
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel lithocholic acid-diindolylmethane hybrids as potent sialyltransferase inhibitors targeting triple-negative breast cancer: a molecular hybridization approach
Christian Angelo P.
Concio
 abc,
Ser John Lynon P.
Perez
ac,
Tzu-Ting
Chang
a,
Chia-Ling
Chen
c,
Yi-Ting
He
cf,
Susan D.
Arco
*b and
Wen-Shan
Li
*acdef
abc,
Ser John Lynon P.
Perez
ac,
Tzu-Ting
Chang
a,
Chia-Ling
Chen
c,
Yi-Ting
He
cf,
Susan D.
Arco
*b and
Wen-Shan
Li
*acdef
aBiomedical Translational Research Center, Academia Sinica, No. 99, Lane 130, Section 1, Yanyuan Road, Nangang District, Taipei City 115, Taiwan. E-mail: wenshan@as.edu.tw
bInstitute of Chemistry, College of Science, University of the Philippines Diliman, Quezon City, Metro Manila, Philippines
cInstitute of Chemistry, Academia Sinica, No. 128, Section 2, Yuanyuan Road, Nangang District, Taipei City 115, Taiwan
dDoctoral Degree Program in Marine Biotechnology, National Sun Yat-Sen University, Kaohsiung 804, Taiwan
ePhD Program in Biotechnology Research and Development, Taipei Medical University, Taipei 115, Taiwan
fDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 807, Taiwan
First published on 28th August 2025
Abstract
Molecular hybridization, an emerging strategy for the discovery of new anticancer therapeutics, shows promise as a powerful tool for the development of new sialyltransferase (ST) inhibitors for cancer treatment. This concept inspired the design of novel ST inhibitors through the hybridization of lithocholic acid and diindolylmethane, leading to the discovery of LCA-DIM hybrids as potential chemical entities targeting STs. Preliminary screening revealed the significance of the DIM moiety and incorporation of Asp linker on enhancing the inhibitory activity and selectivity of the hybrids towards ST6GAL1, inhibiting up to 100% of ST6GAL1 activity at 25 μM with no ST3GAL1 inhibition even at 500 μM. Incorporation of various 5,5′-substituents enhanced the inherent antimigration properties of the hybrids, with IAN-5B (R = Cl) and IAN-15B (R = N3) presenting the highest antimigration activity across several triple-negative breast cancer (TNBC) cell lines (MDA-MB-231, BT549, Hs578T) and considerable antiangiogenetic effect by suppressing HUVEC tube formation. This could be attributed to the excellent ST6GAL1 inhibitory activities of the two hybrids with IC50 values of 6.6 ± 0.2 μM and 3.3 ± 0.1 μM, respectively. Overall, this study highlights LCA-DIM hybrids as novel, potent, and N-glycan-selective ST inhibitors with promising antimigration properties against aggressive TNBC.
1. Introduction
Sialic acids are a diverse group of nine-carbon monosaccharides typically found at the terminal ends of cell surface glycoproteins, where they play essential roles in numerous biological functions. Sialylation induces morphological changes in the cellular surface, resulting in the stabilization of enzymes and hormones, as well as mediating interactions with other neighboring cells. Furthermore, sialic acids serve as ligands for glycan-binding proteins such as selectins, galectins, and siglecs, establishing their critical role in cellular interactions and cell-to-cell recognition.1–3 Although essential for normal cellular functions, this post-translational modification is often exploited by various carcinomas, resulting in hypersialylation — characterized by the excessive accumulation of sialic acids on the cancer cell surface. Extensive studies have established the direct association between hypersialylation and cancer progression and metastasis. For instance, hypersialylation is known to impair cancer cell adhesion at the primary tumor site and promote the epithelial-to-mesenchymal transition (EMT).4–6 Additionally, this process also enables cancer cells to evade immune surveillance and resist apoptosis by mimicking essential glycan motifs.3,6,7 Despite the significant challenges in targeting hypersialylation, the enzyme sialyltransferase plays a pivotal role in this process and remains a critical target for ongoing research.Sialyltransferase (ST) is a key regulator of the sialylation pathway responsible for transferring the sialic acid group from CMP-Neu5Ac to the terminal hydroxyl groups of glycoproteins or glycolipids, thereby its upregulation contribute to the abnormal sialic acid accumulation seen in hypersialylation.1,8 To date, a variety of human STs have already been discovered, demonstrating diversity in their preferred sugar substrates, type of glycosidic linkage formed, and specificity for N-glycans or O-glycans. Interestingly, specific ST isozymes have been shown to be overexpressed and play a key role in promoting hypersialylation in multiple carcinomas including breast,9,10 ovarian,11–13 pancreatic,12,14,15 and colorectal cancer,16,17 positioning the inhibition of ST isozymes with high potency and selectivity as a promising therapeutic strategy for these malignancies.
Although there is a rising attention towards the therapeutic applications of sialyltransferase for cancer treatment, only a few compounds capable of effectively inhibiting ST activity have been recorded,18–20 emphasizing the need for the exploration of novel and more potent ST inhibitors. Notably, molecular hybridization has emerged as a promising strategy in drug discovery given the vast array of available structural motifs with advantageous pharmacological properties. In the context of ST inhibitors, lithocholic acid (LCA) has been highlighted as a key motif, leading to the discovery of potent ST inhibitors such as Lith-O-Asp (see Fig. 1). Both LCA-based structures have demonstrated significant antiangiogenic and antimetastatic effects in lung carcinoma in vitro and in vivo.21–23 Utilization of this steroidal motif also resulted in the development of first ever recorded N-glycan selective ST inhibitors such as FCW34,24 FCW393 (ref. 25) and SPP-002,26 capable of inhibiting melanoma and/or breast cancer progression. Interestingly, recent exploration of the LCA core suggests the importance of indole27,28 and indole-like29 moieties for ST inhibition activity, a trend also observed in other proposed ST inhibitors. Motivated by this, we surveyed various indole-containing metabolites for integration into the LCA core to enhance its inherent ST inhibitory activity.
 | ||
| Fig. 1 Structures of representative lithocholic acid (LCA) and 3,3′-diindolylmethane (3,3′-DIM) derivatives, and the schematic representation of the molecular hybridization approach used in this study for the development of LCA-DIM hybrids. | ||
Among these, the indole alkaloid 3,3′-diindolylmethane (DIM), a metabolic by-product formed during the digestion of cruciferous vegetables, is particularly noteworthy in anticancer research. DIM has been shown to independently suppress metastasis and promote apoptosis across various carcinoma types.30–32 Moreover, DIM-containing compounds, including naturally occurring derivatives (see Fig. 1) such as vibrindole A,33 and trisindoline,34 as well as synthetic analogs like phemindole35 and SK228,36,37 exhibit excellent anticancer properties. Inspired by these insights, herein we present the development of LCA-DIM hybrids (depicted in Fig. 1) as a novel class of LCA-based ST inhibitors with enhanced inhibitory activity and selectivity against the N-glycan sialylating ST6GAL1, presenting potential as antimetastatic agents against the aggressive triple negative breast cancer (TNBC).
2. Results and discussion
2.1 Design and synthesis of LCA-DIM hybrids
On the design of LCA-based ST inhibitors, most modifications were introduced at the C3–OH region of LCA due to the ST activities of C3–OH modified Lith-O-Asp and AL-10.21,23 However, recent advancement have introduced modifications at the COOH of LCA to improve the overall ST inhibitory activity and selectivity of LCA-based inhibitors.24,26 To further utilize this region, we strategically positioned the DIM moiety on the COOH moiety of LCA. For this purpose, we present two LCA-DIM hybrids – the direct LCA-DIM hybrid 4 and the Asp linked LCA-DIM hybrid 11. The synthetic pathways used for the preparation of the hybrids were detailed in Scheme 1.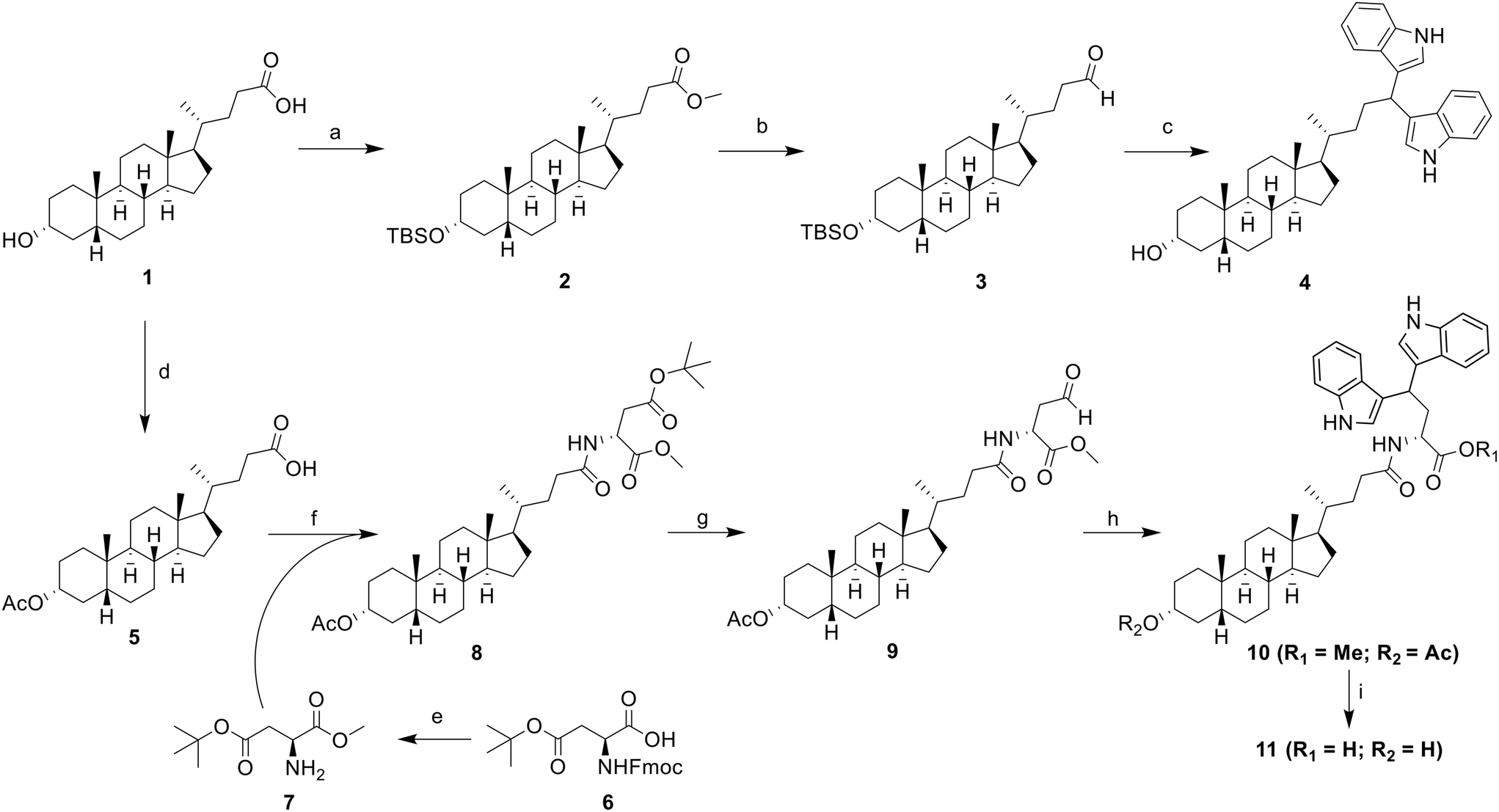 | ||
| Scheme 1 Synthesis of LCA-DIM hybrids 4 and 11. Reagents and conditions: a) i) MeOH, cat. HCl, RT, 8 h; ii) TBSCl, imidazole, DMF, 80 °C, 6 h, 64% (over two steps); b) (i) LiAlH4, THF, 0 °C, 4 h; (ii) PCC, DCM, RT, 12 h, 50%; c) indole, 5% I2 in MeOH, RT, 8 h, 41%; d) Ac2O, DMAP, DCM, RT, 2 h, 99%; e) i) EDC, HOBt, TEA, MeOH, DCM, RT, 8 h; ii) 1-dodecanethiol, DBU, DCM, RT, 0.5 h, 97% (over two steps); f) EDC, HOBt, DIPEA, DCM, RT, 8 h, 90%; g) i) TFA, DCM, RT, 2 h; ii) 1-dodecanthiol, EDC, cat. DMAP, DCM, RT, 4 h; iii) Et3SiH, 10% Pd/C, 4 Å MS, THF/acetone, 2–3 h, 55% (over three steps); h) indole, I2, DCM, RT, 88%; i) Ba(OH)2 8H2O, MeOH, 8 h, RT, 60%. | ||
Successful synthesis of hybrid 4 was achieved through a four-step synthesis process with an overall yield of 24%. This involved the complete protection of LCA (1) through methyl esterification of its COOH group under Fischer conditions, then the silylation of the C3–OH group with tert-butyldimethylsilyl chloride (TBSCl) to successfully afford intermediate 2. Subsequently, the ester group of intermediate 2 was reduced to obtain its alcohol form via LAH reduction. The resulting alcohol was then oxidized to its aldehyde form using pyridinium chlorochromate (PCC) as the oxidizing agent, obtaining the LCA-aldehyde intermediate 3 at 50% yield. Finally, the aldehyde intermediate 3 was transformed into LCA-DIM hybrid 4 using 5% I2 in MeOH. In this reaction, the condition simultaneously catalyzes the nucleophilic addition of two indole groups towards the intermediate's aldehyde moiety forming the diindolyl motif while removing the silyl ether protection on the hybrid's C3–OH,38 affording a yield of 41%.
