reservoir host is the fruit bat, which infects other animals including non-human primates.2 EBOV transmits from these animals to humans, and contact with infected bodily fluids, like saliva, sweat, blood, and semen, causes human-to-human infection.3 Other Orthoebolavirus species that are noted to be human-infectious include Sudan virus (SUDV), Bundibugyo virus (BDBV), and Taï Forest virus (TAFV). Reston virus (RESTV) infections have been reported without evidence of disease progression, while Bombali virus (BOMV) lacks documented cases of human infection.4,5After successful viral transmission, EBOV infection rapidly leads to Ebola virus disease (EVD). EBOV targets a diverse range of host cells,6–8 primarily dendritic, monocyte, and macrophage cells. Immune cell susceptibility disrupts host immune responses by MAPK inhibition and cytokine production.9,10 Endothelial cells are targeted during the late stages of EBOV infection, which causes vascular leakage and hemorrhaging due to reduced levels of blood coagulation factors.6,11,12 As a result, EVD rapidly progresses from mild viral symptoms like fever, fatigue, muscle pain, and sore throat in days 4–7, to more advanced symptoms like internal and external bleeding and organ failure in the latter stages of infection.13–16 The quick onset of severe symptoms associated with EVD often results in high fatality rates, averaging around 40%.17,18
Isolated cases of infection have historically triggered extensive outbreaks, with most taking place in Central and Western African countries.17,19 The 2013–2016 EBOV epidemic was the largest and most fatal outbreak, with nearly 29000 reported cases and approximately 11300 deaths. The main countries impacted by the epidemic included Guinea, Sierra Leone, and Liberia; however, cases were also reported in additional countries like Nigeria, Spain, the United Kingdom, and the United States. The second largest EBOV outbreak occurred shortly after the epidemic, mainly impacting the Democratic Republic of Congo during 2018–2020. Subsequently, there have been several consecutive outbreaks, including those from EBOV in Uganda during 2021; SUDV in Guinea during 2022; Marburg virus (MARV), a related filovirus, in Guinea during 2023; MARV in Rwanda during 2024; and the most recent 2025 SUDV outbreak in Uganda that ended in April. Increased EBOV and related viral outbreaks in recent consecutive years stresses the need for effective filoviral therapeutics.
EBOV biology
EBOV is an enveloped virus containing a negative-sense, single-stranded RNA genome that is approximately 19 kilobases long and encodes seven genes: the glycoprotein (GP), nucleoprotein (NP), VP24, VP30, VP35, VP40, and RNA polymerase (L) (Fig. 1).20 GP is expressed on the viral surface and is an essential protein for mediating viral entry into the host cell. As a homotrimer, each GP monomer is composed of a GP1 and GP2 subunit. GP1 determines host tropism and facilitates viral-cellular attachment by promiscuously binding to various host-cell receptors including β1 integrins, C-type lectins, T-cell immunoglobulin and mucin domain 1, glycosaminoglycans, and tyrosine kinase receptors (Fig. 2).21–25 EBOV uses the heavily glycosylated GP1 subunit to bind to receptors that recognize N- and O-linked oligosaccharides. Once bound to the cell, EBOV is macropinocytosed at the surface and trafficked to the endosome.26 As the endosome progresses toward maturity, the vesicles increase in acidity, which activates low pH-induced cysteine proteases Cathepsin L and B that proteolytically cleave the mucin-like domain and glycan cap from GP1.27 This cleavage reveals the receptor-binding site (RBS) within GP and enables endosomal receptor Niemann–Pick C1 (NPC1) to bind at this region.28 This major binding event, in addition to GP interactions with Ca2+ and two-pore channels (TPCs), triggers fusion, where GP undergoes conformational changes that are mediated by GP2.29,30 During this transformation, the newly folded GP creates a pore within the host endosomal membrane, which allows for the release of the viral ribonucleoprotein complex into the host cytoplasm for viral genome replication, protein synthesis, and the production of viral progeny. | ||
| Fig. 1 Illustration of Ebola virus (EBOV) and viral proteins GP, L, NP, VP24, VP40, VP35, and VP30. | ||
 | ||
| Fig. 2 Schematic overview of Ebola virus (EBOV) life cycle that includes cell attachment, macropinocytosis, proteolytic cleavage, endosomal receptor binding, fusion, transcription, replication, translation, packaging, and release. GP: glycoprotein; NPC1: Niemann–Pick disease type C1; TPC: two-pore channel. | ||
Proteins VP30, VP35, and L form a complex with NP to complete viral transcription and replication. VP30 is an innately phosphorylated transcriptional activator. To initiate transcription, VP30 must be dephosphorylated by host phosphatases;31,32 however, replication can still occur while VP30 is in the phosphorylated state. L is activated by host factors like DNA topoisomerase and heat shock proteins for polymerase activity.33,34 VP35 protects viral dsRNA by capping the ends, preventing recognition and degradation by host cell RIG-I-helicase.35–37 Sufficient numbers of RNA transcripts and replicates lead to the formation of inclusion bodies within the host cytoplasm that are enriched with the viral genome and proteins.38
Both VP35 and VP24 provide viral protection from host factors by antagonizing host interferon (IFN) responses.38 VP35 inhibits host dsRNA sensing, IFN gene expression, and IFN activity.39 MAPK and NF-κB pathway activation are inhibited by VP24.40 In addition, VP24 also assists VP40 in the packaging of the newly synthesized viral genome and proteins. VP40 further regulates the intracellular transport of packaged viral components to the inner leaflet of the plasma membrane for viral egress and the release of new progeny.41,42
In vitro systems to study EBOV and identify inhibitors
High pathogenicity and lethality of EBOV require viral containment in biosafety level 4 (BSL 4) facilities. Heavy restrictions on infectious EBOV can limit the study of filoviruses; however, surrogate systems or pseudotyped viruses are often used to study filoviral entry mechanisms. The pseudotyped viral system uses a glycoprotein of interest and an engineered viral vector that acts as the surrogate. In the case of EBOV study, human immunodeficiency virus (HIV) and vesicular stomatitis virus (VSV) are common surrogates that can incorporate EBOV GP on the viral surface, which allows the pseudovirus to maintain filoviral GP-dependent entry mechanisms.43 During plasmid generation, select coding regions of the surrogate viral genome are deleted so that produced viruses are defective in viral replication,43 while addition of reporter genes allows for the visualization and quantification of pseudoviral entry. For pseudoviral production, producer cells are transfected with plasmids containing the recombinant surrogate viral genome and filoviral GP.44Pseudotyped systems are great models for filoviral entry; however, the inability of the pseudovirus to replicate confines their study to the entry process only, as seen with the HIV pseudovirus (HIV-EBOV GP). Unlike HIV, VSV is not pathogenic to humans; therefore, plasmids containing the full-length VSV genome are used, replacing VSV G with EBOV GP.45 The recombinant vectors are transfected in producer cells to generate replication-competent VSV pseudovirus (rVSV-EBOV GP) that has proven useful in evaluating drug resistance via escape mutants for EBOV.46–48 The challenge of viral similarity still remains as HIV and VSV are morphologically different from filamentous EBOV, and these differences have been shown to impact viral infectivity.49 VSV can also enter at the cell surface and bypass trafficking to the endosome, which is an essential step in filoviral entry.
Alternatively to the pseudovirus system, researchers found that expression of VP40 alone in transfected cells leads to the formation of filamentous EBOV virus-like particles (eVLP).50,51 EBOV VLP formation is solely based on VP40's ability to mediate viral budding in the host cell. Expression of both EBOV VP40 and GP generates eVLPs that model filoviral entry. Further incorporation of a mini-genome has proven useful, as mini-genomes are complementary DNA (cDNA) constructs of full-length or truncated viral RNA genomes. The cDNA is designed to encode a promoter, reporter gene, and viral 3′ and 5′ untranslated regions needed for viral transcription, replication, and packaging.52,53 Use of the mini-genome system that includes EBOV VP40 and GP genes generates transcription- and replication-competent virus-like particles (tr-VLP). Use of tr-VLPs thereby enables the study of all aspects of the EBOV life cycle and provides a screening method to assess therapeutic agents that target filoviral entry, replication, transcription, and egress.52,54
Current EBOV therapeutic agents and value of small molecule treatments
Efforts for EBOV drug development have included siRNA therapeutics, ion channel inhibitors, combination therapies, peptides, antibodies, and small molecules.55–64 In 2019, the FDA approved the prophylactic agent, Ervebo, as it was shown to protect individuals, who were previously exposed to EBOV, from EVD.65 Although effective, Ervebo lacks cross-protective efficacy and is only approved for use against one orthoebolavirus species, Zaire. Furthermore, the EBOV vaccine is only indicated for one-time use, which exemplifies the need for available therapeutics in the event of future EBOV exposure.In addition to the vaccine, there are two FDA-approved monoclonal antibody treatments for EBOV, Inmazeb and Ebanga.66 Both monoclonal antibody treatments target early steps of EBOV entry by neutralizing GP and recruiting immune cells to sites of infection. Due to their ease of synthesis, transport, and storage, small molecules are advantageous compared to antibody therapeutics because they can target several steps throughout the filoviral life cycle including entry, transcription, replication, and budding. In this review, we discuss recent advances of small-molecule therapeutic agents against EBOV to summarize and inspire innovation of novel antifiloviral treatments.
Entry inhibitors
Cell attachment
The first step in EBOV entry is host-cell attachment. Instead of targeting a specific receptor, EBOV promiscuously binds to various host attachment factors including β1 integrins, C-type lectins, T-cell immunoglobulin and mucin domain 1, and Tyrosine kinase receptors.21–24 EBOV uses the heavily glycosylated GP1 subunit to bind to receptors that recognize N- and O-linked oligosaccharides.Heparan sulfate and heparin are cell-surface glycosaminoglycans composed of repeating glucosamine and uronic acid disaccharide units. They are responsible for regulating a range of biological activity, including filoviral/host attachment.67 Extosin 1, a host glycotransferase involved in the biosynthesis of heparan sulfate, was identified as a host factor involved in filoviral entry through the screening of genome-wide RNAi's.25 ELISA was used to demonstrate binding of heparan sulfate and heparin to EBOV GP. Pseudotyped and infectious EBOV entry were inhibited in A549 and human pulmonary artery endothelial cells upon treatment with various glycosaminoglycans. More recently, heparan sulfate was also shown to mediate EBOV infection in Caco-2 cells.68 These results indicate the usefulness of inhibiting viral-host attachment as a therapeutic mechanism.
In the case of C-type lectins, DC-SIGN69 is a known EBOV attachment factor that is specific to dendritic cells. DC-SIGN is a transmembrane receptor with four subunits that each contain a carbohydrate recognition domain (CRD). Thorough DC-SIGN investigation led to the development of multivalent glycoconjugate systems that bind to each CRD with high affinity. In the case of calix[4]arene glycoconjugates, researchers linked α-L-fucose or α-D-mannose with hydroxamic acid or pseudopeptide groups to a calixarene scaffold (Table 1).70 Binding of glycoconjugates1 and 2 to DC-SIGN's extracellular domain was confirmed via SPR, which assisted the ability to inhibit pseudotyped EBOV infection in Jurkat cells. α-L-fucose was the preferred binder, demonstrated by the EC50 of 289 nM for glycoconjugate1, compared to 634 nM for glycoconjugate2. Although these glycoconjugates are water-soluble, previous studies identified cytotoxicity as a major shortcoming of multivalent systems, as they have been shown to accumulate in cellular compartments and cause adverse effects.71 To address this, poly-L-lysine multivalent glycoconjugates that coupled D-mannose residues to lysine linkers were developed.72 In a flow cytometry experiment, glycoconjugate1d inhibited EBOV GP binding to B-THP cells expressing DC-SIGN at 0.198 nM. By labeling the active poly-L-lysine glycoconjugates with the pH-sensitive fluorescent dye rhodamine, researchers could visualize the presence of these active inhibitors in acidic compartments, which suggests that glycoconjugate binding reduces the presence of DC-SIGN on the host surface, further inhibiting attachment of EBOV. The fluorescent glycoconjugates were also cleared from the cells in a time-dependent manner within 24 hours, improving the potential cytotoxic effects.
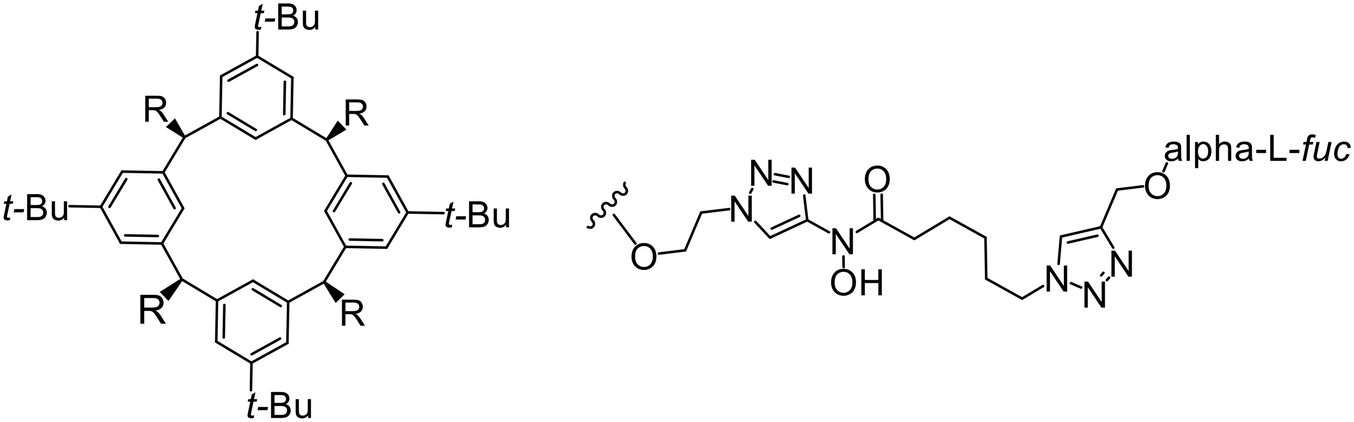
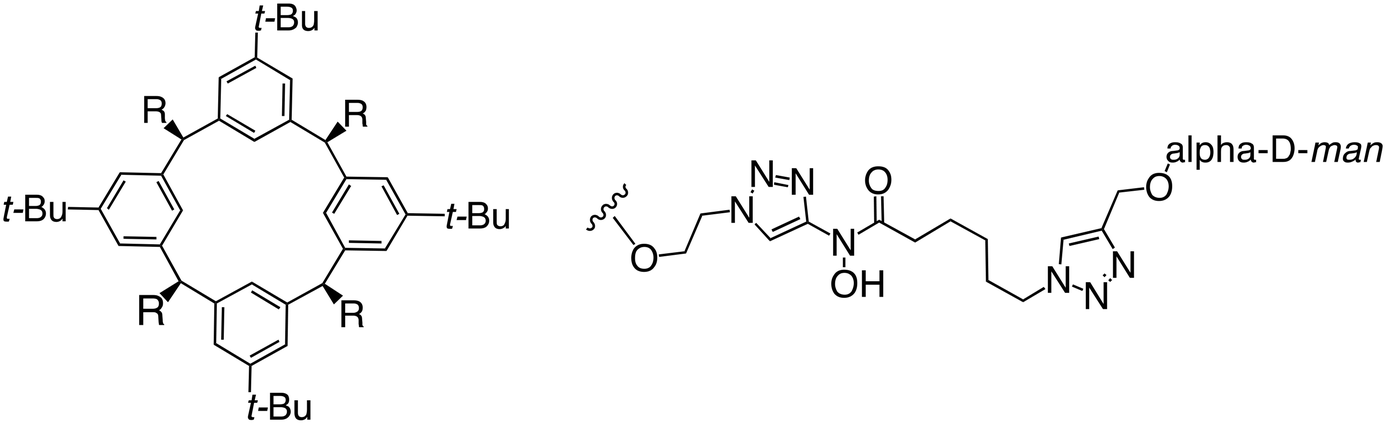
Because EBOV relies on promiscuous binding to glycan-recognizing surface receptors for attachment, small molecules that target host factors at the EBOV attachment step will also target other pathogens that rely on similar surface-cell receptors, including HIV, SARS-CoV-2, and some bacteria. Therefore, developing EBOV/cell attachment inhibitors could be useful for broad-spectrum anti-infective or combination therapeutics.
Macropinocytosis
To determine the full filoviral entry mechanism, researchers have studied how EBOV enters the host cell. EBOV does not use clathrin-, caveolae-, or dynamin-dependent uptake. Instead, EBOV is macropinocytosed into the host cell and trafficked to the endososomal pathway. This mechanism was elucidated in part by use of ethylisopropylamiloride (EIPA), a known macropinocytosis inhibitor, to reduce infectious EBOV entry in Vero cells (Table 2).26 Use of infectious virus was essential for this early discovery stage to ensure clinical relevance. Similar results were recapitulated with EIPA's dose-dependent inhibition of VSV-EBOV GP and VLPs in Vero cells. Other macropinocytosis inhibitors, including LatA, an actin polymerization inhibitor; Rottlerin, a protein kinase C (PKC) inhibitor; and ML9, a light chain kinase inhibitor, also blocked VSV-EBOV GP infection in Vero cells, dendritic cells, and peripheral blood-derived monocytes.73
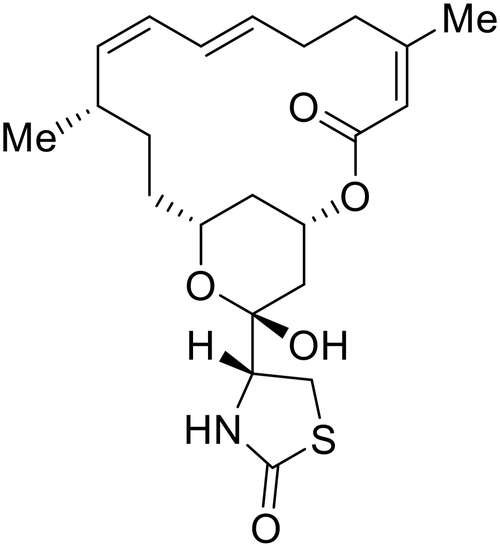
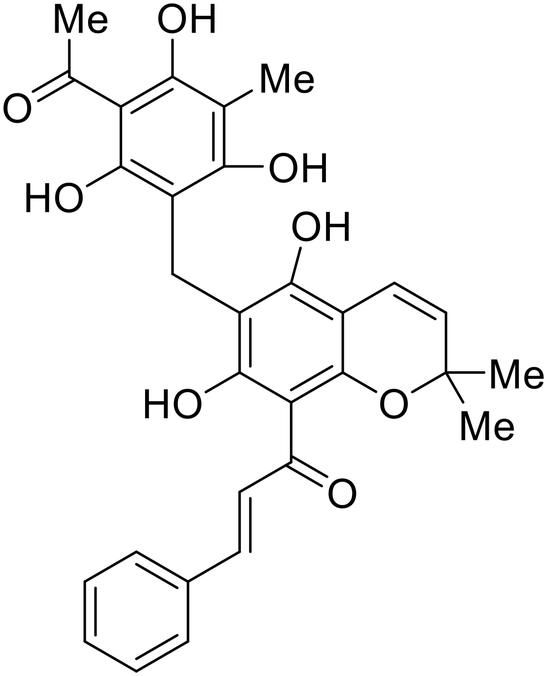
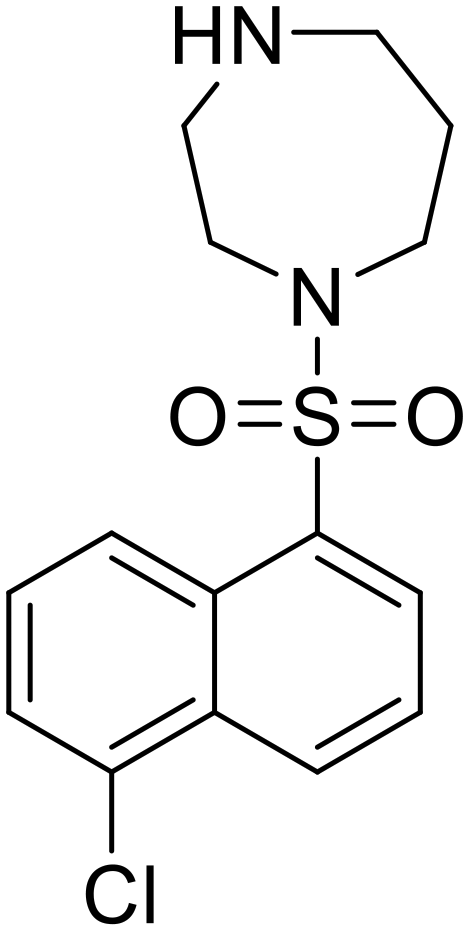
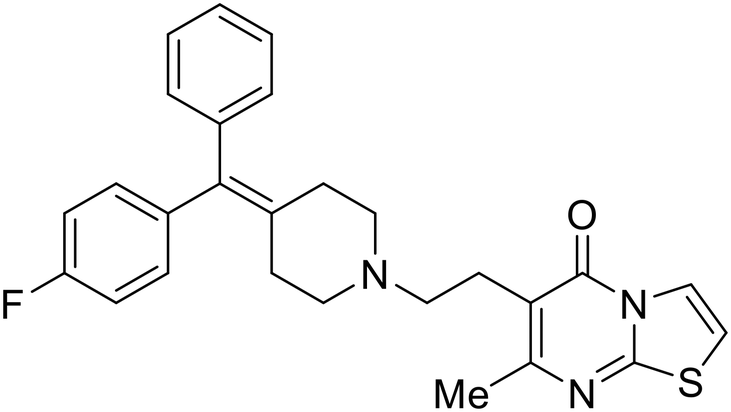
More recent studies discovered diacyl glycerol kinase (DGK) antagonist R-59-022 as a filoviral entry inhibitor.74R-59-022 reduced pseudotyped EBOV entry in Vero cells, as well as VLP entry in Vero and bone marrow-derived macrophages. Time-of-addition studies with R-59-022 showcased EBOV inhibition at an earlier time point compared to NH4Cl, an endolysosomal pH neutralizer that prevents pH-dependent EBOV GP proteolysis. Complete viral entry inhibition within one hour supports use of R-59-022 as a macropinocytosis inhibitor for filoviral entry. R-59-022 is more potent (5 μM) than EIPA (30 μM) and provides a useful starting point and scaffold to develop more potent macropinocytosis inhibitors for EBOV.
