
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of PZ671, a highly potent and in vivo active CRBN-recruiting Bcl-xL degrader†
Peiyi
Zhang‡
a,
Dinesh
Thummuri‡
b,
Wanyi
Hu
a,
Sajid
Khan
b,
Yonghan
He
b,
Xuan
Zhang
a,
Pratik
Pal
a,
Dongwen
Lv
b,
Daohong
Zhou
*bc and
Guangrong
Zheng
 *a
*a
aDepartment of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, FL 32610, USA. E-mail: zhengg@cop.ufl.edu
bDepartment of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, FL 32610, USA. E-mail: zhoud@uthscsa.edu
cDepartment of Biochemistry and Structural Biology, Center for Innovative Drug Discovery, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA. E-mail: zhoud@uthscsa.edu
First published on 23rd May 2025
Abstract
The conversion of conventional inhibitors of Bcl-xL, a key anti-apoptotic protein, to PROTAC degraders has shown significant promise, particularly in mitigating the on-target thrombocytopenia associated with Bcl-xL inhibition but improving their potency. Previously, we reported XZ739, a CRBN-recruiting Bcl-xL PROTAC that was 20-fold more potent than its parent inhibitor ABT-263 against Bcl-xL-dependent MOLT-4 cells while reducing toxicity to human platelets. Building on XZ739, we here report the discovery of PZ671, a more potent Bcl-xL degrader with ∼10-fold improved cellular activity against MOLT-4 cells (IC50 = 1.3 nM) and ∼6-fold enhanced degradation potency against Bcl-xL (DC50 = 0.9 nM) as well as superior potency across multiple SCLC cell lines compared to XZ739. In vivo studies revealed that PZ671 could effectively inhibit MOLT-4 xenograft growth in mice but caused only a moderate and transient reduction in platelet counts following its administration. Our findings highlight the potential of CRBN-recruiting Bcl-xL degraders as promising anticancer agents with improved efficacy and manageable platelet toxicity.
Introduction
Apoptosis, a programmed cell death process, is fundamental for maintaining tissue homeostasis and eliminating damaged or unnecessary cells.1 Dysregulation of apoptosis is a hallmark of cancer, enabling malignant cells to evade death signals and proliferate uncontrollably.2 Targeting the apoptotic machinery has thus emerged as a promising strategy in cancer therapy. The B-cell lymphoma 2 (Bcl-2) protein family plays a central role in regulating apoptosis. This family consists of pro-apoptotic (e.g., BAX, BAK) and anti-apoptotic (e.g., Bcl-2, Bcl-xL, Mcl-1) proteins, which collectively govern mitochondrial integrity and caspase activation. Anti-apoptotic proteins, notably Bcl-2, Bcl-xL, and Mcl-1, are frequently overexpressed in cancers, conferring a survival advantage to malignant cells.3–7 Among them, Bcl-xL is the most frequently overexpressed protein in solid tumors and a subset of leukemias and lymphomas.8 Tumor resistance to anticancer therapies has also been found to be positively correlated with Bcl-xL expression.9 Our recent studies revealed that Bcl-xL inhibitors also possess senolytic properties, enabling the selective elimination of senescent cells implicated in aging and age-related diseases.10–12 This dual activity renders Bcl-xL a target with broader therapeutic implications beyond cancer treatment.Over the past two decades, numerous Bcl-xL selective or Bcl-xL/Bcl-2 dual inhibitors have been developed (Fig. 1).13 Early efforts in targeting the Bcl-2 family led to the development of ABT-737, the first potent Bcl-xL/Bcl-2 dual inhibitor developed via fragment-based drug discovery by NMR approach.14,15 ABT-737 served as a prototype for subsequent Bcl-xL/Bcl-2 dual inhibitors, such as navitoclax (ABT-263), which is orally bioavailable and has been in phase II clinical trials for hematological malignancies and small cell lung cancer (SCLC).16 However, these inhibitors caused dose-limiting thrombocytopenia due to Bcl-xL's role in platelet survival, which cannot be solved by conventional medicinal chemistry, greatly limiting their clinical utility.
 | ||
| Fig. 1 Structures of representative Bcl-xL/Bcl-2 dual (ABT-263 and ABT-737) and Bcl-2 selective (ABT-199) inhibitors. | ||
Efforts to mitigate the on-target platelet toxicity associated with Bcl-xL inhibition have driven the exploration of innovative strategies, including prodrugs,17 antibody–drug conjugates (ADCs),18 and proteolysis-targeting chimeras (PROTACs) (Fig. 2).13,19–28 PROTACs utilize E3 ligases to induce targeted protein degradation, offering the potential for cell/tissue selectivity with reduced on-target effects, even when targeting ubiquitously expressed proteins, as long as the E3 ligase has cell- or tissue-selective/specific expression.29
 | ||
| Fig. 2 Chemical structures of representative Bcl-xL/Bcl-2-targeting PROTACs. | ||
Our group has focused on developing Bcl-xL/Bcl-2 PROTAC degraders using various E3 ligases, including von Hippel–Lindau (VHL) and cereblon (CRBN) (Fig. 2).21–26 Among these, DT2216, a VHL-recruiting Bcl-xL selective degrader derived from ABT-263, demonstrated higher potency against a variety of Bcl-xL-dependent cancer cells with significantly reduced platelet toxicity compared to its parent compound ABT-263.21 DT2216 is currently in clinical trials (https://ClinicalTrials.gov IDs: NCT04886622 and NCT06620302). Building on this, we also developed XZ739, a CRBN-recruiting Bcl-xL degrader, by replacing the VHL-binding moiety in DT2216 with CRBN binding moiety pomalidomide, the piperazine group with an N-methylamine group, and the polymethylene linker with a PEG linker.25 XZ739 exhibited over 7-fold higher potency than DT2216 against MOLT-4 cells (a T cell acute lymphoblastic leukemia (T-ALL) cell line that primarily depends on Bcl-xL for survival) and maintained excellent selectivity (>100-fold) for MOLT-4 cells over human platelets because CRBN E3 ligase and its associated E1 and E2 enzymes are minimally expressed in human platelets.24 Similar to DT2216, no BCL-2 degradation was observed after treating with XZ739 in MOLT-4 cells.25 Our previous studies have revealed that both Bcl-xL and Bcl-2 can form stable ternary complexes with DT2216 and VHL in cell-free conditions, whereas in live cells, only Bcl-xL, but not Bcl-2, can form stable ternary complexes with DT2216 and VHL, which may contribute to the specificity of Bcl-xL degradation.21 These findings demonstrated the feasibility of achieving target specificity through conversion of non-selective inhibitors to PROTACs. However, whether the same mechanism exists for the selectivity of compound XZ739 on Bcl-xL needs to be validated by relevant experiments. In addition, while DT2216 displayed moderate cytotoxicity (IC50 = 278 nM) in NCI-H146 cells (a SCLC cell line that depends on both Bcl-xL and Bcl-2 for survival), XZ739 is more potent than DT2216 against NCI-H146 cells (IC50 = 25 nM), suggesting its potential as a single-agent treatment for tumors dependent on both Bcl-xL and Bcl-2 for survival. These findings laid a solid foundation for the further structural optimization of XZ739 as shown in this report.
Through a series of structure–activity relationship (SAR) studies, we identified PZ671 as a highly potent Bcl-xL degrader. PZ671 exhibited ∼10-fold improved cellular activity against MOLT-4 cells (IC50 = 1.3 nM vs. 10.1 nM), ∼6-fold enhanced degradation potency against Bcl-xL (DC50 = 0.9 nM vs. 5.1 nM) compared to XZ739 and demonstrated superior potency across all tested SCLC cell lines. Importantly, PZ671 showed ∼25-fold higher selectivity for MOLT-4 cells over platelets compared to ABT-263. In vivo, PZ671 effectively inhibited MOLT-4 xenograft tumor growth at a dosing regimen of 1.5 mg kg−1 every 4 days but induced only a transient drop in platelet counts following its administration with rapid recovery observed from the second day onwards. This contrasts with the milder but more prolonged thrombocytopenic effects typically associated with VHL-recruiting degraders. We consider that the rapidly reversible platelet effects induced by the CRBN-recruiting degrader PZ671 may represent a more controllable therapeutic profile.
Results and discussion
Compound design and biological evaluation
As reported,23,30 Glu96 of Bcl-xL and the protonated nitrogen atom on the morpholine ring of ABT-263 form a salt bridge, which plays a crucial role in achieving high Bcl-xL binding affinity. Notably, the morpholine ring can be replaced with a dimethylamino group, as demonstrated in ABT-737. In our previous work, we designed XZ739 by replacing the morpholine ring in ABT-263 with an N-methylamino group as a linker attachment point. To investigate how different substituents at this position influence PROTAC activity, we systematically modified the substituents by either removing the methyl group or introducing various alkyl groups, yielding compounds 1a–1i (Table 1).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | IC50a (nM) | K i | Cmpd | R | IC50a (nM) | K i |
| a IC50 values are the means of at least three independent experiments; MOLT-4 cells were treated for 48 h. b Bcl-xL binding affinity; Ki values (in nM) are the means of at least three independent experiments. c ND: not determined. | |||||||
| ABT-263 | — | 202.3 | 0.26 | ||||
| XZ739 | CH3 | 10.1 | 0.31 | 1e |