Similar approach was used for the synthesis of the Asp-linked hybrid 11 which involved the preparation of an aldehyde intermediate, followed by I2-catalyzed diindolylation of the aldehyde group forming the DIM motif. Before the preparation of the aldehyde moiety in the structure, the Asp linker was initially introduced in the LCA core. Initially, the C3–OH of LCA was protected through acetylation with acetic anhydride and catalytic amount of DMAP to obtain intermediate 5. The Asp linker 7 was then synthesized via a two-step protection-deprotection process. To begin with, the free COOH group of Fmoc-Asp(OtBu)–COOH (6) was coupled with MeOH using EDCI-HCl/HOBt coupling agents and TEA as base. This was followed by Fmoc deprotection, which was carried out using DBU as the deprotection agent and 1-dodecanethiol as the dibenzofulvene scavenging agent to enhance the overall yield of the intermediate. The Asp linker was then introduced on intermediate 5via EDCI-HCl/HOBt/DIPEA-mediated coupling reaction, yielding to the Asp-LCA intermediate 8. Next step involved the selective reduction of the COOtBu side chain of the Asp-LCA conjugate via Fukuyama reduction.39,40 Initially, the tert-butyl protecting group on the linker's side chain was removed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TFA/DCM solvent mixture. The free acid was then coupled with 1-dodecanethiol under Steglich coupling conditions, and the resulting thioester was subsequently reduced to the aldehyde form via Pd-catalyzed reduction, using Et3SiH as the hydride donor. For this reaction, a dry solution of 1:3 acetone/THF was used as solvent and 4 Å molecular sieves were introduced to obtain the LCA-aldehyde 9 with an optimal yield of 55%. The diindolylation of intermediate 9 using I2 as Lewis acid yielded the protected LCA-DIM hybrid 10 at 88% yield. The protected hybrid was then subjected to Ba(OH)2-catalyzed deprotection, simultaneously removing both methyl and acetyl protecting groups. This resulted to the successful synthesis of hybrid 11 at moderate yield of 60%.
:1 TFA/DCM solvent mixture. The free acid was then coupled with 1-dodecanethiol under Steglich coupling conditions, and the resulting thioester was subsequently reduced to the aldehyde form via Pd-catalyzed reduction, using Et3SiH as the hydride donor. For this reaction, a dry solution of 1:3 acetone/THF was used as solvent and 4 Å molecular sieves were introduced to obtain the LCA-aldehyde 9 with an optimal yield of 55%. The diindolylation of intermediate 9 using I2 as Lewis acid yielded the protected LCA-DIM hybrid 10 at 88% yield. The protected hybrid was then subjected to Ba(OH)2-catalyzed deprotection, simultaneously removing both methyl and acetyl protecting groups. This resulted to the successful synthesis of hybrid 11 at moderate yield of 60%.
2.2 Enhanced ST6GAL1 inhibition and selectivity of LCA-DIM hybrids demonstrating antimigratory properties against MDA-MB-231 cells
The inhibitory effects of the hybrid compounds were assessed against two representative ST isozymes—human α2,6-N-sialyltransferase ST6GAL1 and rat α2,3-O-sialyltransferase ST3GAL1—using a UPLC-based ST inhibition assay. The results were compared with those of the scaffold LCA and two established LCA-based ST inhibitors, Lith-O-Asp and AL10, as summarized in Table 1. Our findings indicated that the incorporation of the DIM moiety substantially enhanced the ST6GAL1 inhibitory activity of the LCA core, as demonstrated by the ability of hybrid 4 to inhibit the ST's enzymatic activity by 24% at 25 μM, whereas LCA did not exhibit any inhibition, even at 50 μM. Interestingly, hybrid 11 displayed superior ST6GAL1 inhibition, completely suppressing its activity at 25 μM. Its protected counterpart, hybrid 10, exhibited lower ST6GAL1 inhibition, achieving only 61% inhibition at the same concentration. This suggests that the C3–OH and COOH moieties play a crucial role in the hybrids' ST6GAL1 inhibitory activity. Further analysis of hybrid 11's inhibitory activity showed an IC50 value of 11.3 ± 0.4 μM against the N-glycan-sialylating ST isozyme, indicating relatively stronger inhibition compared to the C3–OH-modified Lith-O-Asp, which exhibited an IC50 of 15.8 ± 0.1 μM. Notably, the Asp-linked hybrids 10 and 11 exhibited minimal inhibition of ST3GAL1—only up to 3% even at 500 μM—while LCA and hybrid 4 showed 53% and 100% inhibition, respectively. In addition to this, hybrid 11 exhibited a significantly higher selectivity index towards ST6GAL1, with an N-to-O selectivity index exceeding 44. This surpasses the selectivity indices of both Lith-O-Asp (N-to-O selectivity index = 3) and AL10 (N-to-O selectivity index = 9), highlighting the apparent selectivity of the extended hybrids towards ST6GAL1.| Compound | R | ST6GAL1a | ST3GAL1a | Selectivity indexb (α2,3-O/α2,6-N) | ||
|---|---|---|---|---|---|---|
| % Inhibition at 25 μM | IC50f (μM) | % Inhibition at 500 μM | IC50f (μM) | |||
| a The selected sialyltransferase (ST) isozymes human α2,6-N-sialyltransferase ST6GAL1 (R&D Systems, catalog #7620-GT) and rat α2,3-O-sialyltransferase ST3GAL1 (Calbiochem, catalog #566227) were used for this study. b The selectivity index is defined as the ratio of the calculated IC50 for ST3GAL1 (or the highest tested concentration in cases without IC50) to the IC50 value for ST6GAL1 inhibition without correction. c Data obtained from.26 d Data obtained from.24 e Inhibition at 50 μM. f The IC50 values are presented as mean ± SD from three independent experiments (n = 3). | ||||||
| 4 | — | 24 | — | 100 | — | — |
| 10 | H | 61 | — | 0 | — | — |
| 11 (IAN-1B) | H | 100 | 11.3 ± 0.4 | 3 | — | >44 |
| 12 | NO2 | 100 | — | 3 | — | — |
| 13 (IAN-5B) | Cl | 95 | 6.6 ± 0.2 | 0 | — | >76 |
| 14 | F | 86 | — | 1 | — | — |
| 15 | OMe | 84 | — | 7 | — | — |
| 16 | OBn | 100 | — | 4 | — | — |
| 17 | NHBoc | 82 | — | 2 | — | — |
| 18 (IAN-15B) | N3 | 89 | 3.3 ± 0.1 | 5 | — | >153 |
| LCA | — | 0e | — | 53 | — | — |
| Lith-O-Asp | — | — | 15.8 ± 0.1 | — | 39 ± 1 | 3 |
| AL10 | — | — | 1.5 ± 0.5 | — | 13.2 ± 0.6 | 9 |
To further expand the structural diversity and therapeutic potential of this newly discovered ST inhibitor, we prepared a new series of 5,5′-substituted LCA-DIM hybrids 12–18 for further biological evaluation. This was inspired by previous studies showing that targeted modifications to the indole rings of DIM-based hybrids can significantly improve their biological activity.41,42 These hybrids were prepared using the optimized synthetic route used for hybrid 10 and 11, employing various 5C-substituted indoles (as summarized in Scheme 2) which were obtained at yields ranging from 19% to 74%. Most of the indoles used in this study were commercially available. However, the indoles required for the preparation of hybrids 16 (R = OBn), 17 (R = NHBoc), and 18 (R = N3) were synthesized using the procedure detailed in SI.
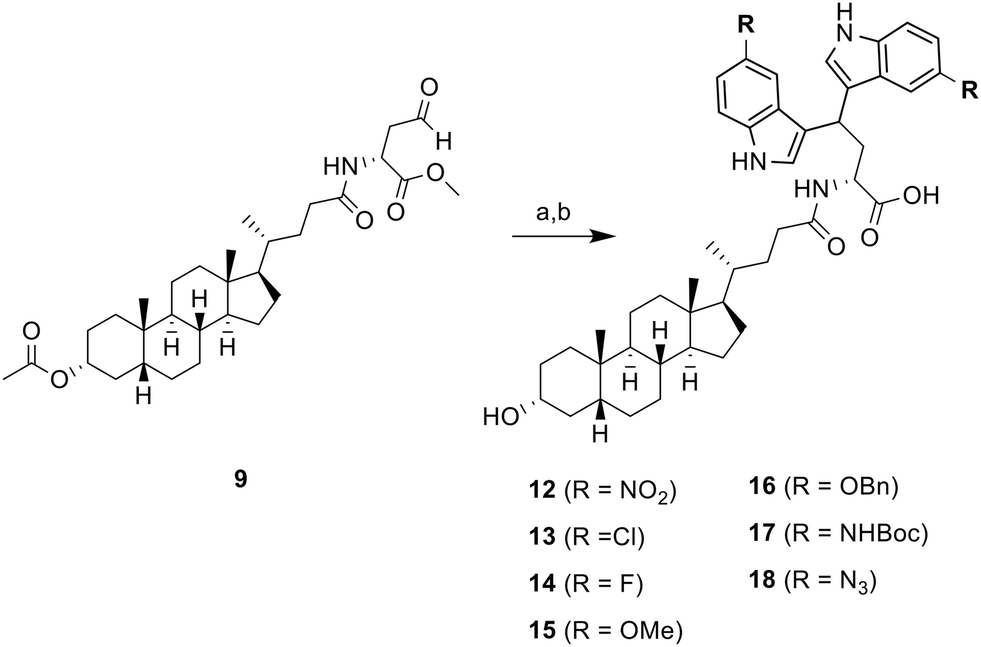 | ||
| Scheme 2 Synthesis of 5,5′-substituted LCA-DIM hybrids 12–18. a) 5R-indole, I2, DCM/THF, RT, for next step; b) Ba(OH)2 8H2O, MeOH, 8 h, RT, 19–74% | ||
Screening of the substituted LCA-DIM hybrids' ST inhibitory activity revealed that most hybrids exhibited robust ST6GAL1 inhibitory activity at 25 μM (≥80% inhibition) and selectivity as shown in Table 1. No clear correlation was observed between the electronic properties of the substituents and the hybrids' ST inhibitory activities, indicating that other factors may drive their intrinsic potency which warrants further investigation.