Proteolytic cleavage
EBOV GP1 contains a mucin-like domain and glycan cap. Chandran et al. found that low pH-dependent proteases Cathepsin L (CatL) and Cathepsin B (CatB) were required for entry, as these proteases remove the GP1 glycan cap that allows for GP-receptor binding in the later steps of entry.27 For this discovery, CatB inhibitor CA074 and CatB/L inhibitor FYdmk (Table 3) were found to dose-dependently interrupt VSV-EBOV GPΔMuc and infectious EBOV entry in Vero cells. Derivative CA074Me was also effective in blocking VSV-EBOV GP and HIV-EBOV GP entry.75,76 Although these studies provided proof of concept for filoviral therapeutic development, CA074 and its derivatives are non-ideal clinical candidates, as EBOV resistance arose within two VSV-EBOV GPΔMuc passages in CA074-treated Vero cells.77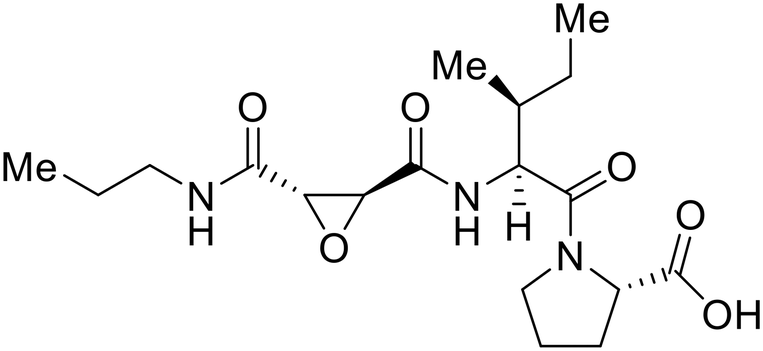
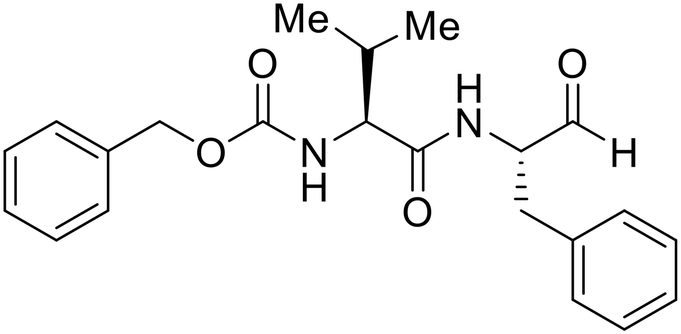
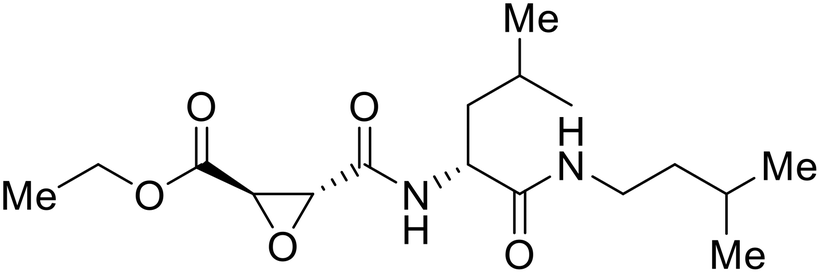
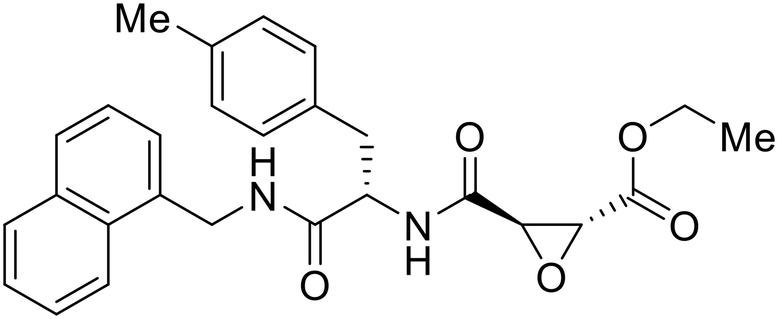
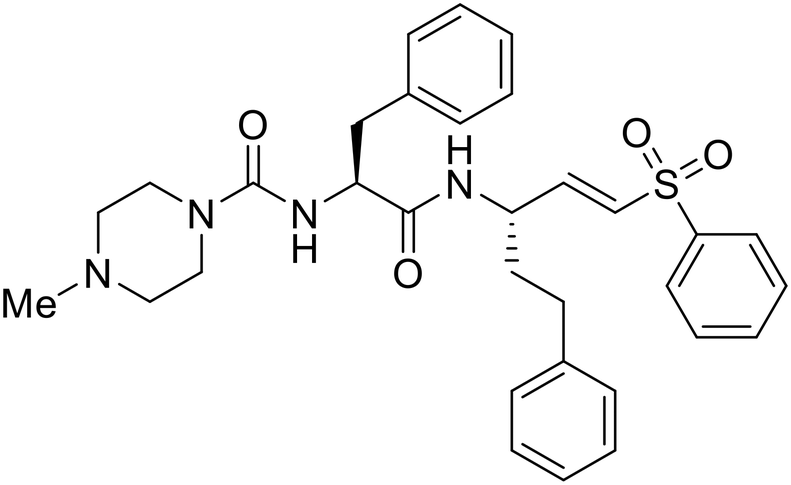
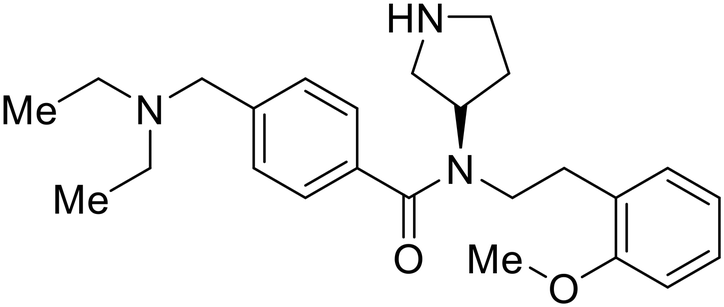
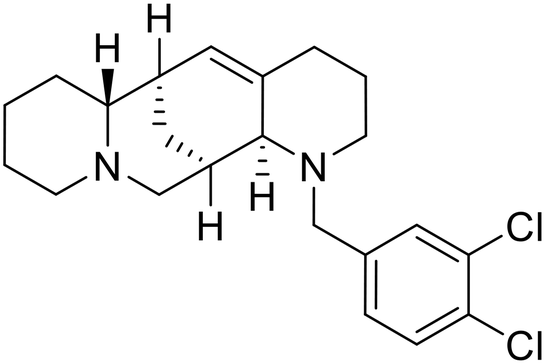
A similar trajectory occurred for MDL28170, a cysteine protease inhibitor.78 Cellular pretreatment with MDL28170 at 0.5 and 10 μM effectively inhibited HIV-EBOV GP entry in 293 T cells;76 however, later studies determined its resistance using replication-competent VSV-EBOV GP.79 In a total of five passages, V37A and S195R mutations developed. GP1 mutation V37A was more aggressive as it occurred within the first passage and contributed more to EBOV resistance to protease inhibitors.
Broad cysteine protease inhibitor E64D has also effectively attenuated EBOV entry;27,76 however, poor permeability of this epoxide-based inhibitor required high doses of up to 300 μM to exert pharmacological effect. Maintaining the use of epoxide-based small molecules, AMS36 was used as a scaffold to develop new cathepsin inhibitors for reduced EBOV entry.80 Various amines containing an aliphatic or aromatic group were coupled to the AMS36 scaffold, while the epoxide stereochemistry was varied. Inhibitors containing an R,R-epoxide motif and a basic amine were most potent, as basic functionality is expected to assist in directing the therapeutic agents to the endolysosomal sites of filoviral entry. Rounds of SAR development of the potent derivative R11Et generated R11P that replaced R11Et's labile ethyl ester with a propylamide. The amide modification improved serum stability and achieved nanomolar entry inhibition against VSV-EBOV GP (EC50 = 1.2 nM) and infectious EBOV (EC50 = 70 nM) in U2OS cells.
SAR development for cysteine protease antagonist K11777 helped establish a pan-filoviral entry inhibitor.81K11777 with sub-nanomolar activity against HIV-EBOV Zaire GP (EC50 = 0.87 nM) displayed additional nanomolar inhibition against pseudotyped SUDV (EC50 = 1.14 nM), TAFV (EC50 = 2.26 nM), RESTV (EC50 = 3.37 nM), BDBV (EC50 = 5.91 nM), and MARV (EC50 = 1.90 nM). Replacement of the 1-methyl piperazine with a 1-cyclopropylmethyl, t-Bu, or ethyl piperazine improved Zaire activity 8.7, 7.9, and 7.3-fold, respectively.
Cysteine protease inhibitor (Z-LL)2-ketone inactivated both CatB and CatL as expected; however, serine protease inhibitor PF429242 was surprisingly active against cysteine protease CatB.82 Instead of inhibiting protease activity, PF429242 blocked CatB-endolysosomal localization for inactivation. Both (Z-LL)2-ketone and PF429242 caused dose-dependent entry inhibition of VSV-EBOV GP at early time points, with additional activity against pseudotyped SUDV, TAFV, BDBV, and MARV.
Natural products have also served as starting points for therapeutics targeting the GP proteolytic step. Aloperine, extracted from the seeds and leaves of Chinese plant Sophora alopecuroides L.,83 displayed activity against HIV-EBOV GP in HEK-293 T cells.84 SAR exploration via N-alkylation, −acylation, and -sulfonylation generated derivative 2e containing an N-coupled 3′,4′-dichlorophenyl group. EBOV entry inhibition of 2e improved 2.6-fold (EC50 = 4.8 μM) compared to aloperine and was effective in reducing the presence of HIV-EBOV GP virus in treated BALB/c mice compared to untreated mice. These in vivo studies exemplify the use of protease inhibitors in more complex systems; however, further studies using clinically relevant infectious virus are needed.
NPC1 binding
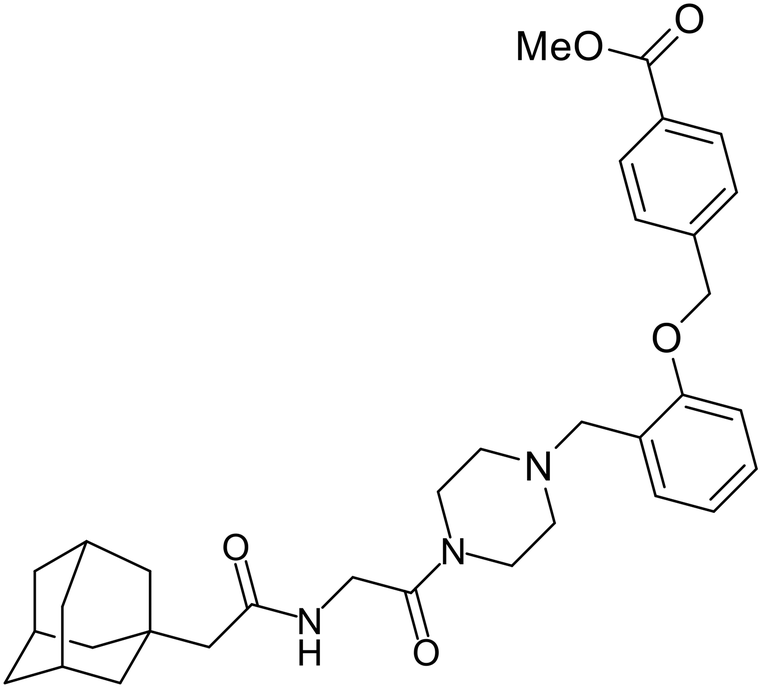
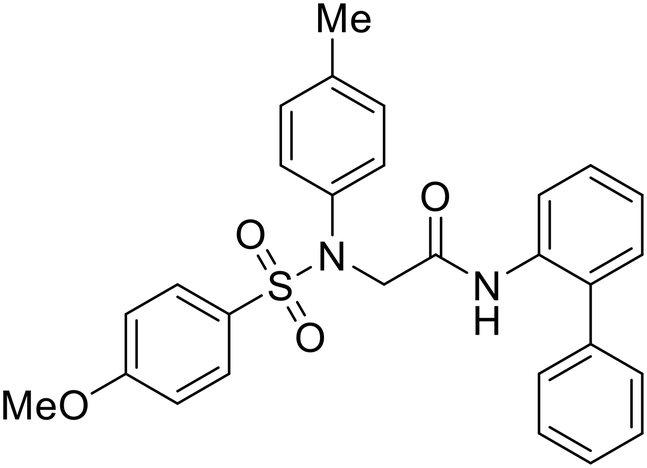
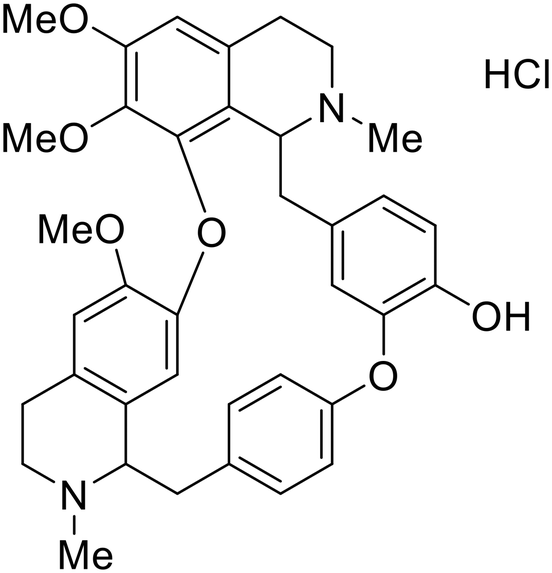
In 2011, researchers found that proteolysis mediated by pH-dependent cathepsins reveals the receptor binding site (RBS) in GP1 that enables the GP-NPC1 binding event required for EBOV entry.85 The screening of a small-molecule library against VSV-EBOV GP identified benzylpiperazine adamantane 3.0 as an entry inhibitor. Initial SAR development generated derivative 3.47 with sub-micromolar EBOV inhibition (Table 4). Both compounds induced cholesterol-accumulation in endolysosomal compartments, helping to identify NPC1 as their target. Immunoprecipitation assays revealed binding between proteolytically cleaved GP1 and NPC1, which was inhibited by dose-dependent administration of 3.0 and 3.47. Though potent, 3.47 contains an adamantane group contributing to an increased logP (7.2) and a labile methyl ester that compromises metabolic stability. Replacement of the adamantane with a difluoro spiro[2.5]octane, and methyl ester with a methyl sulfone generated derivatives 3–22 and 3–25 that improved infectious EBOV entry inhibition from 64 nM, to 19 and 21 nM, respectively.86