|
329.5 | 9.02 |
| 1a | H | 59.7 | 0.27 | 1f |

|
396.9 | NDc |
| 1b | CH2CH3 | 52.8 | 0.38 | 1g |

|
325.8 | NDc |
| 1c | CH(CH3)2 | 59.4 | NDc | 1h |
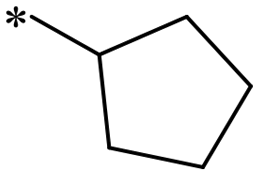
|
559.9 | 9.14 |
| 1d |

|
297.3 | 8.75 | 1i |
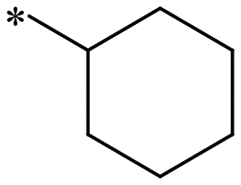
|
>1000 | NDc |

These compounds were evaluated for their effects on the viability of MOLT-4 cells, with XZ739 as the positive control (Table 1 and Fig. S1A†). A clear trend was observed, larger substituents impaired cellular potency against MOLT-4 cells, with the effect becoming more pronounced as the substituent size increased. This is partly due to bulkier substituents disrupting the optimal positioning of the nitrogen atom for salt bridge formation, thereby decreasing Bcl-xL binding affinity (Table 1 and Fig. 3, S2 and S3†). However, other factors, such as the efficiency of forming stable ternary complexes, may also contribute. For example, compounds 1a/1b exhibited similar binding affinity to XZ739 but were 5- to 6-fold less potent in killing MOLT-4 cells. Based on these findings, we retained the methyl-substituted tertiary amine in XZ739 and explored the impact of various linkages to the CRBN-recruiting moiety thalidomide/lenalidomide on cellular activity (Table 2 and Fig. S1B†).
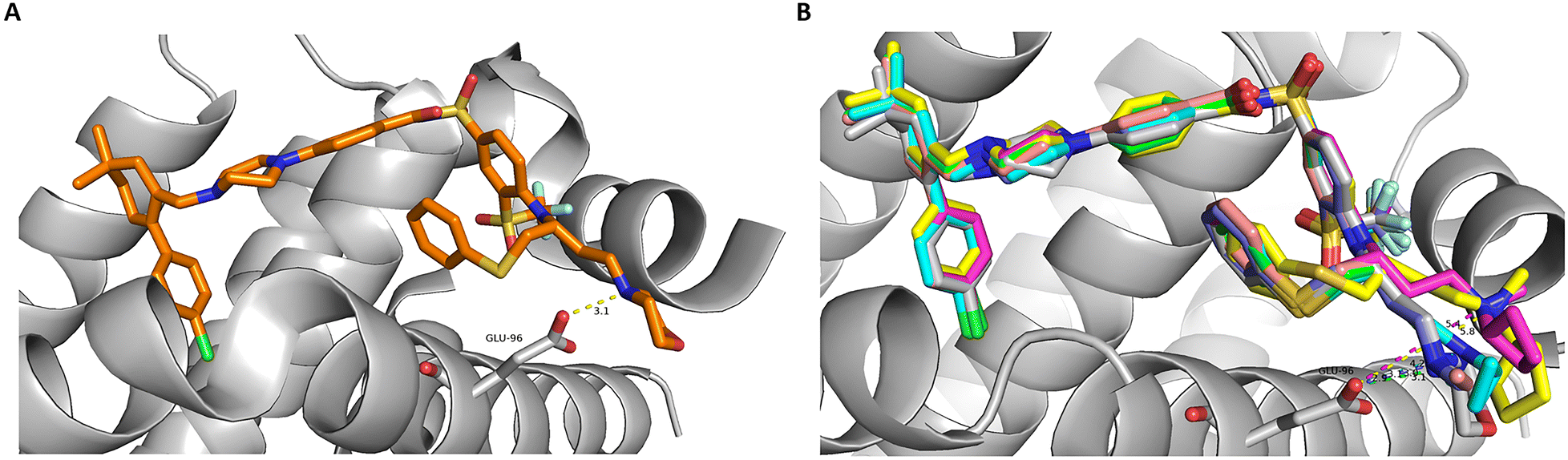 | ||
| Fig. 3 Computational modelling of (A) ABT-263, available from PDB code 4QNQ and (B) derivatives with various alkyl substituents at the salt bridge nitrogen bound to Bcl-xL proteins. The bulky substituents push the nitrogen away from its optimal position to form a salt bridge with the Glu96 residue. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| X | Substitution position | Cmpd | L | IC50a (nM) | Cmpd | L | IC50a (nM) |
| a IC50 values are the means of at least three independent experiments; MOLT-4 cells were treated for 48 h. | |||||||
| ABT-263 | — | 202.3 | XZ739 | — | 10.1 | ||
| CO | C4- | 2a |
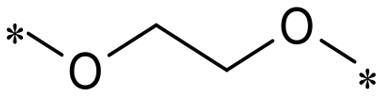
|
10.4 | 2d |