Given the excellent ST6GAL1 inhibitory activity and selectivity of the LCA-DIM hybrids, we sought to explore their potential as promising therapeutic candidates for targeting cancer. For this purpose, we explored the effect of both LCA-DIM hybrids towards the aggressive, hormonal treatment-resistant triple-negative breast cancer (TNBC), a cancer subtype previously linked to ST6GAL1 overexpression.10,25,43,44 To assess their efficacy, we evaluated the cytotoxic and anti-migratory effects of the LCA-DIM hybrids on the MDA-MB-231 TNBC cell line using the MTT and transwell migration assays, respectively. As shown in Fig. S2, the MTT assay demonstrated that most hybrids exhibited minimal cytotoxicity up to concentrations of 80 μM, except for the halogenated derivatives 13 and 14. These derivatives showed moderate cytotoxicity, with MTT IC50 values ranging from 40 to 80 μM—comparable to that of 3,3′-DIM.
Further investigation with the transwell migration assay revealed that the Asp-linked hybrids 11–18 exhibited observable antimigratory effect at the test concentration of 80 μM (Fig. S3). In contrast, no significant migration inhibition was observed for the direct hybrid 4 or the protected hybrid 10. The extended hybrids also showed greater antimetastatic potential than 3,3′-DIM, despite presenting cytotoxicity at the tested concentration. Among all the candidates, hybrids 13 (IAN-5B, R = Cl) and 18 (IAN-15B, R = N3) demonstrated the highest antimetastatic potential against the MDA-MB-231 cell line (Fig. S2). This enhanced activity may be linked to the intrinsic ST6GAL1 inhibitory properties of both hybrids. In the case of IAN-5B, its inherent cytotoxicity could also influence its inherent antimigration properties towards the TNBC cell line. Further assessment of the ST inhibitory activity of the hybrids revealed that IAN-5B and IAN-15B exhibited potent ST6GAL1 inhibition, with IC50 values of 6.6 ± 0.2 μM and 3.3 ± 0.1 μM, respectively (Table 1). Additionally, both IAN-5B and IAN-15B demonstrated enhanced N-to-O selectivity indices (>76 and >153, respectively), significantly surpassing the performance of the unsubstituted hybrid 11(IAN-1B).
2.3 Molecular docking insights into the binding of IAN-5B and IAN-15B towards ST6GAL1
To gain more insights into the excellent ST6GAL1 inhibitory activity of the LCA-DIM hybrids IAN-5B and IAN-15B, molecular docking studies was conducted using Autodock4. For this study, the ST6GAL1 model used was the crystal structure of human ST6GAL1 (PDB ID: 6QVT).45 As summarized in Table S1, IAN-15B exhibited the strongest binding affinity towards the hST6GAL1 model among the two hybrids, with a binding energy of −10.78 kcal mol−1, while IAN-5B showed a slightly lower but comparable binding energy of −10.44 kcal mol−1. These results aligned with the higher experimental ST6GAL1 inhibition observed for IAN-15B. Further analysis of the molecular docking results revealed that both hybrids preferentially bind near the enzyme's acceptor site, located between the two polypeptide chains (as illustrated in Fig. S8). This binding motif were consistent with docking results reported for other LCA-based ST inhibitors.24,46 As shown in Fig. 2, the steroidal core of IAN-5B were involved in multiple hydrophobic interactions with several amino acid residues of hST6GAL1 including PRO259(A), VAL261(A), PRO320(A), TRP302(A), as well as their counterparts PRO259(B), VAL261(B), and PRO320(B). Moreover, the carboxyl group of IAN-5B's Asp linker formed key hydrogen bonds with GLN235(B) and LYS268(A), underscoring the importance of this functional group in binding towards ST6GAL1. The chloro-substituted DIM moiety also shown contribution to the overall interaction of the hybrid towards hST6GAL1, establishing hydrophobic and polar halogen interactions with HIS263(A), PRO320(B), and PRO321(B) as shown in Fig. 2.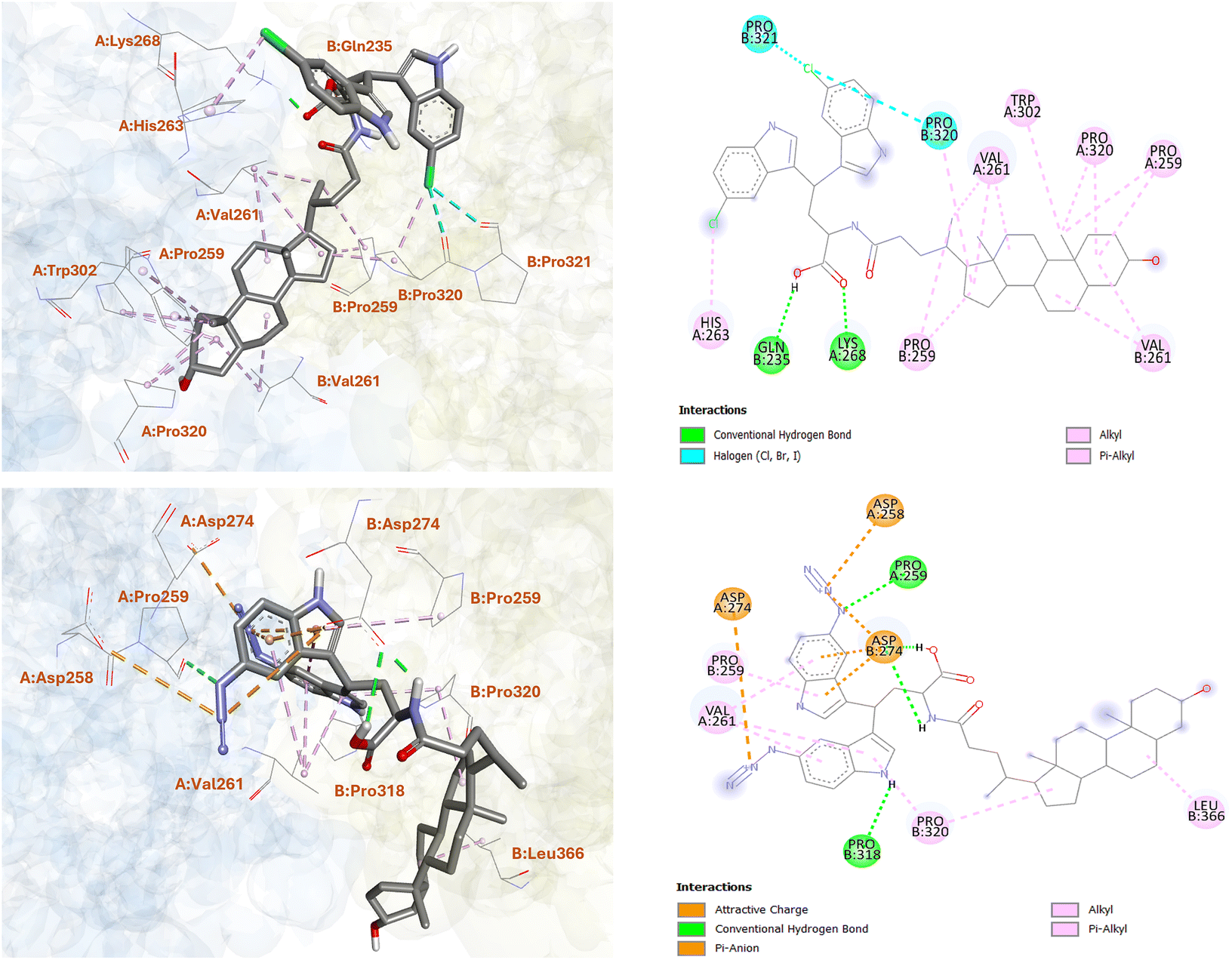 | ||
| Fig. 2 Molecular docking analysis of LCA-DIM hybrids IAN-5B (upper) and IAN-15B (lower) with the hST6GAL1 model. The two- and three-dimensional representations illustrate the binding orientation and key interactions of the hybrids within the active site of hST6GAL1. | ||
On the other hand, the azido-substituted IAN-15B, showed fewer interaction with its steroidal ring structure, forming hydrophobic contacts primarily with PRO320(B) and LEU366(B). However, it exhibited enhanced interactions through its DIM and Asp-linker moieties. For instance, the carboxyl and amide NH groups of the Asp-linker form strong hydrogen bonds with ASP274(B) (as shown in Fig. 2), while the azido groups of the DIM moiety participate in electrostatic interactions with ASP258(A) and ASP274(A), and hydrogen bonding with PRO259(A). Additional π-alkyl and π–anion interactions were also observed between the indole ring of IAN-15B and residues PRO259(B), VAL261(A), and ASP274(B), respectively. Remarkably, these observed binding patterns of the LCA-DIM hybrids toward hST6GAL1 model were particularly significant given that X-ray crystallography studies have highlighted the role of residue PRO320 in stabilizing HIS370, a critical catalytic residue of ST6GAL1.47 Furthermore, ASP274 has been shown to play a crucial role in mediating interactions with the donor substrate CMP-Neu5Ac during the sialylation process. The strong interactions formed by both IAN-5B and IAN-15B—especially through their DIM-Asp linker regions—with PRO320, ASP274, and adjacent residues, likely disrupt the proper structural alignment and catalytic function of HIS370, thereby impairing ST6GAL1 enzymatic activity. In addition to these, it should be noted that C3–OH group of both IAN-5B and IAN-15B did not engage in any interactions with the surrounding amino acid residues of ST6GAL1. This lack of involvement suggests that the C3–OH position could be further structurally modified, offering an opportunity to further enhance the potency and overall efficacy of these ST inhibitors. In total, these findings offer valuable insights into the underlying mechanism of inhibition and emphasize the critical role of the DIM moiety in enhancing the overall ST6GAL1 inhibitory activity of the LCA-DIM hybrids.