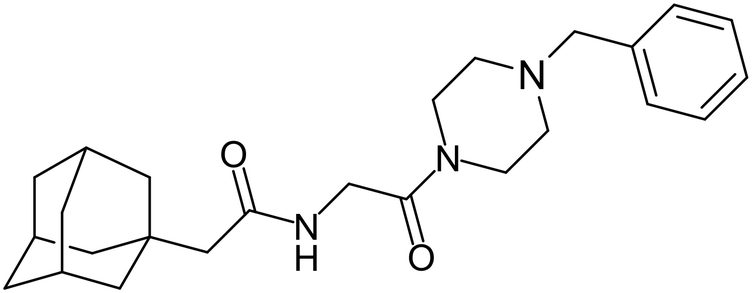
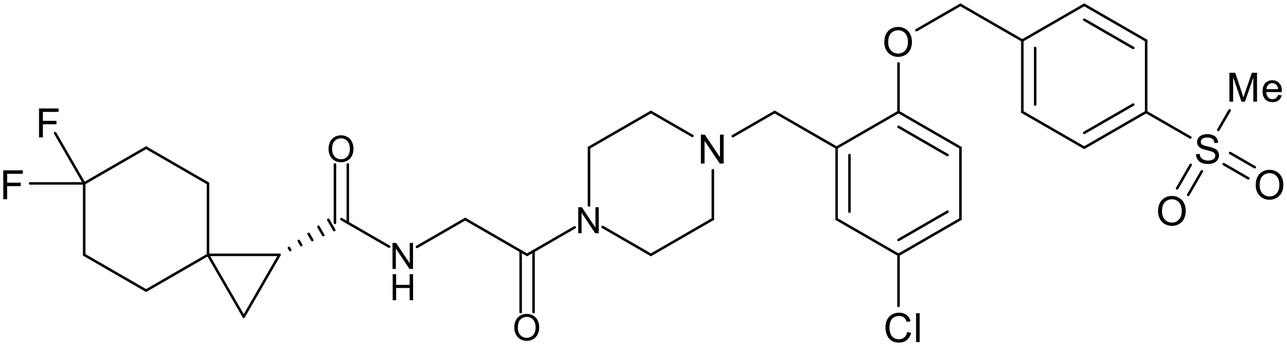
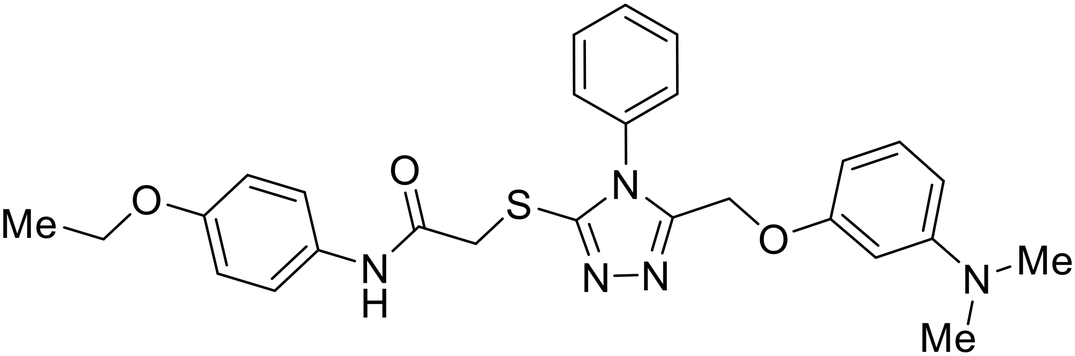
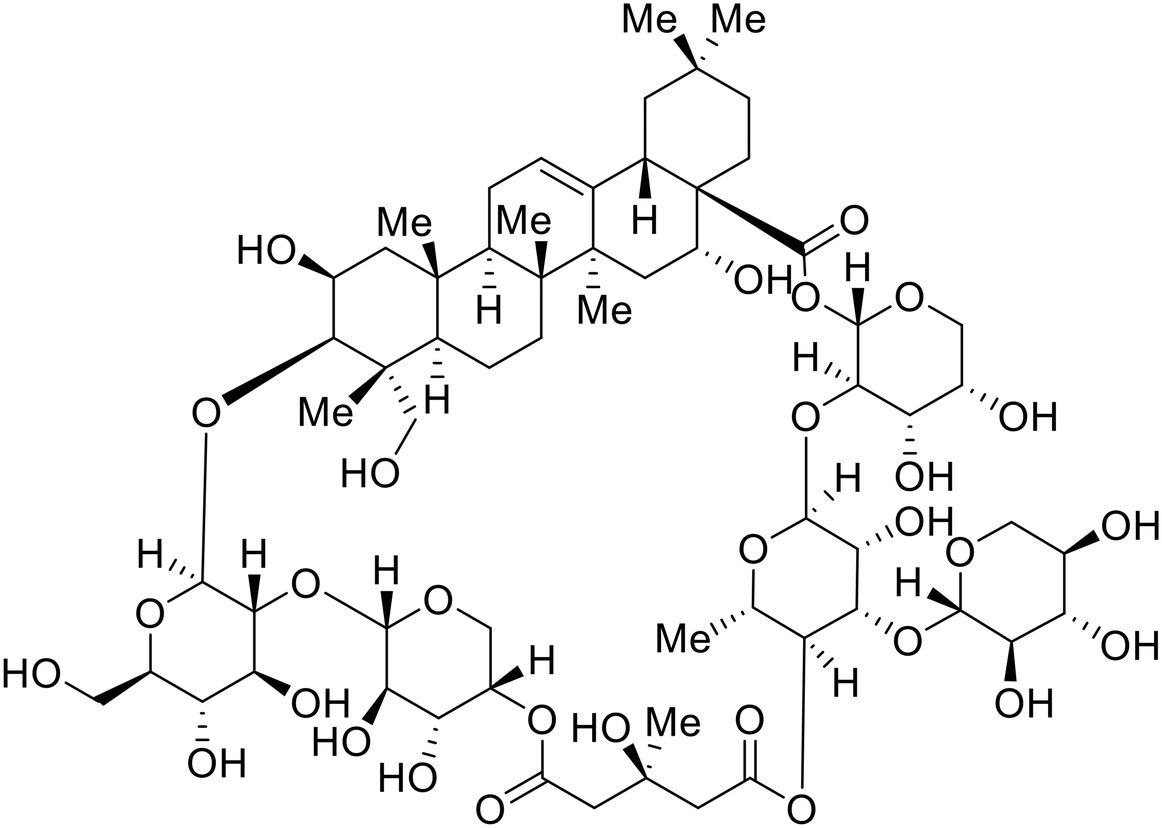
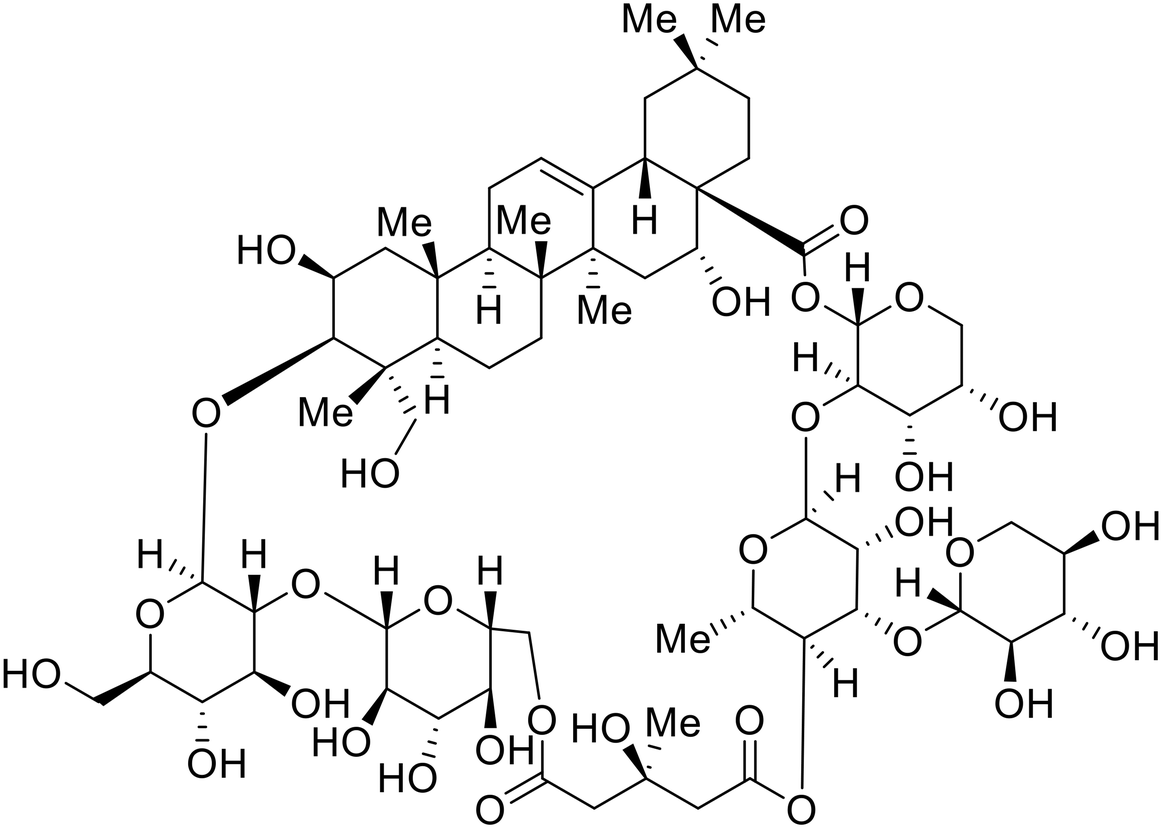
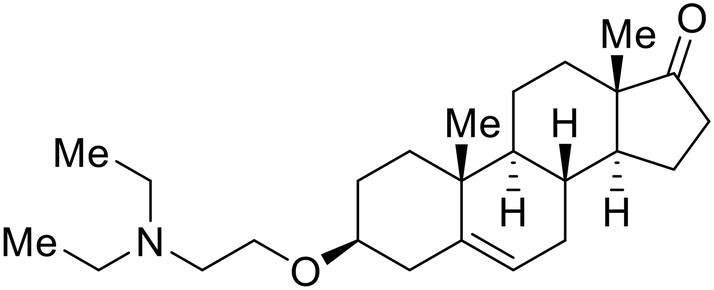
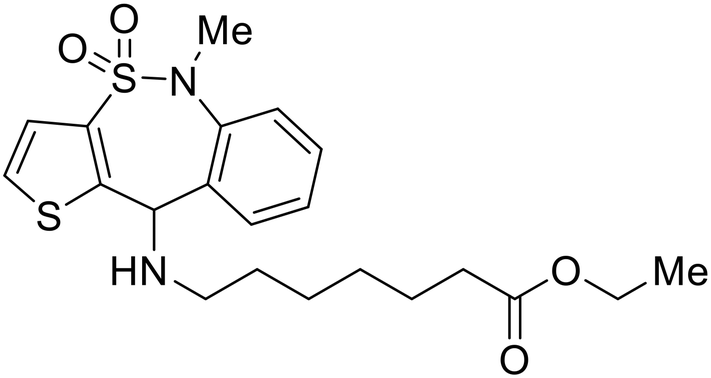
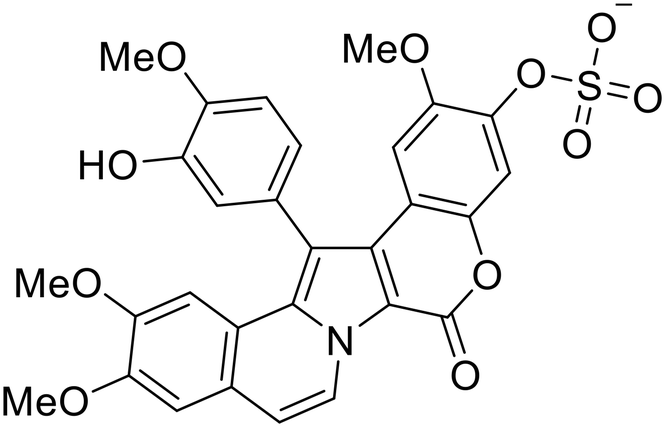
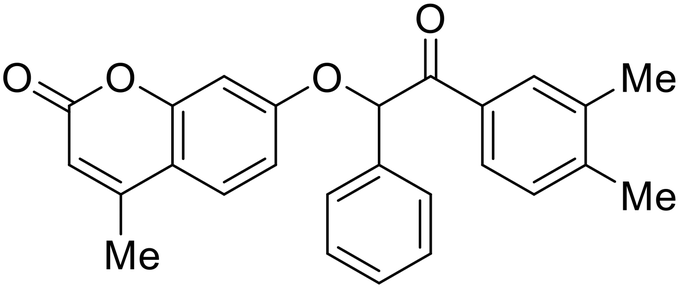
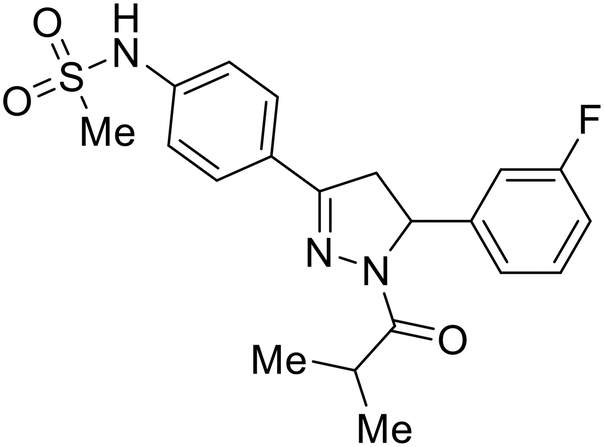
Identification of NPC1 as an integral protein in EBOV entry led to the pursuit of small molecules that inhibit this essential binding interaction. A small-molecule high-throughput screen identified MBX2270 and MBX2254 as HIV-EBOV GP entry inhibitors.87 Endolysosomal pH was not altered by MBX2270 nor MBX2254; however, cholesterol accumulation was induced in A549 cells. Both MBX2270 and MBX2254 additionally blocked the NPC1-GP1 binding event as determined by an AlphaLISA experiment. Potent tubeimosides I, II, and III derived from Bolbostemma paniculatum inhibited HIV-EBOV GP entry in Vero and SNB-19 cells, and EBOV transcription and replication-competent VLP's in HEK-293 T cells (EC50 < 200 nM).88 Docking models suggest that tubeimosides I, II, and III block NPC1's loop 1 from protruding into the EBOV RBS, disrupting the NPC1-EBOV GP1 binding interaction.
Known NPC1 binder itraconazole89 demonstrated infectious EBOV entry inhibition in MoKi, Vero E6, and A549 cells.90 Bio-layer interferometry (BLI) and pull-down assays determined Berberis amurensis natural product berbamine hydrochloride as a thermolysin-treated GP1 binder.91 When administered to BALB/c mice either 1 day pre- or post- mouse-adapted EBOV infection, berbamine hydrochloride treatment resulted in 100% and 83% survival rates, respectively, with 0% survival among the control-treated animals.
U18666A 92,93 was explored due its ability to disrupt endosomal cholesterol export by binding NPC1 at the sterol-sensing domain.94 Both VLP and infectious EBOV entry (EC50 = 8.05 μM) were inhibited by its dose-dependent administration of U18666A in Vero cells. Similarly, benzothiazepine compound9 also inhibits EBOV entry, but lacks evidence for EBOV GP or NPC1 binding.95 Additional studies are needed to elucidate the exact mechanism of action of compound9, although endolysosomal cholesterol accumulation is thought to be a contributing factor.
Virtual screening of suggested NPC1/EBOV GP-targeting inhibitors has proven to be a useful tool in the identification of small molecules with more clearly defined mechanisms of entry inhibition. Natural product lamellarin α sulfate, a marine alkaloid with broad-antiviral activity, decreased HIV-EBOV GP infection in HEK-293 T cells in a dose-dependent manner, albeit at high concentrations (50, 100, and 150 μM).96 Increased doses are needed for this natural product due to its membrane impermeability, attributed to the negatively charged sulfate. Additionally, A (chrome-2-one-based) and U (pyrazole-based) compounds were identified as potential EBOV GP1 binders at the RBS region.97 Moderate HIV-EBOV GP entry inhibition for the initial A and U compounds, 11.9 and 8.77 μM respectively, led to the SAR exploration of each. Unfortunately, chemical modifications to each scaffold either compromised cytotoxicity or lacked potency improvements. Additional SAR developments of lamellarin α sulfate, A, and U compounds are needed, along with binding confirmation to their indicated targets.
Fusion
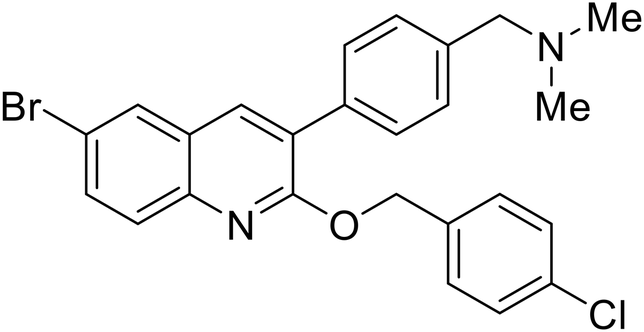
During the early stages of the largest EBOV epidemic in 2013, the need for effective EBOV therapeutics was re-emphasized. An FDA-approved small molecule library was screened against EBOV to identify drugs with repurposing potential.98 Many of the drugs screened were selective estrogen receptor modulators (SERMs), with toremifene (EC50 = 0.162 μM) and clomiphene (EC50 = 2.42 μM) being the most potent against infectious EBOV in Vero cells (Table 5). These SERMs were of interest because they maintained their antifiloviral activity despite the lack of cellular ERα presence, which suggested a distinct antiviral mechanism. Continued studies led to the co-crystallization of toremifene with EBOV GP, identifying the internal fusion loop region as a novel binding site for small molecules.99 Displacement of the GP1 DFF lid by toremifene binding was proposed to destabilize the GP conformation needed for fusion to occur. Reduction of the GP melting temperature in the presence of toremifene also supports this notion. Strong antifiloviral activity of SERMs led to further SERM exploration for EBOV entry inhibitors. The screening of ER ligands identified ridaifen-B as a candidate.100 Rounds of SAR and reverse engineering were used to optimize filoviral potency and reduce ER activity of ridaifen-B. Phenol replacement with an ethyl-linked pyrrolidine (analog 30) improved infectious EBOV (EC50 = 0.64 μM) and MARV entry inhibition, and reduced ER activation compared to ridaifen-B. Future SAR studies of ridaifen-B are needed to improve the cytotoxicity and further reduce ER activation.| Name | Structure | Ref |
|---|---|---|
| Toremifene |
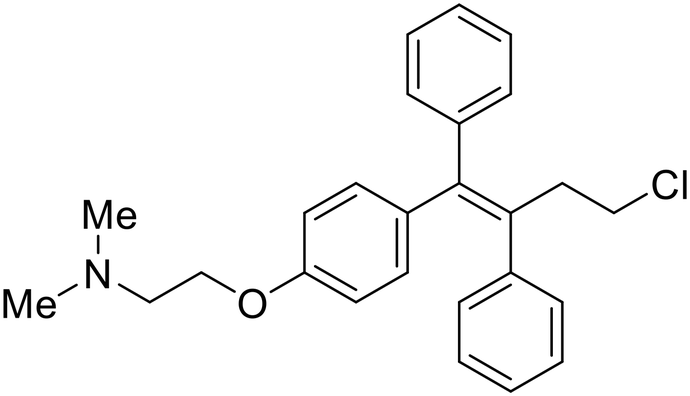