|
10.6 |
| 2b |

|
5.2 | 2e |

|
36.1 | ||
| 2c |

|
6.2 | 2f |

|
45.5 | ||
| C5- | 3a |

|
14.3 | 3b |
|
4.3 | |
| CH2 | C4- | 4a |

|
4.1 | 4b |

|
4.5 |
| C5- | 5a |

|
2.1 | 5b (PZ671) |

|
1.3 | |
Compound 2a, featuring an ether linkage at the C4 position of thalidomide, exhibited comparable potency against MOLT-4 cells to XZ739. Replacing the ether linkage of 2a with a triple bond 2b resulted in a 2-fold increase in potency, while the corresponding saturated C–C linkage containing compound 2c retained similar potency against MOLT-4 cells to 2b. Moving the linker attachment to the C5 position of thalidomide significantly reduced potency for the triple-bond containing compound 3a by almost 3-fold compared to the corresponding C4-substituted 2b, whereas the C–C linkage compound 3b showed cellular activity comparable to that of 2b/2c.
Incorporating conformationally restricted linkers has been shown to stabilize ternary complex formation and improve PK properties of PROTACs.31–35 To test this, we synthesized compounds 2d–2f (Table 2) by inserting conformationally restricted units (piperazine/piperidine/triple bond) into the linker. Compound 2d with a piperazine ring directly connected to thalidomide had similar cellular activity to XZ739 but was 2-fold weaker than 2b. Introducing an additional triple bond between the piperidine ring and thalidomide (compound 2e) reduced activity by ∼4-fold compared to 2d. Reducing the triple bond in 2e to a saturated ethylene (compound 2f) resulted in slightly lower potency in MOLT-4 cells than 2e. These results suggest that introducing ring systems, whether at the linker terminus or in the middle, negatively impact activity.
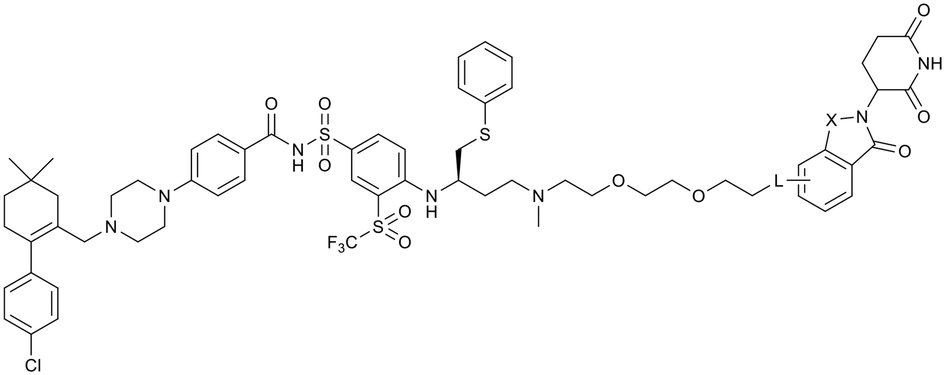
Lenalidomide often offers advantages over thalidomide as a CRBN-recruiting ligand due to its improved metabolic and chemical stability, attributed to the absence of a phthalimide carbonyl group.36,37 Incorporating lenalidomide with the effective C–C linkage at the C4 or C5 positions, we synthesize compounds 4a, 4b and 5b. The cellular activity of these compounds was significantly improved, with the C5-substituted ethylene linkage compound 5b (PZ671, Fig. 4A) showing an 8-fold increase in potency against MOLT-4 cells compared to XZ739.
 | ||
| Fig. 4 PZ671-induced Bcl-xL degradation. (A) The structure of PZ671. (B and D) Representative western blotting analysis of protein levels in MOLT-4 cells after treatment with indicated concentrations of PZ671 and XZ739 for 16 h. Percentage decrease in β-actin levels due to toxicity was normalized to the control sample. Data representative of 3 independent experiments. (C and E) Densitometric analysis of Bcl-xL expression in MOLT-4 cells. The data presented are mean ± SD of three independent experiments. (F) Pretreatment with 1 μM MG-132 for 2 h blocked the degradation of Bcl-xL induced by PZ671. (G) Bcl-xL levels in MOLT-4 cells treated with 100 nM PZ671 at the indicated time points. | ||
PZ671 was selected for further evaluation. We evaluated PZ671 in comparison to XZ739 for their cell killing effects on a panel of SCLC cell lines (Fig. S4†). The results revealed that PZ671 is more potent than XZ739 against all tested SCLC cell lines. Western blot analysis confirmed that PZ671 induced dose-dependent Bcl-xL degradation in MOLT-4 cells (Fig. 4B and S5A†) and had a better degradation potency against Bcl-xL than XZ739, with a DC50 value of 0.9 nM (Fig. 4C) compared to a DC50 value of 5.1 nM for XZ739 (Fig. 4D and E). This degradation was blocked by proteasome inhibitor MG-132 (Fig. 4F and S5C†), indicating a proteasome-dependent mechanism. Furthermore, the Bcl-xL degradation induced by PZ671 in MOLT-4 cells was rapid, with Bcl-xL protein levels significantly reduced within 2 h of treatment, and almost complete degradation observed 4 h after exposure to 100 nM PZ671 (Fig. 4G and S5D and E†).
We next assessed the impact of PZ671 on apoptosis. Western blot analysis showed that PZ671 dose-dependently increased poly(ADP-ribose) polymerase (PARP) and caspase-3 cleavage in MOLT-4 cells (Fig. 5A and S6†), suggesting the apoptotic cell-death mechanism. Notably, we observed a significant reduction in β-actin levels with increasing concentrations of PZ671 in this experiment. MOLT4 T-ALL cells are highly sensitive to Bcl-xL degradation and can rapidly undergo apoptosis upon Bcl-xL loss. In line with our previous observation that 100 nM PZ671 nearly completely degrades Bcl-xL within 4 h (Fig. 4G), we hypothesize that PZ671-induced cell death at higher concentrations may partially account for the observed β-actin decrease. However, since this effect was not consistently observed across all cases, additional factors influencing β-actin levels may be involved and warrant further investigation. The apoptotic cell-death mechanism is further supported by flow cytometry analysis of annexin V and propidium iodide (PI) staining (Fig. 5B), which showed that PZ671-induced apoptosis was attenuated by pretreatment of MOLT-4 cells with the pan-caspase inhibitor Q-VD-OPh (QVD).