2.4 Further biological evaluation of IAN-5B and IAN-15B towards various TNBC and HUVEC cell lines
We further explored their antimigratory properties and cytotoxicity towards MDA-MB-231 cells and two additional TNBC cell lines - BT549 and Hs578T. Initially, the cytotoxicity of the LCA-DIM hybrids was tested against the two additional cell lines via MTT assay. Consistent with their effects on the MDA-MB-231 cell line, both hybrids exhibited comparable cytotoxic activities against BT549 and Hs578T with MTT IC50 values between 40–80 μM (Fig. S7). Afterwards, the effect of IAN-5B and IAN-15B on cancer cell migration were tested in all three TNBC cell lines using transwell migration assay at the 40 and 80 μM test concentrations. As illustrated in Fig. 3, both hybrids effectively suppressed cell motility across the three TNBC cell lines, resulting in up to a 10% reduction in cancer cell migration at 80 μM, with migration IC50 values ranging from 40–80 μM. The effect of both hybrids on tumorigenic angiogenesis was also assessed via HUVEC tube formation assay. Results showed that both hybrids IAN-5B and IAN-15B presented high cytotoxicity towards HUVEC (Fig. S7), with MTT IC50 values of approximately 10 μM and 10–20 μM, respectively. Furthermore, both hybrids inhibited tube formation in HUVEC cells at non-cytotoxic concentrations, with IAN-15B showing the strongest anti-angiogenic effect—reducing tube formation to 67% relative to the control at a 10 μM test concentration.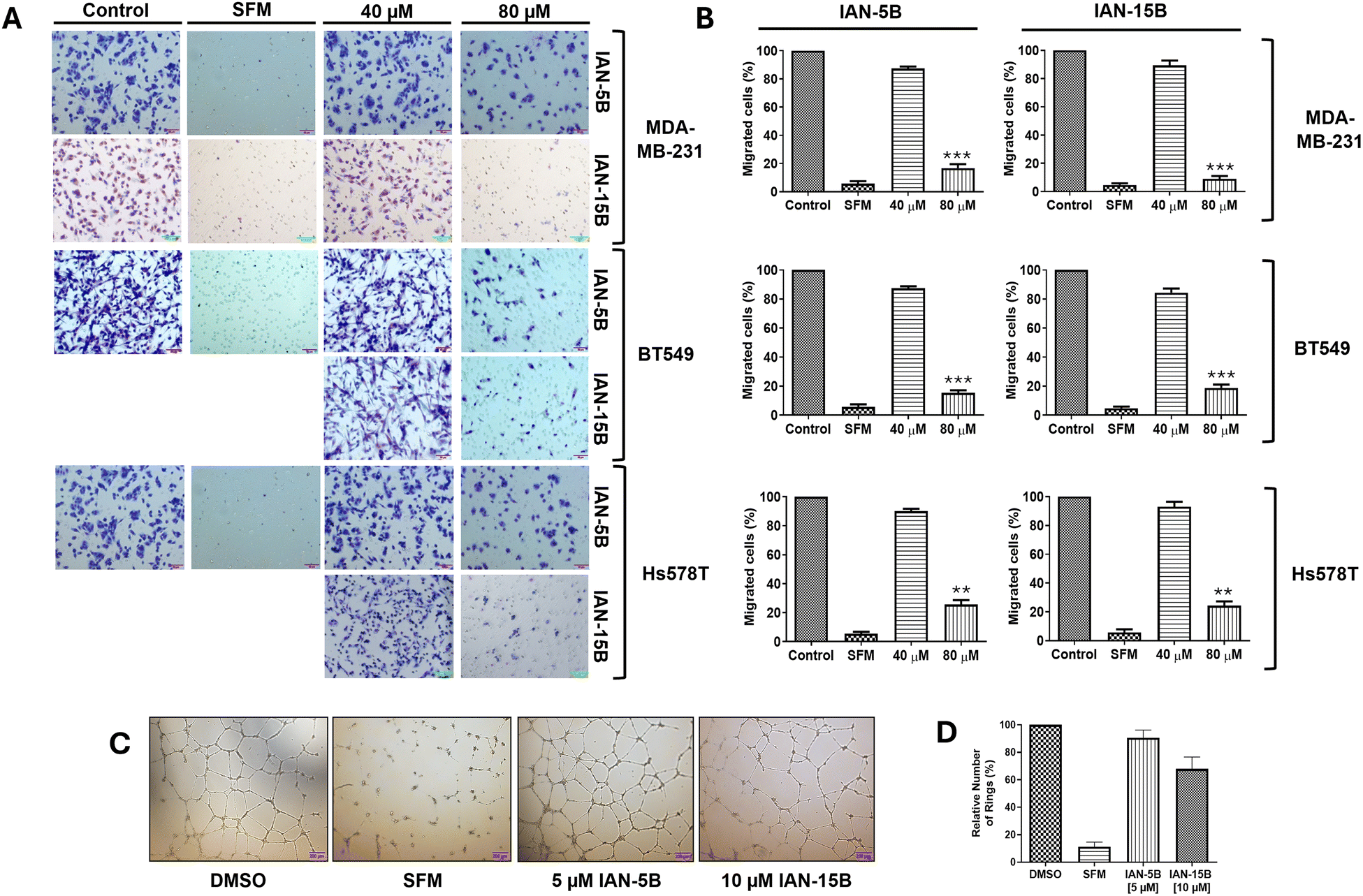 | ||
| Fig. 3 Evaluation of the antimigratory and antiangiogenic effects of IAN-5B and IAN-15B. (A) Representative images of the inhibitory effects of IAN-5B and IAN-15B on the migration of triple-negative breast cancer (TNBC) cell lines BT549, Hs578T, and MDA-MB-231 following 16 hours of incubation with varying concentrations (0, 40, and 80 μM). DMSO and serum-free medium (SFM) were used as control for this study. Several experimental runs were performed using the same control. (B) Quantification of transwell migration assay data against BT549, Hs578T, and MDA-MB-231 TNBC cell lines performed in triplicates. (C) Evaluation of HUVEC tube formation in the presence of non-cytotoxic concentrations of IAN-5B (5 μM) and IAN-15B (10 μM), with DMSO and serum-free medium serving as controls. (D) Quantification of HUVEC tube formation assay performed in triplicates. All data are presented as mean ± SEM from three independent experiments (*: p < 0.05, **: p < 0.01, ***: p < 0.001). | ||
Given the strong antimigratory effects of IAN-5B and IAN-15B, we aimed to investigate the underlying mechanisms further. We suspect that the inherent antimigratory properties of the LCA-DIM hybrids may be attributed to their impact on the sialylation of cancer cells which contributes to its progression. Prior studies have demonstrated integrin sialylation as a key factor in cancer cell migration and metastasis and the ability of ST inhibitors to target this biological pathway.3,25,46,48 Based on these findings, we propose that the strong ST6GAL1 inhibitory activity of IAN-5B and IAN-15B may effectively suppress integrin sialylation, thereby impeding cancer cell motility. To investigate the involvement of both LCA-DIM hybrids towards this mechanism, lectin affinity assays were conducted using the α2,6-sialylation-specific Sambucus nigra agglutinin (SNA) lectin, followed by western blotting to detect integrin subunits β1, β3, and β4 in MDA-MB-231 cells. Remarkably, both hybrids decreased the levels of captured sialylated integrins, in a dose-dependent manner even at non-toxic concentrations, as shown in Fig. 4. The effects of both hybrids were especially prominent on the β1 integrin subunit, which is strongly associated with ST6GAL1-mediated sialylation and has been closely linked to cancer progression.49–51 Overall, these findings highlight the potential antimetastatic and antiangiogenetic potential of LCA-DIM hybrids, offering a promising foundation for further exploration of their therapeutic potential in cancer treatment.
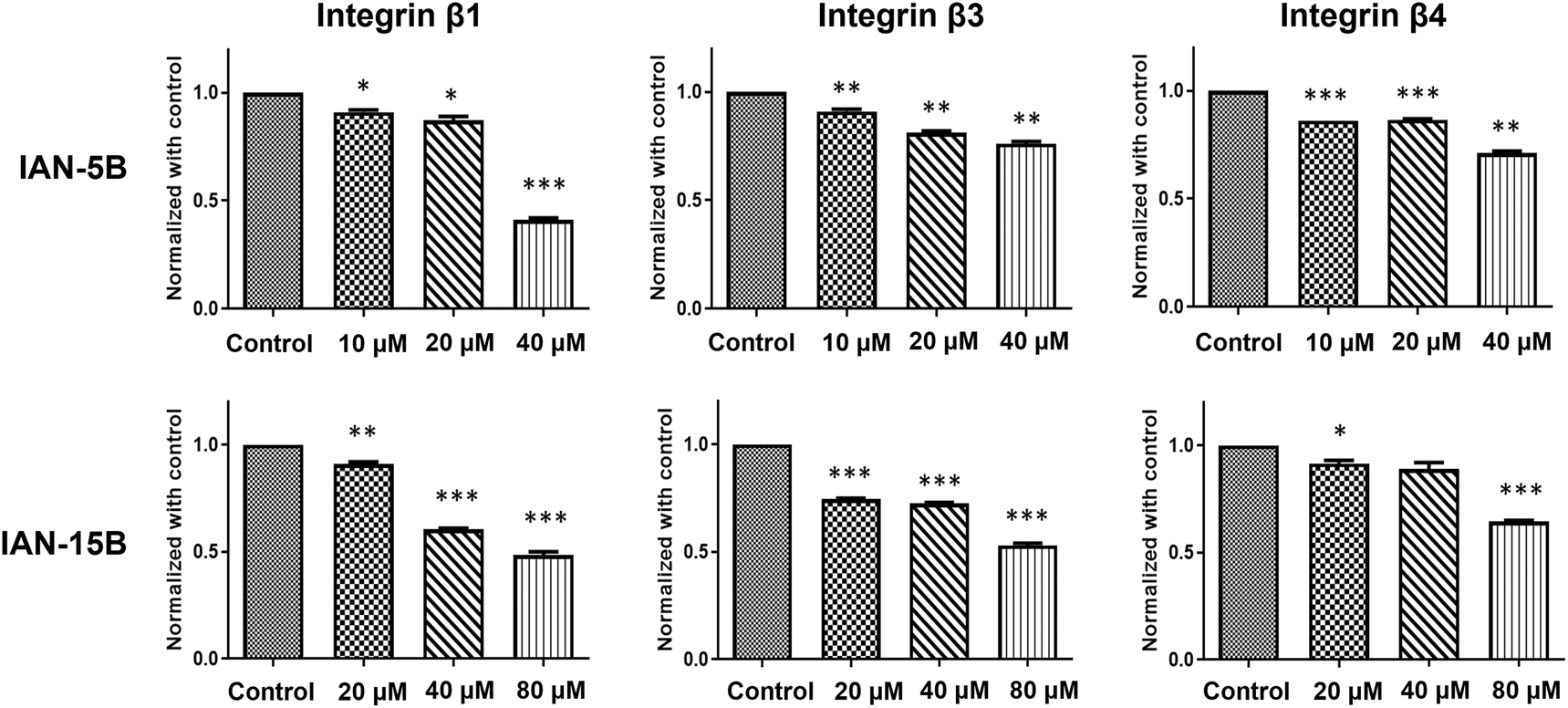 | ||
| Fig. 4 Influence of LCA-DIM hybrids towards integrin sialylation. Lectin affinity assays followed by western blot analysis revealed a concentration-dependent reduction in α2,6-sialylated integrin subunits β1, β3, and β4 upon treatment with IAN-5B and IAN-15B. MDA-MB-231 cells were treated with IAN-5B at 10, 20, and 40 μM, and with IAN-15B at 20, 40, and 80 μM. All data are presented as mean ± SEM from three independent experiments (*: p < 0.05, **: p < 0.01, ***: p < 0.001). | ||
3. Conclusion
In response to the growing demand for compounds capable of inhibiting ST activity as potential therapeutic strategy, this study employed molecular hybridization for the development of the novel LCA-DIM hybrids as potential ST inhibitors. These hybrids presented heightened inhibitory activity and apparent selectivity towards ST6GAL1, with the introduction of an Asp linker between the LCA and DIM moiety further enhanced the hybrids' ST6GAL1 activity. Molecular docking data also supports these findings, indicating the significance of the hybrids' DIM moiety and Asp linker on its binding towards the ST6GAL1 model. Cell-based assays have also revealed that most of the hybrids have low cytotoxicity profiles in exception for the halogen-substituted series. Moreover, the hybrids demonstrate excellent antimigration effects with IAN-5B and IAN-15B demonstrating the highest antimetastatic potential against three TNBC cell lines (MDA-MB-231, BT549, and Hs578T), correlating to their enhanced ST6GAL1 inhibitory activities (IC50 = 6.6 ± 0.2 and 3.3 ± 0.1, respectively). In addition to this, IAN-15B presented antiangiogenetic effect towards HUVEC as presented in the tube formation assay data. We have also revealed the involvement of both hybrids in reducing β integrin sialylation, which could be attributed to its inherent anticancer properties. In summary, this study introduced the LCA-DIM hybrid as a novel and potent ST inhibitor, highlighting the potential of molecular hybridization for the development of targeted therapies for the aggressive TNBC with promising N-glycan selectivity.4. Experimental section
4.1 Chemistry
4.1.2.1. Synthesis of intermediate 2. Lithocholic acid (1) (10 g, 1.0 eq.) was added to a stirring solution of MeOH. After a few minutes, 5 drops of H2SO4 were introduced to the mixture. The reaction was stirred overnight, after which it was neutralized using saturated NaHCO3 solution to quench the reaction. The resulting precipitate was filtered, collected, and dried to yield OMe-protected LCA as a white solid. Afterwards, 1.5 g (1.0 eq.) of the protected LCA was dissolved in DMF together with 0.87 g (1.5 eq.) of tert-butyldimethylsilyl chloride and 0.52 g (2.0 eq) of imidazole. The reaction mixture was heated to 60 °C and was allowed to react for 8 h. Upon completion, the reaction was quenched with ethyl acetate, and the organic layer was extracted with distilled H2O three times to remove most of the DMF and was washed with brine. The resulting solution was then dried over MgSO4, and the solvent was removed under reduced pressure to obtain intermediate 2 as a white powder which was used without further purification. Yield: 64% (1.25 g, for two steps). 1H NMR (400 MHz, CDCl3): δ (ppm) 3.67 (s, 3H), 3.63–3.52 (m, 1H), 2.42–2.32 (m, 1H), 2.29–2.15 (m, 1H), 1.98–1.92 (m, 1H), 1.90–1.71 (m, 5H), 1.65–1.52 (m, 2H), 1.46–1.31 (m, 9H), 1.30–1.20 (m, 3H), 1.20–0.96 (m, 5H), 0.95–0.87 (m, 16H), 0.65 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 174.8, 72.9, 56.5, 56.0, 51.5, 42.8, 42.4, 40.3, 40.2, 37.0, 35.9, 35.6, 35.4, 34.6, 31.1 (3C), 28.2, 27.4, 26.4, 26.0 (3C), 24.3, 23.4, 20.9, 18.4, 18.3, 12.1, −4.5 (2C).