|
98, 99 |
| Clomiphene |
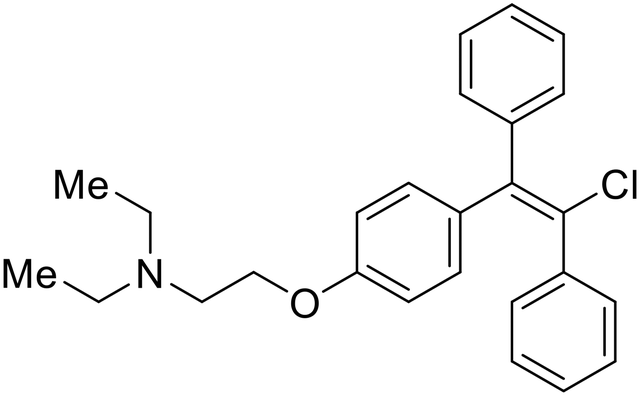
|
98 |
| Ridaifen-B |
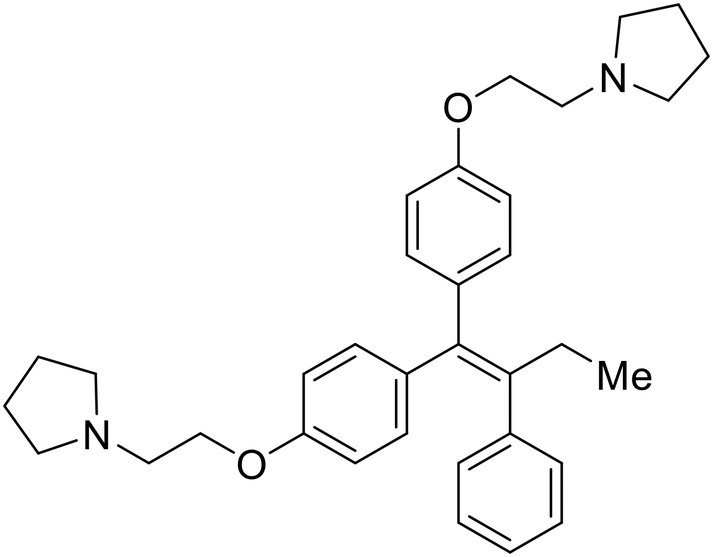
|
100 |
| 30 |
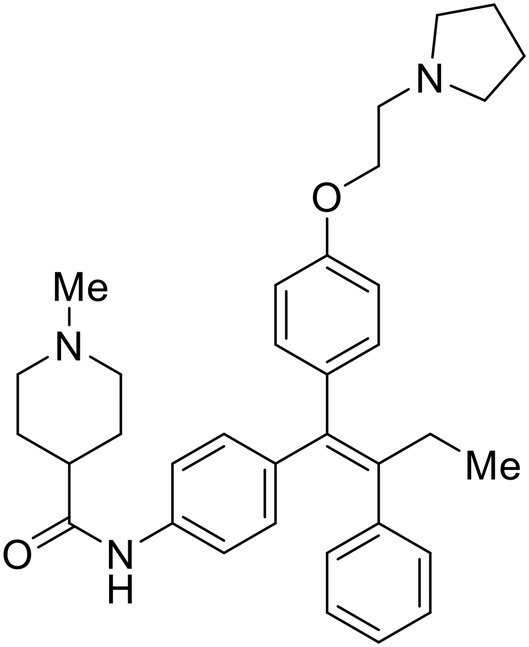
|
100 |
| Imipramine |
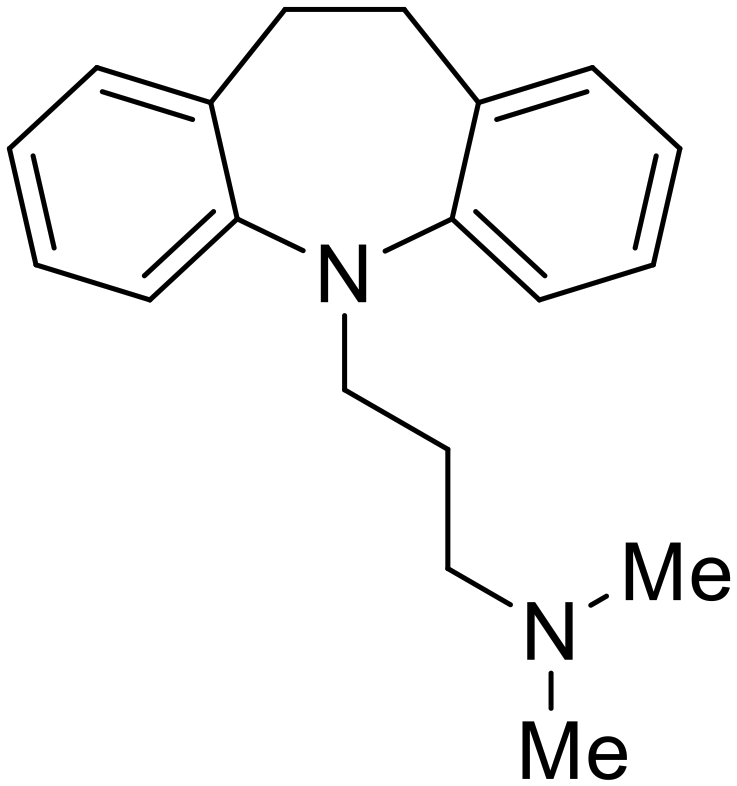
|
101 |
| Clomipramine |
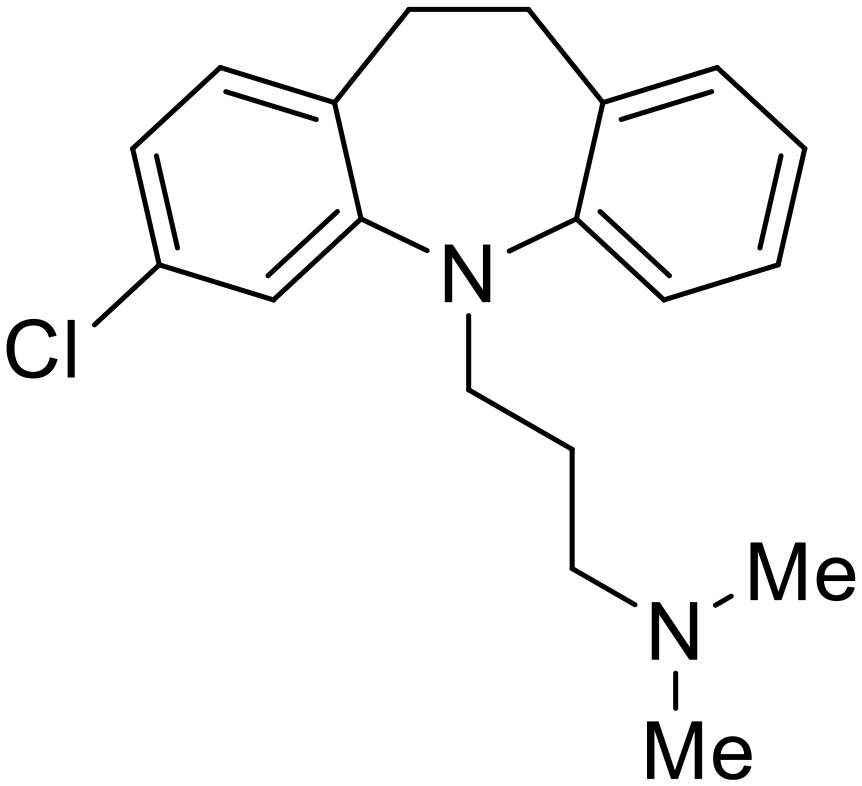
|
101 |
| Thioridazine |
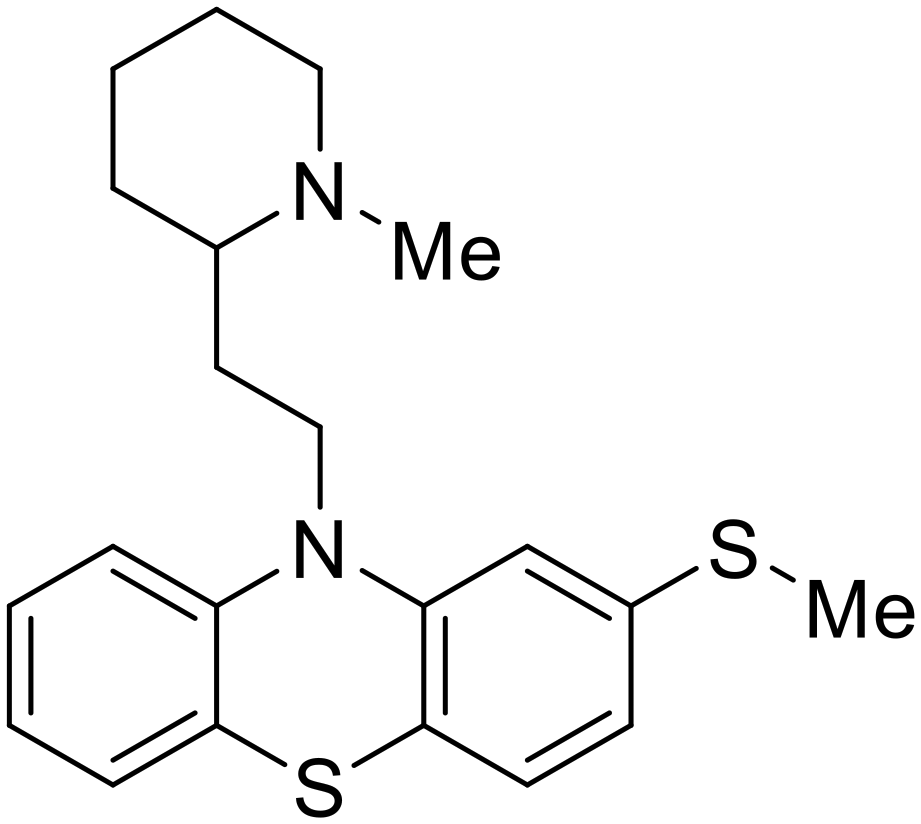
|
101 |
| 118 |
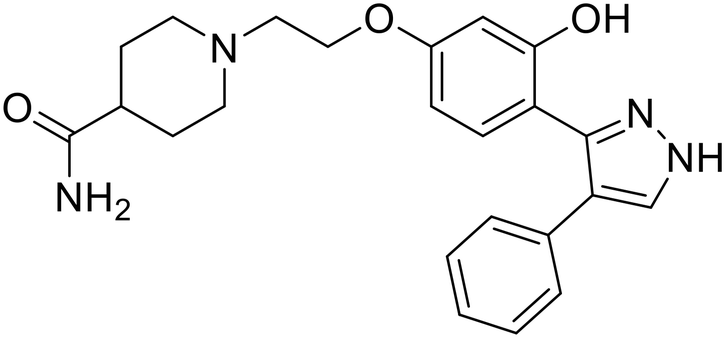
|
102 |
| 118a |
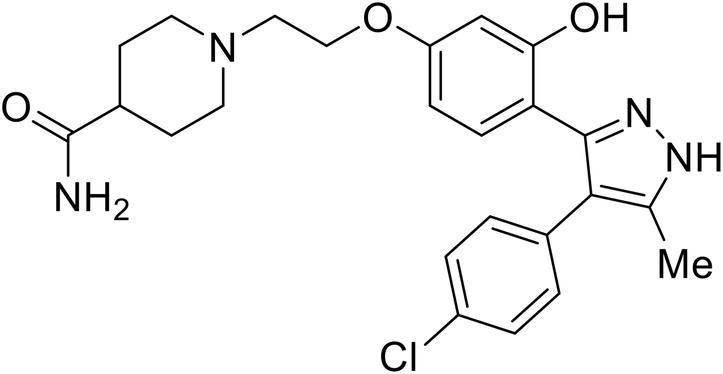
|
102 |
| Procyanidin B2 |
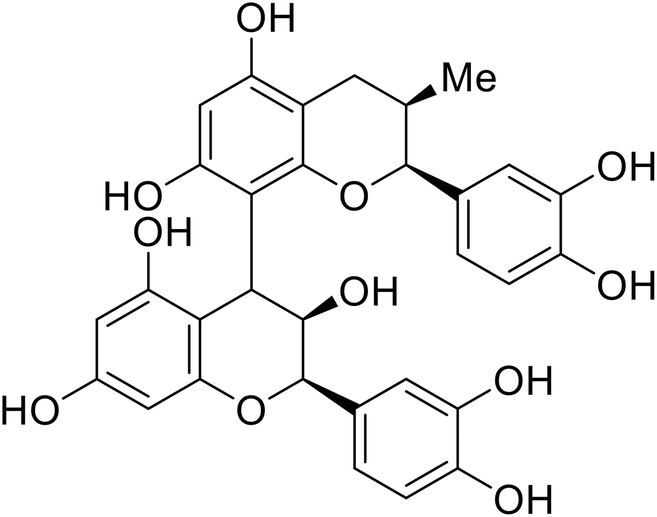
|
103 |
| Chlorcyclizine |
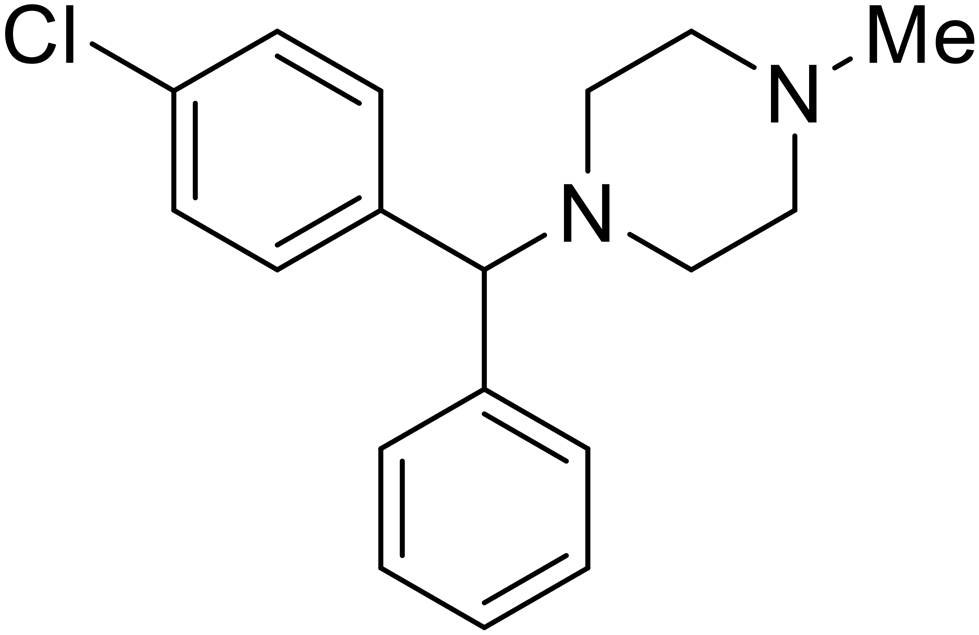
|
104, 105 |
| Diphenhydramine |
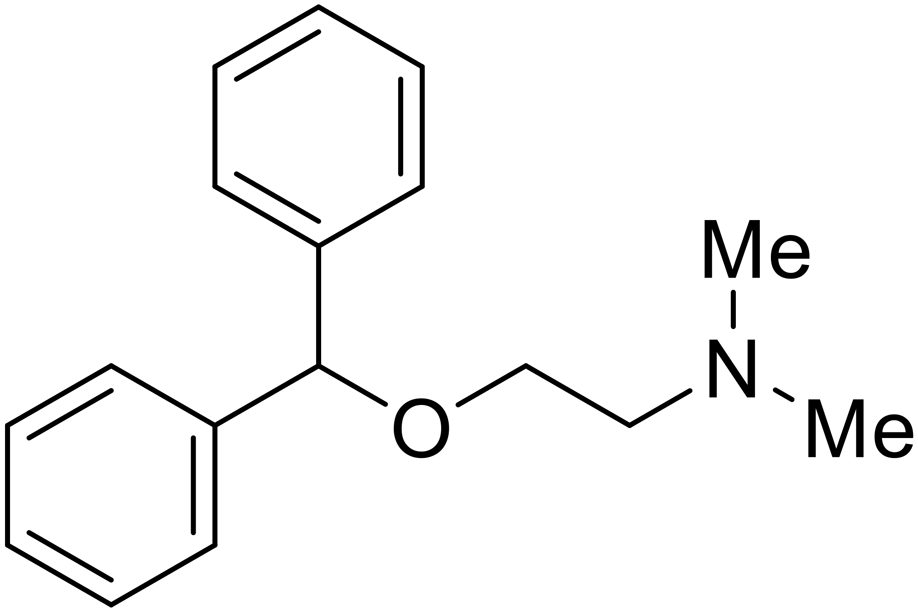
|
104, 105 |
| CP19 |
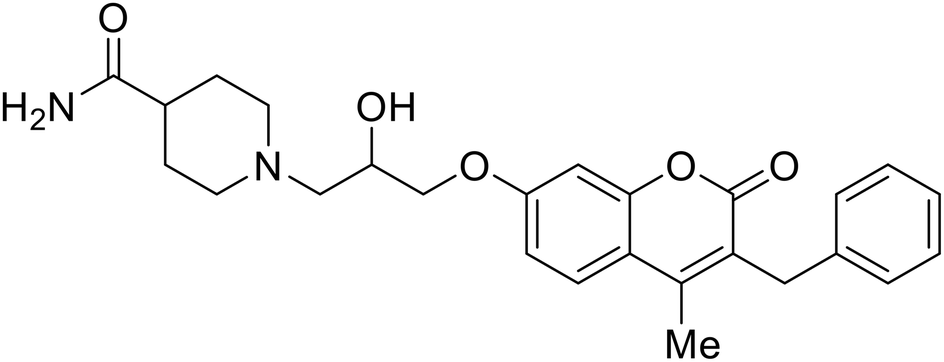
|
104, 105 |
| Tilorone |
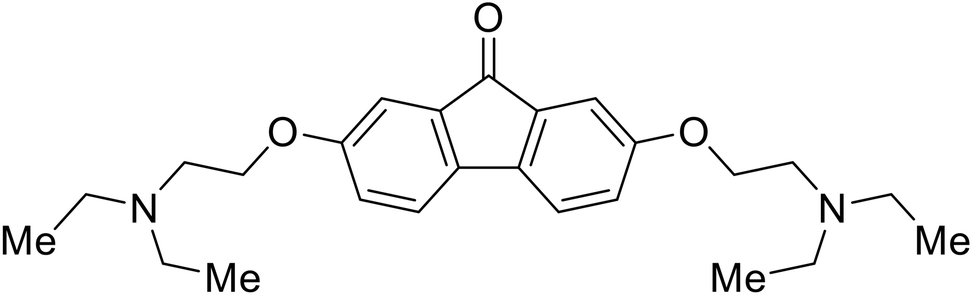
|
106–108 |
| 58 |
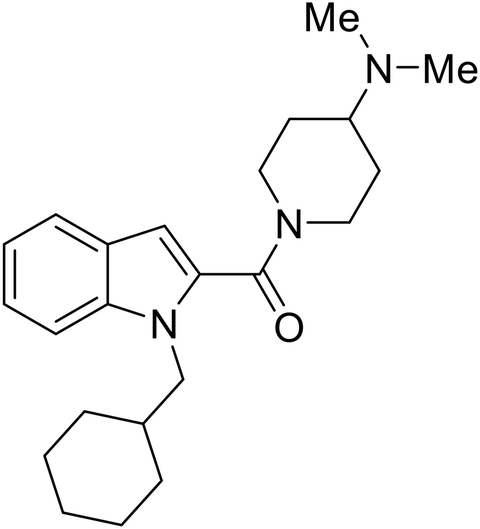
|
44 |
| 28 |
|
109 |
| SYL1712 |
|
110 |
| 60 |
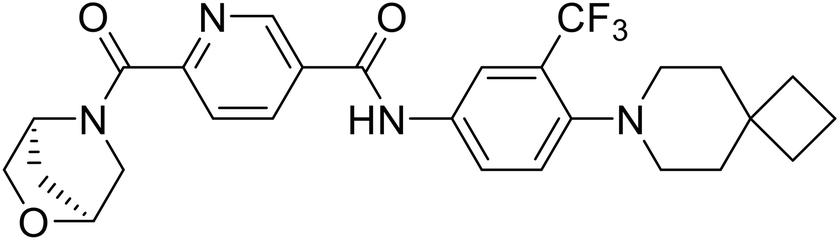
|
111 |
| 4-(Aminomethyl) benzamide (35) |
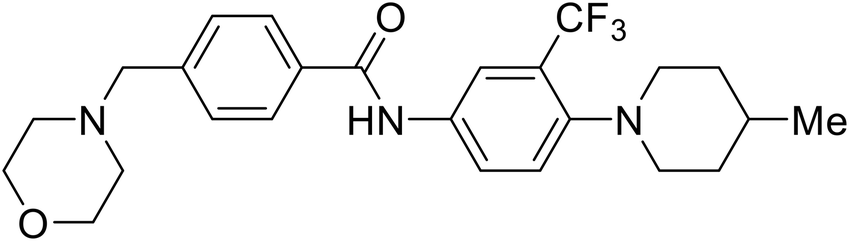
|
112 |
| 38 |
|
113 |
Other hits from the FDA-drug screen included antidepressant and antipsychotic drugs imipramine, clomipramine, and thioridazine. Unlike toremifene, imipramine and clomipramine do not destabilize GP upon increasing temperatures and lack strong binding affinity to GP (imipramineKD = 584 μM; clomipramineKD = 118 μM).101 Nonetheless, X-ray crystallography revealed the GP internal fusion loop region as the imipramine-, clomipramine-, and thioridazine-binding site with DFF lid displacement upon binding, similar to toremifene.
The identification of the internal fusion loop region as a confirmed GP-binding pocket enabled structure-based in silico screening of small molecules in this region. Traditional Chinese medicinal actives 118 (ZINC32540717) and 118a (ZINC09410451) were identified.102 The co-crystal structure of 118a (EC50 = 0.05 μM) bound to EBOV GP showed that two 118a molecules occupy the internal fusion loop binding pocket, further increasing interactions with GP residues. Other traditional Chinese herbs like Maesaperlarius also produce filoviral entry inhibitors.103 The methanolic extract elucidated from this plant contains procyanidin B2 that exhibits anti-EBOV activity and favorable cytotoxicity. Microscale thermophoresis determined procyanidin B2-GP binding (KD = 13 μM), which was comparable to toremifene (KD = 21 μM). Thus, natural products like 118a and procyanidin B2 serve as good starting points for novel entry inhibitors.
EBOV is known to compromise the immune response,9,10 yet antihistamines, which suppress allergic responses, inhibit filoviral entry. H1 receptor antagonists, including chlorcyclizine, diphenhydramine, and CP19, demonstrate moderate entry inhibition; however, H2, H3, and H4 antagonists lack activity.104,105 Docking and mutational analysis studies have suggested fusion inhibition as the antiviral mechanism of action of the antihistamines. Broad-spectrum antiviral tilorone is another approved drug that exhibits potent entry inhibition against infectious EBOV (EC50 = 0.23 μM).106 Microscale thermophoresis suggests that tilorone has 35-fold stronger binding to EBOV GP compared to toremifene.107 Strong binding, coupled with favorable pharmacokinetic and established safe dosing ranges of 2–10 mg kg−1 in mice,108 encourages further study of tilorone.
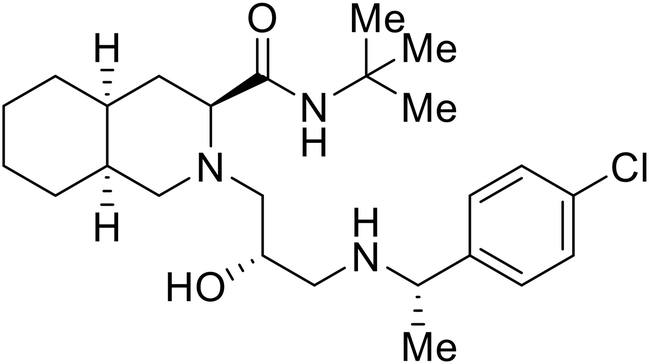
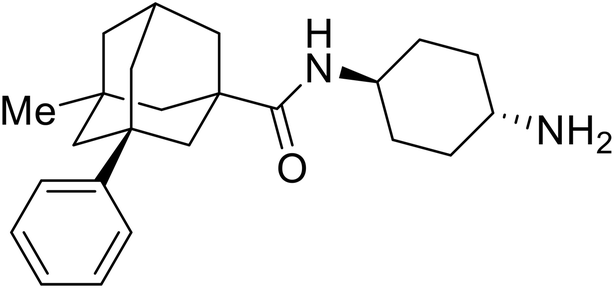
Repurposed drugs are effective starting points for the development of filoviral entry and fusion inhibitors; however, novel small molecules with fewer off-target effects are also needed. An N-substituted furopyrrole discovered in a HTS displayed activity against EBOV.44 Optimization of the amide-amine linker, heterocyclic core, and N-substituent generated compound 58 that maintained sub-micromolar activity in both pseudovirus and infectious EBOV assays (EC50 = 0.29 μM and 0.39 μM, respectively). When tested against MARV and other Ebola virus species including SUDV, BDBV, and TAFV, 58 demonstrated broad-spectrum antifiloviral activity; however, it lacked potent inhibition against non-filoviruses like influenza, showcasing selective filoviral activity. Additionally, isoquinoline (28),109diaryl quinoline (SYL1712),1102,5-pyridinedicarboxamide (60),111 and 4-(aminomethyl)benzamide scaffolds112 were identified in separate HTS's. The 4-(aminomethyl)benzamide derivatives seemed more promising as fusion-specific inhibitors due to the additional mutational analysis studies used. SAR of the benzamides included lipophilic adamantane coupling to the scaffold, which improved activity yet adversely increased logP. Fortunately, this bulky substituent proved useful for an adamantane carboxamide scaffold identified in a HTS.113 SAR development revealed potent inhibitors with the S-configuration with sub-micromolar infectious EBOV entry inhibition (EC50 = 0.24 μM) and favorable cytotoxicity. X-ray co-crystallography with the potent lead compound 38 and EBOV GP revealed the internal fusion loop region as the binding site. Like toremifene and other identified fusion inhibitors, derivative binding in this region displaced the DFF lid. Additional studies can be done to determine GP-stabilization when bound to compound 38 to suggest fusion-specific inhibition.