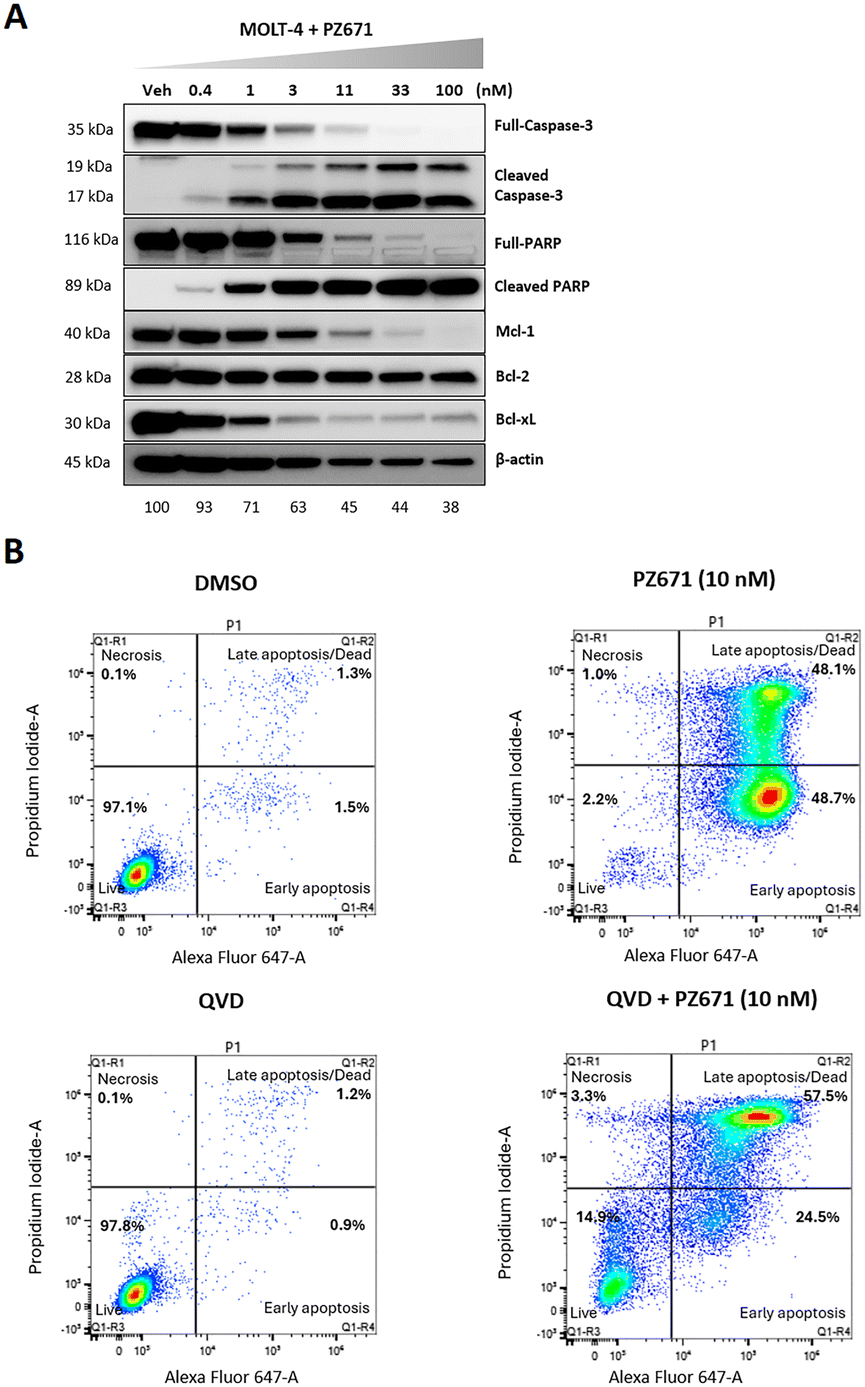 | ||
| Fig. 5 Characterization of PZ671-mediated apoptosis in MOLT-4 cells. (A) Western blot analysis of cleaved PARP and cleaved caspase-3 after 16 h of treatment with indicated concentrations of PZ671. The full-PARP blot was from the stripped membrane and re-probed with cleaved PARP, resulting in the rectangular shape. Percentage decrease in β-actin levels due to toxicity was normalized to the control sample. (B) Flow cytometry analysis of apoptosis using annexin V and PI staining. MOLT-4 cells were treated with PZ671 at 10 nM for 24 h. PZ671 significantly increased the percentage of apoptotic cells and QVD (10 μM) pretreatment for 2 h inhibited the apoptosis induced by PZ671. Data are representative of two independent experiments. | ||
In human platelets, PZ671 degraded Bcl-xL at concentrations above 1 μM (Fig. 6A and S7†), whereas XZ739 showed no degradation at the same concentrations.25 Consistent with the degradation result, the selectivity of PZ671 for MOLT-4 cells over platelets was reduced compared to XZ739 (25-fold vs. 137-fold) (Fig. 6B and S8†). In vivo, PZ671 caused a transient ∼72% reduction in platelet counts after a single dose (1.5 mg kg−1, i.p.) in MOLT-4 xenograft mice, with rapid recovery observed from the second day onward (Fig. 6C–E). Clinically, we believe the transient platelet reduction could be more manageable with optimized dosing regimen. Importantly, there is clear evidence that PZ671 treatment (1.5 mg kg−1, Q4D, i.p.) markedly suppressed the tumor growth (Fig. 6F).
 | ||
| Fig. 6 (A) Western blot showing the Bcl-xL levels in human platelets treated with the indicated concentration of PZ671 for 48 h. (B) A comparison of cellular activity of selected compounds in human platelets and MOLT-4 cells. (C) Illustration of the experiment design of MOLT-4 T-ALL cancer xenograft model. (D) Blood platelet (PLT) counts in mice were measured 1 day after receiving the first dose of vehicle (Veh) or PZ671. The data presented are mean ± SD (n = 3 mice per group). (E) Changes in blood platelet (PLT) counts in mice were measured during one cycle after administration of vehicle (control) and PZ671. The data presented are mean ± SD (n = 3 mice per group). (F) Changes in tumor volume over time after the start of treatment. | ||
Chemistry
The synthesis of PROTAC 1a is outlined in Scheme 1. Briefly, SNAr substitution of 4-fluorothalidomide 6 with azide 7 under microwave irradiation afforded compound 8. Palladium-catalyzed hydrogenation of 8 resulted in compound 9. Coupling of precursor acid 10 with sulfonamide 11 in the presence of EDCI and DMAP afforded compound 12, which was reduced to aldehyde by treating with DIBAL-H to give compound 13. Aldehyde 13 was subjected to reductive amination with 9 to generate the final product 1a. Compounds 7,3810,39 and 1140 were prepared as reported. | ||
| Scheme 1 Reagents and conditions: (a) DIPEA, DMSO, MW, 100 °C; (b) Pd/C, H2, MeOH, rt; (c) EDCI, DMAP, DCM, rt; (d) DIBAL-H, toluene, −78 °C; (e) NaBH(OAc)3, DCM, rt. | ||
Pomalidomide-based PROTACs 1b, 1c, and 1e–1i with various substitutions on the salt bridge nitrogen were synthesized by reductive amination of 1a with the corresponding aldehyde (14a and 14b) or ketone (15a–15e) (Scheme 2). Compound 1d was produced through nucleophilic substitution of 1a with bromocyclopropane 16.
 | ||
| Scheme 2 Reagents and conditions: (a) NaBH(OAc)3, DCE, 50 °C; (b) DIPEA, EtOH, 65 °C. | ||
The syntheses of PROTACs 2a–2d, 3a, 3b, 4a, 4b, 5a, and 5b are outlined in Schemes 3–6. Tosylate PEG linker precursor 20 was synthesized as previously reported.41 It was then reacted with 4-OH-thalidomide 21 in the presence of NaHCO3 and KI at 80 °C to yield compound 22. Removal of the Boc group of 22, followed by reductive amination with aldehyde 13, yielded the final product 2a (Scheme 3). Terminal alkyne linker precursor 25 was prepared by tosylation of the OH group of starting material 23, followed by treating with a methylamine ethanol solution (33 wt%) and Boc protection. Sonogashira coupling of 25 with 4- or 5-bromo-substituted thalidomide/lenalidomide derivatives 26a–26d afforded intermediates 27a–27d, which were subsequently reduced to the corresponding saturated C–C linkage compounds 28a–28d under palladium-catalyzed hydrogenation. Final products 2b, 2c, 3a, 3b, 4a, 4b, 5a, and 5b (PZ671) were synthesized via reductive amination of the corresponding de-Boc intermediates with aldehyde 13 (Scheme 4).