4.1.2.2. Synthesis of intermediate 3. In a dry round-bottom flask, 1.75 g (1.0 eq.) of intermediate 2 was dissolved in anhydrous THF and stirred at 0 °C. After a few minutes, 0.53 g (5.0 eq.) of LiAlH4 was added to the cooled solution, and the reaction was allowed warm to room temperature and was stirred for 3 h. For work-up, saturated Rochelle salt solution was slowly added to the reaction mixture until the vigorous reaction ceased. Additional amounts of Rochelle salt solution and ethyl acetate were then added, and the mixture was stirred overnight. The organic layer was subsequently washed with distilled water and brine, then dried under vacuum to yield the reduced LCA intermediate as a white solid. The obtained solid was dissolved in DCM, and 1.66 g (2.0 eq.) of pyridinium chlorochromate was added to the solution. After stirring for 12 h, the reaction mixture was filtered through celite. The filtrate was washed with distilled H2O and brine, dried over MgSO4, and concentrated to afford the crude product. The crude product was purified by flash chromatography using ethyl acetate/hexane solvent system to yield intermediate 3 as a white crystalline solid. Yield: 50% (0.82 g). 1H NMR (400 MHz, CDCl3): δ (ppm) 9.72 (s, 1H), 3.60–3.50 (m, 1H), 2.48–2.20 (m, 2H), 1.93–1.70 (m, 6H), 1.58–1.48 (m, 2H), 1.41–1.31 (m, 7H), 1.29–1.19 (m, 4H), 1.18–1.11 (m, 2H), 1.08–0.95 (m, 4H), 0.91–0.82 (m, 16H), 0.61 (s, 3H), 0.02 (s, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 202.9, 72.8, 56.4, 56.0, 42.7, 42.3, 40.9, 40.3, 40.2, 36.9, 35.9, 35.6, 35.3, 34.6 (2C), 31.0, 28.2, 28.0, 26.4, 26.0 (3C), 24.2, 23.4, 20.8, 18.4, 18.3, 12.0, −4.6 (2C).
4.1.2.3. Synthesis of LCA-DIM hybrid 4. Intermediate 3 (0.4 g, 1.0 eq.) was dissolved in 5% I2 in MeOH, followed by the addition of 0.30 g (3.0 eq.) of indole. The reaction mixture was stirred for 6 h, then dried and diluted with ethyl acetate. The organic layer was sequentially washed with Na2S2O3, distilled H2O and brine. It was then dried over MgSO4 and concentrated to yield the crude product. The crude was then purified using flash chromatography using ethyl acetate/hexane solvent system to obtain hybrid 4 as brown solid. Yield: 41% (0.20 g). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.97 (br s, 1H), 7.93 (br s, 1H), 7.69–7.60 (m, 2H), 7.34–7.27 (m, 2H), 7.16 (t, J = 7.3 Hz, 2H), 7.07 (d, J = 7.3 Hz, 2H), 6.94 (s, 1H), 6.90 (s, 1H), 4.43 (t, J = 7.3 Hz, 1H), 3.70–3.60 (m, 1H), 2.35–2.25 (m, 1H), 2.21–2.10 (m, 2H), 2.02–1.95 (m, 1H), 1.90–1.78 (m, 3H), 1.78–1.68 (m,2H), 1.62–1.51 (m, 3H), 1.1.46–1.38 (m, 6H), 1.33–1.20 (m, 3H), 1.17–1.08 (m, 4H), 1.03–0.95 (m, 8H), 0.64 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 136.6 (2C), 127.2, 127.1, 121.6 (2C), 121.5, 121.3, 120.8, 120.3, 119.6, 119.5, 118.9 (2C), 111.1 (2C), 71.9, 56.4, 56.0, 42.6, 42.1, 40.4, 40.1, 36.4, 35.8 (2C), 35.3, 34.6, 34.5, 34.4, 32.3, 30.5, 28.2, 27.2, 26.4, 24.2, 23.4, 20.8, 18.8, 12.0. HRMS (ESI) calculated for C40H51N2O (M − H)−, 575.4007; found, 575.3999. HPLC purity: >99%.
4.1.3.1. Synthesis of intermediate 5. Lithocholic acid (1) (10 g, 1.0 eq.) was placed in a dry round bottom flask along with approximately 20 mL (4.0 eq.) of acetic anhydride and a catalytic amount of DMAP (0.05 eq.) in DCM. The mixture was stirred at room temperature for 2 h. The reaction was then quenched with NaHCO3, and the organic layer was then washed with saturated NaHCO3 solution, distilled H2O, and brine. After drying over MgSO4, the solvent was evaporated under reduced pressure to yield intermediate 5 as a white powder solid. Yield: ≥ 99% (11 g). 1H NMR (400 MHz, CDCl3): δ (ppm) 10.43 (br, 1H), 4.71–4.61 (m, 1H), 2.39–2.15 (m, 2H), 1.97 (s, 3H), 1.94–1.88 (m, 1H), 1.85–1.71 (m, 5H), 1.67–1.60 (m, 1H), 1.56–1.45 (m, 2H), 1.44–1.27 (m, 8H), 1.23–1.13 (m, 3H), 1.12–0.92 (m, 6H), 0.90–0.84 (m, 6H), 0.60 (s, 3H); 13C NMR (100 MHz, CDCl3): δ (ppm) 180.3, 170.8, 74.4, 56.5, 56.0, 42.7, 41.9, 40.4, 40.1, 35.8, 35.3, 35.0, 34.5, 32.2, 31.0, 30.8, 28.2, 27.0, 26.6, 26.3, 24.2, 23.3, 21.4, 20.8, 18.2, 12.0.
4.1.3.2. Synthesis of intermediate 7. To a stirring solution of DCM, 10.0 g (1.0 eq.) of Fmoc-Asp(OtBu)–COOH (6) were added together with EDCI-HCl (4.7 g, 1.0 eq.), HOBt (4.1 g, 1.1 eq.), TEA (∼3.5 mL, 1.0 eq.), and 1 mL of MeOH. The mixture was stirred for 8 h, and upon completion, the reaction was diluted with DCM and quenched with dilute HCl. The precipitates were removed via filtration, and the organic layer was sequentially washed with dilute HCl, NaHCO3, distilled H2O, and brine. The organic phase was dried over MgSO4, and the solvent was evaporated under reduced pressure, yielding the fully protected acid as a white crystalline solid. The solid was then dissolved in DCM, and approximately 7.4 g DBU (3.0 eq) was added to the stirring solution alongside 15 mL of 1-dodecanethiol was introduced, and the reaction mixture was stirred for 30 minutes. The solvent was subsequently removed under reduced pressure, and the resulting slurry was immediately purified via flash chromatography using ethyl acetate and hexane solvent system to afford intermediate 7 as a clear yellow liquid. Yield: 97% (4.6 g).1H NMR (400 MHz, CDCl3): δ (ppm) 3.58–3.52 (m, 4H), 2.54–2.44 (m, 2H), 1.67 (s, 2H), 1.25 (s, 9H); 13C NMR (100 MHz, CDCl3): δ (ppm) 174.5, 170.0, 80.7, 51.8, 51.0, 39.9, 27.7 (3C).
4.1.3.3. Synthesis of intermediate 8. In a round-bottom flask, approximately 2.0 g of intermediate 5 (1.0 eq.) and 1.8 g of intermediate 7 (1.5 eq.) were dissolved in DCM. The solution was stirred for a few minutes before adding EDCI-HCl (1.9 g, 1.1 eq.), HOBt (1.5 g, 1.1 eq.), and DIPEA (∼1.5 mL, 1.0 eq.). The reaction mixture was stirred for 8 h, and upon completion, it was diluted with DCM. The organic layer was washed sequentially with dilute HCl, saturated NaHCO3, and brine. After drying over MgSO4, the solvent was removed under reduced pressure to yield the crude product. This was subsequently purified by flash chromatography with solvent system of ethyl acetate and hexane to afford the conjugate 8 as a white foamy solid. Yield: 90% (4.0 g). 1H NMR (400 MHz, CDCl3): δ (ppm) 6.59 (d, J = 7.98 Hz, 1H), 4.72–4.51 (m, 2H), 3.59 (s, 3H), 2.73 (dd, J = 16.7, 4.6 Hz, 1H), 2.59 (dd, J = 16.7, 4.5 Hz, 1H), 2.22–2.09 (m, 1H), 2.06–1.93 (m, 1H), 1.91–1.80 (m, 4H), 1.75–1.62 (m, 5H), 1.58–1.50 (m, 1H), 1.46–1.36 (m, 2H), 1.33–1.22 (m, 16H), 1.16–1.06 (m, 4H), 0.99–0.87 (m, 5H), 0.85–0.75 (m, 7H), 0.51 (s, 3H); 13C NMR (100 MHz, CDCl3): δ (ppm) 173.1, 171.2, 170.2, 169.9, 81.2, 74.1, 56.3, 55.9, 52.2, 48.4, 42.5, 41.7, 40.2, 39.9, 37.3, 35.6, 35.2, 34.8, 34.3, 33.0, 32.0, 31.4, 27.9, 27.7 (3C), 26.8, 26.4, 26.1, 24.0, 23.1, 21.1, 20.6, 18.1, 11.8.