Replication and transcription inhibitors
Various screens were used to identify EBOV replication and transcription inhibitors and host factors involved (Table 6), including a genome-wide siRNA screen that identified host carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) as hits.114 Use of a mini-genome platform found 6-azauridine to reduce EBOV titer growth in Vero cells, as 6-azauridine inhibits orotidylic acid decarboxylation during de novo pyrimidine biosynthesis.115 Additionally, teriflunomide, an FDA-approved drug that blocks the production of orotic acid from the dihydroorotic acid precursor in the uridine monophosphate synthetic pathway, inhibited in vitro transcription and replication in HEK293 cells. By assessing various thymidine, adenosine, cytidine, and guanosine analogs against recombinant EBOV, cytidine analog β-D-N4-hydroxycytidine (NHC) was found to inhibit EBOV genomic replication and dose-dependently attenuated EBOV infection in Vero cells and donor-derived macrophages.116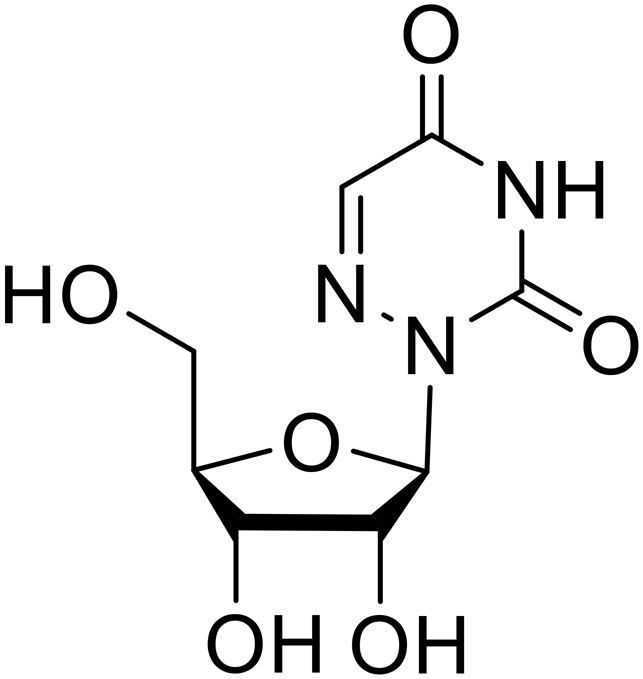
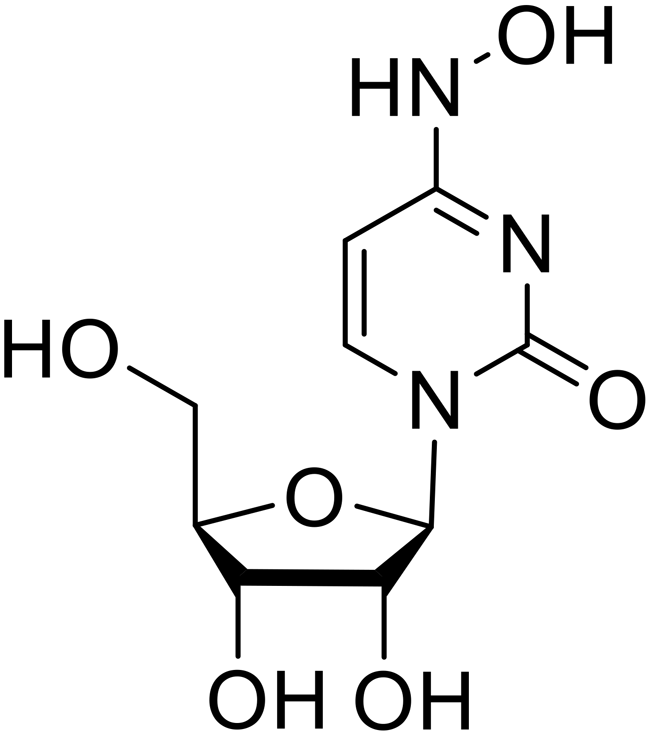
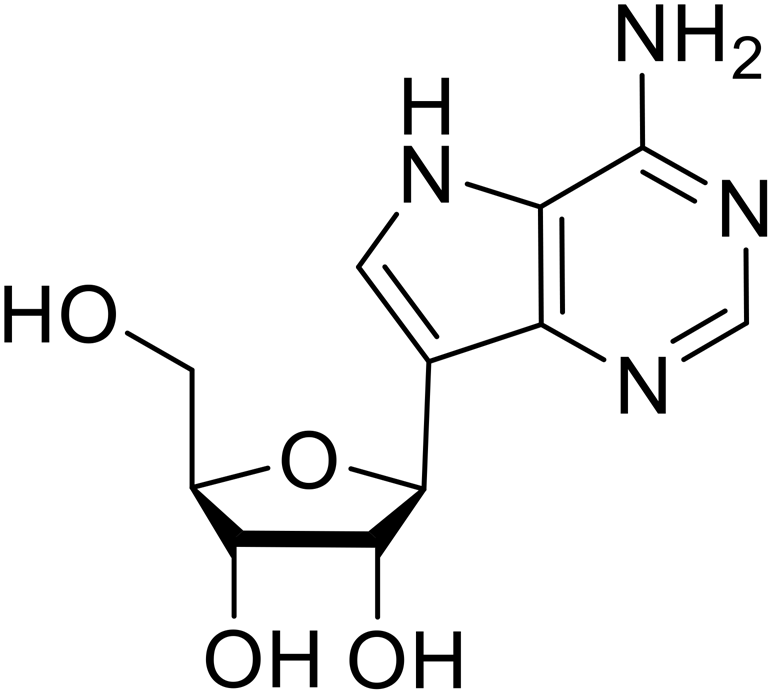
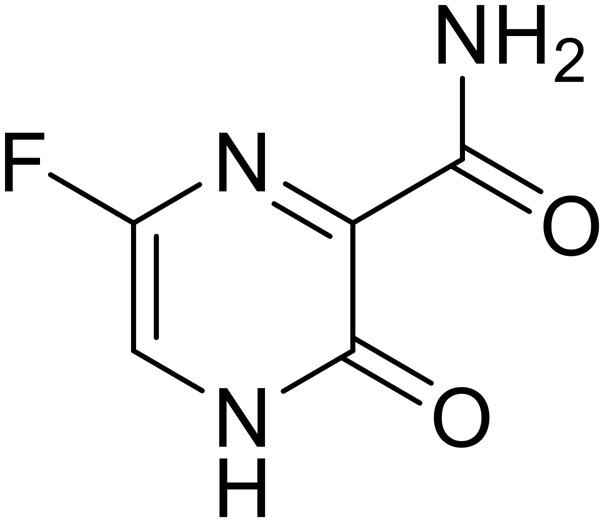
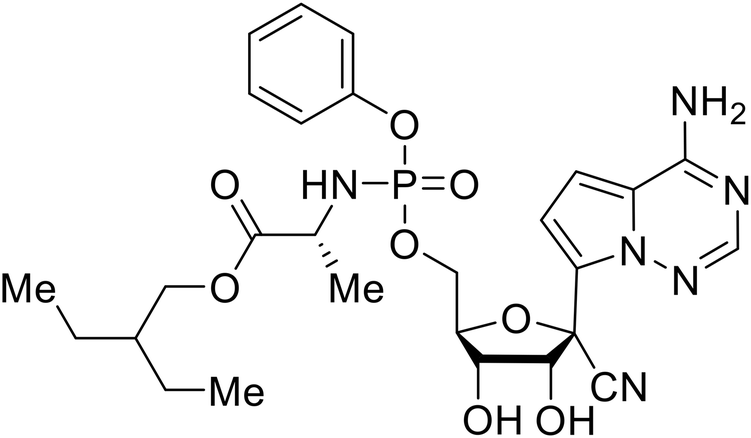
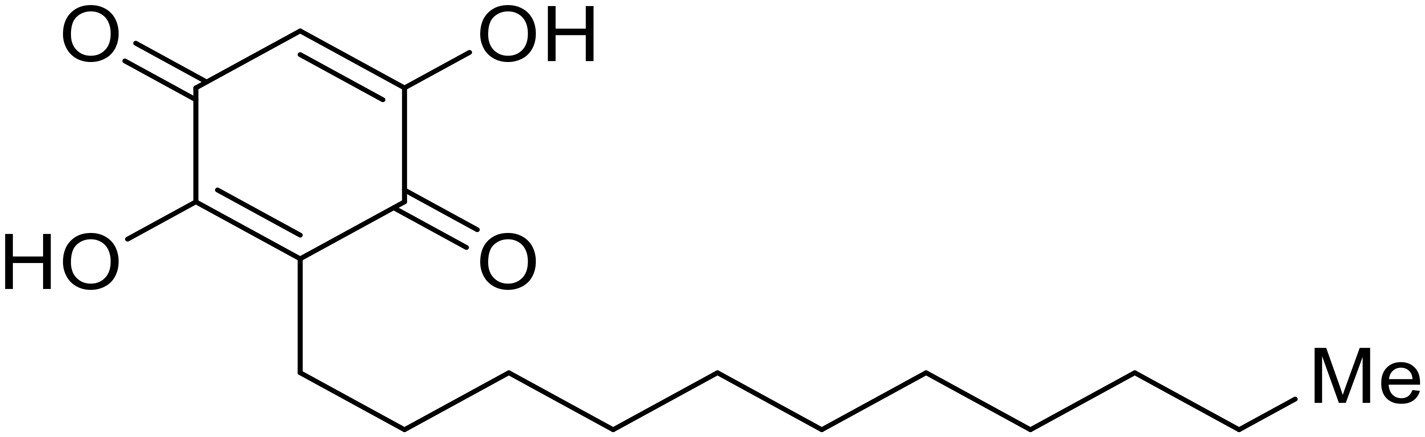
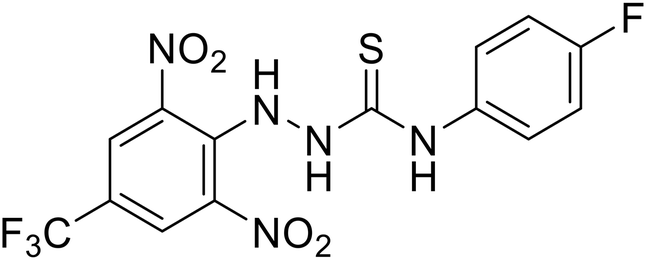
Galidesivir (BCX4430) was designed and synthesized as a novel nucleoside analog to inhibit viral RNA polymerase by premature RNA chain termination during replication and transcription.117,118 High content image-based (HCI) assays demonstrated in vitro inhibition of recombinant EBOV and SUDV by galidesivir (EC50 = 11.8 μM and 3.4 μM, respectively). When administered to mice twice daily via IM injection or orally, galidesivir treatment provided high survival rates among infected mice. Favipiravir (T-705) is a nucleoside prodrug119 previously used as an influenza RNA polymerase inhibitor.120 When assessed for EBOV activity, favipiravir conferred 100% survival when orally administered to A129 alpha/beta knockdown mice challenged with EBOV.121In vivo protection against SUDV-challenged guinea pigs was also demonstrated for favipiravir.122 During the JIKI clinical trial in Guinea, favipiravir failed to achieve targeted plasma concentration in patients infected with EBOV, suggesting the need to establish safe dosing ranges in healthy patients prior to efficacy studies.123 Similar results occurred for brincidofovir, a lipid conjugate of a nucleotide analog prioritized for clinical trials during the 2013–2016 EBOV epidemic.124,125 The phase 2 trial consisting of 4 patients with confirmed EBOV infections was inconclusive, as the small sample size confounded brincidofovir protection of patients against lethality. The results of favipiravir and brincidofovir clinical trials exemplified the importance of thorough in vitro, in vivo, and phase 1 clinical trial data throughout the drug development process.
During SAR development of 1'substituted 4-aza-7,9-dideazaadenosine C-nucleoside analogs, the bis S-acyl-2-thioethyl (SATE) prodrug of nucleoside GS-441524 (9a) was synthesized.126 Maintaining the prodrug approach to the adenosine analog, remdesivir (GS-5734) was generated and demonstrated RNA chain terminator activity in its active triphosphorylated form. Remdesivir's selective inhibition of viral RNA polymerase over host polymerase via the cyano group127 proved advantageous in protecting 100% of rhesus monkeys against infectious EBOV, even when administered 3 days post infection.128 Additionally, remdesivir protected non-human primates against SUDV infection.129 During the EBOV epidemic PALM clinical trial, remdesivir was assessed alongside ZMapp, MAb114, and REGN-EB3; however, antibodies MAb114, and REGN-EB3 were more effective compared to remdesivir.66 Emerged mutations within the EBOV RNA polymerase conferred resistance to remdesivir,130 resulting in the development of RYL-687,131 a synthetic analog of RYL-634 that inhibits dihydroorotate dehydrogenase (DHODH) in de novo pyrimidine synthesis.132 When administered to EBOV ΔVP30 EGFP infected Huh7 cells, RYL-687 (IC50 = 6.65 nM) displayed increased inhibitory EBOV potency compared to remdesivir (IC50 = 46.6 nM). Continued studies of the GS-441524 nucleoside led to the generation of obeldesivir, the isobutyl ester prodrug with improved oral bioavailability,133 a favorable attribute to newly developing therapeutics for epi- and pan-demic pathogens. Potent in vitro activity of obeldesivir against EBOV, SUDV, and MARV led to a non-human primate animal study, where 100% of animals survived lethal SUDV infection upon receiving daily oral obeldesivir doses for 10 days. Recent phase 1 trials of healthy patients receiving various doses of obeldesivir is advantageous for future obeldesivir human efficacy studies.
Most of the replication and transcription inhibitors described above demonstrate an advantageous selectivity for the viral RNA polymerase over host polymerases; however, through their inhibition mechanisms, these small molecules also exhibit activity against other RNA viruses. For filoviral selective replication and transcription inhibitors, an alternative approach to nucleoside analogs or enzyme inhibitors is needed. Studies of the EBOV transcription complex determined a binding event between EBOV's nucleoprotein (NP) and the N-terminus of VP35 at the NPBP binding region. A peptide targeting NPBP was shown to block the NP-VP35 binding event,134 which led to the screening of small molecules that could also inhibit this interaction.135 Small molecule tolcapone was identified as a hit and dose-dependently inhibited EBOV NP-NPBP binding in both fluorescence anisotropy and biolayer interferometry (BLI) experiments. Tolcapone also demonstrated inhibitory activity against SUDV, RESTV, and MARV, showcasing pan-filoviral activity; however, additional studies are needed to determine non-filoviral activity. Similarly, the crystal structure of an EBOV NP-derived peptide in complex with VP30 was determined, indicating an alternative approach to potential transcription inhibition.136 A fluorescence anisotropy high-throughput screen identified Embelin and Kobe2602 as protein-targeting hits with confirmed VP30 binding as determined by SPR (KD = 4.62 μM and 0.88 μM, respectively) and thermal shift assays.137 Importantly, a minigenome assay demonstrated transcription inhibition for Embelin and Kobe2602 (EC50 = 16.9 μM and 22.3 μM, respectively). Both drugs are expected to target the EBOV NP-VP30 binding, yet Embelin and Kobe2602 are proposed to distinctly bind VP30 at alternate binding regions. Diversified VP30-targeting mechanisms is advantageous for discovering novel binding inhibitors. Embelin is an antioxidant while Kobe2602 is a Ras-binding protein; therefore, both drugs have additional biological targets outside of the EBOV transcriptional complex. For EBOV-selective activity, in-depth SAR exploration is needed to reduce off-target effects; however, both Embelin and Kobe2602, as well as tolcapone, serve as promising starting points to develop novel EBOV protein-targeting replication and transcription inhibitors.
Budding inhibitors
As packaged viral components are transported to the cell surface, VP40 mediates the budding process. This step is facilitated by VP40's ability to undergo a conformational change from dimeric to hexameric forms. Inhibition of VP40 function by small molecules can block budding required to produce new progeny (Table 7). Computer-based approaches have proven useful to identify potential budding inhibitors. An in silico screen of approximately 30000 Chinese and African natural product-derived small molecules from Northern African Natural Products Database (NANPDB) and traditional Chinese medicine (TCM) were assessed.138 A total of 42 naturally derived small molecules with favorable ADMET properties were identified as potential VP40 inhibitors that serve as good starting points for additional experimental analysis. Molecular mechanics and binding free energy calculations were determined for five different reported VP40 inhibitors.139,140Vindesine possessed the best binding free energy of −5.0 kcal mol−1. Within the predicted VP40 binding site, vindesine was also hypothesized to from several hydrogen bonding interactions with Gln38, Gln35, and Lys127 that supported its 0.27 μM IC50 against VP40. Additionally, sangivamycin derived from Streptomyces sp. inhibited EBOV VP40 localization to the cell membrane,141 which consequently reduced the production and release of eVLPs. As a nucleoside analog, sangivamycin also inhibited EBOV transcription and replication in a minigenome assay; therefore, additional studies are required to determine the full mechanism of action of this potential VP40 inhibitor.
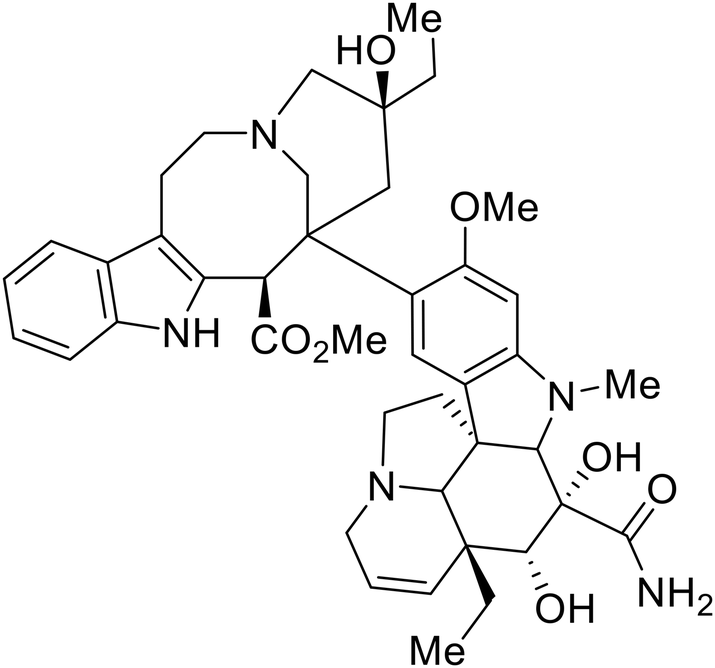
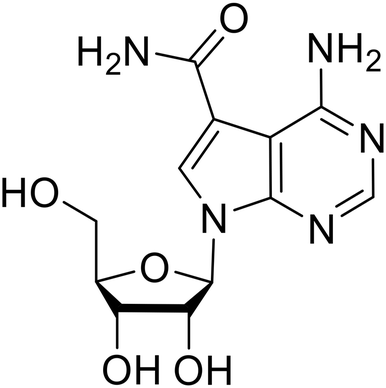
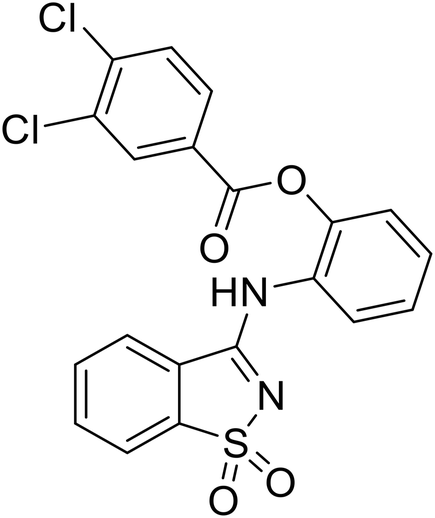
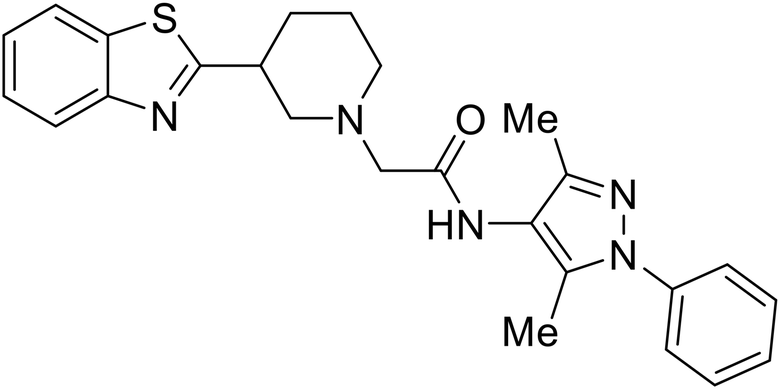
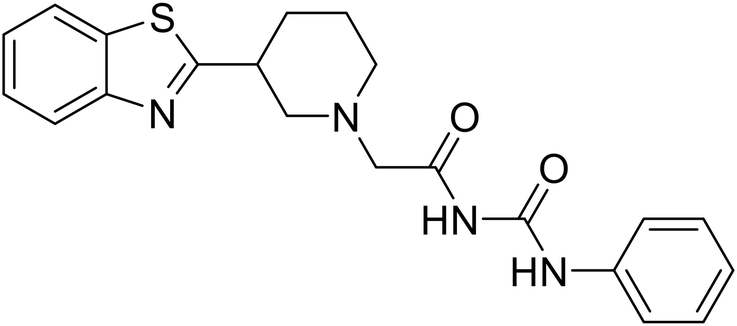
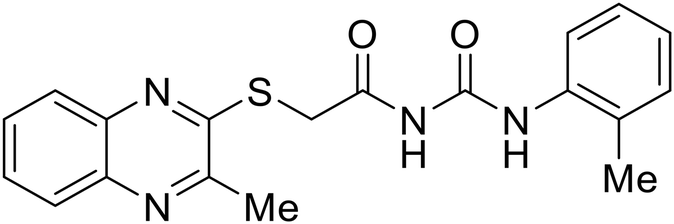
EBOV VP40 function is also dependent on interactions made with host proteins within the endosomal sorting complexes required for transport (ESCRT) family. At the proline-rich late (L) domain, VP40 is ubiquitinated by Nedd4 which regulates its interaction with Tsg101.142,143 Nedd4 and Tsg101 have thus become potential targets for small molecule budding inhibitors. Biomolecular complementation (BiMC) assays have been used to detect and localize formation of the VP40-Tsg101 and VP40-Nedd4 complexes in cells. Compound 5539-0062 reduced the formation of the EBOV VP40-Tsg101 complex by 76% compared to the DMSO control and further inhibited eVLP egress in HEK 293 T cells.144 Similarly, compounds 4 and 5 inhibited the MARV VP40-Nedd4 interaction.145 SAR studies of quinoxalin-2-mercapto-acetyl-urea analogs generated potent derivatives 21 and 24 with 2-chlorophenyl and 2-methylphenyl substitutions at the urea site, respectively.146 Both 21 and 24 inhibited the MARV VP40-Nedd4 interaction and reduced EBOV VLP formation 93% and 83%, respectively, at 30 nM. Additional human liver microsome stability supports the continued study of compounds 21 and 24 as potential EBOV budding inhibitors.