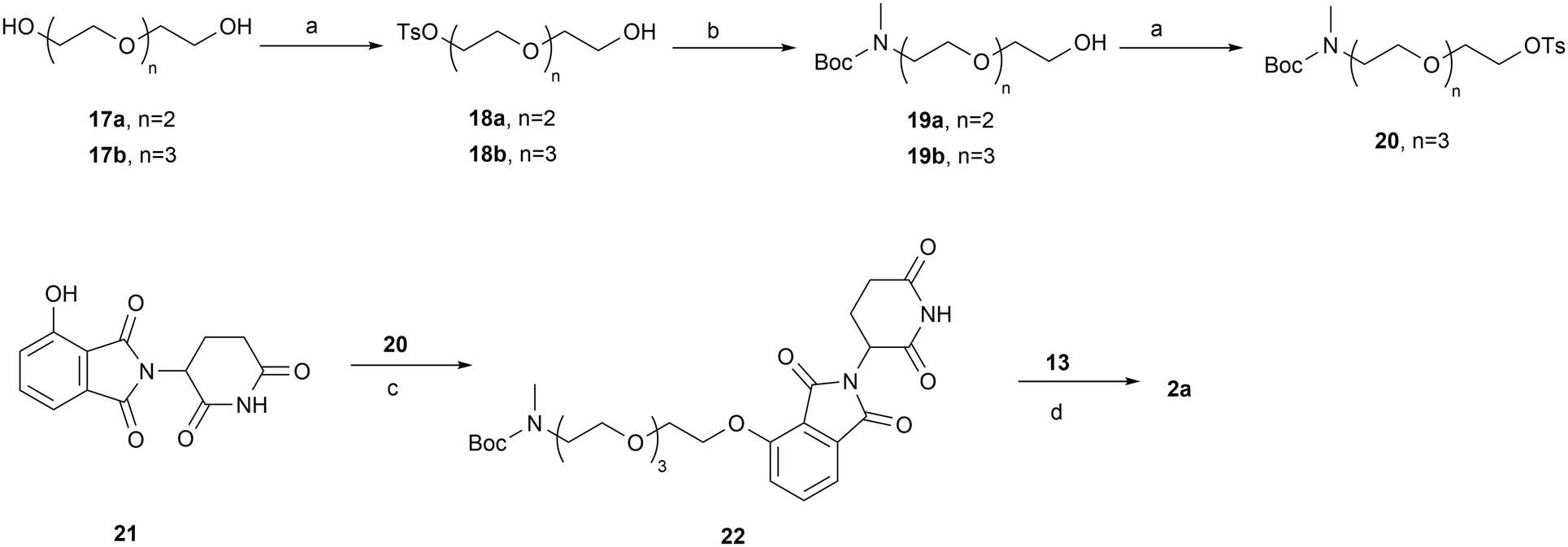 | ||
| Scheme 3 Reagents and conditions: (a) TsCl, TEA, DCM, rt; (b) (i) MeNH2, EtOH, 70 °C; (ii) Boc2O, TEA, DCM, rt; (c) NaHCO3, KI, DMF, 80 °C; (d) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt. | ||
 | ||
| Scheme 4 Reagents and conditions: (a) TsCl, TEA, DCM, rt; (b) (i) MeNH2, EtOH, 90 °C; (ii) Boc2O, TEA, DCM, rt; (c) PdCl2(PPh3)2, PPh3, CuI, TEA, DMF, 90 °C; (d) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt; (e) Pd/C, H2, MeOH, rt. | ||
The synthesis of compound 2d is shown in Scheme 5. Key intermediate 32 was obtained via SNAr substitution of 6 with Boc-protected piperazine 30, followed by Boc deprotection and reductive amination with compound 29. Compound 29 was produced by treating 19a with Dess–Martin periodinane. Subsequent Boc deprotection of 32 and reductive amination with aldehyde 13 gave the final product 2d. Compound 2e was prepared via Sonogashira coupling of 26a with 33, followed by Boc deprotection group and reductive amination with 29 to afford compound 35, then a second round of de-Boc and reductive amination with aldehyde 13. Similarly, compound 2f was synthesized from the triple bond-saturated intermediate 36 (Scheme 6).
 | ||
| Scheme 5 Reagents and conditions: (a) Dess–Martin periodinane, DCM, rt; (b) DIPEA, DMSO, MW, 90 °C; (c) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt; (d) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt. | ||
 | ||
| Scheme 6 Reagents and conditions: (a) PdCl2(PPh3)2, CuI, TEA, DMSO, 90 °C; (b) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt; (c) (i) HCl/1,4-dioxane, DCM, rt; (ii) NaBH(OAc)3, DCM, rt; (d) Pd/C, H2, MeOH, rt. | ||
Conclusions
In this study, based on the lead compound XZ739, we systematically investigated the effects of substituent size on the salt-bridge nitrogen, a crucial interaction for high Bcl-xL binding, and explored alternative CRBN-recruiting moieties connected via various linkages. We identified PZ671, which utilizes lenalidomide as the CRBN-recruiting moiety with an ethylene linkage at the C5-position of the benzene ring of lenalidomide. PZ671 exhibited ∼10-fold higher potency against MOLT-4 cells and ∼6-fold enhanced degradation efficacy against target Bcl-xL compared to XZ739, although its selectivity for MOLT-4 cells over platelets decreased ∼5-fold (112-fold vs. 25-fold). Despite this trade-off, the enhanced cellular activity may translate into a broader anticancer spectrum. In vivo studies revealed that unlike the mild but sustained platelet toxicity caused by VHL-recruiting Bcl-xL/Bcl-2 degrader 753b, the platelet toxicity of CRBN-recruiting degrader PZ671 was more prominent but transient, with maximal effect on the first day post-administration followed by rapid recovery. This transient toxicity profile may be more manageable clinically with appropriately optimized dosing regimens. Further efforts will focus on conjugating novel CRBN-recruiting ligands and exploring alternative linker attachment positions, such as utilizing the P2 pocket of ABT-263, which has proven effective in modifying VHL-recruiting Bcl-xL/Bcl-2 degraders to enhance cellular activity and degradation.Experimental section
Chemistry
The synthetic procedures along with the characterization data of all compounds are reported in the ESI.†Biological assays
Cell lines and culture
MOLT-4 T-ALL (Cat# CRL-1582) cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA). MOLT-4 cells were cultured in RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% penicillin–streptomycin solution (Thermo Fisher Scientific, Waltham, MA, USA). The SCLC cell lines, except H146, were obtained from the NCI-Navy Medical Oncology source supply as stated in our previous publication.42 H146 cells were obtained from ATCC. The cell lines were cultured as reported previously.42 The stocks of the cell lines were STR profiled by the NIH or ATCC. All cultures were tested for mycoplasma negativity using the MycoAlert Mycoplasma Detection Kit (Cat. No. LT07–318, Lonza, Basel, Switzerland). The cell lines were maintained in a humidified incubator at 37 °C and 5% CO2.Cell viability assay