4.1.3.4. Synthesis of intermediate 9. Intermediate 8 (2.4 g, 1.0 eq.) was dissolved in a 1
:1 TFA/DCM solution in a round-bottom flask and allowed to react for 2 h. The reaction mixture was then concentrated using a rotary evaporator to yield the free acid intermediate, which was used directly in the next step without further purification. The deprotected precursor was dissolved in DCM, and 1.4 g of EDCI-HCl (1.5 eq.), 3 mL of 1-dodecanethiol (∼3.0 eq), and 0.024 g of DMAP (0.05 eq.) were added. The mixture was stirred for 4 h, after which the solution was dried under reduced pressure. Excess 1-dodecanethiol was removed by flash chromatography to obtain the thioester precursor as a white crystalline solid. Afterwards, 1 g of the precursor was then dissolved in a 1:1 mixture of THF/acetone with 4 Å molecular sieves. A catalytic amount of 10% Pd/C (100 mg, 5% w/w) was added, and the system was purged with argon gas to maintain an inert atmosphere. After a few minutes, 3.0 eq. of Et3SiH (0.68 mL) were added. Once the effervescence ceased, the reaction mixture was filtered through celite, and the filtrate was concentrated using a rotary evaporator. The crude product was purified by flash chromatography using ethyl acetate and hexane solvent system to yield the desired product 9 as a white solid. Yield: 55% (0.53 g), for three steps. 1H NMR (400 MHz, CDCl3): δ (ppm) 9.68 (s, 1H), 6.45 (d, J = 7.7 Hz, 1H), 4.85–4.62 (m, 2H), 3.71 (s, 3H), 3.14–2.98 (m, 2H), 2.26–2.18 (m, 1H), 2.13–2.05 (m, 1H), 1.99 (s, 3H), 1.95–1.90 (m, 1H), 1.86–1.72 (m, 5H), 1.68–1.60 (m, 1H), 1.56–1.47 (m, 2H), 1.44–1.30 (m, 7H), 1.27–1.18 (m, 4H), 1.13–0.94 (m, 6H), 0.90–0.85 (m, 6H), 0.60 (s, 3H); 13C NMR (100 MHz, CDCl3): δ (ppm) 199.6, 173.5, 171.3, 170.7, 74.5, 56.5, 56.1, 52.9, 47.2, 45.7, 42.8, 41.9, 40.5, 40.2, 35.8, 35.5, 35.1, 34.6, 33.3, 32.3, 31.5, 28.3, 27.1, 26.7, 26.4, 24.2, 23.4, 21.5, 20.9, 18.4, 12.1.
4.1.3.5. Synthesis of protected Asp-linked LCA-DIM hybrids 10. In a round bottom flask, 0.30 g of aldehyde intermediate 9 (1.0 eq.) was dissolved in THF along with 0.20 g (3.0 eq.) of indole. After a few minutes of stirring, catalytic amount of I2 (0.045 g, 0.05 eq.) was added into the solution and the reaction was monitored via TLC. Upon completion, the reaction mixture was concentrated, and ethyl acetate was added, and the resulting solution was washed with Na2S2O3, distilled H2O, and brine. The organic layer was dried over MgSO4 and concentrated in vacuo. The obtained crude product was purified via flash chromatography using ethyl acetate and hexane solvent system to obtain the protected hybrid 10. Obtained as off-white powder. Yield: 88% (0.32 mg). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.29 (s, 2H), 7.54 (d, J = 7.9 Hz, 1H), 7.51 (d, J =7.9 Hz, 1H), 7.26 (d, J = 2.1 Hz, 1H), 7.25 (d, J = 2.1 Hz, 1H), 7.12 (t, J = 7.5 Hz, 2H), 6.87–7.05 (m, 4H), 5.98 (d, J = 7.5 Hz, 1H), 4.70–4.76 (m, 2H), 4.56 (t, J = 7.4 Hz, 1H), 3.51 (s, 3H), 2.58–2.81 (m, 2H), 1.96–2.05 (m, 4H), 1.91–1.95 (m, 1H), 1.75–1.89 (m, 5H), 1.60–1.72 (m, 2H), 1.51–1.57 (m, 2H), 1.32–1.47 (m, 7H), 1.17–1.27(m, 4H), 0.99–1.15 (m, 6H), 0.81–0.93 (m, 6H), 0.6 (s, 3H); 13C NMR (125 MHz, CDCl3): δ (ppm) 173.7, 173.2, 170.9, 136.8, 136.7, 126.7, 126.5, 122.5, 122.2, 121.9, 121.9, 119.5, 119.4, 119.3, 119.2, 118.3, 118.1, 111.5, 111.4, 74.6, 56.5, 56.0, 52.2, 51.8, 42.8, 42.0, 40.5, 40.2, 37.4, 35.9, 35.5, 35.1, 34.7, 33.3, 32.4, 31.6, 31.2, 28.3, 27.1, 26.7, 26.4, 24.3, 23.4, 21.6, 20.9, 18.4, 12.1; HRMS (ESI) calculated for C47H61N3O5Na (M + Na)+, 770.4503; found, 770.4509. HPLC purity: 97%.
4.1.3.6. Synthesis of Asp-linked hybrid 11. For deprotection, the purified intermediate 10 was dissolved in methanol (MeOH) in a round-bottom flask and Ba(OH)2·8H2O (3.0 eq) was added to the solution. The reaction mixture was allowed to react for 8 h and upon completion, the solution was diluted with ethyl acetate and sequentially washed with dilute HCl and brine. The organic layer was dried over MgSO4 and concentrated using rotary evaporator. The crude product was then recrystallized to obtain the LCA-DIM hybrid 11. Obtained as brown solid. Yield: 60% (0.15 g). 1H NMR (500 MHz, acetone-d6): δ (ppm) 10.08 (br s, 1H), 10.02 (br s, 1H), 7.56 (t, J = 9.0 Hz, 2H), 7.49 (d, J = 7.9 Hz, 1H), 7.40–7.20 (m, 4H), 7.05–6.85 (m, 4H), 4.73 (q, J = 5.0 Hz, 1H), 4.55–4.50 (m, 1H), 3.60–3.48 (m, 1H), 2.92–2.80 (m, 1H), 2.65–2.55 (m, 1H), 2.80–2.35 (m, 2H), 1.99–1.93 (m, 1H), 1.92–1.73 (m, 5H), 1.64–1.52 (m, 2H), 1.50–1.32 (m, 8H), 1.32–1.13 (m, 6H), 1.13–0.98 (m, 3H), 0.97–0.89 (m, 7H), 0.65 (s, 3H); 13C NMR (125 MHz, acetone-d6): δ (ppm) 174.7, 174.5, 138.1, 137.9, 127.9, 127.7, 123.6, 122.7, 121.8, 121.8, 120.2, 120.1, 119.6, 119.1, 119.0, 118.2, 112.2, 112.0, 79.1, 71.7, 57.2, 56.8, 52.2, 43.4, 42.9, 41.2, 40.9, 38.2, 37.2, 36.6, 36.2, 35.2, 33.4, 32.6, 31.9, 31.2, 28.9, 28.0, 27.2, 24.9, 23.9, 21.5, 18.8, 12.4; HRMS (ESI) calculated for C44H57N3O4Na (M + Na)+, 714.4241; found, 714.4251. HPLC purity: >99%.
4.1.3.7. Synthesis of Asp-linked hybrid 12. Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-nitro-1H-indole. Obtained as bright yellow solid. Yield: 47% (0.35 g, for two steps). 1H NMR (500 MHz, acetone-d6): δ (ppm) 11.01 (s, 1H), 10.95 (s, 1H), 7.49–8.59 (m, 9H), 4.91 (q, J = 4.8 Hz, 1H), 4.54–4.67 (m, 2H), 3.50–3.58 (s, 1H), 2.90–2.98 (m, 1H), 2.68–2.78 (m, 1H), 2.16–2.42 (m, 2H), 1.86–1.94 (m, 1H), 1.70–1.85 (m, 5H), 1.58–1.64 (m, 1H), 1.39–1.48 (m, 3H), 1.16–1.38 (m, 10H), 0.92–1.15 (m, 6H), 0.85–0.92 (m, 7H), 0.57 (s, 3H); 13C NMR (125 MHz, acetone-d6): δ (ppm) 175.0, 174.3, 141.7 (2C), 141.1, 141.0, 127.7, 127.0, 126.7 (2C), 121.6, 120.1, 117.6, 117.5, 116.9, 116.9, 112.7, 112.6, 71.7, 57.2, 56.8, 51.8, 43.3, 42.9, 41.2, 40.9, 38.0, 37.2, 36.6, 36.2, 35.2, 35.2, 33.5, 32.6, 31.5, 31.2, 28.8, 27.9, 27.1, 24.7, 24.7, 23.8, 21.4, 18.8, 12.3; HRMS (ESI) calculated for C44H55N5O8Na (M + Na)+, 804.3943; found, 804.3950. HPLC purity: >99%.
4.1.3.8. Synthesis of Asp-linked hybrid 13 (IAN-5B). Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-iodo-1H-indole. Obtained as off-white solid. Yield: 19% (0.08 g, for two steps). 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.34 (br s, 1H), 10.28 (br s, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.51–7.34 (m, 6H), 7.02 (t, J = 2.4 Hz, 2H), 7.0 (t, J = 2.4 Hz, 2H), 4.67 (q, J = 5.0 Hz, 1H), 4.58–4. 45 (m, 1H), 3.51–3.48 (m, 1H), 2.89–2.77 (m, 1H), 2.61–2.53 (m, 1H), 2.39–2.29 (m, 1H), 2.24–2.12 (m, 1H), 1.94–1.85 (m, 1H), 1.92–1.73 (m, 5H), 1.64–1.54 (m, 2H), 1.50–1.31 (m, 9H), 1.29–1.02 (m, 8H), 0.98–0.90 (m, 7H), 0.66 (s, 3H); 13C NMR (100 MHz, acetone-d6): δ (ppm) 175.3, 174.7, 136.8, 136.7, 129.3, 129.0, 126.0, 125.0, 124.8 (2C), 122.4, 122.4, 119.6, 119.6, 119.3, 117.8, 114.0, 113.9, 72.08, 57.6, 57.2, 52.2, 43.7, 43.3, 41.6, 41.3, 38.2, 37.5, 36.9, 36.6, 36.5, 35.6, 33.9, 33.0, 32.0, 31.6, 29.3, 28.3, 27.6, 25.2, 24.2, 21.8, 19.2, 12.8; HRMS (ESI) calculated for C44H54N3O4Cl2 (M–H)−, 758.3497; found, 758.3502. HPLC purity: 97%.