Conclusions
The re-emergence of EBOV outbreaks, ease of transmission, EVD prognosis, and lethality continue to stress the need for available EBOV therapeutics. The current FDA-approved monoclonal antibody treatments, although effective, are limited in their use and practicality. Small molecules presented in this literature review have described essential studies that discovered fundamental aspects of the filoviral life cycle, as well as the recent development of effective small-molecule treatments for EBOV.Although great advances have been made, progress remains limited. Many of the antifiloviral treatments described above have focused on defining the mechanism of action and improving the potency of the small molecules discovered, but have not addressed future steps on transforming the active agents into actual drugs. Furthermore, much of the biological assays assessing activity was conducted in vitro, which is far removed from clinically supportive data. For more progressive outcomes in the early drug discovery pipeline, SAR studies should define pharmacokinetics, including metabolic stability, bioavailability, and distribution, and establish strategies for in vivo studies. With improved computational tools for structure-based design, SAR-focused studies could also provide more insights to how proposed or experimental chemical modifications improve activity and drug-like properties. Additionally, future work should also monitor drug resistance through serial passaging experiments to identify potential mutations that may arise during outbreaks. Plans to assess newly designed small molecules and combination therapies to address resistance should be proposed for future studies. With continued efforts and strides to advance the current state of small molecule antifilovirals, these active agents have the promise of becoming effective therapeutics.
Conflicts of interest
The authors declare the following competing financial interest(s): L. R. is the owner of Chicago BioSolutions, Inc. and thus declares potential financial interests, as does I. N. G. who are employed by Chicago BioSolutions, Inc.Data availability
The data in this publication have been previously described in the literature. No new data have been reported in this manuscript.Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (R41 AI126971 and R42 AI126971 to L. R. and R01 AI 168362 to L. R.).References
- N. Biedenkopf, A. Bukreyev, K. Chandran, N. Di Paola, P. B. H. Formenty, A. Griffiths, A. J. Hume, E. Mühlberger, S. V. Netesov, G. Palacios, J. T. Pawęska, S. Smither, A. Takada, V. Wahl and J. H. Kuhn, Renaming of Genera Ebolavirus and Marburgvirus to Orthoebolavirus and Orthomarburgvirus, Respectively, and Introduction of Binomial Species Names within Family Filoviridae, Arch. Virol., 2023, 168(8), 220, DOI:10.1007/s00705-023-05834-2.
- L. K. Koch, S. Cunze, J. Kochmann and S. Klimpel, Bats as Putative Zaire Ebolavirus Reservoir Hosts and Their Habitat Suitability in Africa, Sci. Rep., 2020, 10(1), 14268, DOI:10.1038/s41598-020-71226-0.
- S. T. Jacob, I. Crozier, W. A. Fischer, A. Hewlett, C. S. Kraft, M.-A. de L. Vega, M. J. Soka, V. Wahl, A. Griffiths, L. Bollinger and J. H. Kuhn, Ebola Virus Disease, Nat. Rev. Dis. Primers, 2020, 6(1), 13, DOI:10.1038/s41572-020-0147-3.
- L. Li, T. Gan, Z. Ma, Y. Huang and J. Zhong, Assessing Risk of Bombali Virus Spillover to Humans by Mutagenesis Analysis of Viral Glycoprotein, hLife, 2024, 2(1), 32–42, DOI:10.1016/j.hlife.2023.11.003.
- T. Goldstein, S. J. Anthony, A. Gbakima, B. H. Bird, J. Bangura, A. Tremeau-Bravard, M. N. Belaganahalli, H. L. Wells, J. K. Dhanota, E. Liang, M. Grodus, R. K. Jangra, V. A. DeJesus, G. Lasso, B. R. Smith, A. Jambai, B. O. Kamara, S. Kamara, W. Bangura, C. Monagin, S. Shapira, C. K. Johnson, K. Saylors, E. M. Rubin, K. Chandran, W. I. Lipkin and J. A. K. Mazet, The Discovery of Bombali Virus Adds Further Support for Bats as Hosts of Ebolaviruses, Nat. Microbiol., 2018, 3(10), 1084–1089, DOI:10.1038/s41564-018-0227-2.
- T. W. Geisbert, L. E. Hensley, T. Larsen, H. A. Young, D. S. Reed, J. B. Geisbert, D. P. Scott, E. Kagan, P. B. Jahrling and K. J. Davis, Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques: Evidence That Dendritic Cells Are Early and Sustained Targets of Infection, Am. J. Pathol., 2003, 163(6), 2347–2370, DOI:10.1016/S0002-9440(10)63591-2.
- S. R. Zaki, W.-J. Shieh, P. W. Greer, C. S. Goldsmith, T. Ferebee, J. Katshitshi, F. K. Tshioko, M. A. Bwaka, R. Swanepoel, P. Calain, A. S. Khan, E. Lloyd, P. E. Rollin, T. G. Ksiazek and C. Peters, J.; for the Commission de Lutte contre les Epidémies à Kikwit. A Novel Immunohistochemical Assay for the Detection of Ebola Virus in Skin: Implications for Diagnosis, Spread, and Surveillance of Ebola Hemorrhagic Fever, J. Infect. Dis., 1999, 179(Supplement_1), S36–S47, DOI:10.1086/514319.
- A. Baskerville, S. P. Fisher-Hoch, G. H. Neild and A. B. Dowsett, Ultrastructural Pathology of Experimental Ebola Haemorrhagic Fever Virus Infection, J. Pathol., 1985, 147(3), 199–209, DOI:10.1002/path.1711470308.
- K. I. Diakou, T. Mitsis, K. Pierouli, E. Papakonstantinou, E. Bongcam-Rudloff, M. Wayengera and D. Vlachakis, Ebola Virus Disease and Current Therapeutic Strategies: A Review. in GeNeDis 2020, ed. P. Vlamos, Springer International Publishing, Cham, 2021, pp. 131–137 Search PubMed.
- D.-S. Yu, T.-H. Weng, X.-X. Wu, F. X. C. Wang, X.-Y. Lu, H.-B. Wu, N.-P. Wu, L.-J. Li and H.-P. Yao, The Lifecycle of the Ebola Virus in Host Cells, Oncotarget, 2017, 8(33), 55750–55759, DOI:10.18632/oncotarget.18498.
- H. Ebihara, B. Rockx, A. Marzi, F. Feldmann, E. Haddock, D. Brining, R. A. LaCasse, D. Gardner and H. Feldmann, Host Response Dynamics Following Lethal Infection of Rhesus Macaques With Zaire Ebolavirus, J. Infect. Dis., 2011, 204(suppl_3), S991–S999, DOI:10.1093/infdis/jir336.
- T. W. Geisbert, H. A. Young, P. B. Jahrling, K. J. Davis, E. Kagan and L. E. Hensley, Mechanisms Underlying Coagulation Abnormalities in Ebola Hemorrhagic Fever: Overexpression of Tissue Factor in Primate Monocytes/Macrophages Is a Key Event, J. Infect. Dis., 2003, 188(11), 1618–1629, DOI:10.1086/379724.
- Z. Dembek, S. Hadeed, B. Tigabu, K. Schwartz-Watjen, M. Glass, M. Dressner, D. Frankel, D. Blaney, T. G. Eccles III, T. Chekol, A. Owens and A. Wu, Ebola Virus Disease Outbreaks: Lessons Learned From Past and Facing Future Challenges, Mil. Med., 2024, 189(7–8), e1470–e1478, DOI:10.1093/milmed/usae204.
- W. Furuyama and A. Marzi, Ebola Virus: Pathogenesis and Countermeasure Development, Annu. Rev. Virol., 2019, 6(1), 435–458, DOI:10.1146/annurev-virology-092818-015708.
- M. G. Kortepeter, D. G. Bausch and M. Bray, Basic Clinical and Laboratory Features of Filoviral Hemorrhagic Fever, J. Infect. Dis., 2011, 204(suppl_3), S810–S816, DOI:10.1093/infdis/jir299.
- H. Feldmann and T. W. Geisbert, Ebola Haemorrhagic Fever, Lancet, 2011, 377(9768), 849–862, DOI:10.1016/S0140-6736(10)60667-8.
- Centers for Disease Control and Prevention. History of Ebola Disease Outbreaks, https://www.cdc.gov/vhf/ebola/history/chronology.html, (accessed 2023-07-26).
- B. Beer, R. Kurth and A. Bukreyev, Characteristics of Filoviridae: Marburg and Ebola Viruses, Naturwissenschaften, 1999, 86(1), 8–17, DOI:10.1007/s001140050562.
- Ebola: Overview, History, Origins and Transmission; UK Health Security Agency.
- R. Watanabe, D. Zyla, D. Parekh, C. Hong, Y. Jones, S. L. Schendel, W. Wan, G. Castillon and E. O. Saphire, Intracellular Ebola Virus Nucleocapsid Assembly Revealed by in Situ Cryo-Electron Tomography, Cell, 2024, 187(20), 5587–5603.e19, DOI:10.1016/j.cell.2024.08.044.
- C. P. Alvarez, F. Lasala, J. Carrillo, O. Muñiz, A. L. Corbí and R. Delgado, C-Type Lectins DC-SIGN and L-SIGN Mediate Cellular Entry by Ebola Virus in Cis and in Trans, J. Virol., 2002, 76(13), 6841–6844, DOI:10.1128/jvi.76.13.6841-6844.2002.
- A. S. Kondratowicz, N. J. Lennemann, P. L. Sinn, R. A. Davey, C. L. Hunt, S. Moller-Tank, D. K. Meyerholz, P. Rennert, R. F. Mullins, M. Brindley, L. M. Sandersfeld, K. Quinn, M. Weller, P. B. McCray, J. Chiorini and W. Maury, T-Cell Immunoglobulin and Mucin Domain 1 (TIM-1) Is a Receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(20), 8426–8431, DOI:10.1073/pnas.1019030108.
- A. Takada, S. Watanabe, H. Ito, K. Okazaki, H. Kida and Y. Kawaoka, Downregulation of B1 Integrins by Ebola Virus Glycoprotein: Implication for Virus Entry, Virology, 2000, 278(1), 20–26, DOI:10.1006/viro.2000.0601.
- S. Masayuki, T. Ayato, E. Hideki, N. Gabriele, F. Kouki, I. Tatsuro, J. Steven, F. Heinz and K. Yoshihiro, Tyro3 Family-Mediated Cell Entry of Ebola and Marburg Viruses, J. Virol., 2006, 80(20), 10109–10116, DOI:10.1128/jvi.01157-06.
- A. O'Hearn, M. Wang, H. Cheng, M. Lear-Rooney Calli, K. Koning, E. Rumschlag-Booms, E. Varhegyi, G. Olinger and L. Rong, Role of EXT1 and Glycosaminoglycans in the Early Stage of Filovirus Entry, J. Virol., 2015, 89(10), 5441–5449, DOI:10.1128/jvi.03689-14.
- M. F. Saeed, A. A. Kolokoltsov, T. Albrecht and R. A. Davey, Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-Like Mechanism and Subsequent Trafficking through Early and Late Endosomes, PLoS Pathog., 2010, 6(9), e1001110, DOI:10.1371/journal.ppat.1001110.
- K. Chandran, N. J. Sullivan, U. Felbor, S. P. Whelan and J. M. Cunningham, Endosomal Proteolysis of the Ebola Virus Glycoprotein Is Necessary for Infection, Science, 2005, 308(5728), 1643–1645, DOI:10.1126/science.1110656.
- M. Côté, J. Misasi, T. Ren, A. Bruchez, K. Lee, C. M. Filone, L. Hensley, Q. Li, D. Ory, K. Chandran and J. Cunningham, Small Molecule Inhibitors Reveal Niemann–Pick C1 Is Essential for Ebola Virus Infection, Nature, 2011, 477(7364), 344–348, DOI:10.1038/nature10380.
- N. D. Durham, A. R. Howard, R. Govindan, F. Senjobe, J. M. Fels, W. E. Diehl, J. Luban, K. Chandran and J. B. Munro, Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics, Viruses, 2020, 12(1) DOI:10.3390/v12010103.
- D. K. Das, U. Bulow, W. E. Diehl, N. D. Durham, F. Senjobe, K. Chandran, J. Luban and J. B. Munro, Conformational Changes in the Ebola Virus Membrane Fusion Machine Induced by pH, Ca2+, and Receptor Binding, PLoS Biol., 2020, 18(2), e3000626, DOI:10.1371/journal.pbio.3000626.
- M. J. Martinez, V. A. Volchkova, H. Raoul, N. Alazard-Dany, O. Reynard and V. E. Volchkov, Role of VP30 Phosphorylation in the Ebola Virus Replication Cycle, J. Infect. Dis., 2011, 204(suppl_3), S934–S940, DOI:10.1093/infdis/jir320.
- J. Modrof, E. Mühlberger, H.-D. Klenk and S. Becker, Phosphorylation of VP30 Impairs Ebola Virus Transcription *, J. Biol. Chem., 2002, 277(36), 33099–33104, DOI:10.1074/jbc.M203775200.
- K. Takahashi, P. Halfmann, M. Oyama, H. Kozuka-Hata, T. Noda and Y. Kawaoka, DNA Topoisomerase 1 Facilitates the Transcription and Replication of the Ebola Virus Genome, J. Virol., 2013, 87(16), 8862–8869, DOI:10.1128/jvi.03544-12.
- I. García-Dorival, W. Wu, S. D. Armstrong, J. N. Barr, M. W. Carroll, R. Hewson and J. A. Hiscox, Elucidation of the Cellular Interactome of Ebola Virus Nucleoprotein and Identification of Therapeutic Targets, J. Proteome Res., 2016, 15(12), 4290–4303, DOI:10.1021/acs.jproteome.6b00337.
- S. Bale, J.-P. Julien, A. Bornholdt Zachary, S. Krois Alexander, A. Wilson Ian and E. O. Saphire, Ebolavirus VP35 Coats the Backbone of Double-Stranded RNA for Interferon Antagonism, J. Virol., 2013, 87(18), 10385–10388, DOI:10.1128/JVI.01452-13.
- M. R. Edwards, G. Liu, C. E. Mire, S. Sureshchandra, P. Luthra, B. Yen, R. S. Shabman, D. W. Leung, I. Messaoudi, T. W. Geisbert, G. K. Amarasinghe and C. F. Basler, Differential Regulation of Interferon Responses by Ebola and Marburg Virus VP35 Proteins, Cell Rep., 2016, 14(7), 1632–1640, DOI:10.1016/j.celrep.2016.01.049.
- C. R. Kimberlin, Z. A. Bornholdt, S. Li, V. L. Woods, I. J. MacRae and E. O. Saphire, Ebolavirus VP35 Uses a Bimodal Strategy to Bind dsRNA for Innate Immune Suppression, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(1), 314–319, DOI:10.1073/pnas.0910547107.
- A. L. Rasmussen, Host Factors Involved in Ebola Virus Replication. in Roles of Host Gene and Non-coding RNA Expression in Virus Infection, ed. R. A. Tripp and S. M. Tompkins, Springer International Publishing, Cham, 2018, pp. 113–150. DOI:10.1007/82_2017_27.
- M. Schümann, T. Gantke and E. Mühlberger, Ebola Virus VP35 Antagonizes PKR Activity through Its C-Terminal Interferon Inhibitory Domain, J. Virol., 2009, 83(17), 8993–8997, DOI:10.1128/jvi.00523-09.
- P. Halfmann, G. Neumann and Y. Kawaoka, The Ebolavirus VP24 Protein Blocks Phosphorylation of P38 Mitogen-Activated Protein Kinase, J. Infect. Dis., 2011, 204(suppl_3), S953–S956, DOI:10.1093/infdis/jir325.
- E. Adu-Gyamfi, S. P. Soni, Y. Xue, M. A. Digman, E. Gratton and R. V. Stahelin, The Ebola Virus Matrix Protein Penetrates into the Plasma Membrane, J. Biol. Chem., 2013, 288(8), 5779–5789, DOI:10.1074/jbc.M112.443960.
- E. Adu-Gyamfi, K. Johnson, M. Fraser, J. Scott, S. Soni, K. Jones, M. Digman, E. Gratton, C. Tessier and R. Stahelin, Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus, J. Virol., 2015, 89(18), 9440–9453, DOI:10.1128/jvi.01087-15.
- J. Wang, H. Cheng, K. Ratia, E. Varhegyi, W. Hendrickson, J. Li and L. Rong, A Comparative High-Throughput Screening Protocol to Identify Entry Inhibitors of Enveloped Viruses, J. Biomol. Screening, 2014, 19(1), 100–107 CrossRef CAS PubMed.
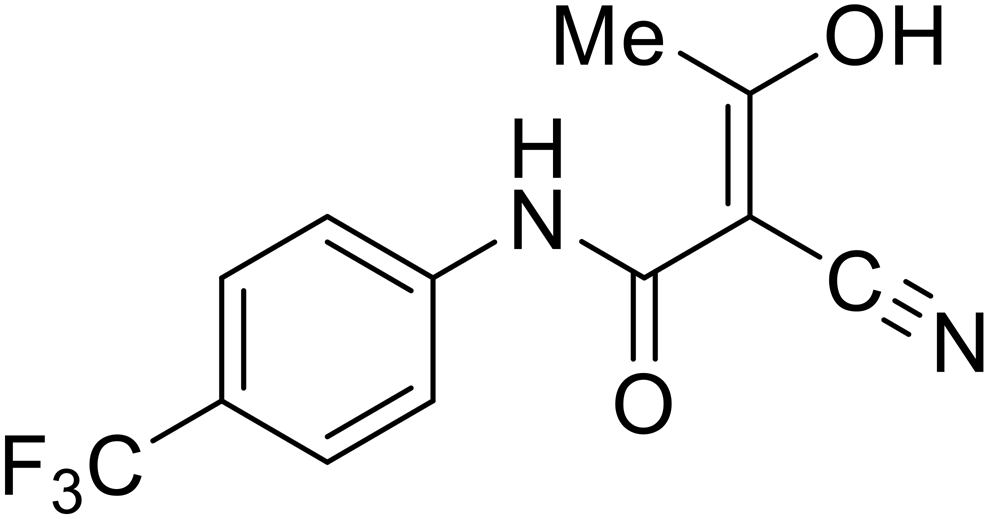
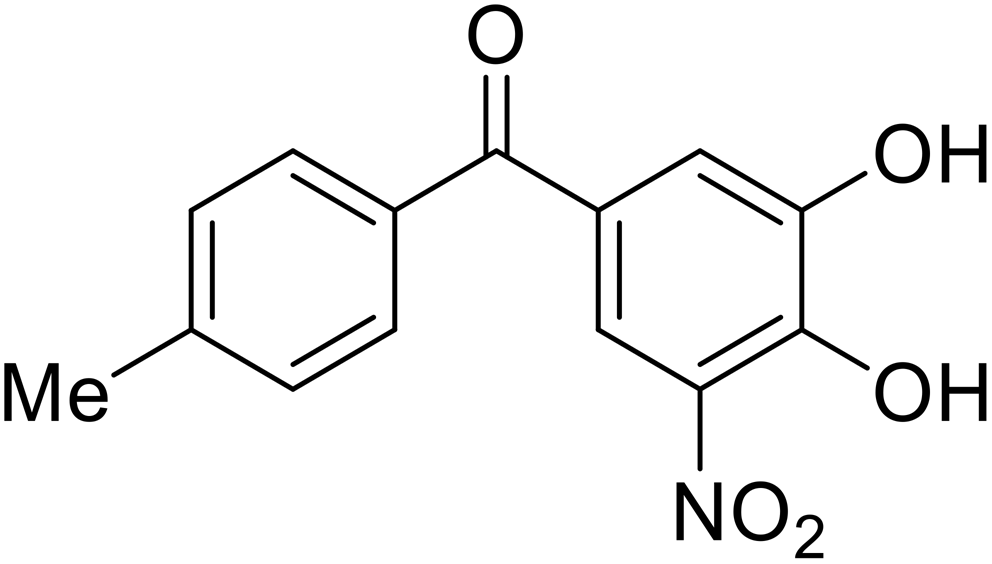
- D. Durante, R. Bott, L. Cooper, C. Owen, K. M. Morsheimer, J. J. Patten, C. Zielinski, N. P. Peet, R. A. Davey, I. N. Gaisina, L. Rong and T. W. Moore, N-Substituted Pyrrole-Based Heterocycles as Broad-Spectrum Filoviral Entry Inhibitors, J. Med. Chem., 2024, 67(16), 13737–13764, DOI:10.1021/acs.jmedchem.4c00527.