Cell viability was measured by tetrazolium-based MTS assay (Promega, Madison, WI, USA). 5 × 104 to 1 × 105 suspension cells were seeded and treated in 96-well plates for 48 h or 72 h. The IC50 values of individual agents were calculated with GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA).Immunoblotting
Cells were collected and lysed in lysis buffer (Boston Bio Products, Ashland, MA, USA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO, USA). An equal amount of protein lysates was separated on pre-casted 4–20% Tris-glycine gels (Bio-Rad, Hercules, CA, USA). Thereafter, the proteins were transferred to PVDF membranes (MilliporeSigma, Billerica, MA, USA). The membranes were blocked with 5% (w/v) non-fat dry milk in TBS + Tween 20 (0.1% (v/v)), and then probed with primary antibodies overnight at 4 °C. Next day, the membranes were washed and incubated with appropriate HRP-conjugated secondary antibodies. The signal was detected using ECL substrate (MilliporeSigma) and captured on X-ray films or ChemiDoc MP Imaging System (Bio-Rad). The band intensities were calculated on ImageJ software and normalized to equal loading control β-actin.Human platelet isolation and viability assays
Platelet-rich plasma (PRP) was purchased from Life South Community Blood Center (Gainesville, FL, USA). PRP was transferred into a 50 mL tube containing 5 mL acid citrate buffer (Cat. No. sc-214![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 744, Santa Cruz Biotechnology). To prevent clotting, prostaglandin E1 (PGE1, Cat. No. sc-201223A, Santa Cruz Biotechnology) and apyrase (Cat. No. A6237, Sigma-Aldrich) were added to final concentrations of 1 μM and 0.2 units per mL, respectively. After gently mixing the solution, platelets were pelleted by centrifugation at 1200g for 10 min without break. Pelleted platelets were gently washed without disrupting the platelets in 2 mL HEPES Tyrode's buffer (Cat. No. PY-921WB, Boston BioProducts, Ashland, MA, USA) containing 1 μM PGE1 and 0.2 units per mL apyrase. After washing, pellets were slowly suspended in 10 mL HEPES Tyrode's buffer containing 1 μM PGE1 and 0.2 units per mL apyrase. Then platelets number was counted using a HEMAVET 950FS hematology analyzer (Drew Scientific, Inc., Oxford, CT, USA). For viability assays, platelet number was adjusted to 2 × 108 per mL in HEPES Tyrode's buffer containing 1 μM PGE1 and 0.2 units per mL apyrase. Each treatment was given in 2 mL platelet suspensions in 15 mL polypropylene tubes. The tubes were placed on a rotating platform at room temperature and the viability of platelets was measured after 24 h of treatment by using MTS reagent (Cat. No. G1111, Promega, Madison, WI, USA). The data were analyzed using GraphPad Prism 7 software for the calculation of IC50 values.
744, Santa Cruz Biotechnology). To prevent clotting, prostaglandin E1 (PGE1, Cat. No. sc-201223A, Santa Cruz Biotechnology) and apyrase (Cat. No. A6237, Sigma-Aldrich) were added to final concentrations of 1 μM and 0.2 units per mL, respectively. After gently mixing the solution, platelets were pelleted by centrifugation at 1200g for 10 min without break. Pelleted platelets were gently washed without disrupting the platelets in 2 mL HEPES Tyrode's buffer (Cat. No. PY-921WB, Boston BioProducts, Ashland, MA, USA) containing 1 μM PGE1 and 0.2 units per mL apyrase. After washing, pellets were slowly suspended in 10 mL HEPES Tyrode's buffer containing 1 μM PGE1 and 0.2 units per mL apyrase. Then platelets number was counted using a HEMAVET 950FS hematology analyzer (Drew Scientific, Inc., Oxford, CT, USA). For viability assays, platelet number was adjusted to 2 × 108 per mL in HEPES Tyrode's buffer containing 1 μM PGE1 and 0.2 units per mL apyrase. Each treatment was given in 2 mL platelet suspensions in 15 mL polypropylene tubes. The tubes were placed on a rotating platform at room temperature and the viability of platelets was measured after 24 h of treatment by using MTS reagent (Cat. No. G1111, Promega, Madison, WI, USA). The data were analyzed using GraphPad Prism 7 software for the calculation of IC50 values.
Flow cytometry
MOLT-4 cells were seeded in 12-well plates at a density of 4 × 105 cells per well and pretreated with or without Q-VD-OPh (QVD, Cat. No. S7311, Selleckchem, Houston, TX, USA) for 4 h. After pretreatment, cells were treated with PZ671 (10 nM) for 48 h. Caspase activity was blocked using QVD pretreatment for 2 h. After treatment, cells were stained with Alexa Fluor 647-annexin V (1:50, Cat. No. 640912, BioLegend, San Diego, CA, USA) and PI (10 μg mL−1, Cat. No. 421301, BioLegend, San Diego, CA, USA) at room temperature for 30 min. For apoptosis analysis, the stained cells were analyzed on an Aurora flow cytometer (Cytek Aurora, Fremont, CA, USA).