4.1.3.9. Synthesis of Asp-linked hybrid 14. Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-fluoro-1H-indole. Obtained as off-white solid. Yield: 22% (0.07 g, for two steps).1H NMR (400 MHz, acetone-d6): δ (ppm) 10.24 (br s, 1H), 10.18 (br s, 1H), 6.82 (d, J = 3.2 Hz, 1H), 7.45–7.13 (m, 6H), 6.86–6.78 (m, 2H), 4.74–4.45 (m, 2H), 3.58–3.50 (m, 1H), 2.78–2.64 (m, 1H), 2.61–2.53 (m, 1H), 2.37–2.28 (m, 1H), 2.23–2.11 (m, 1H), 2.02–1.95 (m, 1H), 1.91–1.72 (m, 5H), 1.64–1.45 (m, 4H), 1.44–1.24 (m, 10H), 1.21–1.03 (m, 5H), 0.98–0.90 (m, 7H), 0.65 (s, 3H); 13C NMR (100 MHz, acetone-d6): δ (ppm) 175.3, 174.8, 159.4, 159.3, 157.1, 157.0, 135.0, 134.8, 128.5, 128.4, 128.2, 128.1, 126.2, 125.2, 119.7, 119.6, 118.1, 118.1, 113.5, 113.4, 113.4, 113.3, 110.5, 110.2, 105.2, 105.1, 104.9, 104.8, 72.1, 57.6, 57.2, 52.3, 43.7, 43.3, 41.6, 41.3, 38.0, 37.5, 37.0, 36.6, 33.9, 33.0, 32.2, 31.5, 29.2, 28.3, 27.5, 25.2, 24.2, 19.2, 12.8. C–F splitting present; 19F NMR: δ (ppm) −126.8, −126.9; HRMS (ESI) calculated for C44H54N3O4 (M − H)−, 726.4088; found, 726.4096. HPLC purity: >99%.
4.1.3.10. Synthesis of Asp-linked hybrid 15. Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-methoxy-1H-indole. Obtained as off-white solid. Yield: 42% (0.17 g, for two steps). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 12.35 (b, 1H), 10.66 (br s, 1H), 10.61 (br s, 1H), 8.21 (d, J = 7.67 Hz, 1H), 7.28–6.65 (m, 8H), 4.50–4.40 (m, 2H), 4.13–4.05 (m, 1H), 3.66 (d, J = 2.2 Hz, 6H), 3.43–3.35 (m, 2H), 2.69–2.60 (m, 1H), 2.43–2.35 (m, 1H), 2.32–2.27 (m, 1H), 2.02–1.91 (m, 2H), 1.87–1.65 (m, 4H), 1.56–1.32 (m, 8H), 1.28–1.13 (m, 7H), 1.12–0.98 (m, 4H), 0.96–0.86 (m, 7H), 0.62 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 174.8, 172.6, 152.6, 152.5, 131.9, 131.7, 127.0, 126.9, 123.4, 122.6, 118.2, 116.5, 111.9, 111.8, 110.3, 101.6, 101.4, 69.9, 56.1, 55.7, 55.3, 55.3, 51.0, 42.3, 41.6, 36.8, 36.3, 35.4, 35.2, 35.0, 34.2, 32.4, 31.7, 30.7, 30.4, 30.3, 27.8, 26.9, 26.2, 23.9, 23.3, 20.5, 18.4, 11.9 (some peaks overlapped with the solvent peak.); HRMS (ESI) calculated for C46H60N3O6 (M − H)−, 750.4488; found, 750.4493. HPLC purity: >99%.
4.1.3.11. Synthesis of Asp-linked hybrid 16. Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-(benzyloxy)-1H-indole (see SI). Obtained as off-white solid. Yield: 51% (0.29 g, for two steps). 1H NMR (400 MHz, acetone-d6): δ (ppm) 9.94 (br s, 1H), 9.87 (br s, 1H), 7.50–7.15 (m, 17H), 6.84–8.73 (m, 2H), 5.15–4.90 (m, 4H), 4.65 (q, J = 4.9 Hz, 1H), 4.57 (m, 1H), 3.60–3.52 (m, 1H), 2.89–2.81 (m, 1H), 2.65–2.57 (m, 1H), 2.36–2.28 (m, 1H), 2.18–2.10 (m, 1H), 1.97–1.90 (m, 1H), 1.89–1.70 (m, 5H), 1.66–1.58 (m, 1H), 1.55–1.22 (m, 12H), 1.20–0.97 (m, 6H), 0.96–0.88 (m, 7H), 0.62 (s, 3H); 13C NMR (100 MHz, acetone-d6): δ (ppm) 174.6, 174.4, 153.4, 153.3, 139.0 (2C), 133.4, 133.3, 129.1 (4C), 128.5 (4C), 128.4 (3C), 128.3, 124.4, 123.5, 119.5, 118.1, 112.8, 112.7 (2C), 112.6, 103.9, 103.9, 71.7, 71.2, 71.2, 57.2, 56.9, 52.3, 43.4, 43.0, 41.2, 40.9, 38.1, 37.3, 36.6, 36.3, 36.3, 36.2, 35.3, 33.7, 32.7, 31.8, 31.3, 28.9, 28.0, 27.2, 24.9, 23.9, 21.5, 18.9, 12.4; HRMS (ESI) calculated for C58H68N3O6 (M − H)−, 902.5114; found, 902.5107. HPLC purity: >99%.
4.1.3.12. Synthesis of Asp-linked hybrid 17. Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-tert-butyl (1H-indol-5-yl) carbamate (see SI). Obtained as off-white solid. Yield: 74% (0.38 g, for two steps). 1H NMR (400 MHz, acetone-d6): δ (ppm) 9.94 (br s, 1H), 9.87 (br s, 1H), 8.81–7.68 (m, 4H), 7.46–7.12 (m, 7H), 4.69–4.48 (m, 2H), 3.62–3.50 (m, 1H), 2.90–2.80 (m, 1H), 2.65–2.54 (m, 1H), 2.40–2.30 (m, 1H), 2.24–2.13 (m, 1H), 1.98–1.71 (m, 6H), 1.66–1.54 (m, 2H), 1.53–1.44 (m, 20H), 1.40–1.20 (m, 10H), 1.17–0.99 (m, 5H), 0.98–0.88 (m, 7H), 0.63 (s, 3H). 13C NMR (100 MHz, acetone-d6): δ (ppm) 174.7, 174.4, 154.3, 154.3, 134.7134.5, 131.8, 131.8, 127.8, 124.3, 123.5, 119.5, 118.0, 115.7, 112.0, 111.9, 110.5, 110.2, 79.2, 79.2, 71.7, 57.2, 56.8, 52.4, 43.4, 43.0, 41.2, 40.9, 38.2, 37.2, 36.9, 36.3, 35.3, 33.4, 32.5, 31.9, 31.3, 28.9, 28.8 (6C), 28.7 (3C), 28.0, 27.2, 24.9, 23.9, 21.5, 18.9, 12.4; HRMS (ESI) calculated for C54H74N5O8 (M − H)−, 920.5543; found, 920.5543. HPLC purity: >99%.
4.1.3.13. Synthesis of Asp-linked hybrid 18 (IAN-15B). Prepared following the general diindolylation and deprotection procedure previously described for hybrids 10 and 11 using 5-azido-1H-indole (see SI). Obtained as beige solid. Yield: 41% (0.18 g, for two steps). 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.31 (br s, 1H), 10.25 (br s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.43–7.14 (m, 6H), 6.82–6.75 (m, 2H), 4.69 (q, J = 5.0 Hz, 1H), 4.60–4.52 (m, 1H), 3.61–3.51 (m, 1H), 2.90–2.80 (m, 1H), 2.69–2.52 (m, 1H), 2.40–2.30 (m, 1H), 2.25–2.14 (m, 1H), 1.98–1.62 (m, 1H), 1.90–1.71 (m, 5H), 1.67–1.59 (m, 1H), 1.55–1.40 (m, 4H), 1.39–1.30 (m, 6H), 1.26–0.98 (m, 8H), 0.95–0.86 (m, 7H), 0.63 (s, 3H); 13C NMR (100 MHz, acetone-d6): δ (ppm) 174.5, 174.0, 135.6, 135.4, 131.0, 130.9, 128.4, 128.2, 125.2, 124.2, 118.9, 117.4, 113.5, 113.4, 113.4, 113.2, 109.5, 109.5, 71.4, 57.0, 56.6, 51.7, 51.6, 43.1, 42.7, 41.0, 40.7, 37.5, 36.9, 36.4, 35.9, 35.0, 33.3, 32.4, 31.4, 31.0, 28.6, 27.7, 26.9, 24.5, 23.6, 21.2, 18.6, 12.1; HRMS (ESI) calculated for C44H54N9O4 (M − H)−, 772.4304; found, 772.4310. HPLC purity: >99%.
4.2 Biology
All cells used for the cell-based assays were purchased commercially. The human breast carcinoma cell line MDA-MB-231 was obtained from the Bioresource Collection and Research Center (BCRC 60425). The other two TNBC cell lines, BT549 (HTB-122) and Hs578T (HTB-126), were acquired from the American Type Culture Collection (ATCC). HUVEC were obtained from Lonza (#C2519A). All cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) under humidified conditions at 37 °C with 5% CO2.
All reactions were incubated at 37 °C for 15 minutes, followed by thermal inactivation at 101 °C for 10 minutes. The resulting sialylated products were quantified via UPLC analysis to assess inhibitory activity. For IC50 determination, selected compounds were tested across 5 to 8 different concentrations, spanning the full range of inhibition (0–100%). Residual enzymatic activity was normalized using an internal standard, and the inhibition curves were plotted against inhibitor concentration. IC50 values were calculated by fitting the data to a sigmoidal dose–response model (4-parameter logistic model) using GraphPad Prism. Enzyme kinetics were further evaluated by applying nonlinear regression to the initial rate data.
000 TNBC (MDA-MB-231, BT549, and Hs578T) cells were seeded into 96-well plates and treated with varying concentrations of 3,3-DIM and the LCA-DIM hybrids ranging from 0.5–80 μM. The cells were then incubated for 48 h, with a 0.1% DMSO-containing medium used as a control. Following incubation, MTT reagent (1 mg mL−1; Sigma) was added, and cells were further incubated at 37 °C for 4 hours to allow for formazan crystal formation. The medium was then replaced with DMSO to solubilize the crystals, and plates were kept at room temperature for 30 minutes. Absorbance was measured at 540 nm using a Molecular Devices SPECTRAmax PLUS384 microplate reader to assess the cytotoxicity of the test compounds.
Author contributions
C. A. P. Concio - methodology, investigation, validation, visualization, formal analysis, writing – original draft; Y. T. He - methodology, investigation, validation; S. J. L. P. Perez - methodology, formal analysis, writing – review & editing; T. T. Chang - investigation, formal analysis, visualization; C. L. Cheng - investigation, formal analysis, visualization; S. D. Arco - supervision, project administration; W. S. Li - supervision, project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information is available: The SI contains – synthesis methods for selected indoles, MTT data, Transwell migration data, IC50 plots, western blots analyses, NMR and Mass spectra, and HPLC profiles. See DOI: https://doi.org/10.1039/D5MD00390C.The data supporting this article have been included as part of the SI.