- M. Garbutt, R. Liebscher, V. Wahl-Jensen, S. Jones, P. Möller, R. Wagner, V. Volchkov, H.-D. Klenk, H. Feldmann and U. Ströher, Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses, J. Virol., 2004, 78(10), 5458–5465, DOI:10.1128/jvi.78.10.5458-5465.2004.
- J. B. Ruedas, C. E. Arnold, G. Palacios and J. H. Connor, Growth-Adaptive Mutations in the Ebola Virus Makona Glycoprotein Alter Different Steps in the Virus Entry Pathway, J. Virol., 2018, 92(19) DOI:10.1128/jvi.00820-18 DOI:10.1128/jvi.00820-18.
- V. Rayaprolu, B. O. Fulton, A. Rafique, E. Arturo, D. Williams, C. Hariharan, H. Callaway, A. Parvate, S. L. Schendel, D. Parekh, S. Hui, K. Shaffer, K. E. Pascal, E. Wloga, S. Giordano, N. Negron, M. Ni, R. Copin, G. S. Atwal, M. Franklin, R. M. Boytz, C. Donahue, R. Davey, A. Baum, C. A. Kyratsous and E. O. Saphire, Structure of the Inmazeb Cocktail and Resistance to Ebola Virus Escape, Cell Host Microbe, 2023, 31(2), 260–272.e7, DOI:10.1016/j.chom.2023.01.002.
- J. B. Ruedas, J. T. Ladner, C. R. Ettinger, S. Gummuluru, G. Palacios and J. H. Connor, Spontaneous Mutation at Amino Acid 544 of the Ebola Virus Glycoprotein Potentiates Virus Entry and Selection in Tissue Culture, J. Virol., 2017, 91(15) DOI:10.1128/jvi.00392-17.
- T. Li, Z. Li, E. E. Deans, E. Mittler, M. Liu, K. Chandran and T. Ivanovic, The Shape of Pleomorphic Virions Determines Resistance to Cell-Entry Pressure, Nat. Microbiol., 2021, 6(5), 617–629, DOI:10.1038/s41564-021-00877-0.
- L. D. Jasenosky, G. Neumann, I. Lukashevich and Y. Kawaoka, Ebola Virus VP40-Induced Particle Formation and Association with the Lipid Bilayer, J. Virol., 2001, 75(11), 5205–5214, DOI:10.1128/jvi.75.11.5205-5214.2001.
- J. Timmins, S. Scianimanico, G. Schoehn and W. Weissenhorn, Vesicular Release of Ebola Virus Matrix Protein VP40, Virology, 2001, 283(1), 1–6, DOI:10.1006/viro.2001.0860.
- T. Hoenen, A. Groseth, F. de Kok-Mercado, J. H. Kuhn and V. Wahl-Jensen, Minigenomes, Transcription and Replication Competent Virus-like Particles and beyond: Reverse Genetics Systems for Filoviruses and Other Negative Stranded Hemorrhagic Fever Viruses, Antiviral Res., 2011, 91(2), 195–208, DOI:10.1016/j.antiviral.2011.06.003.
- J. Su, Y. Dou, Y. You and X. Cai, Application of Minigenome Technology in Virology Research of the Paramyxoviridae Family, J. Microbiol., Immunol. Infect., 2015, 48(2), 123–129, DOI:10.1016/j.jmii.2014.02.008.
- C. Salata, A. Calistri, G. Alvisi, M. Celestino, C. Parolin and G. Palú, Ebola Virus Entry: From Molecular Characterization to Drug Discovery, Viruses, 2019, 11(3), 274–293 CrossRef CAS PubMed.
- E. Picazo and F. Giordanetto, Small Molecule Inhibitors of Ebola Virus Infection, Drug Discovery Today, 2015, 20(2), 277–286, DOI:10.1016/j.drudis.2014.12.010.
- Z. Janeba, Development of Small-Molecule Antivirals for Ebola, Med. Res. Rev., 2015, 35(6), 1175–1194, DOI:10.1002/med.21355.
- M. U. Mirza, M. Vanmeert, A. Ali, K. Iman, M. Froeyen and M. Idrees, Perspectives towards Antiviral Drug Discovery against Ebola Virus, J. Med. Virol., 2019, 91(12), 2029–2048, DOI:10.1002/jmv.25357.
- E. K. Schneider-Futschik, D. Hoyer, A. A. Khromykh, J. B. Baell, G. A. Marsh, M. A. Baker, J. Li and T. Velkov, Contemporary Anti-Ebola Drug Discovery Approaches and Platforms, ACS Infect. Dis., 2019, 5(1), 35–48, DOI:10.1021/acsinfecdis.8b00285.
- C. Chakraborty, Therapeutics Development for Ebola Virus Disease: A Recent Scenario, Curr. Opin. Pharmacol., 2021, 60, 208–215, DOI:10.1016/j.coph.2021.07.020.
- R. W. Cross, C. E. Mire, H. Feldmann and T. W. Geisbert, Post-Exposure Treatments for Ebola and Marburg Virus Infections, Nat. Rev. Drug Discovery, 2018, 17(6), 413–434, DOI:10.1038/nrd.2017.251.
- Z. A. Shyr, Y.-S. Cheng, D. C. Lo and W. Zheng, Drug Combination Therapy for Emerging Viral Diseases, Drug Discovery Today, 2021, 26(10), 2367–2376, DOI:10.1016/j.drudis.2021.05.008.
- M. M. Levine, Monoclonal Antibody Therapy for Ebola Virus Disease, N. Engl. J. Med., 2019, 381(24), 2365–2366, DOI:10.1056/NEJMe1915350.
- E. H. Miller, J. S. Harrison, S. R. Radoshitzky, C. D. Higgins, X. Chi, L. Dong, J. H. Kuhn, S. Bavari, J. R. Lai and K. Chandran, Inhibition of Ebola Virus Entry by a C-Peptide Targeted to Endosomes*, J. Biol. Chem., 2011, 286(18), 15854–15861, DOI:10.1074/jbc.M110.207084.
- Q. Li, L. Ma, D. Yi, H. Wang, J. Wang, Y. Zhang, Y. Guo, X. Li, J. Zhou, Y. Shi, G. F. Gao and S. Cen, Novel Cyclo-Peptides Inhibit Ebola Pseudotyped Virus Entry by Targeting Primed GP Protein, Antiviral Res., 2018, 155, 1–11, DOI:10.1016/j.antiviral.2018.04.020.
- S. Meakin, J. Nsio, A. Camacho, R. Kitenge, R. M. Coulborn, E. Gignoux, J. Johnson, E. Sterk, E. M. Musenga, S. H. B. Mustafa, F. Finger and S. Ahuka-Mundeke, Effectiveness of rVSV-ZEBOV Vaccination during the 2018–20 Ebola Virus Disease Epidemic in the Democratic Republic of the Congo: A Retrospective Test-Negative Study, Lancet Infect. Dis., 2024, 24(12), 1357–1365, DOI:10.1016/S1473-3099(24)00419-5.
- S. Mulangu, L. E. Dodd, R. T. Davey, O. Tshiani Mbaya, M. Proschan, D. Mukadi, M. Lusakibanza Manzo, D. Nzolo, A. Tshomba Oloma, A. Ibanda, R. Ali, S. Coulibaly, A. C. Levine, R. Grais, J. Diaz, H. C. Lane and J.-J. Muyembe-Tamfum, The Palm Writing Group. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics, N. Engl. J. Med., 2019, 381(24), 2293–2303, DOI:10.1056/NEJMoa1910993.
- Z. Shriver, I. Capila, G. Venkataraman and R. Sasisekharan, Heparin and Heparan Sulfate: Analyzing Structure and Microheterogeneity. in Heparin - A Century of Progress, ed. R. Lever, B. Mulloy and C. P. Page, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 159–176. DOI:10.1007/978-3-642-23056-1_8.
- M. Tamhankar, D. M. Gerhardt, R. S. Bennett, N. Murphy, P. B. Jahrling and J. L. Patterson, Heparan Sulfate Is an Important Mediator of Ebola Virus Infection in Polarized Epithelial Cells, Virol. J., 2018, 15(1), 135, DOI:10.1186/s12985-018-1045-0.
- P.-Y. Lozach, L. Burleigh, I. Staropoli and A. Amara, The C Type Lectins DC-SIGN and L-SIGN. in Glycovirology Protocols, ed. R. J. Sugrue, Humana Press, Totowa, NJ, 2007, pp. 51–68. DOI:10.1007/978-1-59745-393-6_4.
- K. Chakroun, M. Taouai, V. Porkolab, J. Luczkowiak, R. Sommer, C. Cheneau, D. Mathiron, M. A. Ben Maaouia, S. Pilard, R. Abidi, C. Mullié, F. Fieschi, P. J. Cragg, F. Halary, R. Delgado and M. Benazza, Low-Valent Calix[4]Arene Glycoconjugates Based on Hydroxamic Acid Bearing Linkers as Potent Inhibitors in a Model of Ebola Virus Cis-Infection and HCMV-gB-Recombinant Glycoprotein Interaction with MDDC Cells by Blocking DC-SIGN, J. Med. Chem., 2021, 64(19), 14332–14343, DOI:10.1021/acs.jmedchem.1c00818.
- S. Bhatia, L. C. Camacho and R. Haag, Pathogen Inhibition by Multivalent Ligand Architectures, J. Am. Chem. Soc., 2016, 138(28), 8654–8666, DOI:10.1021/jacs.5b12950.
- J. Cramer, B. Aliu, X. Jiang, T. Sharpe, L. Pang, A. Hadorn, S. Rabbani and B. Ernst, Poly-l-Lysine Glycoconjugates Inhibit DC-SIGN-Mediated Attachment of Pandemic Viruses, ChemMedChem, 2021, 16(15), 2345–2353, DOI:10.1002/cmdc.202100348.
- N. Mulherkar, M. Raaben, J. C. de la Torre, S. P. Whelan and K. Chandran, The Ebola Virus Glycoprotein Mediates Entry via a Non-Classical Dynamin-Dependent Macropinocytic Pathway, Virology, 2011, 419(2), 72–83, DOI:10.1016/j.virol.2011.08.009.
- C. M. Stewart, S. S. Dorion, M. A. F. Ottenbrite, N. D. LeBlond, T. K. T. Smith, S. Qiu, M. D. Fullerton, D. Kobasa and M. Côté, A Diacylglycerol Kinase Inhibitor, R-59-022, Blocks Filovirus Internalization in Host Cells, Viruses, 2019, 11(3) DOI:10.3390/v11030206.
- K. Schornberg, S. Matsuyama, K. Kabsch, S. Delos, A. Bouton and J. White, Role of Endosomal Cathepsins in Entry Mediated by the Ebola Virus Glycoprotein, J. Virol., 2006, 80(8), 4174–4178, DOI:10.1128/jvi.80.8.4174-4178.2006.
- K. Gnirß, A. Kühl, C. Karsten, I. Glowacka, S. Bertram, F. Kaup, H. Hofmann and S. Pöhlmann, Cathepsins B and L Activate Ebola but Not Marburg Virus Glycoproteins for Efficient Entry into Cell Lines and Macrophages Independent of TMPRSS2 Expression, Virology, 2012, 424(1), 3–10, DOI:10.1016/j.virol.2011.11.031.
- A. C. Wong, R. G. Sandesara, N. Mulherkar, S. Whelan and K. Chandran, A Forward Genetic Strategy Reveals Destabilizing Mutations in the Ebolavirus Glycoprotein That Alter Its Protease Dependence during Cell Entry, J. Virol., 2010, 84(1), 163–175, DOI:10.1128/jvi.01832-09.
- S. Mehdi, Cell-Penetrating Inhibitors of Calpain, Trends Biochem. Sci., 1991, 16, 150–153, DOI:10.1016/0968-0004(91)90058-4.
- M. Hoffmann, S. V. Kaufmann, C. Fischer, W. Maurer, A.-S. Moldenhauer and S. Pöhlmann, Analysis of Resistance of Ebola Virus Glycoprotein-Driven Entry Against MDL28170, An Inhibitor of Cysteine Cathepsins, Pathogens, 2019, 8(4) DOI:10.3390/pathogens8040192.
- W. A. van der Linden, C. J. Schulze, A. S. Herbert, T. B. Krause, A. A. Wirchnianski, J. M. Dye, K. Chandran and M. Bogyo, Cysteine Cathepsin Inhibitors as Anti-Ebola Agents, ACS Infect. Dis., 2016, 2(3), 173–179, DOI:10.1021/acsinfecdis.5b00130.
- Y. Zhou, P. Vedantham, K. Lu, J. Agudelo, R. Carrion, J. W. Nunneley, D. Barnard, S. Pöhlmann, J. H. McKerrow, A. R. Renslo and G. Simmons, Protease Inhibitors Targeting Coronavirus and Filovirus Entry, Antiviral Res., 2015, 116, 76–84, DOI:10.1016/j.antiviral.2015.01.011.
- T. Plegge, M. Spiegel, N. Krüger, I. Nehlmeier, M. Winkler, M. González Hernández and S. Pöhlmann, Inhibitors of Signal Peptide Peptidase and Subtilisin/Kexin-Isozyme 1 Inhibit Ebola Virus Glycoprotein-Driven Cell Entry by Interfering with Activity and Cellular Localization of Endosomal Cathepsins, PLoS One, 2019, 14(4), e0214968, DOI:10.1371/journal.pone.0214968.
- H. Zhou, J. Li, F. Sun, F. Wang, M. Li, Y. Dong, H. Fan and D. Hu, A Review on Recent Advances in Aloperine Research: Pharmacological Activities and Underlying Biological Mechanisms, Front. Pharmacol., 2020, 11 DOI:10.3389/fphar.2020.538137.
- X. Zhang, Q. Liu, N. Zhang, Q. Li, Z. Liu, Y. Li, L. Gao, Y. Wang, H. Deng and D. Song, Discovery and Evolution of Aloperine Derivatives as Novel Anti-Filovirus Agents through Targeting Entry Stage, Eur. J. Med. Chem., 2018, 149, 45–55, DOI:10.1016/j.ejmech.2018.02.061.
- M. Côté, J. Misasi, T. Ren, A. Bruchez, K. Lee, C. M. Filone, L. Hensley, Q. Li, D. Ory, K. Chandran and J. Cunningham, Small Molecule Inhibitors Reveal Niemann–Pick C1 Is Essential for Ebola Virus Infection, Nature, 2011, 477(7364), 344–348, DOI:10.1038/nature10380.
- H. Liu, Y. Tian, K. Lee, P. Krishnan, M. K.-M. Wang, S. Whelan, E. Mevers, V. Soloveva, B. Dedic, X. Liu and J. M. Cunningham, Identification of Potent Ebola Virus Entry Inhibitors with Suitable Properties for in Vivo Studies, J. Med. Chem., 2018, 61(14), 6293–6307, DOI:10.1021/acs.jmedchem.8b00704.
- A. Basu, D. M. Mills, D. Mitchell, E. Ndungo, J. D. Williams, A. S. Herbert, J. M. Dye, D. T. Moir, K. Chandran, J. L. Patterson, L. Rong and T. L. Bowlin, Novel Small Molecule Entry Inhibitors of Ebola Virus, J. Infect. Dis., 2015, 212(suppl_2), S425–S434, DOI:10.1093/infdis/jiv223.
- I. Khan, S. Li, L. Tao, C. Wang, B. Ye, H. Li, X. Liu, I. Ahmad, W. Su, G. Zhong, Z. Wen, J. Wang, R.-H. Hua, A. Ma, J. Liang, X.-P. Wan, Z.-G. Bu and Y.-H. Zheng, Tubeimosides Are Pan-Coronavirus and Filovirus Inhibitors That Can Block Their Fusion Protein Binding to Niemann-Pick C1, Nat. Commun., 2024, 15(1), 162, DOI:10.1038/s41467-023-44504-4.
- T. Long, X. Qi, A. Hassan, Q. Liang, J. K. De Brabander and X. Li, Structural Basis for Itraconazole-Mediated NPC1 Inhibition, Nat. Commun., 2020, 11(1), 152, DOI:10.1038/s41467-019-13917-5.
- S. Kummer, A. Lander, J. Goretzko, N. Kirchoff, U. Rescher and S. Schloer, Pharmacologically Induced Endolysosomal Cholesterol Imbalance through Clinically Licensed Drugs Itraconazole and Fluoxetine Impairs Ebola Virus Infection in Vitro, Emerging Microbes Infect., 2022, 11(1), 195–207, DOI:10.1080/22221751.2021.2020598.
- D. Yi, Q. Li, H. Wang, K. Lv, L. Ma, Y. Wang, J. Wang, Y. Zhang, M. Liu, X. Li, J. Qi, Y. Shi, G. F. Gao and S. Cen, Repurposing of Berbamine Hydrochloride to Inhibit Ebola Virus by Targeting Viral Glycoprotein, Acta Pharm. Sin. B, 2022, 12(12), 4378–4389, DOI:10.1016/j.apsb.2022.05.023.
- R. J. Cenedella, Cholesterol Synthesis Inhibitor U18666A and the Role of Sterol Metabolism and Trafficking in Numerous Pathophysiological Processes, Lipids, 2009, 44(6), 477–487, DOI:10.1007/s11745-009-3305-7.
- C. J. Shoemaker, K. L. Schornberg, S. E. Delos, C. Scully, H. Pajouhesh, G. G. Olinger, L. M. Johansen and J. M. White, Multiple Cationic Amphiphiles Induce a Niemann-Pick C Phenotype and Inhibit Ebola Virus Entry and Infection, PLoS One, 2013, 8(2), e56265, DOI:10.1371/journal.pone.0056265.
- F. Lu, Q. Liang, L. Abi-Mosleh, A. Das, J. K. De Brabander, J. L. Goldstein and M. S. Brown, Identification of NPC1 as the Target of U18666A, an Inhibitor of Lysosomal Cholesterol Export and Ebola Infection, eLife, 2015, 4, e12177, DOI:10.7554/eLife.12177.
- F. Lasala, A. García-Rubia, C. Requena, I. Galindo, M. A. Cuesta-Geijo, I. García-Dorival, P. Bueno, N. Labiod, J. Luczkowiak, A. Martinez, N. E. Campillo, C. Alonso, R. Delgado and C. Gil, Identification of Potential Inhibitors of Protein-Protein Interaction Useful to Fight against Ebola and Other Highly Pathogenic Viruses, Antiviral Res., 2021, 186, 105011, DOI:10.1016/j.antiviral.2021.105011.
- M. Izumida, O. Kotani, H. Hayashi, C. Smith, T. Fukuda, K. Suga, M. Iwao, F. Ishibashi, H. Sato and Y. Kubo, Unique Mode of Antiviral Action of a Marine Alkaloid against Ebola Virus and SARS-CoV-2, Viruses, 2022, 14(4), 816, DOI:10.3390/v14040816.
- L. L. Wang, N. Palermo, L. Estrada, C. Thompson, J. J. Patten, M. Anantpadma, R. A. Davey and S.-H. Xiang, Identification of Filovirus Entry Inhibitors Targeting the Endosomal Receptor NPC1 Binding Site, Antiviral Res., 2021, 189, 105059, DOI:10.1016/j.antiviral.2021.105059.
- L. M. Johansen, J. M. Brannan, S. E. Delos, C. J. Shoemaker, A. Stossel, C. Lear, B. G. Hoffstrom, L. E. DeWald, K. L. Schornberg, C. Scully, J. Lehár, L. E. Hensley, J. M. White and G. G. Olinger, FDA-Approved Selective Estrogen Receptor Modulators Inhibit Ebola Virus Infection, Sci. Transl. Med., 2013, 5(190), 190ra79, DOI:10.1126/scitranslmed.3005471.
- Y. Zhao, J. Ren, K. Harlos, D. Jones, A. Zeltina, T. Bowden, S. Padilla-Parra, E. Fry and D. Stuart, Toremifene Interacts with and Destabilizes the Ebola Virus Glycoprotein, Nature, 2016, 535(7610), 169–172 CrossRef CAS.
- L. Cooper, A. Schafer, Y. Li, H. Cheng, B. Medegan Fagla, Z. Shen, R. Nowar, K. Dye, M. Anantpadma, R. A. Davey, G. R. J. Thatcher, L. Rong and R. Xiong, Screening and Reverse-Engineering of Estrogen Receptor Ligands as Potent Pan-Filovirus Inhibitors, J. Med. Chem., 2020, 63(19), 11085–11099, DOI:10.1021/acs.jmedchem.0c01001.
- Y. Zhao, J. Ren, E. E. Fry, J. Xiao, A. R. Townsend and D. I. Stuart, Structures of Ebola Virus Glycoprotein Complexes with Tricyclic Antidepressant and Antipsychotic Drugs, J. Med. Chem., 2018, 61(11), 4938–4945, DOI:10.1021/acs.jmedchem.8b00350.
- F. Shaikh, Y. Zhao, L. Alvarez, M. Iliopoulou, C. Lohans, C. J. Schofield, S. Padilla-Parra, S. W. I. Siu, E. E. Fry, J. Ren and D. I. Stuart, Structure-Based in Silico Screening Identifies a Potent Ebolavirus Inhibitor from a Traditional Chinese Medicine Library, J. Med. Chem., 2019, 62(6), 2928–2937, DOI:10.1021/acs.jmedchem.8b01328.
- N. Y. Tsang, W.-F. Li, E. Varhegyi, L. Rong and H.-J. Zhang, Ebola Entry Inhibitors Discovered from Maesa Perlarius, Int. J. Mol. Sci., 2022, 23(5), 2620, DOI:10.3390/ijms23052620.
- Y. Gao, H. Cheng, S. Khan, G. Xiao, L. Rong and C. Bai, Development of Coumarine Derivatives as Potent Anti-Filovirus Entry Inhibitors Targeting Viral Glycoprotein, Eur. J. Med. Chem., 2020, 204, 112595, DOI:10.1016/j.ejmech.2020.112595.
- A. Schafer, H. Cheng, R. Xiong, V. Soloveva, C. Retterer, F. Mo, S. Bavari, G. Thatcher and L. Rong, Repurposing Potential of 1st Generation H1-Specific Antihistamines as Anti-Filovirus Therapeutics, Antiviral Res., 2018, 157, 47–56, DOI:10.1016/j.antiviral.2018.07.003.
- S. Ekins, J. Freundlich, A. Clark, M. Anantpadma, R. Davey and P. Madrid, Machine Learning Models Identify Molecules Active against the Ebola Virus in Vitro, F1000Research, 2017, 4(1091) DOI:10.12688/f1000research.7217.3.
- T. R. Lane and S. Ekins, Toward the Target: Tilorone, Quinacrine, and Pyronaridine Bind to Ebola Virus Glycoprotein, ACS Med. Chem. Lett., 2020, 11(8), 1653–1658, DOI:10.1021/acsmedchemlett.0c00298.
- S. Ekins, M. A. Lingerfelt, J. E. Comer, A. N. Freiberg, J. C. Mirsalis, K. O'Loughlin, A. Harutyunyan, C. McFarlane, C. E. Green and P. B. Madrid, Efficacy of Tilorone Dihydrochloride against Ebola Virus Infection, Antimicrob. Agents Chemother., 2018, 62(2) DOI:10.1128/aac.01711-17.
- S. Han, H. Li, W. Chen, L. Yang, X. Tong, J. Zuo and Y. Hu, Discovery of Potent Ebola Entry Inhibitors with (3S,4aS,8aS)-2-(3-Amino-2-Hydroxypropyl) Decahydroisoquinoline-3-Carboxamide Scaffold, Eur. J. Med. Chem., 2022, 240, 114608, DOI:10.1016/j.ejmech.2022.114608.
- Q. Cui, H. Cheng, R. Xiong, G. Zhang, R. Du, M. Anantpadma, R. A. Davey and L. Rong, Identification of Diaryl-Quinoline Compounds as Entry Inhibitors of Ebola Virus, Viruses, 2018, 10(12), 678, DOI:10.3390/v10120678.
- M. D. Argade, J. G. Achi, R. Bott, K. M. Morsheimer, C. D. Owen, C. A. Zielinski, A. M. Gaisin, M. Alvarez, T. W. Moore, F. Bu, F. Li, M. Cameron, M. Anantpadma, R. A. Davey, N. P. Peet, L. Rong and I. N. Gaisina, Guardians at the Gate: Optimization of Small Molecule Entry Inhibitors of Ebola and Marburg Viruses, J. Med. Chem., 2024, 68(1), 135–155, DOI:10.1021/acs.jmedchem.4c01646.
- I. N. Gaisina, N. P. Peet, L. Wong, A. M. Schafer, H. Cheng, M. Anantpadma, R. A. Davey, G. R. J. Thatcher and L. Rong, Discovery and Structural Optimization of 4-(Aminomethyl)Benzamides as Potent Entry Inhibitors of Ebola and Marburg Virus Infections, J. Med. Chem., 2020, 63(13), 7211–7225, DOI:10.1021/acs.jmedchem.0c00463.
- M. B. Plewe, N. V. Sokolova, V. R. Gantla, E. R. Brown, S. Naik, A. Fetsko, D. D. Lorimer, D. M. Dranow, H. Smutney, J. Bullen, R. Sidhu, A. Master, J. Wang, E. A. Kallel, L. Zhang, B. Kalveram, A. N. Freiberg, G. Henkel and K. McCormack, Discovery of Adamantane Carboxamides as Ebola Virus Cell Entry and Glycoprotein Inhibitors, ACS Med. Chem. Lett., 2020, 11(6), 1160–1167, DOI:10.1021/acsmedchemlett.0c00025.
- S. Martin, A. I. Chiramel, M. L. Schmidt, Y.-C. Chen, N. Whitt, A. Watt, E. C. Dunham, K. Shifflett, S. Traeger, A. Leske, E. Buehler, C. Martellaro, J. Brandt, L. Wendt, A. Müller, S. Peitsch, S. M. Best, J. Stech, S. Finke, A. Römer-Oberdörfer, A. Groseth, H. Feldmann and T. Hoenen, A Genome-Wide siRNA Screen Identifies a Druggable Host Pathway Essential for the Ebola Virus Life Cycle, Genome Med., 2018, 10(1), 58, DOI:10.1186/s13073-018-0570-1.
- L. S. Uebelhoer, C. G. Albariño, L. K. McMullan, A. K. Chakrabarti, J. P. Vincent, S. T. Nichol and J. S. Towner, High-Throughput, Luciferase-Based Reverse Genetics Systems for Identifying Inhibitors of Marburg and Ebola Viruses, Antiviral Res., 2014, 106, 86–94, DOI:10.1016/j.antiviral.2014.03.018.
- O. Reynard, X.-N. Nguyen, N. Alazard-Dany, V. Barateau, A. Cimarelli and V. E. Volchkov, Identification of a New Ribonucleoside Inhibitor of Ebola Virus Replication, Viruses, 2015, 7(12), 6233–6240, DOI:10.3390/v7122934.
- T. K. Warren, J. Wells, R. G. Panchal, K. S. Stuthman, N. L. Garza, S. A. Van Tongeren, L. Dong, C. J. Retterer, B. P. Eaton, G. Pegoraro, S. Honnold, S. Bantia, P. Kotian, X. Chen, B. R. Taubenheim, L. S. Welch, D. M. Minning, Y. S. Babu, W. P. Sheridan and S. Bavari, Protection against Filovirus Diseases by a Novel Broad-Spectrum Nucleoside Analogue BCX4430, Nature, 2014, 508(7496), 402–405, DOI:10.1038/nature13027.
- J. G. Julander, J. F. Demarest, R. Taylor, B. B. Gowen, D. M. Walling, A. Mathis and Y. S. Babu, An Update on the Progress of Galidesivir (BCX4430), a Broad-Spectrum Antiviral, Antiviral Res., 2021, 195, 105180, DOI:10.1016/j.antiviral.2021.105180.
- T. Baranovich, S.-S. Wong, J. Armstrong, H. Marjuki, R. Webby, R. Webster and E. Govorkova, T-705 (Favipiravir) Induces Lethal Mutagenesis in Influenza A H1N1 Viruses In Vitro, J. Virol., 2013, 87(7), 3741–3751, DOI:10.1128/jvi.02346-12.
- Y. Furuta, K. Takahashi, M. Kuno-Maekawa, H. Sangawa, S. Uehara, K. Kozaki, N. Nomura, H. Egawa and K. Shiraki, Mechanism of Action of T-705 against Influenza Virus, Antimicrob. Agents Chemother., 2005, 49(3), 981–986, DOI:10.1128/aac.49.3.981-986.2005.
- S. J. Smither, L. S. Eastaugh, J. A. Steward, M. Nelson, R. P. Lenk and M. S. Lever, Post-Exposure Efficacy of Oral T-705 (Favipiravir) against Inhalational Ebola Virus Infection in a Mouse Model, Antiviral Res., 2014, 104, 153–155, DOI:10.1016/j.antiviral.2014.01.012.
- M. N. Rahim, Z. Zhang, S. He, W. Zhu, L. Banadyga, D. Safronetz and X. Qiu, Postexposure Protective Efficacy of T-705 (Favipiravir) Against Sudan Virus Infection in Guinea Pigs, J. Infect. Dis., 2018, 218(suppl_5), S649–S657, DOI:10.1093/infdis/jiy303.
- T. H. T. Nguyen, J. Guedj, X. Anglaret, C. Laouénan, V. Madelain, A.-M. Taburet, S. Baize, D. Sissoko, B. Pastorino, A. Rodallec, G. Piorkowski, S. Carazo, M. N. Conde, J.-L. Gala, J. A. Bore, C. Carbonnelle, F. Jacquot, H. Raoul, D. Malvy, X. de Lamballerie and F. Mentré, on behalf of the JIKI study group. Favipiravir Pharmacokinetics in Ebola-Infected Patients of the JIKI Trial Reveals Concentrations Lower than Targeted, PLoS Neglected Trop. Dis., 2017, 11(2), e0005389, DOI:10.1371/journal.pntd.0005389.
- Potential Ebola Therapies and Vaccines; World Health Organization, 2014, https://www.who.int/publications/i/item/WHO_EVD_HIS_EMP_14.1.
- J. Dunning, S. B. Kennedy, A. Antierens, J. Whitehead, I. Ciglenecki, G. Carson, R. Kanapathipillai, L. Castle, R. Howell-Jones, R. Pardinaz-Solis, J. Grove, J. Scott, T. Lang, P. Olliaro and P. W. Horby, Experimental Treatment of Ebola Virus Disease with Brincidofovir, PLoS One, 2016, 11(9), e0162199, DOI:10.1371/journal.pone.0162199.
- A. Cho, O. L. Saunders, T. Butler, L. Zhang, J. Xu, J. E. Vela, J. Y. Feng, A. S. Ray and C. U. Kim, Synthesis and Antiviral Activity of a Series of 1′-Substituted 4-Aza-7,9-Dideazaadenosine C-Nucleosides, Bioorg. Med. Chem. Lett., 2012, 22(8), 2705–2707, DOI:10.1016/j.bmcl.2012.02.105.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][Triazin-4-Amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses, J. Med. Chem., 2017, 60(5), 1648–1661, DOI:10.1021/acs.jmedchem.6b01594.
- T. K. Warren, R. Jordan, M. K. Lo, A. S. Ray, R. L. Mackman, V. Soloveva, D. Siegel, M. Perron, R. Bannister, H. C. Hui, N. Larson, R. Strickley, J. Wells, K. S. Stuthman, S. A. Van Tongeren, N. L. Garza, G. Donnelly, A. C. Shurtleff, C. J. Retterer, D. Gharaibeh, R. Zamani, T. Kenny, B. P. Eaton, E. Grimes, L. S. Welch, L. Gomba, C. L. Wilhelmsen, D. K. Nichols, J. E. Nuss, E. R. Nagle, J. R. Kugelman, G. Palacios, E. Doerffler, S. Neville, E. Carra, M. O. Clarke, L. Zhang, W. Lew, B. Ross, Q. Wang, K. Chun, L. Wolfe, D. Babusis, Y. Park, K. M. Stray, I. Trancheva, J. Y. Feng, O. Barauskas, Y. Xu, P. Wong, M. R. Braun, M. Flint, L. K. McMullan, S.-S. Chen, R. Fearns, S. Swaminathan, D. L. Mayers, C. F. Spiropoulou, W. A. Lee, S. T. Nichol, T. Cihlar and S. Bavari, Therapeutic Efficacy of the Small Molecule GS-5734 against Ebola Virus in Rhesus Monkeys, Nature, 2016, 531(7594), 381–385, DOI:10.1038/nature17180.
- R. W. Cross, Z. A. Bornholdt, A. N. Prasad, C. Woolsey, V. Borisevich, K. N. Agans, D. J. Deer, D. M. Abelson, D. H. Kim, W. S. Shestowsky, L. A. Campbell, E. Bunyan, J. B. Geisbert, N. S. Dobias, K. A. Fenton, D. P. Porter, L. Zeitlin and T. W. Geisbert, Combination Therapy with Remdesivir and Monoclonal Antibodies Protects Nonhuman Primates against Advanced Sudan Virus Disease, JCI insight, 2022, 7(10) DOI:10.1172/jci.insight.159090.
- M. K. Lo, C. G. Albariño, J. K. Perry, S. Chang, E. P. Tchesnokov, L. Guerrero, A. Chakrabarti, P. Shrivastava-Ranjan, P. Chatterjee, L. K. McMullan, R. Martin, R. Jordan, M. Götte, J. M. Montgomery, S. T. Nichol, M. Flint, D. Porter and C. F. Spiropoulou, Remdesivir Targets a Structurally Analogous Region of the Ebola Virus and SARS-CoV-2 Polymerases, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(43), 26946–26954, DOI:10.1073/pnas.2012294117.
- M. Gong, Y. Yang, Y. Huang, T. Gan, Y. Wu, H. Gao, Q. Li, J. Nie, W. Huang, Y. Wang, R. Zhang, J. Zhong, F. Deng, Y. Rao and Q. Ding, Novel Quinolone Derivatives Targeting Human Dihydroorotate Dehydrogenase Suppress Ebola Virus Infection in Vitro, Antiviral Res., 2021, 194, 105161, DOI:10.1016/j.antiviral.2021.105161.
- Y. Yang, L. Cao, H. Gao, Y. Wu, Y. Wang, F. Fang, T. Lan, Z. Lou and Y. Rao, Discovery, Optimization, and Target Identification of Novel Potent Broad-Spectrum Antiviral Inhibitors, J. Med. Chem., 2019, 62(8), 4056–4073, DOI:10.1021/acs.jmedchem.9b00091.
- R. L. Mackman, R. V. Kalla, D. Babusis, J. Pitts, K. T. Barrett, K. Chun, V. Du Pont, L. Rodriguez, J. Moshiri, Y. Xu, M. Lee, G. Lee, B. Bleier, A.-Q. Nguyen, B. M. O'Keefe, A. Ambrosi, M. Cook, J. Yu, K. E. Dempah, E. Bunyan, N. C. Riola, X. Lu, R. Liu, A. Davie, T.-Y. Hsiang, J. Dearing, M. Vermillion, M. Gale Jr., A. Niedziela-Majka, J. Y. Feng, C. Hedskog, J. P. Bilello, R. Subramanian and T. Cihlar, Discovery of GS-5245 (Obeldesivir), an Oral Prodrug of Nucleoside GS-441524 That Exhibits Antiviral Efficacy in SARS-CoV-2-Infected African Green Monkeys, J. Med. Chem., 2023, 66(17), 11701–11717, DOI:10.1021/acs.jmedchem.3c00750.
- D. W. Leung, D. Borek, P. Luthra, J. M. Binning, M. Anantpadma, G. Liu, I. B. Harvey, Z. Su, A. Endlich-Frazier, J. Pan, R. S. Shabman, W. Chiu, R. A. Davey, Z. Otwinowski, C. F. Basler and G. K. Amarasinghe, An Intrinsically Disordered Peptide from Ebola Virus VP35 Controls Viral RNA Synthesis by Modulating Nucleoprotein-RNA Interactions, Cell Rep., 2015, 11(3), 376–389, DOI:10.1016/j.celrep.2015.03.034.
- G. Liu, P. J. Nash, B. Johnson, C. Pietzsch, M. X. G. Ilagan, A. Bukreyev, C. F. Basler, T. L. Bowlin, D. T. Moir, D. W. Leung and G. K. Amarasinghe, A Sensitive in Vitro High-Throughput Screen To Identify Pan-Filoviral Replication Inhibitors Targeting the VP35–NP Interface, ACS Infect. Dis., 2017, 3(3), 190–198, DOI:10.1021/acsinfecdis.6b00209.
- W. Xu, P. Luthra, C. Wu, J. Batra, D. W. Leung, C. F. Basler and G. K. Amarasinghe, Ebola Virus VP30 and Nucleoprotein Interactions Modulate Viral RNA Synthesis, Nat. Commun., 2017, 8(1), 15576, DOI:10.1038/ncomms15576.
- Y. Ma, X. Hong, F. Wu, X. Xu, R. Li, J. Zhong, Y. Zhou, S. Liu, J. Zhan and W. Xu, Inhibiting the Transcription and Replication of Ebola Viruses by Disrupting the Nucleoprotein and VP30 Protein Interaction with Small Molecules, Acta Pharmacol. Sin., 2023, 44(7), 1487–1499, DOI:10.1038/s41401-023-01055-0.
- E. Broni, C. Ashley, J. Adams, H. Manu, E. Aikins, M. Okom, W. A. Miller, M. D. Wilson and S. K. Kwofie, Cheminformatics-Based Study Identifies Potential Ebola VP40 Inhibitors, Int. J. Mol. Sci., 2023, 24(7), 6298, DOI:10.3390/ijms24076298.
- S. J. Capuzzi, W. Sun, E. N. Muratov, C. Martínez-Romero, S. He, W. Zhu, H. Li, G. Tawa, E. G. Fisher, M. Xu, P. Shinn, X. Qiu, A. García-Sastre, W. Zheng and A. Tropsha, Computer-Aided Discovery and Characterization of Novel Ebola Virus Inhibitors, J. Med. Chem., 2018, 61(8), 3582–3594, DOI:10.1021/acs.jmedchem.8b00035.
- S. Khan, Z. Fakhar and A. Ahmad, Targeting Ebola Virus VP40 Protein through Novel Inhibitors: Exploring the Structural and Dynamic Perspectives on Molecular Landscapes, J. Mol. Model., 2021, 27(2), 49, DOI:10.1007/s00894-021-04682-8.
- R. P. Bennett, C. L. Finch, E. N. Postnikova, R. A. Stewart, Y. Cai, S. Yu, J. Liang, J. Dyall, J. D. Salter, H. C. Smith and J. H. Kuhn, A Novel Ebola Virus VP40 Matrix Protein-Based Screening for Identification of Novel Candidate Medical Countermeasures, Viruses, 2021, 13(1), 52, DOI:10.3390/v13010052.
- S. Urata, T. Noda, Y. Kawaoka, S. Morikawa, H. Yokosawa and J. Yasuda, Interaction of Tsg101 with Marburg Virus VP40 Depends on the PPPY Motif, but Not the PT/SAP Motif as in the Case of Ebola Virus, and Tsg101 Plays a Critical Role in the Budding of Marburg Virus-Like Particles Induced by VP40, NP, and GP, J. Virol., 2007, 81(9), 4895–4899, DOI:10.1128/jvi.02829-06.
- S. Urata and J. Yasuda, Regulation of Marburg Virus (MARV) Budding by Nedd4.1: A Different WW Domain of Nedd4.1 Is Critical for Binding to MARV and Ebola Virus VP40, J. Gen. Virol., 2010, 91, 228–234, DOI:10.1099/vir.0.015495-0.
- Y. Liu, M. S. Lee, M. A. Olson and R. N. Harty, Bimolecular Complementation to Visualize Filovirus VP40-Host Complexes in Live Mammalian Cells: Toward the Identification of Budding Inhibitors, Adv. Virol., 2011, 2011, 341816, DOI:10.1155/2011/341816.
- Z. Han, J. Lu, Y. Liu, B. Davis, M. Lee, M. Olson, G. Ruthel, B. Freedman, M. Schnell, J. Wrobel, A. Reitz and R. Harty, Small-Molecule Probes Targeting the Viral PPxY-Host Nedd4 Interface Block Egress of a Broad Range of RNA Viruses, J. Virol., 2014, 88(13), 7294–7306, DOI:10.1128/jvi.00591-14.
- H. M. Loughran, Z. Han, J. E. Wrobel, S. E. Decker, G. Ruthel, B. D. Freedman, R. N. Harty and A. B. Reitz, Quinoxaline-Based Inhibitors of Ebola and Marburg VP40 Egress, Bioorg. Med. Chem. Lett., 2016, 26(15), 3429–3435, DOI:10.1016/j.bmcl.2016.06.053.
| This journal is © The Royal Society of Chemistry 2025 |