MOLT-4 T-ALL xenograft mouse model
To test the effect of PZ671 on tumor growth in MOLT-4 T-ALL xenografts, MOLT-4 T-ALL cells were collected and suspended in regular RPMI medium and mixed with Matrigel (1:1) (cat. no. 356231, Corning). The cells (5 × 106 cells) suspended in 100 μL of RPMI medium–Matrigel mixture were subcutaneously (s.c.) implanted in the right flank of 5 week-old CB-17 SCID mice purchased from the Charles River Laboratories (Wilmington, MA, USA). Tumor growth was monitored daily, and tumors were measured twice per week using a vernier caliper or digital calipers. Tumor volume was determined using the formula (L × W2) × 0.5, where L is the length in mm and W is the width in mm. The treatment started once the average tumor volume reached ∼150 mm3. The animals were randomly assigned into separate groups (n = 6 animals per group) such that each group had nearly equal starting average tumor volume and started undergoing treatment with vehicle (i.p., 100 μL per injection) and PZ671 (1.5 mg kg−1, every 4 days, i.p., 100 μL per injection). PZ671 for i.p. administration was formulated in 50% Phosal 50 PG, 45% Miglyol 810 N and 5% polysorbate 80.
In vivo platelet toxicity assays
Female 5–6 weeks old CB-17 SCID-beige mice were treated with single i.p. doses of PZ671 (1.5 mg per kg body weight). Approximately 50 μL blood was collected every day for 4 days via submandibular plexus route in EDTA tubes (RAM Scientific), and platelets were enumerated using an automated hematology analyzer (HEMAVET 950FS, Drew Scientific Inc., Miami Lakes, FL, USA).Ethical statement
All animal procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Florida and approved by the animal ethics committee of the University of Florida under protocol # 201710069.Data availability
ESI† to this article can be found online. Additional data will be made available upon request as reasonably possible.Conflicts of interest
The authors declare the following competing financial interest(s): P. Zhang, S. Khan, X. Zhang, D. Zhou, and G. Zheng are co-inventors for Bcl-xL PROTACs disclosed in this study. D. Zhou and G. Zheng are co-founders and shareholders of Dialectic Therapeutics, a company that is developing Bcl-xL PROTACs to treat cancers.Acknowledgements
This study was supported in part by NIH grants R01CA241191 and R01CA260239.Notes and references
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- J. M. Adams and S. Cory, Oncogene, 2007, 26, 1324–1337 CrossRef CAS PubMed.
- R. J. Youle and A. Strasser, Nat. Rev. Mol. Cell Biol., 2008, 9, 47–59 CrossRef CAS PubMed.
- J. K. Brunelle and A. Letai, J. Cell Sci., 2009, 122, 437–441 CrossRef CAS PubMed.
- P. E. Czabotar, G. Lessene, A. Strasser and J. M. Adams, Nat. Rev. Mol. Cell Biol., 2014, 15, 49–63 CrossRef CAS PubMed.
- P. D. Bhola and A. Letai, Mol. Cell, 2016, 61, 695–704 CrossRef CAS PubMed.
- M. Vogler, Adv. Med., 2014, 2014, 943648 Search PubMed.
- S. A. Amundson, T. G. Myers, D. Scudiero, S. Kitada, J. C. Reed and A. J. Fornace, Cancer Res., 2000, 60, 6101–6110 CAS.
- J. Chang, Y. Wang, L. Shao, R. M. Laberge, M. Demaria, J. Campisi, K. Janakiraman, N. E. Sharpless, S. Ding, W. Feng, Y. Luo, X. Wang, N. Aykin-Burns, K. Krager, U. Ponnappan, M. Hauer-Jensen, A. Meng and D. Zhou, Nat. Med., 2016, 22, 78–83 CrossRef CAS PubMed.
- R. Yosef, N. Pilpel, R. Tokarsky-Amiel, A. Biran, Y. Ovadya, S. Cohen, E. Vadai, L. Dassa, E. Shahar, R. Condiotti, I. Ben-Porath and V. Krizhanovsky, Nat. Commun., 2016, 7, 11190 CrossRef CAS PubMed.
- Y. Zhu, T. Tchkonia, H. Fuhrmann-Stroissnigg, H. M. Dai, Y. Y. Ling, M. B. Stout, T. Pirtskhalava, N. Giorgadze, K. O. Johnson, C. B. Giles, J. D. Wren, L. J. Niedernhofer, P. D. Robbins and J. L. Kirkland, Aging Cell, 2016, 15, 428–435 CrossRef CAS PubMed.
- P. Pal, P. Zhang, S. Poddar and G. Zheng, Expert Opin. Ther. Pat., 2022, 32, 1003–1026 CrossRef CAS PubMed.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. Ng, P. M. Nimmer, J. M. O'Connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. Wang, M. D. Wendt, H. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677–681 CrossRef CAS PubMed.
- M. Bruncko, T. K. Oost, B. A. Belli, H. Ding, M. K. Joseph, A. Kunzer, D. Martineau, W. J. McClellan, M. Mitten, S. Ng, P. M. Nimmer, T. Oltersdorf, C. Park, A. M. Petros, A. R. Shoemaker, X. Song, X. Wang, M. D. Wendt, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2007, 50, 641–662 CrossRef CAS PubMed.
- C. Tse, A. R. Shoemaker, J. Adickes, M. G. Anderson, J. Chen, S. Jin, E. F. Johnson, K. C. Marsh, M. J. Mitten, P. Nimmer, L. Roberts, S. K. Tahir, Y. Xiao, X. Yang, H. Zhang, S. Fesik, S. H. Rosenberg and S. W. Elmore, Cancer Res., 2008, 68, 3421–3428 CrossRef CAS PubMed.
- L. Bai, J. Chen, L. Liu, D. McEachern, A. Aguilar, H. Zhou, C. Y. Yang, H. Wang, J. Wen, G. Wang, Y. Zhai, M. Guo, D. Yang and S. Wang, Eur. J. Cancer., 2014, 50, 109–110 CrossRef.
- A. W. Tolcher, B. A. Carneiro, A. Dowlati, A. R. A. Razak, Y. K. Chae, J. A. Villella, S. Coppola, S. Englert, A. C. Phillips, A. J. Souers, Z. Salman, S. Penugonda, J. D. Powderly and P. LoRusso, J. Clin. Oncol., 2021, 39, 3015 Search PubMed.
- X. Zhang, D. Thummuri, Y. He, X. Liu, P. Zhang, D. Zhou and G. Zheng, Chem. Commun., 2019, 55, 14765–14768 RSC.
- X. Zhang, Y. He, P. Zhang, V. Budamagunta, D. Lv, D. D. Thummuri, Y. Yang, J. Pei, Y. Yuan, D. Zhou and G. Zheng, Eur. J. Med. Chem., 2020, 199, 112397 CrossRef CAS PubMed.
- S. Khan, X. Zhang, D. Lv, Q. Zhang, Y. He, P. Zhang, X. Liu, D. Thummuri, Y. Yuan, J. S. Wiegand, J. Pei, W. Zhang, A. Sharma, C. R. McCurdy, V. M. Kuruvilla, N. Baran, A. A. Ferrando, Y. Kim, A. Rogojina, P. J. Houghton, G. Huang, R. Hromas, M. Konopleva, G. Zheng and D. Zhou, Nat. Med., 2019, 25, 1938–1947 CrossRef CAS PubMed.
- D. Lv, P. Pal, X. Liu, Y. Jia, D. Thummuri, P. Zhang, W. Hu, J. Pei, Q. Zhang, S. Zhou, S. Khan, X. Zhang, N. Hua, Q. Yang, S. Arango, W. Zhang, D. Nayak, S. K. Olsen, S. T. Weintraub, R. Hromas, M. Konopleva, Y. Yuan, G. Zheng and D. Zhou, Nat. Commun., 2021, 12, 6896 CrossRef CAS PubMed.
- D. Nayak, Y. Lv, P. Yuan, W. Zhang, A. Hu, E. A. Nayak, Z. Ruben, P. Lv, R. Sung, G. Hromas, D. Zhou Zheng and S. K. Olsen, Nat. Commun., 2024, 15, 2743 CrossRef CAS PubMed.
- Y. He, X. Zhang, J. Chang, H. Kim, P. Zhang, Y. Wang, S. Khan, X. Liu, X. Zhang, D. Lv, L. Song, W. Li, D. Thummuri, Y. Yuan, J. S. Wiegand, Y. T. Ortiz, V. Budamagunta, J. H. Elisseeff, J. Campisi, M. Almeida, G. Zheng and D. Zhou, Nat. Commun., 2020, 11, 1996 CrossRef CAS PubMed.
- X. Zhang, D. Thummuri, X. Liu, W. Hu, P. Zhang, S. Khan, Y. Yuan, D. Zhou and G. Zheng, Eur. J. Med. Chem., 2020, 192, 112186 CrossRef CAS PubMed.
- P. Pal, D. Thummuri, D. Lv, X. Liu, P. Zhang, W. Hu, S. K. Poddar, N. Hua, S. Khan, Y. Yuan, X. Zhang, D. Zhou and G. Zheng, J. Med. Chem., 2021, 64, 14230–14246 CrossRef CAS PubMed.
- S. Zhang, Y. Chen, Z. Xu, J. Yang, R. Sun, J. Wang, Y. Sun, B. Jiang, X. Yang and W. Tan, Theranostics, 2022, 12, 7476–7490 CrossRef CAS PubMed.
- X. Zhang, Y. Tao, Z. Xu, B. Jiang, X. Yang, T. Huang and W. Tan, Biochem. Pharmacol., 2024, 230, 116542 CrossRef CAS PubMed.
- P. Zhang, X. Zhang, X. Liu, S. Khan, D. Zhou and G. Zheng, Explor. Target. Antitumor Ther., 2020, 1, 259–272 Search PubMed.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C. M. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Target Antitumor Ther., 2020, 1, 273–312 Search PubMed.
- A. Zagidullin, V. Milyukov, A. Rizvanov and E. Bulatov, Explor. Target Antitumor Ther., 2020, 1, 381–390 Search PubMed.
- Y. Dong, T. Ma, T. Xu, Z. Feng, Y. Li, L. Song, X. Yao, C. R. Ashby and G. Hao, Acta Pharm. Sin. B, 2024, 14, 4266–4295 CrossRef CAS PubMed.
- N. A. Zografou-Barredo, A. J. Hallatt, J. Goujon-Ricci and C. Cano, Bioorg. Med. Chem., 2023, 88-89, 117334 CrossRef CAS PubMed.
- M. Hoffmann, C. Kasserra, J. Reyes, P. Schafer, J. Kosek, L. Capone, A. Parton, H. Kim-Kang, S. Surapaneni and G. Kumar, Cancer Chemother. Pharmacol., 2013, 71, 489–501 CrossRef CAS PubMed.
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed.
- J. Davila, A. Chassepot, J. Longo, F. Boulmedais, A. Reisch, B. Frisch, F. Meyer, J. Voegel, P. J. Mesini, B. Senger, M. Metz-Boutigue, J. Hemmerle, P. Lavalle, P. Schaaf and L. Jierry, J. Am. Chem. Soc., 2012, 134, 83–86 CrossRef CAS PubMed.
- P. Cheol-Min, M. Bruncko, J. Adickes, J. Bauch, H. Ding, A. Kunzer, K. C. Marsh, P. Nimmer, A. R. Shoemaker, X. Song, S. K. Tahir, C. Tse, X. Wang, M. D. Wendt, X. Yang, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2008, 51, 6902–6915 CrossRef PubMed.
- G. Zheng, D. Zhou, W. Hu, D. Thummuri, P. Zhang, P. Pratik, D. Lv and Y. Yuan, WO2023107606A1, 2023.
- A. P. Crew, K. R. Hornberger, J. Wang, H. Dong, M. Berlin and C. M. Crews, WO2019195609A2, 2019.
- S. Khan, P. Kellish, N. Connis, D. Thummuri, J. Wiegand, P. Zhang, X. Zhang, V. Budamagunta, N. Hua, Y. Yang, U. De, L. Jin, W. Zhang, G. Zheng, R. Hromas, C. Hann, M. Zajac-Kaye, F. J. Kaye and D. Zhou, Cell Death Discovery, 2023, 9, 1 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00119f |
| ‡ Equal contribution as first author to this work. |
| This journal is © The Royal Society of Chemistry 2025 |