Acknowledgements
We are thankful to the Institute of Chemistry at Academia Sinica, Taiwan, for providing access to their NMR and Mass Spectrometry facilities. We also acknowledge the Chemical Biology Facility at the Institute of Chemistry, Academia Sinica, Taiwan, which also provided instrumentation support. We're also giving our full gratitude for financial support of this work given by the Academia Sinica (AS-α-110-01-16 and AS-IR–112-08-A) and National Science and Technology Council (NSTC, Taiwan, NSTC 112-2113-M-001-009 and NSTC 113-2113-M-001-025).References
- C. Traving and R. Schauer, Cell. Mol. Life Sci., 1998, 54, 1330–1349 CrossRef CAS PubMed.
- S. Ghosh, in Sialic Acids and Sialoglycoconjugates in the Biology of Life, Health and Disease, ed. S. Ghosh, Academic Press, 2020, pp. 1–61 Search PubMed.
- C. Dobie and D. Skropeta, Br. J. Cancer, 2021, 124, 76–90 CrossRef CAS PubMed.
- J. Du, S. Hong, L. Dong, B. Cheng, L. Lin, B. Zhao, Y.-G. Chen and X. Chen, J. Biol. Chem., 2015, 290, 12000–12013 CrossRef CAS PubMed.
- M. Hugonnet, P. Singh, Q. Haas and S. von Gunten, Front. Immunol., 2021, 12, 1–16 Search PubMed.
- S. Pietrobono and B. Stecca, Cancers, 2021, 13, 2014 CrossRef CAS PubMed.
- F. Li and J. Ding, Protein Cell, 2018, 10, 550–565 CrossRef PubMed.
- Y. Li and X. Chen, Appl. Microbiol. Biotechnol., 2012, 94, 887–905 CrossRef CAS PubMed.
- H. X. Cui, H. Wang, Y. Wang, J. Song, H. Tian, C. Xia and Y. Shen, Oncol. Rep., 2016, 36, 3317–3324 CrossRef CAS PubMed.
- N. C. Hait, A. Maiti, R. Wu, V. L. Andersen, C.-C. Hsu, Y. Wu, D. G. Chapla, K. Takabe, M. E. Rusiniak, W. Bshara, J. Zhang, K. W. Moremen and J. T. Y. Lau, Cancer Gene Ther., 2022, 29, 1662–1675 CrossRef CAS PubMed.
- X. Wu, J. Zhao, Y. Ruan, L. Sun, C. Xu and H. Jiang, Cell Death Dis., 2018, 9, 1102 Search PubMed.
- R. B. Jones, K. A. Dorsett, A. B. Hjelmeland and S. L. Bellis, J. Biol. Chem., 2018, 293, 5659–5667 Search PubMed.
- R. B. Jones, A. D. Silva, K. E. Ankenbauer, C. M. Britain, A. Chakraborty, J. A. Brown, S. W. Ballinger and S. L. Bellis, Glycobiology, 2023, 33, 626–636 CrossRef CAS PubMed.
- N. Bhalerao, A. Chakraborty, M. P. Marciel, J. Hwang, C. M. Britain, A. D. Silva, I. E. Eltoum, R. B. Jones, K. L. Alexander, L. E. Smythies, P. D. Smith, D. K. Crossman, M. R. Crowley, B. Shin, L. E. Harrington, Z. Yan, M. M. Bethea, C. S. Hunter, C. A. Klug, D. J. Buchsbaum and S. L. Bellis, JCI insight, 2023, 8, 1–23 CrossRef PubMed.
- M. Pérez-Garay, B. Arteta, L. Pagès, R. de Llorens, C. de Bolòs, F. Vidal-Vanaclocha and R. Peracaula, PLoS One, 2010, 5, e12524 CrossRef PubMed.
- G. Venturi, I. Gomes Ferreira, M. Pucci, M. Ferracin, N. Malagolini, M. Chiricolo and F. Dall'Olio, Glycobiology, 2019, 29, 684–695 CrossRef CAS PubMed.
- M. Smithson, R. Irwin, G. Williams, K. L. Alexander, L. E. Smythies, M. Nearing, M. C. McLeod, S. Al Diffalha, S. L. Bellis and K. M. Hardiman, J. Biol. Chem., 2022, 298, 1–7 CrossRef PubMed.
- S. J. L. P. Perez, C.-W. Fu and W.-S. Li, Molecules, 2021, 26, 5673 CrossRef CAS PubMed.
- R. Al Saoud, A. Hamrouni, A. Idris, W. K. Mousa and T. Abu Izneid, Biomed. Pharmacother., 2023, 165, 115091 CrossRef CAS PubMed.
- R. Szabo and D. Skropeta, Med. Res. Rev., 2017, 37, 219–270 CrossRef PubMed.
- J.-Y. Chen, Y.-A. Tang, S.-M. Huang, H.-F. Juan, L.-W. Wu, Y.-C. Sun, S.-C. Wang, K.-W. Wu, G. Balraj, T.-T. Chang, W.-S. Li, H.-C. Cheng and Y.-C. Wang, Cancer Res., 2011, 71, 473–483 CrossRef CAS PubMed.
- K.-H. Chang, L. Lee, J. Chen and W.-S. Li, Chem. Commun., 2006, 629–631, 10.1039/B514915K.
- C.-H. Chiang, C.-H. Wang, H.-C. Chang, S. V. More, W.-S. Li and W.-C. Hung, J. Cell. Physiol., 2010, 223, 492–499 CrossRef CAS PubMed.
- C.-W. Fu, H.-E. Tsai, W.-S. Chen, T.-T. Chang, C.-L. Chen, P.-W. Hsiao and W.-S. Li, J. Med. Chem., 2021, 64, 527–542 CrossRef CAS PubMed.
- H.-E. Tsai, C.-L. Chen, T.-T. Chang, C.-W. Fu, W.-C. Chen, S. J. L. P. Perez, P.-W. Hsiao, M.-H. Tai and W.-S. Li, Int. J. Mol. Sci., 2024, 25, 4283 CrossRef CAS PubMed.
- S. J. L. P. Perez, C.-L. Chen, T.-T. Chang and W.-S. Li, Bioorg. Med. Chem. Lett., 2024, 105, 129760 CrossRef CAS PubMed.
- Y. Kang, Z.-T. Mai, L.-F. Yau, R.-F. Li, T.-T. Tong, C.-G. Yang, K.-M. Chan, Z.-H. Jiang, Y. Wang, Z.-F. Yang and J.-R. Wang, J. Virol., 2022, 96(6), 1–19 CrossRef PubMed.
- K.-Y. Lee, H. G. Kim, M. R. Hwang, J. I. Chae, J. M. Yang, Y. C. Lee, Y. K. Choo, Y. I. Lee, S.-S. Lee and S.-I. Do, J. Biol. Chem., 2002, 277, 49341–49351 CrossRef CAS PubMed.
- A. P. Montgomery, C. Dobie, R. Szabo, L. Hallam, M. Ranson, H. Yu and D. Skropeta, Bioorg. Med. Chem., 2020, 28, 115561 CrossRef CAS PubMed.
- S. M. Kim, Int. J. Mol. Sci., 2016, 17, 1155 CrossRef PubMed.
- E. J. Kim, M. Shin, H. Park, J. E. Hong, H.-K. Shin, J. Kim, D. Y. Kwon and J. H. Y. Park, J. Nutr., 2009, 139, 2373–2379 CrossRef CAS PubMed.
- Y. Jiang, Y. Fang, Y. Ye, X. Xu, B. Wang, J. Gu, M. Aschner, J. Chen and R. Lu, Front. Pharmacol., 2019, 10, 1–23 Search PubMed.
- S.-Y. Fang, S.-Y. Chen, Y.-Y. Chen, T.-J. Kuo, Z.-H. Wen, Y.-H. Chen, T.-L. Hwang and P.-J. Sung, Nat. Prod. Commun., 2021, 16, 1934578X211033735 CrossRef CAS.
- F. A. Wati, M. Santoso, Z. Moussa, S. Fatmawati, A. Fadlan and Z. M. A. Judeh, RSC Adv., 2021, 11, 25381–25421 RSC.
- S. Chakraborty, S. Ghosh, B. Banerjee, A. Santra, A. Adhikary, A. K. Misra and P. C. Sen, Front. Pharmacol., 2016, 7, 1–21 CrossRef PubMed.
- S.-M. Huang, P.-C. Hsu, M.-Y. Chen, W.-S. Li, S. V. More, K.-T. Lu and Y.-C. Wang, Int. J. Cancer, 2012, 131, 722–732 CrossRef CAS PubMed.
- C.-H. Wang, C.-L. Chen, S. V. More, P.-W. Hsiao, W.-C. Hung and W.-S. Li, PLoS One, 2014, 9, e101088 CrossRef PubMed.
- B. H. Lipshutz and J. Keith, Tetrahedron Lett., 1998, 39, 2495–2498 CrossRef CAS.
- T. Fukuyama, S. C. Lin and L. Li, J. Am. Chem. Soc., 1990, 112, 7050–7051 CrossRef CAS.
- T. Miyazaki, Y. Han-ya, H. Tokuyama and T. Fukuyama, Synlett, 2004, 2004, 477–480 Search PubMed.
- B. Biersack, Cancer Drug Resist., 2020, 3, 867–878 CAS.
- O. D. Reyes-Hernández, G. Figueroa-González, L. I. Quintas-Granados, S. C. Gutiérrez-Ruíz, H. Hernández-Parra, A. Romero-Montero, M. L. Del Prado-Audelo, S. A. Bernal-Chavez, H. Cortés, S. I. Peña-Corona, L. Kiyekbayeva, D. A. Ateşşahin, T. Goloshvili, G. Leyva-Gómez and J. Sharifi-Rad, Cancer Cell Int., 2023, 23, 180 CrossRef PubMed.
- S.-Y. Chen, P.-L. He, L.-Y. Lu, M.-C. Lin, S.-H. Chan, J.-S. Tsai, W.-T. Luo, L.-H. Wang and H.-J. Li, Cancer Res., 2025, 85, 1199–1218 CrossRef CAS PubMed.
- J. Lu, T. Isaji, S. Im, T. Fukuda, N. Hashii, D. Takakura, N. Kawasaki and J. Gu, J. Biol. Chem., 2014, 289, 34627–34641 CrossRef PubMed.
- D. Harrus, A. Harduin-Lepers and T. Glumoff, J. Struct. Biol., 2020, 212, 107628 CrossRef CAS PubMed.
- W.-S. Chen, C. A. P. Concio, T.-T. Chang, C.-L. Chen, S. J. L. P. Perez and W.-S. Li, Bioorg. Chem., 2025, 159, 108401 CrossRef CAS PubMed.
- B. Kuhn, J. Benz, M. Greif, A. M. Engel, H. Sobek and M. G. Rudolph, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2013, 69, 1826–1838 CrossRef CAS PubMed.
- S. J. L. P. Perez, Z.-F. Hsu, T.-T. Chang, C.-L. Chen and W.-S. Li, Eur. J. Med. Chem., 2025, 292, 117674 CrossRef CAS PubMed.
- T. Isaji, Y. Sato, T. Fukuda and J. Gu, J. Biol. Chem., 2009, 284, 12207–12216 CrossRef CAS PubMed.
- J. Liu, X. Dong, R. Xie, Y. Tang, A. M. Thomas, S. Li, S. Liu, M. Yu and H. Qin, Placenta, 2024, 149, 18–28 CrossRef CAS PubMed.
- D. R. Christie, F. M. Shaikh, J. A. Lucas, J. A. Lucas and S. L. Bellis, J. Ovarian Res., 2008, 1, 3 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |