
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePegylation approach applied to erlotinib–carbonic anhydrase inhibitors hybrids towards anticancer agents†
Serena
Filiberti
 a,
Gioele
Renzi
*b,
Fabrizio
Carta
b,
Marialuigia
Fantacuzzi
c,
Ilaria
D'Agostino
d,
German
Benito
b,
Andrea
Angeli
b,
Maria Luisa
Massardi
a,
Rahime
Simsek
e,
Clemente
Capasso
f,
Simone
Carradori
c,
Roberto
Ronca
a and
Claudiu T.
Supuran
b
a,
Gioele
Renzi
*b,
Fabrizio
Carta
b,
Marialuigia
Fantacuzzi
c,
Ilaria
D'Agostino
d,
German
Benito
b,
Andrea
Angeli
b,
Maria Luisa
Massardi
a,
Rahime
Simsek
e,
Clemente
Capasso
f,
Simone
Carradori
c,
Roberto
Ronca
a and
Claudiu T.
Supuran
b
aDepartment of Molecular and Translational Medicine, University of Brescia, v.le Europa 11, 25121 Brescia, Italy. E-mail: s.filiberti@studenti.unibs.it; marialuisa.massardi@unibs.it; roberto.ronca@unibs.it
bNEUROFARBA Department, Pharmaceutical and Nutraceutical section, University of Florence, via Ugo Schiff 6, 50019 Sesto Fiorentino, Italy. E-mail: gioele.renzi@unifi.it; fabrizio.carta@unifi.it; andrea.angeli@unifi.it; claudiu.supuran@unifi.it
cDepartment of Pharmacy, “G. d'Annunzio” University of Chieti and Pescara, via dei Vestini 31, 66100, Chieti, Italy. E-mail: marialuigia.fantacuzzi@unich.it; simone.carradori@unich.it
dDepartment of Pharmacy, University of Pisa, via Bonanno Pisano 6, 56126 Pisa, Italy. E-mail: ilaria.dagostino@unipi.it
eHacettepe University, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, 06100, Sihhiye, Ankara, Turkey. E-mail: rsimsek@hacettepe.edu.tr
fDepartment of Biology, Agriculture and Food Sciences, National Research Council, Institute of Biosciences & Bioresources, Naples, 80131, Italy. E-mail: clemente.capasso@ibbr.cnr.it
First published on 28th April 2025
Abstract
Herein we report a first study on single molecular entities bearing both epidermal growth factor receptor (EGFR) and carbonic anhydrase (CA) inhibiting moieties as new tools for the management of hypoxic cancers. Specifically, we designed and synthesized a library of erlotinib (ERL)-based compounds bearing both the primary sulfonamide/coumarin moieties with the intent to selectively interfere with EGFR and CA targets respectively. The compounds obtained were investigated in silico and in vitro for their ability to interact with the appropriate targets followed by the assessment on selected compounds for the anti-proliferative activity using human (h) TNBC cell line MDA-MB-231. We are confident that the data provided in this study are fundamental for paving the way toward the development of multi-targeting molecular structures useful for the management of chronic diseases such as hypoxic tumors.
1. Introduction
Over the years, the definition of cancer has evolved to reflect an increasing understanding of its overall complexity and heterogeneity,1,2 although data on its morbidity and mortality consistently underscore the ongoing Public Health emergency.3,4 The current gold standard in cancer treatment, which combines surgery, radiotherapy, and chemotherapy, often faces significant challenges, including limited effectiveness and the associated high failure rates.5 Recently, the development of immunotherapy and gene-based therapeutics has emerged in response to the urgent need for new effective treatment options, aligning with the rising trend towards personalized medicine.6 Conversely, the more traditional approach of small molecules in cancer treatment still focuses on modulating enzymatic activities that are crucial for the disease progression and the novelty of the mode of action, in the attempt to avoid cross-resistance phenomena.7–9 In particular, a valuable strategy consists of anticancer agents endowed with multi-targeted mode of action10–12 and relies on hybrid compounds13–17 that combine multiple pharmacophoric features, enabling them to target various pathways and mechanisms simultaneously,18,19 often spaced by simple flexible linkers as methylene units.20 In this context, several known anticancer drugs undergo this chemical modification21–24 and, among them, one of the most employed is erlotinib (ERL, Fig. 1).25–32 This small molecule selectively inhibits the epidermal growth factor receptor (EGFR) tyrosine kinase, which is a key player in cell proliferation and disease progression in various cancers, such as non-small cell lung cancer (NSCLC) and pancreatic cancer.33,34 Mechanistically, ERL binds reversibly the tyrosine kinase domain of EGFR, specifically to the ATP-binding site in a competitive manner,35 thereby, blocking the phosphorylation of EGFR and, consequently, the signaling pathways of PI3K–AKT and RAS–RAF–MEK–ERK,36–38 mainly involved in tumor growth, survival, and metastasis, along with increased aggressiveness and poor prognosis.39–41 Interestingly, although the binding to the targeted receptor is governed by relevant networks of interactions also including hydrophobic contacts between the triad of residues (Thr766, leu764, and Lys721) and the acetylene terminal function,42,43 the latter has often been modified to generate various libraries of compounds, being often employed as starting point for derivatizations29,30,44–46 with only slight decrease in the inhibitory potency towards EGFR.44 Specifically, it served for the copper-catalyzed azide-alkyne cycloaddition (CuAAC), a cornerstone of click chemistry, to form the characteristic 1,4-disubstituted 1,2,3-triazole. The introduction of the triazole ring, a known surrogate for amides, esters, and carboxylic acids,45,47,48 is reported to often increase the affinity for the target, allowing the establishment of additional interactions, such as hydrogen bonds and dipole interactions.49 | ||
| Fig. 1 Structures of ERL and clicked derivatives from the 1st (1–6) and 2nd (pegylated 7–14) series. | ||
On the other hand, recent findings highlighted the high potential of another family of enzymes in the frame of anticancer treatment development: carbonic anhydrases (CAs, EC: 4.2.1.1), being widely reported during the last century due to their ubiquitary nature and their pivotal role in cell life.50 These metalloenzymes catalyze the interconversion of carbon dioxide (CO2) to bicarbonate (HCO3−) and proton (H+), thereby regulating pH and CO2 homeostasis, electrolyte secretion in various tissues and organs, and other crucial biological processes.51–53 In particular, the α-CA subfamily is the only one among the identified eight genetically distinct ones (α-, β-, γ- δ-, ζ-, η-, θ-, and ι-CAs) found in humans and is composed of 16 members (I–IV, VA, VB, VI–XV).50,54 Both the isoforms I and II are physiologically abundant in red blood cells, playing a role in the CO2 transport, whereas hCA II levels are high also in kidneys, assisting in HCO3−-resorption and diuresis.55,56 Conversely, hCAs overexpression is often associated with different diseases,10,52,57 such as glaucoma for hCA II58 and hypoxic cancer for isoforms IX59 and XII,60 to name but a few. In particular, the latter isozymes were highly exploited and several libraries of CA inhibitors (CAIs), especially bearing the sulfonamide or the coumarin CA inhibiting chemotypes, have been described till now.61–63
2. Material and methods
2.1. Chemistry
3-(3-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)ureido)benzenesulfonamide (19a). Yellow oil. Yield: 65%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.00 (1H, s, NH, exchange with D2O), 7.54 (1H, d, J = 7.50 Hz), 7.42 (t, 1H, J = 7.55 Hz, NH, exchange with D2O), 7.19 (2H, t, J = 7.76 Hz), 6.79 (2H, bs, SO2NH2, exchange with D2O), 6.28 (1H, t, J = 5.37 Hz), 3.62 (2H, t, J = 3.77 Hz), 3.58 (8H, m), 3.50 (2H, t, J = 5.37 Hz), 3.41 (2H, m), 3.30 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 154.3, 139.9, 136.2, 127.0, 124.8, 122.9, 119.8, 70.4, 70.3, 70.2, 70.1, 70.0, 69.7, 50.0, 41.5; MS (ESI positive) m/z: 417.15 [M + H]+. Elemental analysis: calculated C, 43.26; H, 5.81; N, 20.18; found C, 43.25; H, 5.82; N, 20.16.
4-(3-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)ureido)benzenesulfonamide (19b). Yellow oil. Yield: 60%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.02 (1H, s, NH, exchange with D2O), 7.72 (2H, d, J = 7.89 Hz), 7.57 (2H, d, J = 7.89 Hz), 6.80 (2H, bs, SO2NH2, exchange with D2O), 6.36 (1H, t, J = 5.56 Hz, NH, exchange with D2O), 3.63 (3H, t, J = 4.79 Hz), 3.59 (8H, s), 3.42 (3H, m), 3.30 (2H, q, J = 6.12 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 154.3, 142.6, 136.5, 129.5, 129.4, 118.1, 118.0, 70.4, 70.3, 70.2, 70.1, 70.0, 69.7, 50.0, 41.5; MS (ESI positive) m/z: 417.15 [M + H]+. Elemental analysis: calculated C, 43.26; H, 5.81; N, 20.18; found C, 43.25; H, 5.82; N, 20.19.
3-(3-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)thioureido)benzenesulfonamide (20a). Yellow oil. Yield: 95%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.94 (1H, s, NH, exchange with D2O), 7.99 (2H, d, J = 18.6 Hz), 7.76 (1H, d, J = 7.11 Hz), 7.55 (2H, bs, SO2NH2, exchange with D2O), 7.40 (1H, s, NH, exchange with D2O), 4.06 (2H, d, J = 7.22 Hz), 3.60 (14H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.5, 140.0, 137.4, 129.7, 127.1, 124.7, 123.3, 70.6, 70.5, 70.4, 70.2, 70.1, 70.0, 50.0, 45.3; MS (ESI positive) m/z: 433.13 [M + H]+. Elemental analysis: calculated C, 41.66; H, 5.59; N, 19.43; found C, 41.64; H, 5.58; N, 19.45.
4-(3-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)thioureido)benzenesulfonamide (20b). Yellow oil. Yield: 95%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.97 (1H, s, NH, exchange with D2O), 8.01 (1H, s, NH, exchange with D2O), 7.74 (4H, q, J = 8.07 Hz), 7.31 (2H, bs, SO2NH2, exchange with D2O), 3.61 (16H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.5, 141.7, 136.9, 129.6, 129.5, 122.9, 122.8, 70.5, 70.4, 70.3, 70.2, 70.1, 70.0, 50.0, 45.3; MS (ESI positive) m/z: 433.13 [M + H]+. Elemental analysis: calculated C, 41.66; H, 5.59; N, 19.43; found C, 41.65; H, 5.58; N, 19.44.
4-(15-Azido-3-thioxo-7,10,13-trioxa-2,4-diazapentadecyl)benzenesulfonamide (20c). Yellow oil. Yield: 97%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.10 (1H, s, NH, exchange with D2O), 7.80 (2H, d, J = 7.59 Hz), 7.67 (1H, s, NH, exchange with D2O), 7.47 (2H, d, J = 7.59 Hz), 7.35 (2H, bs, SO2NH2, exchange with D2O), 4.77 (2H, s), 4.07 (2H, d, J = 6.51 Hz), 3.58 (14H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.3, 141.7, 141.1, 128.3, 128.2, 127.3, 127.2, 70.6, 70.5, 70.4, 70.2, 70.1, 70.0, 50.8, 50.0, 45.3; MS (ESI positive) m/z: 447.14 [M + H]+. Elemental analysis: calculated C, 43.04; H, 5.87; N, 18.82; found C, 43.05; H, 5.86; N, 18.83.
4-(16-Azido-4-thioxo-8,11,14-trioxa-3,5-diazahexadecyl)benzenesulfonamide (20d). Yellow oil. Yield: 97%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.79 (2H, d, J = 7.59 Hz), 7.60 (1H, s, NH, exchange with D2O), 7.54 (1H, s, NH, exchange with D2O), 7.45 (2H, d, J = 7.59 Hz), 7.34 (2H, bs, SO2NH2, exchange with D2O), 3.64 (5H, m), 3.57 (14H, m), 2.92 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.8, 142.6, 140.9, 128.4, 128.3, 128.1, 128.0, 70.5, 70.4, 70.3, 70.2, 70.1, 70.0, 50, 47.8, 45.3, 35.3; MS (ESI positive) m/z: 461.16 [M + H]+. Elemental analysis: calculated C, 44.33; H, 6.13; N, 18.25; found C, 44.34; H, 6.12; N, 18.26.
4-(3-(2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl)thioureido)-3-bromo-2-methylbenzenesulfonamide (21). Yellow oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.40 (1H, s, NH, exchange with D2O), 8.44 (1H, s), 8.05 (1H, s), 7.51 (2H, bs, SO2NH2, exchange with D2O), 7.28 (1H, s, NH, exchange with D2O), 4.14 (2H, q, J = 5.19 Hz), 3.61 (8H, m), 3.21 (5H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.5, 140.3, 138.3, 137.1, 126.5, 126.0, 120.9, 70.5, 70.4, 70.1, 70.4, 70.0, 50.0, 45.3, 13.6; MS (ESI positive) m/z: 525.05 [M + H]+. Elemental analysis: calculated C, 36.57; H, 4.80; N, 15.99; found C, 36.58; H, 4.81; N, 15.97.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95), to yield compounds 7a–b, 8a–d, and 9.
:95), to yield compounds 7a–b, 8a–d, and 9.
3-(3-(2-(2-(2-(2-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl)ureido)benzenesulfonamide (7a). Yellow oil. Yield: 5%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.58 (1H, s, NH, exchange with D2O), 8.90 (1H, s), 8.54 (1H, s), 8.48 (1H, s), 8.28 (1H, s, NH, exchange with D2O), 7.95 (2H, d, J = 10.52 Hz), 7.88 (1H, d, J = 7.94 Hz), 7.55 (1H, d, J = 7.57 Hz), 7.48 (2H, t, J = 8.31 Hz), 7.38 (1H, t, J = 7.79 Hz), 7.33 (1H, d, J = 7.74 Hz), 7.29 (2H, bs, SO2NH2, exchange with D2O), 7.23 (1H, s), 6.24 (1H, t, J = 5.23 Hz), 4.58 (2H, t, J = 4.63 Hz), 4.31 (4H, m), 3.88 (2H, t, J = 4.72 Hz), 3.79 (2H, m), 3.75 (2H, m), 3.45 (16H, m), 3.21 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 156.4, 154.9, 153.6, 152.9, 148.1, 146.9, 146.2, 144.5, 140.9, 140.0, 131.0, 129.2, 129.0, 121.8, 121.7, 120.3, 118.7, 118.0, 114.4, 108.9, 108.1, 103.2, 101.8, 70.1, 70.0, 69.7, 69.6, 68.6, 68.3, 68.0, 58.4, 58.3, 49.6, 28.9; MS (ESI positive) m/z: 810.32 [M + H]+. Elemental analysis: calculated C, 54.87; H, 5.85; N, 15.57; found C, 54.88; H, 5.84; N, 15.55.
4-(3-(2-(2-(2-(2-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl)ureido)benzenesulfonamide (7b). Yellow oil. Yield: 19%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.59 (1H, s, NH, exchange with D2O), 8.96 (1H, s), 8.55 (1H, s), 8.48 (1H, s), 8.27 (1H, s), 7.93 (1H, s, NH, exchange with D2O), 7.88 (1H, d, J = 7.65 Hz), 7.65 (2H, d, J = 8.29 Hz), 7.55 (1H, d, J = 7.8 Hz), 7.51 (2H, d, J = 8.76 Hz), 7.46 (1H, t, J = 7.87 Hz), 7.23 (1H, s), 7.17 (2H, bs, SO2NH2, exchange with D2O), 6.31 (1H, t, J = 5.4 Hz), 4.58 (2H, t, J = 4.94 Hz), 4.30 (4H, m), 3.87 (2H, t, J = 5.0 Hz), 3.79 (2H, m), 3.75 (2H, m), 3.52 (8H, m), 3.36 (6H, m), 3.21 (2H, m), 3.16 (2H, d, J = 5.17 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 156.4, 154.7, 153.6, 152.9, 148.1, 146.9, 146.2, 143.6, 140.0, 136.0, 131.0, 129.0, 126.8, 121.8, 121.7, 120.3, 118.7, 116.7, 108.9, 108.1, 103.2, 70.1, 70.0, 69.7, 69.6, 68.6, 68.3, 68.0, 58.4, 58.3, 49.6, 48.6; MS (ESI positive) m/z: 810.32 [M + H]+. Elemental analysis: calculated C, 54.87; H, 5.85; N, 15.57; found C, 54.89; H, 5.86; N, 15.56.
3-(3-(2-(2-(2-(2-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl)thioureido)benzenesulfonamide (8a). Yellow oil. Yield: 27%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.86 (1H, m, NH, exchange with D2O), 9.57 (1H, s), 8.54 (1H, s), 8.48 (1H, m), 8.28 (1H, s, NH, exchange with D2O), 7.96 (1H, m), 7.93 (1H, s), 7.88 (1H, d, J = 7.18 Hz), 7.70 (1H, d, J = 7.33 Hz), 7.53 (2H, t, J = 7.98 Hz), 7.46 (2H, m), 7.36 (2H, bs, SO2NH2, exchange with D2O), 7.23 (1H, m), 4.59 (2H, t, J = 4.93 Hz), 4.30 (4H, m), 3.88 (2H, t, J = 5.19 Hz), 3.79 (3H, t, J = 4.94 Hz), 3.75 (3H, m), 3.50 (12H, m), 3.37 (4H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.4, 180.6, 165.4, 156.4, 153.6, 153.0, 148.1, 147.0, 146.8, 146.2, 144.3, 140.0, 131.1, 134.7, 129.0, 125.7, 121.9, 121.8, 120.3, 118.8, 108.1, 103.2, 70.1, 70.0, 69.7, 69.6, 68.7, 68.4, 68.0, 58.4, 58.3, 49.6, 48.6, 43.6; MS (ESI positive) m/z: 826.30 [M + H]+. Elemental analysis: calculated C, 53.81; H, 5.74; N, 15.26; found C, 53.82; H, 5.73; N, 15.24.
4-(3-(2-(2-(2-(2-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl)thioureido)benzenesulfonamide (8b). Yellow oil. Yield: 35%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.64 (1H, s, NH, exchange with D2O), 8.55 (1H, s), 8.50 (1H, s), 8.28 (1H, s), 8.09 (1H, s NH, exchange with D2O), 7.96 (1H, s), 7.88 (1H, d, J = 7.34 Hz), 7.76 (2H, d, J = 9.15 Hz), 7.66 (1H, m), 7.55 (1H, m), 7.47 (1H, d, J = 7.92 Hz), 7.41 (1H, d, J = 7.92 Hz), 7.32 (2H, bs, SO2NH2, exchange with D2O), 7.24 (1H, s), 4.72 (2H, m), 4.59 (2H, m) 4.30 (4H, m), 4.13 (2H, m), 3.88 (2H, t, J = 5.09 Hz), 3.79 (2H, m), 3.75 (2H, m), 3.55 (3H, m), 3.52 (10H, m), 3.16 (3H, d, J = 4.85 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 185.4, 156.3, 154.8, 146.2, 140.0, 136.0, 131.0, 129.4, 129.0, 127.0, 126.7, 121.8, 120.7, 118.7, 116.7, 103.3, 90.2, 70.1, 69.9, 69.7, 69.6, 69.1, 68.6, 68.0, 65.3, 61.9, 60.2, 58.4, 49.7, 49.6, 29.0; MS (ESI positive) m/z: 826.30 [M + H]+. Elemental analysis: calculated C, 53.81; H, 5.74; N, 15.26; found C, 53.79; H, 5.75; N, 15.28.
4-(15-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3-thioxo-7,10,13-trioxa-2,4-diazapentadecyl)benzenesulfonamide (8c). Yellow oil. Yield: 42%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.64 (1H, m, NH, exchange with D2O), 8.59 (1H, s), 8.32 (1H, s), 8.00 (1H, s, NH, exchange with D2O), 7.90 (1H, d, J = 7.38 Hz), 7.80 (2H, d, J = 7.72 Hz), 7.59 (1H, d, J = 7.72 Hz), 7.51 (1H, d, J = 8.13 Hz), 7.46 (2H, d, J = 7.72 Hz), 7.35 (2H, bs, SO2NH2, exchange with D2O), 7.33 (1H, m), 4.62 (2H, m), 4.34 (5H, m), 3.92 (2H, m), 3.81 (5H, m), 3.54 (14H, m), 3.41 (8H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 163.2, 156.4, 155.8, 153.6, 152.8, 148.1, 146.8, 146.2, 142.5, 140.0, 137.6, 131.1, 129.0, 127.8, 127.4, 125.6, 121.8, 120.3, 118.8, 108.1, 103.3, 70.1, 70.0, 69.7, 69.6, 68.8, 68.6, 68.4, 68.0, 58.4, 58.4, 49.6, 48.6; MS (ESI positive) m/z: 840.31 [M + H]+. Elemental analysis: calculated C, 53.98; H, 5.78; N, 15.18; found C, 53.99; H, 5.79; N, 15.16.
4-(16-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-4-thioxo-8,11,14-trioxa-3,5-diazahexadecyl)benzenesulfonamide (8d). Yellow oil. Yield: 72%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.69 (1H, m, NH, exchange with D2O), 8.55 (2H, m), 8.28 (1H, s), 7.94 (1H, s, NH, exchange with D2O), 7.84 (1H, m), 7.75 (3H, m), 7.57 (1H, d, J = 7.51 Hz), 7.48 (1H, m), 7.42 (3H, m), 7.32 (2H, bs, SO2NH2, exchange with D2O), 7.32 (1H, s), 4.58 (2H, m), 4.30 (4H, m), 3.87 (2H, m), 3.79 (2H, m), 3.75 (2H, m), 3.49 (16H, m), 3.35 (4H, m), 2.88 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 190.0, 158.9, 158.3, 153.7, 148.3, 148.2, 146.5, 146.2, 143.4, 142.2, 131.1, 129.7, 129.3, 129.2, 126.3, 126.0, 125.7, 122.5, 121.9, 103.2, 80.2, 70.1, 70.0, 69.7, 69.6, 69.3, 68.9, 68.7, 68.4, 68.1, 58.4, 58.3, 50.0, 49.6; MS (ESI positive) m/z: 854.33 [M + H]+. Elemental analysis: calculated C, 54.85; H, 6.02; N, 14.76; found C, 54.84; H, 6.01; N, 14.78.
4-(3-(2-(2-(2-(2-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl)thioureido)-3-bromo-2-methylbenzenesulfonamide (9). Yellow oil. Yield: 46%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.57 (1H, s, NH, exchange with D2O), 8.53 (1H, s), 8.46 (2H, m), 8.28 (1H, s), 8.11 (1H, m), 7.93 (1H, s, NH, exchange with D2O), 7.88 (1H, d, J = 8.26 Hz), 7.65 (1H, d, J = 8.68 Hz), 7.54 (1H, d, J = 7.44 Hz), 7.46 (2H, m), 7.23 (1H, s), 7.20 (2H, bs, SO2NH2, exchange with D2O), 4.58 (2H, m), 4.31 (4H, m), 3.87 (2H, t, J = 5.37 Hz), 3.79 (2H, t, J = 4.22 Hz), 3.75 (2H, t, J = 4.21 Hz), 3.54 (8H, m), 3.45–3.40 (9H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.2, 180.4, 175.7, 173.9, 172.0, 168.6, 152.9, 147.3, 144.8, 141.6, 140.1, 138.7, 134.5, 130.3, 121.8, 120.2, 119.0, 117.3, 116.2, 112.4, 107.8, 105.5, 103.3, 75.3, 70.0, 69.6, 68.5, 68.3, 68.0, 67.3, 63.4, 58.3, 55.3, 46.8, 31.9; MS (ESI positive) m/z: 918.22 [M + H]+. Elemental analysis: calculated C, 52.17; H, 5.63; N, 14.22; found C, 52.18; H, 5.64; N, 14.21.
:95), to yield compounds 25a–b and 26.
1-Azido-N-(4-sulfamoylbenzyl)-3,6,9,12,15-pentaoxaoctadecan-18-amide (25a). Yellow oil. Yield: 61%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.45 (1H, t, J = 5.32 Hz, NH, exchange with D2O), 7.75 (2H, d, J = 8.20 Hz), 7.41 (2H, d, J = 7.91 Hz), 7.30 (2H, bs, SO2NH2, exchange with D2O), 4.33 (2H, d, J = 5.73 Hz), 3.63 (2H, t, J = 7.09 Hz), 3.60 (2H, t, J = 4.84 Hz), 3.50 (16H, m), 3.38 (2H, t, J = 4.91 Hz), 2.39 (2H, t, J = 6.09 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 174.3, 142.3, 138.2, 130.3, 129.2, 70.6, 70.4, 69.5, 68.4, 49.1, 45.8, 25.9; MS (ESI positive) m/z: 504.2 [M + H]+. Elemental analysis: calculated C, 47.70; H, 6.61; N, 13.91; found C, 47.71; H, 6.62; N, 13.90.
1-Azido-N-(4-sulfamoylphenethyl)-3,6,9,12,15-pentaoxaoctadecan-18-amide (25b). Yellow oil. Yield: 49%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.93 (1H, t, J = 5.70 Hz, NH, exchange with D2O), 7.73 (2H, d, J = 8.15 Hz), 7.38 (2H, d, J = 8.15 Hz), 7.28 (2H, bs, SO2NH2, exchange with D2O), 3.58 (4H, m), 3.53 (4H, m), 3.49 (8H, m), 3.47 (4H, m), 3.38 (2H, t, J = 5.14 Hz), 3.27 (2H, t, J = 6.14 Hz), 2.77 (2H, t, J = 7.14 Hz), 2.28 (2H, t, J = 6.42 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.3, 142.5, 133.8, 130.9, 128.5, 71.4, 70.6, 70.4, 69.1, 67.3, 45.4, 42.7, 37.4, 30.4; MS (ESI positive) m/z: 517.22 [M + H]+. Elemental analysis: calculated C, 48.73; H, 6.82; N, 13.53; found C, 48.72; H, 6.83; N, 13.54.
1-Azido-N-(2-((2-oxo-2H-chromen-6-yl)oxy)ethyl)-3,6,9,12,15-pentaoxaoctadecan-18-amide (26). Yellow oil. Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.12 (1H, t, J = 5.82 Hz, NH, exchange with D2O), 8.00 (1H, d, J = 9.41 Hz), 7.34 (1H, d, J = 9.09 Hz), 7.30 (1H, d, J = 2.76 Hz), 7.20 (1H, dd, J = 8.08, 9.02 Hz), 6.49 (1H, d, J = 9.61 Hz), 4.02 (2H, t, J = 5.64 Hz), 3.58 (4H, m), 3.52 (4H, m), 3.49 (4H, m), 3.45 (10H, m), 3.38 (2H, t, J = 4.73 Hz), 2.34 (2H, t, J = 6.20 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 171.9, 165.2, 158.1, 154.7, 142.0, 119.3, 115.2, 113.4, 111.5, 71.3, 70.8, 70.6, 70.3, 69.8, 65.5, 61.4, 48.1, 39.3, 29.2; MS (ESI positive) m/z: 523.2 [M + H]+. Elemental analysis: calculated C, 55.16; H, 6.56; N, 10.72; found C, 55.17; H, 6.55; N, 10.71.
:95), to yield compounds 29a–c.
3-(3-(17-Azido-3,6,9,12,15-pentaoxaheptadecyl)ureido)benzenesulfonamide (29a). Yellow oil. Yield: 67%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.88 (1H, s, NH, exchange with D2O), 7.96 (1H, s, NH, exchange with D2O), 7.50 (1H, d, J = 7.51 Hz), 7.39 (1H, t, J = 7.93 Hz), 7.33 (1H, d, J = 7.51 Hz), 7.28 (2H, bs, SO2NH2, exchange with D2O), 6.24 (1H, t, J = 5.18 Hz), 3.59 (2H, t, J = 4.47 Hz), 3.52 (16H, m), 3.46 (2H, t, J = 5.35 Hz), 3.38 (2H, t, J = 4.07 Hz), 3.26 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 155.6, 145.1, 141.4, 130.0, 121.2, 118.9, 115.1, 70.4, 70.3, 70.2, 69.9, 50.7, 31.3; MS (ESI positive) m/z: 505.1 [M + H]+. Elemental analysis: calculated C, 45.23; H, 6.39; N, 16.66; found C, 45.22; H, 6.38; N, 16.67.
4-(3-(17-Azido-3,6,9,12,15-pentaoxaheptadecyl)ureido)benzenesulfonamide (29b). Yellow oil. Yield: 32%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.95 (1H, s, NH, exchange with D2O), 7.66 (2H, d, J = 8.27 Hz), 7.52 (2H, d, J = 9.05 Hz), 7.14 (2H, bs, SO2NH2, exchange with D2O), 6.32 (1H, t, J = 5.51 Hz, NH, exchange with D2O), 3.58 (2H, t, J = 5.11 Hz), 3.52 (16H, m), 3.46 (2H, t, J = 5.38 Hz), 3.38 (2H, t, J = 4.69 Hz), 3.25 (2H, dt, J = 10.71, 11.34 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.8, 143.7, 137.4, 130.6, 128.1, 70.6, 70.4, 70.5, 69.4, 50.2, 46.3; MS (ESI positive) m/z: 527.2 [M + H]+. Elemental analysis: calculated C, 45.23; H, 6.39; N, 16.66; found C, 45.21; H, 6.40; N, 16.68.
4-(21-Azido-3-oxo-7,10,13,16,19-pentaoxa-2,4-diazahenicosyl)benzenesulfonamide (29c). Yellow oil. Yield: 52%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.75 (2H, d, J = 8.27 Hz), 7.39 (2H, d, J = 8.27 Hz), 7.26 (2H, bs, SO2NH2, exchange with D2O), 6.51 (1H, t, J = 6.39 Hz, NH, exchange with D2O), 6.02 (1H, t, J = 4.88 Hz, NH, exchange with D2O), 4.25 (2H, d, J = 5.26 Hz), 3.59 (2H, t, J = 5.13 Hz), 3.52 (16H, m), 3.38 (4H, m), 3.17 (2H, t, J = 5.47 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 163.7, 142.3, 137.4, 129.4, 129.1, 128.5, 71.7, 70.8, 70.6, 70.4, 67.3, 65.9, 50.2, 45.6, 44.9; MS (ESI positive) m/z: 519.2 [M + H]+. Elemental analysis: calculated C, 45.60; H, 6.47; N, 16.50; found C, 45.61; H, 6.48; N, 16.49.
:95), to yield compounds 18b–d.
4-(3-(17-Azido-3,6,9,12,15-pentaoxaheptadecyl)thioureido)benzensulfonamide (32b). Yellow oil. Yield: 67%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.92 (1H, s, NH, exchange with D2O), 8.02 (1H, s, NH, exchange with D2O), 7.72 (2H, d, J = 8.69 Hz), 7.67 (2H, d, J = 8.99 Hz), 7.26 (2H, bs, SO2NH2, exchange with D2O), 3.64 (2H, m), 3.59 (5H, m), 3.54 (15H, m), 3.38 (2H, t, J = 4.71 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.2, 140.1, 138.7, 129.1, 119.6, 119.5, 71.1, 70.8, 70.6, 69.8, 65.8, 50.2, 44.7; MS (ESI positive) m/z: 521.2 [M + H]+. Elemental analysis: calculated C, 43.83; H, 6.20; N, 16.14; found C, 43.82; H, 6.21; N, 16.12.
4-(21-Azido-3-thioxo-7,10,13,16,19-pentaoxa-2,4-diazahenicosyl)benzenesulfonamide (32c). Yellow oil. Yield: 63%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.03 (1H, s, NH, exchange with D2O), 7.75 (2H, d, J = 8.39 Hz), 7.63 (1H, s, NH, exchange with D2O), 7.42 (2H, d, J = 8.44 Hz), 7.31 (2H, bs, SO2NH2, exchange with D2O), 4.73 (2H, m), 3.54 (22H, m), 3.38 (2H, t, J = 5.26 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 168.9, 146.7, 136.2, 127.4, 124.7, 70.9, 70.7, 70.6, 69.3, 63.5, 47.5, 46.3, 44.4; MS (ESI positive) m/z: 535.1 [M + H]+. Elemental analysis: calculated C, 44.39; H, 6.30; N, 15.93; found C, 44.38; H, 6.31; N, 15.94.
4-(22-Azido-4-thioxo-8,11,14,17,20-pentaoxa-3,5-diazadocosyl)benzenesulfonamide (32d). Yellow oil. Yield: 60%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.74 (2H, d, J = 8.20 Hz), 7.53 (1H, s, NH, exchange with D2O), 7.47 (1H, s, NH, exchange with D2O), 7.41 (2H, d, J = 8.11 Hz), 7.29 (2H, bs, SO2NH2, exchange with D2O), 3.59 (3H, t, J = 4.22 Hz), 3.51 (21H, m), 3.38 (2H, t, J = 4.87 Hz), 2.87 (2H, t, J = 7.06 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 144.4, 142.5, 129.8, 128.7, 126.4, 70.4, 70.3, 69.9, 69.6, 50.7, 35.2; MS (ESI positive) m/z: 549.3 [M + H]+. Elemental analysis: calculated C, 45.97; H, 6.61; N, 15.32; found C, 45.98; H, 6.62; N, 15.33.
:1) and stirred overnight at 60 °C. Then, the reaction mixture was filtered through a cake of Celite 521© and the filtrate was treated with slush. The crude was extracted with EtOAc thrice. The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated at reduced pressure. The crude material was purified by flash column chromatography (MeOH/DCM: 5:95), to yield compounds 10a–b, 11, 12a–c, 13, and 14b–d.
1-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-N-(4-sulfamoylbenzyl)-3,6,9,12,15-pentaoxaoctadecan-18-amide (10a). Yellow powder. Yield: 47%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.55 (1H, s, NH, exchange with D2O), 8.53 (1H, s), 8.48 (1H, s), 8.42 (1H, t, J = 5.90 Hz), 8.27 (1H, s), 7.93 (1H, s), 7.89 (1H, d, J = 9.02 Hz), 7.75 (2H, d, J = 7.98 Hz), 7.54 (1H, d, J = 7.63 Hz), 7.47 (1H, d, J = 8.06 Hz), 7.40 (2H, d, J = 8.29 Hz), 7.29 (2H, bs, SO2NH2, exchange with D2O), 7.23 (1H, s), 4.58 (2H, t, J = 5.12 Hz), 4.30 (6H, m), 3.88 (2H, t, J = 5.12 Hz), 3.79 (2H, t, J = 4.44 Hz), 3.75 (2H, t, J = 4.44 Hz), 3.61 (2H, t, J = 6.15 Hz), 3.56 (2H, m), 3.50 (2H, m), 3.45 (14H, m), 3.38 (3H, s), 3.36 (3H, s), 2.38 (2H, t, J = 6.01 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 170.8, 156.6, 153.9, 153.1, 148.4, 147.0, 146.4, 143.8, 142.5, 140.0, 131.2, 129.3, 127.5, 125.8, 122.0, 121.9, 120.6, 119.0, 109.1, 108.3, 103.5, 70.3, 70.2, 69.9, 69.8, 69.7, 68.8, 68.6, 68.3, 67.0, 58.6, 58.5, 49.8, 41.9, 41.8, 36.3; MS (ESI positive) m/z: 897.4 [M + H]+. Elemental analysis: calculated C, 52.34; H, 6.41; N, 13.56; found C, 52.33; H, 6.42; N, 13.55.
1-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-N-(4-sulfamoylphenethyl)-3,6,9,12,15-pentaoxanonadecan-19-amide (10b). Green powder. Yield: 44%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.58 (1H, s, NH, exchange with D2O), 8.56 (1H, s), 8.51 (1H, s), 8.45 (1H, t, J = 5.90 Hz), 8.30 (1H, s), 7.96 (1H, s), 7.92 (1H, d, J = 9.02 Hz), 7.78 (2H, d, J = 7.98 Hz), 7.57 (1H, d, J = 7.63 Hz), 7.50 (1H, d, J = 8.06 Hz), 7.43 (2H, d, J = 8.29 Hz), 7.32 (2H, bs, SO2NH2, exchange with D2O), 7.26 (1H, s), 4.61 (2H, t, J = 5.12 Hz), 4.33 (6H, m), 3.91 (2H, t, J = 5.12 Hz), 3.82 (2H, t, J = 4.44 Hz), 3.78 (2H, t, J = 4.44 Hz), 3.64 (2H, t, J = 6.15 Hz), 3.59 (2H, m), 3.53 (2H, m), 3.48 (16H, m), 3.41 (3H, s), 3.39 (3H, s), 2.41 (2H, t, J = 6.01 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 175.9, 172.1, 157.4, 154.6, 153.7, 149.0, 147.0, 144.7, 142.0, 140.3, 131.5, 130.1, 130.0, 126.4, 123.0, 122.8, 121.6, 120.0, 109.6, 108.3, 103.8, 70.8, 70.7, 70.3, 70.2, 70.1, 69.2, 69.2, 68.9, 67.3, 62.7, 59.1, 56.1, 50.5, 49.3, 40.4, 36.7, 35.2; MS (ESI positive) m/z: 911.4 [M + H]+. Elemental analysis: calculated C, 54.21; H, 6.46; N, 13.00; found C, 54.20; H, 6.47; N, 13.01.
1-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazoline-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-N-(2-((2-oxo-2H-chromen-6-yl)oxy)ethyl)-3,6,9,12,15-pentaoxaoctadecan-18-amide (11). Green oil. Yield: 45%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.68 (1H, s, NH, exchange with D2O), 8.54 (1H, s), 8.51 (1H, s), 8.26 (1H, s), 8.11 (1H, t, J = 4.93 Hz), 7.98 (1H, d, J = 9.42 Hz), 7.94 (1H, s), 7.87 (1H, d, J = 8.35 Hz), 7.56 (1H, d, J = 7.59 Hz), 7.46 (1H, t, J = 7.84 Hz), 7.32 (1H, d, J = 8.85 Hz), 7.27 (1H, d, J = 2.85 Hz), 7.23 (1H, s), 7.19 (1H, dd, J = 8.87 Hz, 9.27 Hz), 6.48 (1H, d, J = 9.50 Hz), 4.58 (2H, t, J = 5.51 Hz), 4.30 (4H, m), 4.00 (2H, t, J = 5.55 Hz), 3.88 (2H, t, J = 5.10 Hz), 3.79 (2H, t, J = 4.80 Hz), 3.75 (2H, t, J = 4.35 Hz), 3.57 (4H, m), 3.49 (2H, m), 3.42 (12H, m), 3.37 (8H, m), 2.32 (2H, t, J = 6.37 Hz). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.7, 170.4, 160.0, 156.5, 154.7, 153.8, 152.5, 148.2, 147.9, 146.1, 144.0, 139.7, 131.1, 129.0, 121.9, 121.8, 120.5, 119.8, 119.1, 118.9, 117.3, 116.5, 111.5, 108.9, 107.5, 103.4, 70.1, 70.0, 69.7, 69.6, 69.4, 68.6, 68.4, 68.1, 67.0, 66.7, 58.4, 58.3, 49.6, 47.2, 38.1, 36.0, 30.6; MS (ESI positive) m/z: 916.9 [M + H]+. Elemental analysis: calculated C, 55.91; H, 6.41; N, 12.36; found C, 55.92; H, 6.42; N, 12.34.
3-(3-(17-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazoline-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3,6,9,12,15-pentaoxaheptadecyl)ureido)benzenesulfonamide (12a). Yellow powder. Yield: 40%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.56 (1H, s, NH, exchange with D2O), 8.87 (1H, s, NH, exchange with D2O), 8.53 (1H, s), 8.48 (1H, s), 8.27 (1H, s), 7.96 (1H, m), 7.93 (1H, s), 7.89 (1H, d, J = 8.71 Hz), 7.54 (1H, d, J = 7.87 Hz), 7.48 (2H, m), 7.34 (2H, m), 7.28 (2H, bs, SO2NH2, exchange with D2O), 7.22 (1H, s), 6.23 (1H, t, J = 5.58 Hz), 4.58 (2H, t, J = 5.27 Hz), 4.30 (4H, m), 3.88 (2H, t, J = 4.96 Hz), 3.79 (2H, t, J = 4.34 Hz), 3.75 (2H, t, J = 4.23 Hz), 3.55 (2H, m), 3.46 (16H, m), 3.38 (3H, s), 3.36 (3H, s), 3.24 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 156.4, 154.9, 153.6, 152.9, 148.1, 146.9, 146.2, 144.5, 140.9, 140.0, 131.0, 129.2, 128.9, 121.8, 121.7, 120.3, 120.2, 118.7, 118.0, 114.4, 109.0, 108.2, 103.3, 76.4, 72.9, 70.6, 70.1, 70.0, 69.7, 69.6, 68.6, 68.4, 68.0, 59.5, 58.4, 58.3, 56.6, 49.6, 47.2, 30.6; MS (ESI positive) m/z: 898.4 [M + H]+. Elemental analysis: calculated C, 54.84; H, 6.17; N, 14.04; found C, 54.86; H, 6.15; N, 14.03.
4-(3-(17-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3,6,9,12,15-pentaoxaheptadecyl)ureido)benzenesulfonamide (12b). Reddish oil. Yield: 25%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.59 (1H, s, NH, exchange with D2O), 8.56 (1H, s, NH, exchange with D2O), 8.50 (1H, s), 8.29 (1H, s), 7.95 (1H, s), 7.91 (1H, d, J = 8.23 Hz), 7.77 (2H, d, J = 7.84 Hz), 7.56 (1H, d, J = 8.07 Hz), 7.48 (1H, t, J = 7.81 Hz), 7.41 (2H, d, J = 8.22 Hz), 7.30 (2H, bs, SO2NH2, exchange with D2O), 7.25 (1H, s), 6.54 (1H, t, J = 4.86 Hz), 6.05 (1H, t, J = 5.43 Hz), 4.61 (2H, t, J = 4.94 Hz), 4.32 (4H, m), 4.27 (2H, d, J = 5.87 Hz), 3.90 (2H, t, J = 5.10 Hz), 3.81 (2H, t, J = 4.34 Hz), 3.77 (2H, t, J = 3.06 Hz), 3.57 (2H, m), 3.47 (14H, m), 3.39 (8H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.3, 155.7, 154.9, 149.5, 148.6, 145.6, 143.7, 137.4, 135.9, 135.2, 131.4, 129.3, 126.7, 124.2, 123.1, 122.0, 121.3, 116.6, 107.2, 105.0, 100.6, 72.6, 69.9, 69.7, 69.6, 69.0, 68.8, 68.6, 58.4, 57.3, 51.3, 49.7, 44.0, 36.3, 35.4, 29.0; MS (ESI positive) m/z: 898.4 [M + H]+. Elemental analysis: calculated C, 54.84; H, 6.17; N, 14.04; found C, 54.86; H, 6.16; N, 14.02.
4-(21-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazoline-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3-oxo-7,10,13,16,19-pentaoxa-2,4-diazahenicosyl)benzenesulfonamide (12c). Yellow powder. Yield: 52%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.57 (1H, s, NH, exchange with D2O), 8.54 (1H, s, NH, exchange with D2O), 8.48 (1H, s), 8.27 (1H, s), 7.93 (1H, s), 7.89 (1H, d, J = 8.23 Hz), 7.75 (2H, d, J = 7.84 Hz), 7.54 (1H, d, J = 8.07 Hz), 7.46 (1H, t, J = 7.81 Hz), 7.39 (2H, d, J = 8.22 Hz), 7.28 (2H, bs, SO2NH2, exchange with D2O), 7.23 (1H, s), 6.52 (1H, t, J = 4.86 Hz), 6.03 (1H, t, J = 5.43 Hz), 4.59 (2H, t, J = 4.94 Hz), 4.30 (4H, m), 4.25 (2H, d, J = 5.87 Hz), 3.88 (2H, t, J = 5.10 Hz), 3.79 (2H, t, J = 4.34 Hz), 3.75 (2H, t, J = 3.06 Hz), 3.55 (2H, m), 3.45 (14H, m), 3.37 (8H, m), 3.15 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 162.6, 160.9, 158.3, 154.9, 152.8, 149.5, 146.9, 144.8, 140.1, 138.2, 135.2, 133.1, 129.3, 126.7, 124.2, 123.2, 122.1, 121.3, 116.6, 112.1, 108.7, 102.3, 98.2, 69.9, 69.7, 69.6, 69.0, 68.8, 68.6, 58.4, 57.3, 49.7, 44.0, 29.0; MS (ESI positive) m/z: 912.6 [M + H]+. Elemental analysis: calculated C, 55.31; H, 6.30; N, 13.82; found C, 55.30; H, 6.31; N, 13.83.
1-(17-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3,6,9,12,15-pentaoxa heptadecyl)-3-(2-oxo-2H-chromen-6-yl)urea (13). White powder. Yield: 54%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.56 (1H, s, NH, exchange with D2O), 8.81 (1H, s, NH, exchange with D2O), 8.53 (1H, s), 8.48 (1H, s), 8.27 (1H, s), 8.01 (1H, d, J = 9.6 Hz), 7.93 (1H, s), 7.89 (1H, d, J = 7.8 Hz), 7.79 (1H, d, J = 2.5 Hz), 7.54 (1H, d, J = 7.6 Hz), 7.49 (1H, dd, J = 9.2 Hz, J = 2.7 Hz), 7.45 (1H, t, J = 7.9 Hz) 7.27 (1H, d, J = 8.9 Hz), 7.22 (1H, s), 6.43 (1H, d, J = 9.5 Hz), 6.28 (1H, t, J = 5.4 Hz), 4.58 (2H, t, J = 4.9 Hz), 4.30 (4H, m), 3.88 (2H, t, J = 5.0 Hz), 3.79 (2H, t, J = 4.7 Hz), 3.75 (2H, t, J = 4.5 Hz), 3.55 (2H, m), 3.46 (16H, m), 3.37 (3H, s), 3.36 (3H, s), 3.23 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.7, 170.4, 160.0, 156.5, 154.7, 153.8, 152.5, 148.2, 147.9, 146.1, 144.0, 139.7, 131.1, 129.0, 121.9, 121.8, 120.5, 119.8, 119.1, 118.9, 117.3, 116.5, 111.5, 108.9, 107.5, 103.4, 70.1, 70.0, 69.7, 69.6, 69.4, 68.6, 68.4, 68.1, 67.0, 66.7, 58.4, 58.3, 49.6, 38.1, 36.0; MS (ESI positive) m/z: 887.6 [M + H]+. Elemental analysis: calculated C, 59.58; H, 6.14; N, 12.63; found C, 59.56; H, 6.15; N, 12.64.
4-(3-(17-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3,6,9,12,15-pentaoxaheptadecyl)thioureido)benzenesulfonamide (14b). Yellow powder. Yield: 36%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.95 (1H, s, NH, exchange with D2O), 8.55 (1H, s, NH, exchange with D2O), 8.49 (1H, s), 8.28 (1H, s), 7.94 (1H, s), 7.90 (1H, d, J = 8.23 Hz), 7.76 (2H, d, J = 7.84 Hz), 7.55 (1H, d, J = 8.07 Hz), 7.47 (1H, t, J = 7.81 Hz), 7.40 (2H, d, J = 8.22 Hz), 7.29 (2H, bs, SO2NH2, exchange with D2O), 7.24 (1H, s), 6.53 (1H, t, J = 4.86 Hz), 6.04 (1H, t, J = 5.43 Hz), 4.60 (2H, t, J = 4.94 Hz), 4.31 (4H, m), 4.26 (2H, d, J = 5.87 Hz), 3.89 (2H, t, J = 5.10 Hz), 3.80 (2H, t, J = 4.34 Hz), 3.76 (2H, t, J = 3.06 Hz), 3.56 (2H, m), 3.47 (14H, m), 3.39 (8H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 162.9, 148.8, 147.3, 146.5, 144.3, 141.9, 140.0, 138.9, 131.3, 129.5, 129.4, 128.4, 126.0, 125.9, 125.7, 122.1, 120.8, 119.2, 117.6, 112.1, 104.5, 70.3, 70.2, 69.9, 68.8, 68.7, 68.5, 60.3, 58.6, 56.5, 55.4, 49.9 48.7, 36.0, 29.1, 28.8, 22.2; MS (ESI positive) m/z: 914.4 [M + H]+. Elemental analysis: calculated C, 53.87; H, 6.07; N, 13.79; found C, 53.85; H, 6.06; N, 13.78.
4-(21-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazoline-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-3-thioxo-7,10,13,16,19-pentaoxa-2,4-diazahenicosyl)benzenesulfonamide (14c). Yellow powder. Yield: 41%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.15 (1H, s, NH, exchange with D2O), 8.59 (1H, s, NH, exchange with D2O), 8.53 (1H, s), 8.32 (1H, s), 7.98 (1H, s), 7.94 (1H, d, J = 8.23 Hz), 7.80 (2H, d, J = 7.84 Hz), 7.59 (1H, d, J = 8.07 Hz), 7.51 (1H, t, J = 7.81 Hz), 7.44 (2H, d, J = 8.22 Hz), 7.33 (2H, bs, SO2NH2, exchange with D2O), 7.28 (1H, s), 6.57 (1H, t, J = 4.86 Hz), 6.08 (1H, t, J = 5.43 Hz), 4.64 (2H, t, J = 4.94 Hz), 4.35 (4H, m), 4.30 (2H, d, J = 5.87 Hz), 3.93 (2H, t, J = 5.10 Hz), 3.84 (2H, t, J = 4.34 Hz), 3.80 (2H, t, J = 3.06 Hz), 3.60 (2H, m), 3.50 (14H, m), 3.42 (8H, m), 3.20 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 155.4, 148.8, 146.5, 145.1, 144.3, 141.8, 140.0, 133.0, 131.3, 129.6, 129.3, 128.5, 126.0, 125.9, 125.7, 122.1, 120.8, 119.1, 115.8, 112.0, 104.5, 70.4, 70.3, 69.9, 68.8, 68.7, 68.4, 66.4, 61.9, 60.3, 58.6, 49.9, 48.7, 36.0, 29.1, 28.8, 22.3; MS (ESI positive) m/z: 928.4 [M + H]+. Elemental analysis: calculated C, 54.35; H, 6.19; N, 13.58; found C, 54.32; H, 6.21; N, 13.56.
4-(22-(4-(3-((6,7-Bis(2-methoxyethoxy)quinazolin-4-yl)amino)phenyl)-1H-1,2,3-triazol-1-yl)-4-thioxo-8,11,14,17,20-pentaoxa-3,5-diazadocosyl)benzenesulfonamide (14d). Yellow powder. Yield: 42%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.84 (1H, s, NH, exchange with D2O), 8.52 (1H, s, NH, exchange with D2O), 8.46 (1H, s), 8.25 (1H, s), 7.91 (1H, s), 7.87 (1H, d, J = 8.23 Hz), 7.73 (2H, d, J = 7.84 Hz), 7.52 (1H, d, J = 8.07 Hz), 7.44 (1H, t, J = 7.81 Hz), 7.37 (2H, d, J = 8.22 Hz), 7.26 (2H, bs, SO2NH2, exchange with D2O), 7.21 (1H, s), 6.50 (1H, t, J = 4.86 Hz), 6.01 (1H, t, J = 5.43 Hz), 4.57 (2H, t, J = 4.94 Hz), 4.28 (4H, m), 4.23 (2H, d, J = 5.87 Hz), 3.86 (2H, t, J = 5.10 Hz), 3.77 (2H, t, J = 4.34 Hz), 3.73 (2H, t, J = 3.06 Hz), 3.54 (2H, m), 3.45 (14H, m), 3.35 (8H, m), 3.13 (2H, m), 2.97 (2H, m). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 153.3, 148.8, 145.2, 144.3, 141.9, 140.0, 138.3, 131.3, 129.7, 129.5, 129.4, 128.4, 125.9, 125.7, 122.1, 120.8, 119.2, 119.2, 112.1, 111.0, 104.3, 72.8, 70.3, 70.2, 70.1, 69.9, 68.8, 68.7, 68.5, 60.3, 58.6, 52.0, 49.9, 48.7, 36.0, 29.1, 28.8, 22.2; MS (ESI positive) m/z: 942.2 [M + H]+. Elemental analysis: calculated C, 54.82; H, 6.31; N, 13.38; found C, 54.81; H, 6.33; N, 13.36.
:98), to yield compound 25. Brownish oil, Yield: 47%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.81 (1H, s, NH, exchange with D2O), 8.07 (1H, d, J = 9.9 Hz), 7.85 (1H, s, NH, exchange with D2O), 7.54 (1H, d, J = 8.7 Hz), 7.33 (1H, d, J = 9.0 Hz), 6.49 (1H, d, J = 8.9 Hz), 6.29 (1H, t, J = 4.9 Hz), 3.57 (21H, m), 3.30 (3H, m). MS (ESI positive) m/z: 494.2 [M + H]+. Elemental analysis: calculated C, 53.54; H, 6.33; N, 14.19; found C, 53.52; H, 6.32; N, 14.18.
2.2. Pan assays interference compounds
The behaviour of final compounds as pan-assay interference compounds (PAINS)72 was evaluated through two different web tools SwissADME (http://www.swissadme.ch, accessed 2024-01-07)73 and False Positive Remover (https://www.cbligand.org/PAINS/login.php, accessed 2024-01-07).74 JChem for Office (21.15.704, 2023) by ChemAxon http://www.chemaxon.com) was used for structure management, SMILES generation, and file conversion. All the analyzed compounds were “accepted” by both the web services.2.3. In vitro carbonic anhydrase inhibition assay
The CA-catalyzed CO2 hydration activity measurement was performed on an Applied Photophysics stopped-flow instrument using phenol red, at a concentration of 0.2 mM, as a pH indicator with 20 mM HEPES (pH 7.5) as the buffer, 20 mM Na2SO4, and following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s and working at the maximum absorbance of 557 nm. The CO2 concentrations ranged from 1.7 to 17 mM.75 For each inhibitor, six traces of the initial 5–10% of the reaction have been used to determine the initial velocity. The uncatalyzed reaction rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled water, and dilutions up to 0.01 nM were prepared. Solutions containing inhibitor and enzyme were preincubated for 15 min at room temperature before assay to allow the formation of the E–I complex. The inhibition constants were obtained as non-linear least-squares protocols using PRISM 3 and are the mean from at least three different measurements. All CAs were recombinant ones and were obtained in-house.762.4. Molecular docking
The two-dimensional structures of the compounds were generated using the Maestro module of the Schrödinger Life-Sciences Suite 2024-1 software.77 The LigPrep utility was then employed to prepare the compounds to obtain the three-dimensional geometry and identify all possible tautomers and protonation states at pH 7.0 ± 0.4, as determined by Epik.78 The sulfonamide group was considered as deprotonated to evaluate the interaction with zinc ion, but after observing no interaction (even considering the zinc constraint and the core constraint), the sulfonamide group was used as unprotonated. The crystal structures of hCA I, hCA II, hCA IX, hCA XI, and EGFR were retrieved from the Protein Data Bank (PDB ID: 6I0J, 3K34, 8Q1A, 5LL5, and 3W2S, respectively).79–83Protein Preparation Workflow was used to correct, optimize and minimize the crystal structures. Molecular docking analyses were performed using the Glide software. Each enclosing box grid was generated using the centroid of the proper crystallographic ligand and the generated grid file was used for molecular docking using the SP-peptide module of Maestro.84 In all CA isoforms, a water molecule was positioned in front of the zinc ion after no interaction between the zinc ion and the sulfonamide group was found. SP-peptide docking protocol was used by setting 5000 poses per ligand for the initial phase and 400 poses per ligand for energy minimization with the OPLS4 forcefield. The docking results were analyzed for the best-docked pose based on the Glide SP score.
2.5. In vitro cell proliferation assays
Human triple-negative breast cancer MDA-MB-231 cells (ATCC HTB-262) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% glutamine. Cells were kept at the low passage, returning to their original frozen stocks every 3–4 months. For hypoxic culture conditions cells were grown in 1% O2 and 5% CO2. Cell proliferation assays were performed seeding 10.000 cells per well in 48-well plates, and treatments were performed in 1% FBS with increasing concentrations of compounds 8b–d and 7a, SLC-0111, or ERL. After 72 h of incubation at 37 °C, cells detached using trypsin/2 mM EDTA, and cell counting was performed with a MACSQuant® Analyzer (Miltenyi Biotec).3. Results and discussion
3.1. Synthetic approach
We recently reported the click chemistry approach applied to the hybridization of ERL, as an innovative strategy to tackle infections promoted by Helicobacter pylori.85 Specifically, the hybrids obtained accounted for the ERL linked to the prototypic CA-inhibiting chemotype86 (compounds 1–4, Fig. 1, Table 1) and for the pyrimidine-2,4(1H,3H)-dione moiety as in the antiviral azidothymidine (AZT) drug (namely 5, Fig. 1, Table 1) which is also endowed with antimicrobial features.87,88| Cpd | Structure | K I (nM) | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
HpαCA* | hCA I* | hCA II* | hCA VA | hCA VI | hCA IX | hCA XII | |
| K I values are reported as means of three independent experiments. Errors are in the range of ±5–10% of the reported values. AAZ was used as a reference control in these assays. n.a.: not active at the highest concentration tested (100 μM). Data on HpαCA and hCAs I and II (indicated with an asterisk, *) are already reported by Benito et al.85 | ||||||||
| 1a |

|
63.3 | 70.0 | 2.0 | 13.2 | 360 | 18.5 | n.a. |
| 1b |

|
70.1 | 85.4 | 22.0 | 11.6 | 174 | 18.1 | n.a. |
| 1c |

|
252 | 3503 | 5.0 | n.a. | 60.1 | 18.3 | 28.0 |
| 1d |
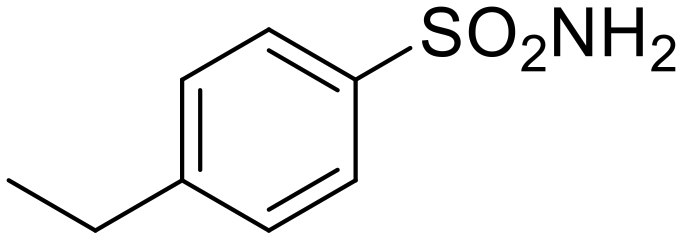
|
19.3 | 9.3 | 4.5 | 13.0 | 51.3 | 20.0 | 6.0 |
| 2a |

|
56.3 | 3.3 | 10.4 | 8.3 | 39.7 | 443 | 7.0 |
| 2b |

|
71.6 | 382 | 7.4 | 12.4 | 99.0 | 393 | n.a. |
| 3a |

|
48.0 | 35.2 | 5.4 | 98.0 | 2601 | 40.0 | n.a. |
| 3b |

|
43.6 | 31.2 | 3.0 | 107 | 192 | 561 | n.a. |
| 4 |

|
805 | 2.6 | 5.0 | 6.2 | 28.1 | 425 | n.a. |
| 5 |

|
19.3 | 7.1 | 3.0 | 145 | 103 | 1445 | n.a. |
| AZT |

|
n.a. | n.a. | 684 | n.a. | 505 | n.a. | 590 |
| ERL |

|
n.a. | n.a. | 3976 | n.a. | 453 | n.a. | n.a. |
| AAZ | 21.0 | 250 | 12.1 | 63.0 | 16.0 | 25.7 | 5.7 | |
The main structural features contained in 1–5 were thought to inhibit the pathogenic expressed CA (HpαCA) which is fundamental for the microorganism survival at the harsh stomach pH values,89,90 and to induce overexpression of EGFR in the host cells91,92 along with its inhibition by the ERL portion.
The synthesized compounds allowed us to define the tolerance of introduced modifications, thus including a preliminary exploration on spacers connecting the two active portions. The enzymatic results on a panel of bacterial CAs were highly promising, highlighting some compounds to potently and selectively inhibit HpαCA (Table 1) over other bacterial isoenzymes and exhibit a moderate anti-H. pylori activity, as shown by minimal bacterial concentration (MIC) values.85 Interestingly, the emerged compound 2b was also investigated in silico and the modification of the ERL structure into the triazole-containing tail was proved not to severely impair the binding to EGFR.85
Herein, as a follow-up of our research, this first series of ERL-clicked derivatives were tested on a selected panel of human (h) CAs such as the I, II, VA, VI, IX, and XII to assess their potency and selectivity profiles in comparison to ERL and AZT (Table 1).
Based on data in Table 1 relevant structure–activity relationships (SARs) can be drawn. The presence of the 4-benzenesulfonamide group (1a) seems to be responsible for a selective inhibitory profile for hCA II (KI = 2.0 nM), whereas a low nanomolar inhibition was also detected towards hCA VA and hCA IX (KI values of 13.2 and 18.5 nM, respectively) (selectivity index (SI) values for the tumor-associated hCAs are reported in Table S1 in the ESI† file). Interestingly, no activity was found against the XII isoform. By moving the sulfonamide function in the meta-position of the phenyl ring, as in compound 1b, the potency towards isoforms VA and IX was maintained (KI values of 11.6 and 18.1 nM, respectively), with no significant difference, such as the lack of activity against hCA XII. Similar activities were also found on HpαCA and hCA I, whereas an 11-fold decrease in potency was noticed versus hCA II (KI values of 22.0 nM) and a 2-fold lower KI on hCA VI (= 360 and 174 nM for 1a and 1b, respectively). On the other hand, the ortho-isomer 1c showed a very different inhibitory profile. In fact, the compound proved to be highly selective for hCA II, with a low nanomolar inhibition (KI = 5.0 nM), and still maintained the potency against hCA IX (KI = 18.3 nM). However, 1c reverted the trend against the isoforms VA and XII; indeed, the complete loss of activity towards the former corresponded to a relevant gain in potency on hCA XII (KI = 28.0 nM). Thereby, data for 1a–c highlighted that the position of the zinc-binding group is not relevant in the inhibition of hCA IX, but fundamental to discriminate potency towards hCAs VA and XII.
A dramatic change was observed by introducing a methylene unit between the triazole ring and the para-benzenesulfonamide ring, as in compound 1d: a net increase in the inhibitory potency is highlighted and low nanomolar KI values were found for HpαCA, hCA I, hCA VI and hCA XII (= 19.3, 9.3, 51.3, and 6.0 nM), although the potency against the isoforms II, VA, and IX were not relevantly affected with respect to 1a. Introducing an amido moiety between the methylene unit and the benzenesulfonamide group in 2a the inhibition of hCA XII was maintained (KI = 7.0 nM), while resulting in a 2.9- and 22-fold weaker inhibitor of hCAs II and IX (KI = 56.3 and 443 nM), respectively. Shifting the sulfonamide function in meta-position leading to compound 2b resulted in a complete loss of activity towards hCA XII along with a general decrease in potency with the maximum example in the 115-fold reduced inhibition of hCA I (KI = 382 nM), whereas the chemical modification was better tolerated by hCA II and hCA IX. The affinity towards the hCA XII active site was clearly reset by an additional structural complication, i.e., the insertion of a benzyl group in the tail, resulting in distancing the ERL core from the CA inhibiting function, as in 3a and 3b. Also, inhibition of hCA II was quite unaffected (KI values of 5.4 and 3.0 nM), whereas higher discrepancy was highlighted by enzymatic data on hCA VI (KI = 2601 and 192 nM for 3a and 3b, respectively) and hCA IX (KI = 40.0 and 561 nM for 3a and 3b, respectively). Surprisingly, despite the isomerism, the compounds shared a similar inhibitory profile against HpαCA and hCAs I, II, and VA. Additional tail elongation as in compound 4 severely worsened the KI value till the submicromolar range (= 805 nM) against HpαCA, while improving the potency against hCAs I, II, VA, and VI. As regards the tumor-associated hCAs IX and XII, while for the former a KI of 425 nM was detected, inactivity emerged for the latter. In the end, when the hybrid 5 was tested on hCAs IX and XII, again no worthy-of-note inhibition can be highlighted. Moreover, CA inhibition profiling of AZT and ERL did not provide satisfactory results.
The dataset for this series of compounds highlighted that the shorter the linker the higher the inhibitory potency towards the tumor-associated hCAs, with 1a–d being the most active on hCA IX. Thus, the introduction of additional moieties (amido groups as in 2a and 2b) and/or hydrophobic portions (phenyl rings as in 2a, 2b, and 4) in the spacer seemed to cause a loss in activity. However, we tried to slightly increase the linker without implementing the chemical complexity, thereby designing compound 6, being a homologue compound that bears only a methylene unit more (Table 2, synthesis in Scheme S1 in the ESI† file).
| Cpd | Structure | K I (nM) | SI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | I/IX | II/IX | I/XII | II/XII | ||
| K I values are reported as means of three independent experiments. Errors are in the range of ±5–10% of the reported values. AAZ was used as a reference control in these assays. n.a.: not active at the highest concentration tested (100 μM). Data indicated with an asterisk (*) were already reported by Benito et al.85 SI values are calculated as the ratio between the KI values of the physiologically relevant CA isoform I or II (as indicated) and the CA isoform of interest (hCA IX or hCA XII, as indicated). The higher the SI value, the higher the isoform preference. | |||||||||
| 1a |

|
63.3* | 70.0* | 18.5 | n.a. | 3.42 | 3.78 | — | — |
| 1d |
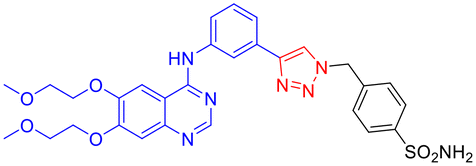
|
19.3* | 9.3* | 20.0 | 6.0 | 0.96 | 0.47 | 3.21 | 1.55 |
| 6 |

|
1031 | 18.4 | 9.3 | 353 | 111 | 1.98 | 2.92 | 0.05 |
Data in Table 2 showed compound 6 being a weak inhibitor of hCA I, with a micromolar inhibitory potency versus this isoenzyme (KI = 1031 nM), and possesses a stronger affinity towards hCAs II and IX, showing KI values of 18.4 and 9.3 nM, respectively. Interestingly, the compound exhibited a higher preference for hCA IX with respect to its homologues 1a and 1d, as highlighted by the SI values. However, the activity towards hCA XII decreased reaching the medium nanomolar range (KI = 353 nM), also impacting the SI.
However, representative compounds of the ERL-clicked series (1a–c) were selected along with the reference hCAs IX and XII inhibitor SLC-0111, and ERL to evaluate their anti-proliferative effect in vitro on non-small cell lung carcinoma A549 and pancreatic epithelioid carcinoma PANC-1 cell lines, both characterized by overexpression of the tumor-associated hCA IX93,94 and EGFR95,96 (Fig. S1 in the ESI† file). The reference compound SLC-0111 was found to be ineffective in reducing the proliferation of both the cell lines and only ERL and compound 1c displayed an antiproliferative effect, with the latter being the most effective and possessing IC50 values of 16.3 and 57.1 μM on A549 and PANC-1 cells, respectively.
The overall promising results encouraged further investigation and the design of a second series of hybrids aimed at enhancing the compounds solubility. Thereby, in place of the traditional methylene chains,20,97 we introduced non-cleavable monodispersed poly(ethylene glycol) (PEG) spacers between the pharmacophores interacting with hCAs and EGFR. Thus, we selected 4- and 6-unit PEG spacers to hold a precise length and good flexibility, which should ensure the two active portions could better fit into their targeted binding sites. In fact, due to their non-toxicity and amphiphilic nature, PEG polymers have garnered attention in pharmaceutical technologies, being included in formulations as excipients or vehicles/carriers of small molecules, biopharmaceuticals (e.g., peptides, antibody-drug conjugates), and drug delivery systems (e.g., liposomes and nanoparticles) or covalently bound to the drug of interest.98 In this case, PEGylated biopharmaceuticals99 and small molecule drugs100 resulted in decreased immunogenicity and improved half-life in blood and showed increased metabolic stability, solubility, and overall enhanced pharmacokinetic properties. Known examples are the FDA-approved PEGylated naloxegol,101 PEG-docetaxel,102 PEG-camptothecin,103 and many others.104 However, few small molecules-based hybrid compounds have been reported to possess PEG spacer105 with respect to the widely explored PROTACs106 and antibody-drug conjugates.107
Thus, we generated seventeen new compounds as Pegylated ERL–CAI hybrids (7–14, Fig. 1) through the Huisgen click chemistry reaction. After synthesis, the compounds were then tested on hCAs I and II along with the tumor-associated isoforms IX and XII, and docking simulations helped the rationalization of the enzymatic results, and the affinity towards EGFR was investigated in silico. In the end, to assess the compounds antiproliferative effect, we moved towards the triple-negative breast cancer (TNBC) cell line MDA-MB-231, characterized by the upregulation of both EGFR and the tumor-associated hCAs.108
3.2. Synthesis of the ERL-based library
The synthetic pattern aimed at introducing PEG (= 4 or 6 oxyethylene units) spacers between the CA-interacting functionalities, such as benzenesulfonamide and coumarin, and ERL. The first series of derivatives (7–9) was successfully obtained by a two-step procedure involving the insertion of the PEG-4 linker by means of the nucleophilic attack of the amino head of 15 to the electrophilic carbamates 16a–b or isothiocyanates 17a–d and 18 on the benzenesulfonamide tails. The obtained intermediates 19a–b, 20a–d, and 21 were then reacted with ERLvia a CuAAC by using nanosized copper in presence of tetramethylammonium chloride (TMACl) in DMF, affording the final compounds 7–9 (Scheme 1). | ||
| Scheme 1 Synthesis of hybrids 7–9. | ||
The second series of derivatives (10–14) was synthesized starting from the N-hydroxysuccinimide ester-functionalized PEG-6 (22) and suitable amines 23a–b and 24, gaining intermediates 25a–b and 26, tailored with benzenesulfonamide and coumarin cores, respectively (Scheme 2). Similarly, the amino group of the PEG-6 chemical 27 reacted with carbamates 16a–c and 28 and thiocarbamates 17b–d to obtain intermediates 29–32. In the end, all the intermediates were clicked on ERL by using copper sulfate and sodium ascorbate in presence of TMACl, yielding the final compounds 10–14 (Scheme 2).
 | ||
| Scheme 2 Synthesis of hybrids 10–14. | ||
3.3. In vitro enzymatic inhibition on CAs
This library of ERL-derived compounds was tested for the capability to exert selective inhibition of a panel of four human CAs using the stopped-flow CO2 hydration assay.109 Data obtained were also compared with those of the reference AAZ and reported in Table 3 as KI values.| Cpd | Structure | K I (nM) | SI | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
hCA I | hCA II | hCA IX | hCA XII | I/IX | II/IX | I/XII | II/XII | |
| K I values are reported as means of three independent experiments. Errors are in the range of ±5–10% of the reported values. AAZ was used as a reference control in these assays. n.a.: not active at the highest concentration tested (100 μM). Data indicated with an asterisk (*) were already reported by Benito et al.85 SI values are calculated as the ratio between the KI values of the physiologically relevant CA isoform I or II (as indicated) and the CA isoform of interest (hCA IX or hCA XII, as indicated). The higher the SI value, the higher the isoform preference. | |||||||||
| 7a |
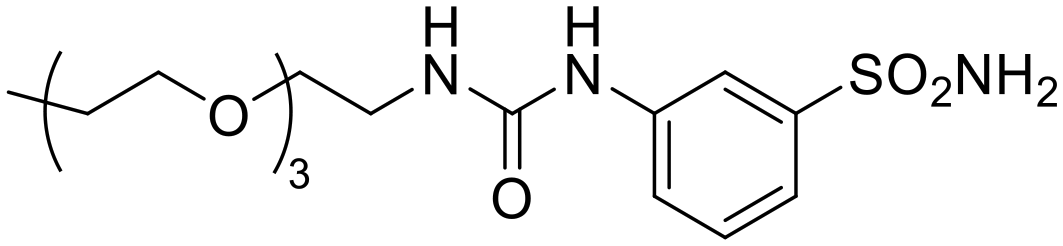
|
46546 |
723 | 285 | 58.9 | 164 | 2.54 | 790 | 12.3 |
| 7b |

|
36052 |
950 | 1124 | 57.4 | 32.1 | 0.85 | 628 | 16.6 |
| 8a |

|
45796 |
3169 | 1924 | 72 | 23.8 | 1.65 | 636 | 44.0 |
| 8b |

|
3144 | 135 | 112 | 5.33 | 28 | 1.2 | 590 | 25.4 |
| 8c |

|
3277 | 1150 | 26 | 69.7 | 127 | 44.57 | 47.0 | 16.5 |
| 8d |

|
923 | 106 | 28 | 6.49 | 32.6 | 3.73 | 142 | 16.3 |
| 9 |

|
4909 | 531.0 | 1345 | 69.8 | 3.65 | 0.39 | 70.3 | 7.61 |
| 10a |

|
903 | 253 | 4158 | 3305 | 0.22 | 0.06 | 0.27 | 0.08 |
| 10b |

|
632.4 | 33.5 | 3673 | 1363 | 0.17 | 0.01 | 0.46 | 0.02 |
| 11 |
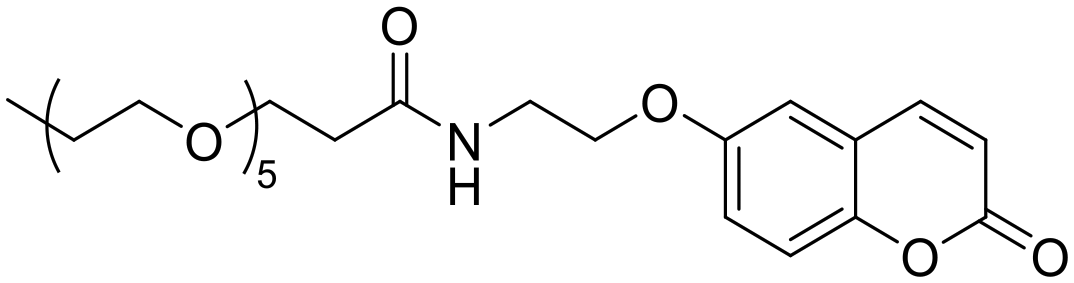
|
70140 |
33700 |
4677 | n.a. | 15.0 | 7.21 | — | — |
| 12a |
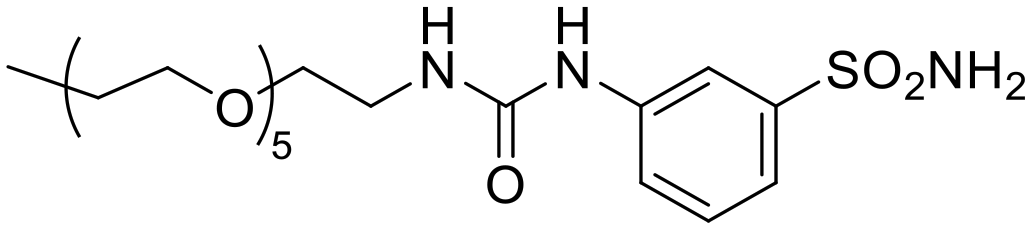
|
70090 |
1023 | 4473 | 6403 | 15.7 | 0.23 | 11.0 | 0.16 |
| 12b |

|
24790 |
90.8 | 5620 | 75.3 | 4.41 | 0.02 | 329 | 1.21 |
| 12c |
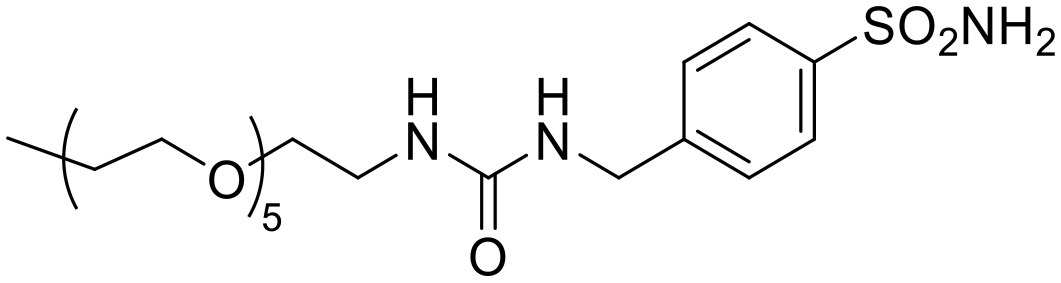
|
160 | 0.80 | 387 | 940 | 0.41 | 0.002 | 0.17 | 0.0008 |
| 13 |

|
28301 |
11364 |
242 | 8059 | 117 | 46.9 | 3.51 | 1.41 |
| 14b |
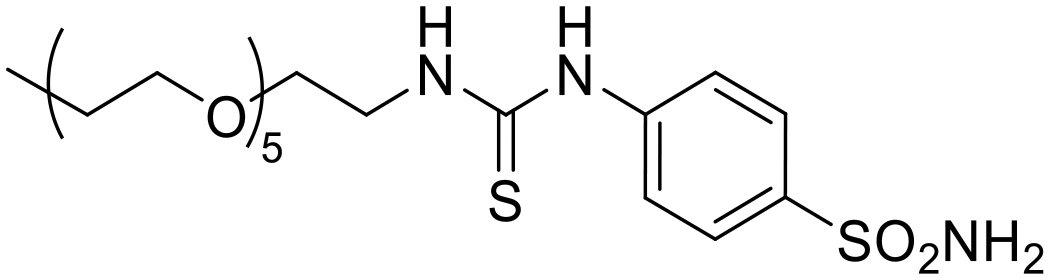
|
43340 |
9712 | 4928 | 1523 | 8.79 | 1.97 | 28.4 | 6.38 |
| 14c |

|
4976 | 1011 | 3727 | 665 | 1.33 | 0.27 | 7.48 | 1.52 |
| 14d |
|
957 | 32703 |
1877 | 95.2 | 0.51 | 17.4 | 10.0 | 344 |
| ERL |
|
n.a.* | 3976* | n.a. | n.a. | — | — | — | — |
| AZT | 250 | 12.1 | 25.7 | 5.7 | 9.73 | 0.47 | 43.86 | 2.12 | |

As reported in Table 3, interesting SARs for each CA isoform can be deduced.
i) As for the hCA I, all the derivatives are not strongly effective inhibitors, being the associated KI values in the micromolar range (except for nanomolar inhibitors 8d, 10a, 10b, 14d, and above all 12c). Compounds showed less potency when the PEG units are 6 (10–14), while the inhibitory activity is improved with PEG-4 (7–9). The CA inhibiting warhead must be the benzenesulfonamide with respect to the coumarin ring (11 and 13). In the presence of PEG-4, thiourea is preferred over urea especially if not close to the ArSO2NH2 (n = 2 > 1 > 0). 3-SO2NH2 and 4-SO2NH2 contributed equally to the inhibitory activity. If the linker is PEG-6, thiourea is like urea especially if not close to the ArSO2NH2 (n = 2 > 1 > 0). The amide was a good option to substitute the (thio)urea moiety but only if n = 2 > 1.
ii) Overall, the kinetic trend for the hCA II is better for almost all the compounds (23c is even in the picomolar range KI = 0.8 nM). Again, PEG-6 is preferred over PEG-4 and the CA inhibiting warhead must be the benzenesulfonamide with respect to the coumarin ring (11 and 13). If the linker is PEG-4, thiourea is more favored than urea, especially if n = 2 > 0 > 1. Conversely, if the linker is PEG-6, urea is better (n = 1 > 0 > 2) than thiourea, whereas the amide moiety can be a valid alternative.
iii) For hCA IX, the general situation is preferred for PEG-4. In this series, 4-SO2NH2 impacts positively the inhibitory values with respect to 3-SO2NH2, and thiourea and urea contributed equally (n = 1 ≥ 2 > 0). In the subset PEG-6, compounds are almost in the same micromolar range (except for 12 and 13).
iv) The most interesting data were registered for hCA XII: if the linker is PEG-4 (better than 6), 4-NH2SO2Ar improves the inhibitory values with respect to 3-NH2SO2Ar and, again, thiourea is preferred to urea with n = 0–1 > 2. In the subset PEG-6, coumarins are still not important for the activity (high micromolar activity for compound 13 and no activity for compound 11) and amide is more tolerated than (thio)urea especially if the 4-SO2NH2 moiety is present and with n = 2 > 0 > 1 in the thiourea series and n = 0 > 1 for ureas.
Isoform selectivity could be easily assessed by analyzing SI values reported in Table 3. Taking into consideration the first subset of PEG-4 derivatives (7–9), the higher affinity towards hCA XII was demonstrated by a large number of compounds with respect to isoforms I and II, with SI (I/XII) values ranging from 47.0 up to 790. Conversely, hCA IX inhibitory data almost overlapped with the values determined against hCA II in terms of potency range, not showing remarkable selectivity. However, the PEG-6 derivatives series (10–14) required a more accurate analysis of SI values since each compound seem to show a different isoform preference.
3.4. In silico studies on targeted enzymes
To rationalize the enzyme inhibition data, structure-based computational studies were conducted on all studied CA isoforms. The crystallographic structures of hCAs I, II, IX, and XII were obtained from the Protein Data Bank (PDB ID: 6I0J, 3K34, 8Q1A, and 5LL5, respectively). The selection of the PDB structures, validated in our previous works as well, was made taking into account the good crystallographic resolution and the absence of structural defects in the chosen structures. The investigation focused on compounds demonstrating the highest activity toward hCAs IX and XII while exhibiting selectivity over hCAs I and II. Thereby, the in silico study included compounds 8b, 8c, and 8d (Table 3).Given their chemical structure, dimension, and flexibility, the Glide SP-peptide docking method was employed. In the initial trial, the sulfonamide group was assumed to be deprotonated in order to ascertain the potential for interaction with the zinc ion in the active site. This binding mode has not yet been observed in any X-ray crystal structure of CA-sulfonamide adducts54 and should be checked experimentally in order to be validated. Unexpectedly, the docking poses obtained without any constraint do not exhibit the sulfonamide functionality of the compounds coordinated with the zinc ion. In some cases, the ERL-derived portion of the three compounds interacts with some residues in the active site. The protocol was then modified by introducing a constraint on the zinc ion, yet no poses in which this ion coordinates the sulfonamide were obtained. Subsequently, the core constraint approach was employed to force the sulfonamide group to coordinate the metal center, but the results were again negative. Because of the evidence that there is no direct interaction between the zinc ion and the sulfonamide group of compounds in this theoretical in silico approach, a coordination water molecule was introduced to the zinc ion in the hCA isoforms, with the sulfonamide group assumed to be in its undeprotonated form at physiological pH. The docking poses obtained with the docking SP-peptide were clustered, and the representative geometry of the most abundant cluster for each compound and each enzyme was considered. In hCA XII, compounds 8b and 8d (Fig. 2a and b) are positioned outside the active site and form a cation–π interaction between Lys69 and the phenyl ring of the ERL fragment. Additionally, the sulfonamide NH2 establishes two H-bonds with the carbonyl of the Gly169 backbone and the carbonyl oxygen of Glu171. In the case of 8d, in addition to the H-bond between the amide nitrogen of Asn71 and the proximal oxygen of the 2-methoxyethoxy tail of ERL, the two nitrogen atoms of the thiourea directly linked to benzenesulfonamide mediate two H-bonds with Asn64.
 | ||
| Fig. 2 Predicted 3D binding mode within hCA XII (green surface) of compounds (a) 8b (yellow sticks), (b) 8d (magenta sticks), and (c) 8c (cyan sticks). H-bond interacting residues are shown as green lines while H-bonding and cation–π interactions as yellow and green dotted lines, respectively. | ||
The arrangement of compound 8c (Fig. 2c), which possesses 10 times less activity, differs from that of the other compounds. It enters the active site with the thiourea group, whose two nitrogen atoms mediate two H-bonds with the oxygen of Thr199. Additionally, the first oxygen atom of the ethoxy linker forms an H-bond with the nitrogen of Gln89. At the edge of the active site, the sulfonamide forms an H-bond with the amine group of Lys3, while H-bonds between the nitrogen of the triazole and Lys69 and between Leu60 and the oxygen of the ERL tail are observed outside the active site.
Compounds 8c and 8d (Fig. 3b and c), exhibiting a potent inhibition of hCA IX, gain access to the active site of hCA IX, despite their differing geometries. The sulfonamide moiety of 8b or 8d (Fig. 3a and b) forms an H-bond with the zinc-coordinated water molecule, while the aromatic ring of 8d engages in a π–π interaction with the zinc-coordinated His94. The nitrogen of the thiourea moiety of the three compounds forms an H-bond with His68.
 | ||
| Fig. 3 Predicted 3D binding mode within hCA IX (pink surface) of compounds (a) 8b (yellow sticks), (b) 8d (magenta sticks), and (c) 8c (cyan sticks). H-bond interacting residues are shown as magenta lines, while H-bonding, π–π interactions, and cation–π interactions as yellow, cyan, and green dotted lines, respectively. | ||
In the region external to the active site, the quinazoline moiety of the ERL portion of 8c (Fig. 3c) engages in a cation–π interaction with Arg62, whereas the terminal methoxy of the 6-(2-methoxyethoxy) substituent of 8d forms an H-bond with Arg89. Furthermore, two oxygen atoms of the PEG linker of 8d form H-bonds with Arg64. As regards compound 8b, no strong interactions are observed outside the active site.
All three compounds are positioned on the surface of hCA I without entering the active site, justifying the lack of activity toward this isoform. However, they display distinct binding modes within the hCA II isoform. Interestingly, 8c remains outside the active site, exhibiting no evidence of bonding that could justify its observed activity. On the other hand, 8d (Fig. 4b) enters the active site with the ERL moiety, and the two methoxy groups form H-bonds with Asn62 and Gln92. The triazole moiety engages in a π–π interaction with Phe131, the nitrogen atom of the thiourea moiety forms an H-bond with Asp130, whereas the nitrogen and oxygen atoms of the sulfonamide moiety form H-bonds with Asp72 and the backbone of Phe131, respectively. Conversely, 8b (Fig. 4a) enters the active site with the ERL portion, and the only notable interactions outside the active site are H-bonds between the triazole nitrogen and Trp5, and between the oxygen and nitrogen atoms belonging to the sulfonamide moiety and Arg58 and Gly171, respectively. Furthermore, a π–π interaction between the triazole and Phe20 can be observed.
 | ||
| Fig. 4 Predicted 3D binding mode within hCA II (grey surface) of compounds (a) 8b (yellow sticks) and (b) 8d (magenta sticks). H-bond interacting residues are shown as green lines while H-bonding and π–π interactions as yellow and cyan dotted lines, respectively. | ||
In summary, compounds 8b, 8c, and 8d were proven to establish favorable interactions within and outside the active site of the hCAs IX and XII isoforms, thereby justifying their observed activity and selectivity.
Although the activity toward EGFR kinases has yet to be evaluated, we conducted a docking study on the EGFR kinase domain with the same compounds to evaluate a potential dual activity. Among the available protein in the Protein Data Bank, the PDB ID 3W2S was selected on the basis of the dimensions of the crystal ligand, which is comparable to those of compounds under investigation. The three compounds were observed to dock within the ATP binding site, forming hydrophobic interactions. Furthermore, compounds 8c and 8d (Fig. 5a and b) establish H-bonds between the sulfonamide and Lys875 (8c with Val876), while 8b interacts with Ala859. Lys745 forms an H-bond with the oxygen of the oxyethylene linker of 8d and with two oxygens of 8c. The aromatic ring of the benzenesulfonamide of 8c forms a cation–π interaction with Arg841, while the nitrogen atoms of the thiourea of 8b and 8d engage in H bond interactions with Gly857 and Asp855, respectively. These observations are consistent with the docking score and conserved key interactions, which indicate that ERL derivatives retain the structural features necessary for binding to the EGFR kinase domain.
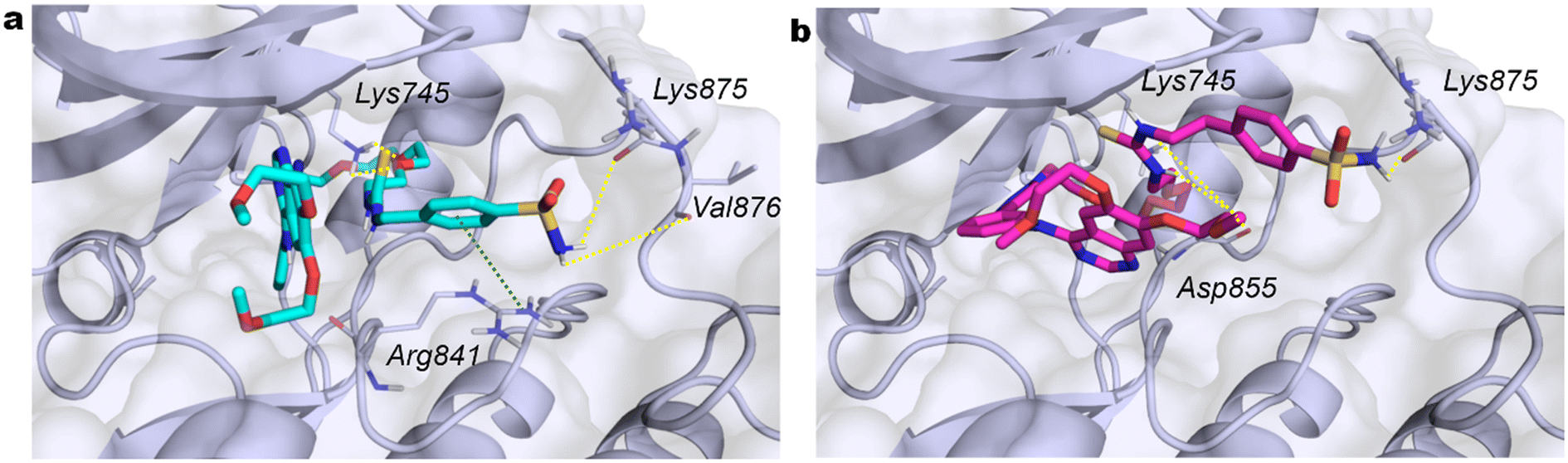 | ||
| Fig. 5 Predicted 3D binding mode within EGFR kinase domain (blue-grey surface and cartoon) of compounds (a) 8c (cyan sticks) and (b) 8d (magenta sticks). H-bond interacting residues are shown as grey lines while H-bonding and cation–π interactions as yellow and green dotted lines, respectively. | ||
3.5. Anti-proliferative effect on TNBC cells
TNBC is one of the most reported causes of death in the female population worldwide110 and one of its distinguishing features compared to other breast cancers is the overexpression of EGFR.111–114 Interestingly, this receptor was proven to be activated in several TNBC cell lines115 and its inhibition was demonstrated to arrest tumor growth and metastasis in TNBC xenograft models116,117 by controlling the initial progression of the disease.118 However, drug resistance to ERL has been recorded in some patients119–121 with an unclear mechanism.108 Also, ERL seems to inhibit the non-cancer stem cells, responsible for stemness and drug-resistance, or bulk TNBC cells.122Thus, we selected representative compounds of the new series (8b–d and 7a) to evaluate their anti-proliferative effect in vitro on the human TNBC cell line MDA-MB-231 that expresses both the tumor-associated hCA isoenzymes and EGFR.60,123 We also tested the known hCAs IX and XII inhibitor SLC-0111, and ERL, as reference compounds (Fig. 6). Cancer cells were treated with the compounds at increasing concentrations under hypoxic culture conditions, and the anti-proliferative readout was assessed after 72 hours by flow cytometry-based cell counting. As shown in Fig. 6, the reference compound SLC-0111 was found to be effective in reducing the proliferation of MDA-MB-231, with an IC50 of 23.4 μM. Interestingly, compounds 8b and 8d displayed the most promising anti-proliferative effect, with IC50 values ranging from 30 and 48.51 μM, respectively. A significantly lower effect on tumor cells was observed for compound 8c (IC50 ∼190 μM). Notably, treatment with ERL resulted only in a mild reduction of cell proliferation observed only at the higher 100 μM concentration tested, in accordance to our previous studies underlining its lack of efficacy on this cell line,122 thus supporting the increased effect obtained with the novel ERL-derived compounds.
 | ||
| Fig. 6 Cell proliferation of TNBC MDA-MB-231 cells treated for 72 hours in hypoxic conditions with compounds 8b–d and 7a, SLC-0111, or ERL. Cell count is referred to the untreated/control considered as 100%, mean ± SEM, and the IC50 are reported. | ||
4. Conclusions
In summary, a second series of seventeen new Pegylated ERL–CAI hybrids was synthesized through the Huisgen click chemistry reaction, bearing the benzenesulfonamide or the coumarin moiety as pharmacophores. Two different linker lengths were selected, i.e. 4- and 6-unit PEG spacers, to hold good flexibility and simultaneously explore the best length for coordination with the zinc ion within the CA active cleft, thus enhancing pharmacokinetic properties. Most of the derivatives were found to strongly inhibit hCAs IX and XII at low-to-high nanomolar concentration, showing isoform selectivity in a large number of compounds. Concerning hCA IX the general situation shows a remarkable preference for PEG-4 derivatives. Moreover, no differences between thiourea and urea emerged, while the sulfonamide moiety position showed a high impact in KI values (4-SO2NH2 > 3-SO2NH2). The same linker length preference is confirmed against hCA XII, in which the PEG-4 linker showed the best results. As well as previous isoform, thiourea is preferred to urea for hCA XII inhibition, with n = 0–1 > 2, and, similarly to hCA IX, 4-SO2NH2 improved inhibitory features with respect to 3-SO2NH2. Selectivity index (SI) values were then used to assess isoform selectivity: compounds 7–9 (PEG-4) showed a high preference for hCA XII (SI up to 790.25) compared to hCA I and II, while compounds 10–14 (PEG-6) had similar activity against hCA IX and II, with some selectivity over hCA I. The lower SI values (down to 345.52) for the latter group reflect their stronger inhibition of hCA IX and XII.Computational molecular docking studies on hCA isoforms rationalize the inhibitory activity and selectivity of compounds 8b, 8c, and 8d. Concerning hCA XII, 8b and 8d bind outside the active site, with key cation–π and hydrogen bonding interactions. Conversely, 8c enters the active site differently. For hCA IX all three compounds showed binding within the active site, with the sulfonamide group of 8b and 8d forming an H-bond with a zinc-coordinated water molecule, and the aromatic ring of 8c interacting with His94. Moreover, minimal interactions in the active sites of hCA I and II do support compounds' selectivity profile. Notably, docking study to the EGFR kinase domain indicated potential for dual activity. Thus, compounds 8b, 8c, and 8d docked within the ATP binding site and retained key binding features, indicating the conservation of structural elements needed for binding to the EGFR domain, as well. We further evaluated new synthesized hybrids compounds for TNBC treatment, using the MDA-MB-231 cell line which expresses EGFR and hCAs. Compounds 8b and 8d demonstrated strong anti-proliferative activity (IC50 30 and 48.51 μM respectively), thus comparable to the single target hCA inhibitor SLC-0111 (IC50 = 23.4 μM) and in contrast to the low efficacy observed for the EGFR inhibitor ERL. These results highlight the potential of such compounds for further development within TNBC therapy and more.
Data availability
The data supporting this article have been included as part of the ESI.† The following data are made available in the ESI:† selectivity index (SI) values for the first ERL-clicked derivatives (Table S1†); synthesis of compound 6 (Scheme S1†); procedure for the synthesis of compound 6); antiproliferative effect of the first series of ERL clicked derivatives (Fig. S1†); 1H and 13C NMR of representative compounds. The ESI† file contains the NMR spectra of the compounds reported in this article.Author contributions
Conceptualization, review and editing, S. C., I. D. A., A. A., F. C., and C. T. S.; methodology and formal analysis, G. R., S. F., M. F., G. B., M. L. M., R. S., C. C., R. R.; funding acquisition, F. C., R. R.; writing – original draft preparation, F. C., R. R., C. S., and I. D. A. All authors have read and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
FC and CTS are grateful to the European Union – Next Generation EU. National Recovery and Resilience Plan (NRRP) – M4C2 Investment 1.5 – Research Programme ECS_00000017 – CUP B83C22003920001 for financial support.References
- J. S. Brown, S. R. Amend, R. H. Austin, R. A. Gatenby, E. U. Hammarlund and K. J. Pienta, Updating the Definition of Cancer, Mol. Cancer Res., 2023, 21, 1142–1147, DOI:10.1158/1541-7786.MCR-23-0411.
- L. Magrassi, G. Pinton, S. Luzzi, S. Comincini, A. Scravaglieri and V. Gigliotti, et al., A New Vista of Aldehyde Dehydrogenase 1A3 (ALDH1A3): New Specific Inhibitors and Activity-Based Probes Targeting ALDH1A3 Dependent Pathways in Glioblastoma, Mesothelioma and Other Cancers, Cancers, 2024, 16, 2397, DOI:10.3390/cancers16132397.
- Global Cancer Observatory, https://gco.iarc.fr/en, (accessed January 6, 2025).
- Global cancer burden growing, amidst mounting need for services, https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services, (accessed January 6, 2025).
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra and D. K. Pandey, et al., Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2023, 10, 1367–1401, DOI:10.1016/j.gendis.2022.02.007.
- A. Zafar, M. J. Khan, J. Abu and A. Naeem, Revolutionizing cancer care strategies: immunotherapy, gene therapy, and molecular targeted therapy, Mol. Biol. Rep., 2024, 51, 219, DOI:10.1007/s11033-023-09096-8.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu and T. Yuan, et al., Small molecules in targeted cancer therapy: advances, challenges, and future perspectives, Signal Transduction Targeted Ther., 2021, 6, 1–48, DOI:10.1038/s41392-021-00572-w.
- G. Liu, T. Chen, X. Zhang, X. Ma and H. Shi, Small molecule inhibitors targeting the cancers, MedComm, 2022, 3, e181, DOI:10.1002/mco2.181.
- R. Li, X.-L. Ma, C. Gou and W. K. F. Tse, Editorial: Novel small molecules in targeted cancer therapy, Front. Pharmacol., 2023, 14, 1272523, DOI:10.3389/fphar.2023.1272523.
- A. Talevi, Multi-target pharmacology: possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective, Front. Pharmacol., 2015, 6, 205, DOI:10.3389/fphar.2015.00205.
- X. H. Makhoba, C. Viegas Jr., R. A. Mosa, F. P. D. Viegas and O. J. Pooe, Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases, Drug Des., Dev. Ther., 2020, 14, 3235–3249, DOI:10.2147/DDDT.S257494.
- A. Doostmohammadi, H. Jooya, K. Ghorbanian, S. Gohari and M. Dadashpour, Potentials and future perspectives of multi-target drugs in cancer treatment: the next generation anti-cancer agents, Cell Commun. Signaling, 2024, 22, 228, DOI:10.1186/s12964-024-01607-9.
- Q. Zhao and G. Huang, 7 - Anticancer Hybrids, in Des. Hybrid Mol. Drug Dev., ed. M. Decker, Elsevier, 2017, pp. 193–218, DOI:10.1016/B978-0-08-101011-2.00007-6.
- M. Szumilak, A. Wiktorowska-Owczarek and A. Stanczak, Hybrid Drugs-A Strategy for Overcoming Anticancer Drug Resistance?, Molecules, 2021, 26, 2601, DOI:10.3390/molecules26092601.
- Shalini and V. Kumar, Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on?, Expert Opin. Drug Discovery, 2021, 16(4), 335–363, DOI:10.1080/17460441.2021.1850686.
- A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, Design and synthesis of hybrid compounds as novel drugs and medicines, RSC Adv., 2022, 12, 19470–19484, 10.1039/d2ra03281c.
- A. I. Shagufta, Therapeutic significance of molecular hybrids for breast cancer research and treatment, RSC Med. Chem., 2023, 14, 218–238, 10.1039/D2MD00356B.
- X. H. Ma, Z. Shi, C. Tan, Y. Jiang, M. L. Go and B. C. Low, et al., In-silico approaches to multi-target drug discovery : computer aided multi-target drug design, multi-target virtual screening, Pharm. Res., 2010, 27, 739–749, DOI:10.1007/s11095-010-0065-2.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, H. Paliwal and S. Thareja, et al., Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids, Pharmaceuticals, 2022, 15, 1071, DOI:10.3390/ph15091071.
- H. M. Sampath Kumar, L. Herrmann and S. B. Tsogoeva, Structural hybridization as a facile approach to new drug candidates, Bioorg. Med. Chem. Lett., 2020, 30, 127514, DOI:10.1016/j.bmcl.2020.127514.
- G. Zhang, L. Fang, L. Zhu, D. Sun and P. G. Wang, Syntheses and biological activity of bisdaunorubicins, Bioorg. Med. Chem., 2006, 14, 426–434, DOI:10.1016/j.bmc.2005.08.014.
- N. Kerru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Recent advances (2015–2016) in anticancer hybrids, Eur. J. Med. Chem., 2017, 142, 179–212, DOI:10.1016/j.ejmech.2017.07.033.
- A. A. Moiseeva, O. I. Artyushin, L. V. Anikina and V. K. Brel, Synthesis and antitumor activity of daunorubicin conjugates with of 3,4-methylendioxybenzaldehyde, Bioorg. Med. Chem. Lett., 2019, 29, 126617, DOI:10.1016/j.bmcl.2019.08.021.
- L.-S. Feng, W.-Q. Su, J.-B. Cheng, T. Xiao, H.-Z. Li and D.-A. Chen, et al., Benzimidazole hybrids as anticancer drugs: An updated review on anticancer properties, structure–activity relationship, and mechanisms of action (2019–2021), Arch. Pharm., 2022, 355, 2200051, DOI:10.1002/ardp.202200051.
- T. Beckers, S. Mahboobi, A. Sellmer, M. Winkler, E. Eichhorn and H. Pongratz, et al., Chimerically designed HDAC- and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases, MedChemComm, 2012, 3, 829–835, 10.1039/C2MD00317A.
- Y. Zhang, M. D. Tortorella, J. Liao, X. Qin, T. Chen and J. Luo, et al., Synthesis and Evaluation of Novel Erlotinib–NSAID Conjugates as More Comprehensive Anticancer Agents, ACS Med. Chem. Lett., 2015, 6, 1086–1090, DOI:10.1021/acsmedchemlett.5b00286.
- Y. Wei, D. C. Poon, R. Fei, A. S. M. Lam, S. C. F. Au-Yeung and T. KKW, A platinum-based hybrid drug design approach to circumvent acquired resistance to molecular targeted tyrosine kinase inhibitors, Sci. Rep., 2016, 6, 25363, DOI:10.1038/srep25363.
- M. M. Alam, A. H. E. Hassan, K. W. Lee, M. C. Cho, J. S. Yang and J. Song, et al., Design, synthesis and cytotoxicity of chimeric erlotinib-alkylphospholipid hybrids, Bioorg. Chem., 2019, 84, 51–62, DOI:10.1016/j.bioorg.2018.11.021.
- E. Ortega, A. Zamora, U. Basu, P. Lippmann, V. Rodríguez and C. Janiak, et al., An Erlotinib gold(I) conjugate for combating triple-negative breast cancer, J. Inorg. Biochem., 2020, 203, 110910, DOI:10.1016/j.jinorgbio.2019.110910.
- P. Biegański, M. Godel, C. Riganti, D. F. Kawano, J. Kopecka and K. Kowalski, Click ferrocenyl-erlotinib conjugates active against erlotinib-resistant non-small cell lung cancer cells in vitro, Bioorg. Chem., 2022, 119, 105514, DOI:10.1016/j.bioorg.2021.105514.
- J. Murányi, C. Duró, B. Gurbi, I. Móra, A. Varga and K. Németh, et al., Novel Erlotinib–Chalcone Hybrids Diminish Resistance in Head and Neck Cancer by Inducing Multiple Cell Death Mechanisms, Int. J. Mol. Sci., 2023, 24, 3456, DOI:10.3390/ijms24043456.
- P. Biegański, M. Gazecka, R. Nowak, A. Gorski, N. Dutkiewicz and D. F. Kawano, et al., Organometallic–Erlotinib Conjugates Active against Lung Cancer Cells and as Emerging Virus Entry Inhibitors, Organometallics, 2024, 43, 2505–2519, DOI:10.1021/acs.organomet.4c00145.
- D. Singh, B. K. Attri, R. K. Gill and J. Bariwal, Review on EGFR Inhibitors: Critical Updates, Mini-Rev. Med. Chem., 2016, 16, 1134–1166, DOI:10.2174/1389557516666160321114917.
- M. Steins, M. Thomas and M. Geißler, Erlotinib, in Small Mol. Oncol., ed. U. M. Martens, Springer International Publishing, Cham, 2018, pp. 1–17, DOI:10.1007/978-3-319-91442-8_1.
- J. D. Minna and J. Dowell, Erlotinib hydrochloride, Nat. Rev. Drug Discovery, 2005, S14–S15, DOI:10.1038/nrd1612.
- A. F. Gazdar, Personalized medicine and inhibition of EGFR signaling in lung cancer, N. Engl. J. Med., 2009, 361, 1018–1020, DOI:10.1056/NEJMe0905763.
- U. Baumgartner, F. Berger, A. Hashemi Gheinani, S. S. Burgener, K. Monastyrskaya and E. Vassella, miR-19b enhances proliferation and apoptosis resistance via the EGFR signaling pathway by targeting PP2A and BIM in non-small cell lung cancer, Mol. Cancer, 2018, 17, 44, DOI:10.1186/s12943-018-0781-5.
- E. Levantini, G. Maroni, M. Del Re and D. G. Tenen, EGFR signaling pathway as therapeutic target in human cancers, Semin. Cancer Biol., 2022, 85, 253–275, DOI:10.1016/j.semcancer.2022.04.002.
- J. R. Sainsbury, J. R. Farndon, G. K. Needham, A. J. Malcolm and A. L. Harris, Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer, Lancet, 1987, 1, 1398–1402, DOI:10.1016/s0140-6736(87)90593-9.
- D. S. Salomon, R. Brandt, F. Ciardiello and N. Normanno, Epidermal growth factor-related peptides and their receptors in human malignancies, Crit. Rev. Oncol. Hematol., 1995, 19, 183–232, DOI:10.1016/1040-8428(94)00144-i.
- M. L. Burness, T. A. Grushko and O. I. Olopade, Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker?, Cancer J., 2010, 16, 23–32, DOI:10.1097/PPO.0b013e3181d24fc1.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor *, J. Biol. Chem., 2002, 277, 46265–46272, DOI:10.1074/jbc.M207135200.
- J. H. Park, Y. Liu, M. A. Lemmon and R. Radhakrishnan, Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain, Biochem. J., 2012, 448, 417–423, DOI:10.1042/BJ20121513.
- G. Sun, L. Mao, W. Deng, S. Xu, J. Zhao and J. Yang, et al., Discovery of a Series of 1,2,3-Triazole-Containing Erlotinib Derivatives With Potent Anti-Tumor Activities Against Non-Small Cell Lung Cancer, Front. Chem., 2022, 9, 789030, DOI:10.3389/fchem.2021.789030.
- L. Mao, Z.-Z. Wang, Q. Wu, X. Chen, J.-X. Yang and X. Wang, et al., Design, Synthesis, and Antitumor Activity of Erlotinib Derivatives, Front. Pharmacol., 2022, 13, 849364, DOI:10.3389/fphar.2022.849364.
- P. Deng, G. Sun, J. Zhao, K. Yao, M. Yuan and L. Peng, et al., Synthesis and Antitumor Activity of Erlotinib Derivatives Linked With 1,2,3-Triazole, Front. Pharmacol., 2022, 12, 793905, DOI:10.3389/fphar.2021.793905.
- Y. Gao, H. Zhang, Y. Zhang, T. Lv, L. Zhang and Z. Li, et al., Erlotinib-Guided Self-Assembled Trifunctional Click Nanotheranostics for Distinguishing Druggable Mutations and Synergistic Therapy of Nonsmall Cell Lung Cancer, Mol. Pharmaceutics, 2018, 15, 5146–5161, DOI:10.1021/acs.molpharmaceut.8b00561.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, The 1,2,3-triazole ring as a bioisostere in medicinal chemistry, Drug Discovery Today, 2017, 22, 1572–1581, DOI:10.1016/j.drudis.2017.05.014.
- R. Khandelwal, M. Vasava, R. B. Abhirami and M. Karsharma, Recent advances in triazole synthesis via click chemistry and their pharmacological applications: A review, Bioorg. Med. Chem. Lett., 2024, 112, 129927, DOI:10.1016/j.bmcl.2024.129927.
- C. T. Supuran, A simple yet multifaceted 90 years old, evergreen enzyme: Carbonic anhydrase, its inhibition and activation, Bioorg. Med. Chem. Lett., 2023, 93, 129411, DOI:10.1016/j.bmcl.2023.129411.
- C. Baroni, I. D'Agostino, G. Renzi, J. T. Kilbile, Y. Tamboli and M. Ferraroni, et al., Lasamide, a Potent Human Carbonic Anhydrase Inhibitor from the Market: Inhibition Profiling and Crystallographic Studies, ACS Med. Chem. Lett., 2024, 15, 1749–1755, DOI:10.1021/acsmedchemlett.4c00341.
- I. D'Agostino, A. Bonardi, M. Ferraroni, P. Gratteri, A. Angeli and C. T. Supuran, Exploring the Polypharmacological Potential of PCI-27483: A Selective Inhibitor of Carbonic Anhydrases IX and XII, ACS Med. Chem. Lett., 2024, 15, 2042–2045, DOI:10.1021/acsmedchemlett.4c00443.
- C. B. Mishra, M. Tiwari and C. T. Supuran, Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today?, Med. Res. Rev., 2020, 40, 2485–2565, DOI:10.1002/med.21713.
- K. D'Ambrosio, A. Di Fiore, V. Alterio, E. Langella, S. M. Monti and C. T. Supuran, et al., Multiple Binding Modes of Inhibitors to Human Carbonic Anhydrases: An Update on the Design of Isoform-Specific Modulators of Activity, Chem. Rev., 2025, 125(1), 150–222, DOI:10.1021/acs.chemrev.4c00278.
- T. C. Denner, A. Angeli, M. Ferraroni, C. T. Supuran and R. Csuk, Ureidobenzenesulfonamides as Selective Carbonic Anhydrase I, IX, and XII Inhibitors, Molecules, 2023, 28, 7782, DOI:10.3390/molecules28237782.
- C. T. Supuran, Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas, Expert Opin. Ther. Pat., 2018, 28, 709–712, DOI:10.1080/13543776.2018.1523897.
- G. De Simone and C. T. Supuran, Anticancer drugs: where are we now?, Expert Opin. Ther. Pat., 2024, 34, 525–527, DOI:10.1080/13543776.2024.2353625.
- J. T. Kilbile, S. B. Sapkal, G. Renzi, I. D'Agostino, M. Boudjelal and Y. Tamboli, et al., Lasamide Containing Sulfonylpiperazines as Effective Agents for the Management of Glaucoma Associated Symptoms, ChemMedChem, 2024, 19, e202400601, DOI:10.1002/cmdc.202400601.
- R. Ronca and C. T. Supuran, Carbonic anhydrase IX: An atypical target for innovative therapies in cancer, Biochim. Biophys. Acta, Rev. Cancer, 2024, 1879, 189120, DOI:10.1016/j.bbcan.2024.189120.
- H. Aslan, G. Renzi, A. Angeli, I. D'Agostino, R. Ronca and M. Massardi, et al., Benzenesulfonamide Decorated Dihydropyrimidin(thi)ones: Carbonic Anhydrase Profiling and Antiproliferative Activity, RSC Med. Chem., 2024, 15, 1929–1941, 10.1039/D4MD00101J.
- M. Abdoli, A. Bonardi, C. T. Supuran and R. Žalubovskis, Synthesis and Carbonic Anhydrase I, II, IX, and XII Inhibition Studies with a Series of Cyclic Sulfonyl Guanidines, ChemMedChem, 2024, 19, e202400197, DOI:10.1002/cmdc.202400197.
- A. Angeli, I. Chelli, L. Lucarini, S. Sgambellone, S. Marri and S. Villano, et al., Novel Carbonic Anhydrase Inhibitors with Dual-Tail Core Sulfonamide Show Potent and Lasting Effects for Glaucoma Therapy, J. Med. Chem., 2024, 67, 3066–3089, DOI:10.1021/acs.jmedchem.3c02254.
- A. Angeli, A. Petrou, V. G. Kartsev, A. Zubenko, L. N. Divaeva and V. Chekrisheva, et al., Phthalazine Sulfonamide Derivatives as Carbonic Anhydrase Inhibitors. Synthesis, Biological and in silico Evaluation, ChemMedChem, 2024, e202400147, DOI:10.1002/cmdc.202400147.
- M. Ali, M. Bozdag, U. Farooq, A. Angeli, F. Carta and P. Berto, et al., Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases, Int. J. Mol. Sci., 2020, 21, 2560, DOI:10.3390/ijms21072560.
- M. Ali, A. Angeli, M. Bozdag, F. Carta, C. Capasso and U. Farooq, et al., Benzylaminoethylureido-Tailed Benzenesulfonamides Show Potent Inhibitory Activity against Bacterial Carbonic Anhydrases, ChemMedChem, 2020, 15, 2444–2447, DOI:10.1002/cmdc.202000680.
- A. Bonardi, A. Nocentini, S. Giovannuzzi, N. Paoletti, A. Ammara and S. Bua, et al., Development of Penicillin-Based Carbonic Anhydrase Inhibitors Targeting Multidrug-Resistant Neisseria gonorrhoeae, J. Med. Chem., 2024, 67, 9613–9627, DOI:10.1021/acs.jmedchem.4c00740.
- M. Bozdag, A. M. Alafeefy, F. Carta, M. Ceruso, A.-M. S. Al-Tamimi and A. A. Al-Kahtani, et al., Synthesis 4-[2-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-ethyl]-benzenesulfonamides with subnanomolar carbonic anhydrase II and XII inhibitory properties, Bioorg. Med. Chem., 2016, 24, 4100–4107, DOI:10.1016/j.bmc.2016.06.052.
- A. Kumar, K. Siwach, C. T. Supuran and P. K. Sharma, A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors, Bioorg. Chem., 2022, 126, 105920, DOI:10.1016/j.bioorg.2022.105920.
- A. Bonardi, A. Nocentini, S. Giovannuzzi, N. Paoletti, A. Ammara and S. Bua, et al., Development of Penicillin-Based Carbonic Anhydrase Inhibitors Targeting Multidrug-Resistant Neisseria gonorrhoeae, J. Med. Chem., 2024, 67, 9613–9627, DOI:10.1021/acs.jmedchem.4c00740.
- F. Carta, A. Akdemir, A. Scozzafava, E. Masini and C. T. Supuran, Xanthates and Trithiocarbonates Strongly Inhibit Carbonic Anhydrases and Show Antiglaucoma Effects in Vivo, J. Med. Chem., 2013, 56, 4691–4700, DOI:10.1021/jm400414j.
- B. Marinacci, I. D'Agostino, A. Angeli, S. Carradori, F. Melfi and R. Grande, et al., Inhibition of Pseudomonas aeruginosa Carbonic Anhydrases, Exploring Ciprofloxacin Functionalization Toward New Antibacterial Agents: An In-Depth Multidisciplinary Study, J. Med. Chem., 2024, 67(21), 19077–19102, DOI:10.1021/acs.jmedchem.4c01555.
- J. B. Baell and J. W. M. Nissink, Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations, ACS Chem. Biol., 2018, 13, 36–44, DOI:10.1021/acschembio.7b00903.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- L. J. I. Balestri, I. D'Agostino, E. Rango, C. Vagaggini, R. Marchitiello and M. Mariotti, et al., Focused library of phenyl-fused macrocyclic amidinoureas as antifungal agents, Mol. Diversity, 2022, 26, 3399–3409, DOI:10.1007/s11030-022-10388-7.
- I. D'Agostino, G. E. Mathew, P. Angelini, R. Venanzoni, G. Angeles Flores and A. Angeli, et al., Biological investigation of N-methyl thiosemicarbazones as antimicrobial agents and bacterial carbonic anhydrases inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37, 986–993, DOI:10.1080/14756366.2022.2055009.
- A. Redij, S. Carradori, A. Petreni, C. T. Supuran and M. P. Toraskar, Coumarin-pyrazoline Hybrids as Selective Inhibitors of the Tumor-associated Carbonic Anhydrase IX and XII, Anti-Cancer Agents Med. Chem., 2023, 23, 1217–1223, DOI:10.2174/1871520623666230220162506.
- Schrödinger Release 2024–1, Maestro, Glide, Protein Preparation Wizard, Epik, MacroModel, Prime, New York (NY), Schrödinger, LLC, 2024 Search PubMed.
- J. R. Greenwood, D. Calkins, A. P. Sullivan and J. C. Shelley, Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution, J. Comput.-Aided Mol. Des., 2010, 24, 591–604, DOI:10.1007/s10822-010-9349-1.
- M. Bozdag, M. Ferraroni, C. Ward, F. Carta, S. Bua and A. Angeli, et al., Carbonic anhydrase inhibitors based on sorafenib scaffold: Design, synthesis, crystallographic investigation and effects on primary breast cancer cells, Eur. J. Med. Chem., 2019, 182, 111600, DOI:10.1016/j.ejmech.2019.111600.
- J. Leitans, A. Kazaks, J. Bogans, C. T. Supuran, I. Akopjana and J. Ivanova, et al., Structural Basis of Saccharin Derivative Inhibition of Carbonic Anhydrase IX, ChemMedChem, 2023, 18, e202300454, DOI:10.1002/cmdc.202300454.
- C. A. Behnke, I. Le Trong, J. W. Godden, E. A. Merritt, D. C. Teller and J. Bajorath, et al., Atomic resolution studies of carbonic anhydrase II, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 616–627, DOI:10.1107/S0907444910006554.
- E. Čapkauskaitė, V. Linkuvienė, A. Smirnov, G. Milinavičiūtė, D. D. Timm and A. Kasiliauskaitė, et al., Combinatorial Design of Isoform-Selective N-Alkylated Benzimidazole-Based Inhibitors of Carbonic Anhydrases, ChemistrySelect, 2017, 2, 5360–5371, DOI:10.1002/slct.201700531.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki and D. R. Cary, et al., Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors, ACS Med. Chem. Lett., 2013, 4, 201–205, DOI:10.1021/ml300327z.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic and D. T. Mainz, et al., Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy, J. Med. Chem., 2004, 47, 1739–1749, DOI:10.1021/jm0306430.
- G. Benito, I. D'Agostino, S. Carradori, M. Fantacuzzi, M. Agamennone and V. Puca, et al., Erlotinib-containing benzenesulfonamides as anti-Helicobacter pylori agents through carbonic anhydrase inhibition, Future Med. Chem., 2023, 15, 1865–1883, DOI:10.4155/fmc-2023-0208.
- A. Gumus, I. D'Agostino, V. Puca, V. Crocetta, S. Carradori and L. Cutarella, et al., Cyclization of acyl thiosemicarbazides led to new Helicobacter pylori α-carbonic anhydrase inhibitors, Arch. Pharm., 2024, 357, e2400548, DOI:10.1002/ardp.202400548.
- A. Doléans-Jordheim, E. Bergeron, F. Bereyziat, S. Ben-Larbi, O. Dumitrescu and M.-A. Mazoyer, et al., Zidovudine (AZT) has a bactericidal effect on enterobacteria and induces genetic modifications in resistant strains, Eur. J. Clin. Microbiol. Infect. Dis., 2011, 30, 1249–1256, DOI:10.1007/s10096-011-1220-3.
- L. P. Elwell, R. Ferone, G. A. Freeman, J. A. Fyfe, J. A. Hill and P. H. Ray, et al., Antibacterial activity and mechanism of action of 3′-azido-3′-deoxythymidine (BW A509U), Antimicrob. Agents Chemother., 1987, 31, 274–280, DOI:10.1128/AAC.31.2.274.
- B. L. Bernardoni, C. La Motta, S. Carradori and I. D'Agostino, Helicobacter pylori CAs inhibition, Enzymes, 2024, 55, 213–241, DOI:10.1016/bs.enz.2024.05.013.
- C. T. Supuran, Novel carbonic anhydrase inhibitors for the treatment of Helicobacter pylori infection, Expert Opin. Invest. Drugs, 2024, 1–10, DOI:10.1080/13543784.2024.2334714.
- J. Kim, N. Kim, J. H. Park, H. Chang, J. Y. Kim and D. H. Lee, et al., The Effect of Helicobacter pylori on Epidermal Growth Factor Receptor-Induced Signal Transduction and the Preventive Effect of Celecoxib in Gastric Cancer Cells, Gut Liver, 2013, 7, 552–559, DOI:10.5009/gnl.2013.7.5.552.
- B. E. Chichirau, S. Diechler, G. Posselt and S. Wessler, Tyrosine Kinases in Helicobacter pylori Infections and Gastric Cancer, Toxins, 2019, 11, 591, DOI:10.3390/toxins11100591.
- F. Liguori, S. Carradori, R. Ronca, S. Rezzola, S. Filiberti and F. Carta, et al., Benzenesulfonamides with different rigidity-conferring linkers as carbonic anhydrase inhibitors: an insight into the antiproliferative effect on glioblastoma, pancreatic, and breast cancer cells, J. Enzyme Inhib. Med. Chem., 2022, 37, 1857–1869, DOI:10.1080/14756366.2022.2091557.
- V. Ciccone, A. Filippelli, A. Angeli, C. T. Supuran and L. Morbidelli, Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness, Int. J. Mol. Sci., 2020, 21, 2983, DOI:10.3390/ijms21082983.
- S. Ali, B. F. El-Rayes, F. H. Sarkar and P. A. Philip, Simultaneous targeting of the epidermal growth factor receptor and cyclooxygenase-2 pathways for pancreatic cancer therapy, Mol. Cancer Ther., 2005, 4, 1943–1951, DOI:10.1158/1535-7163.MCT-05-0065.
- C. Deng, J. Xiong, X. Gu, X. Chen, S. Wu and Z. Wang, et al., Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro, Onco Targets Ther, 2017, 8, 38568–38580, DOI:10.18632/oncotarget.16930.
- C. Vagaggini, D. Petroni, I. D'Agostino, F. Poggialini, C. Cavallini and A. Cianciusi, et al., Early investigation of a novel SI306 theranostic prodrug for glioblastoma treatment, Drug Dev. Res., 2024, 85, e22158, DOI:10.1002/ddr.22158.
- W. Li, P. Zhan, E. De Clercq, H. Lou and X. Liu, Current drug research on PEGylation with small molecular agents, Prog. Polym. Sci., 2013, 38, 421–444, DOI:10.1016/j.progpolymsci.2012.07.006.
- F. M. Veronese and A. Mero, The impact of PEGylation on biological therapies, BioDrugs, 2008, 22, 315–329, DOI:10.2165/00063030-200822050-00004.
- R. B. Greenwald, PEG drugs: an overview, J. Control. Release, 2001, 74, 159–171, DOI:10.1016/s0168-3659(01)00331-5.
- W. D. Chey, L. Webster, M. Sostek, J. Lappalainen, P. N. Barker and J. Tack, Naloxegol for Opioid-Induced Constipation in Patients with Noncancer Pain, N. Engl. J. Med., 2014, 370, 2387–2396, DOI:10.1056/NEJMoa1310246.
- J. Liu, P. Zahedi, F. Zeng and C. Allen, Nano-sized assemblies of a PEG-docetaxel conjugate as a formulation strategy for docetaxel, J. Pharm. Sci., 2008, 97, 3274–3290, DOI:10.1002/jps.21245.
- R. Omar, Y. L. Bardoogo, E. Corem-Salkmon and B. Mizrahi, Amphiphilic star PEG-Camptothecin conjugates for intracellular targeting, J. Control. Release, 2017, 257, 76–83, DOI:10.1016/j.jconrel.2016.09.025.
- E. J. Park, J. Choi, K. C. Lee and D. H. Na, Emerging PEGylated non-biologic drugs, Expert Opin. Emerging Drugs, 2019, 24, 107–119, DOI:10.1080/14728214.2019.1604684.
- D. Bebbington, C. E. Dawson, S. Gaur and J. Spencer, Prodrug and covalent linker strategies for the solubilization of dual-Action antioxidants/Iron chelators, Bioorg. Med. Chem. Lett., 2002, 12, 3297–3300, DOI:10.1016/S0960-894X(02)00698-4.
- R. I. Troup, C. Fallan and M. G. J. Baud, Current strategies for the design of PROTAC linkers: a critical review, Explor. Targeted Anti-Tumor Ther., 2020, 1, 273–312, DOI:10.37349/etat.2020.00018.
- Q. Li, W. Li, K. Xu, Y. Xing, H. Shi and Z. Jing, et al., PEG Linker Improves Antitumor Efficacy and Safety of Affibody-Based Drug Conjugates, Int. J. Mol. Sci., 2021, 22, 1540, DOI:10.3390/ijms22041540.
- Y. Ma, Z. Fang, H. Zhang, Y. Qi, Y. Mao and J. Zheng, PDZK1 suppresses TNBC development and sensitizes TNBC cells to erlotinib via the EGFR pathway, Cell Death Dis., 2024, 15, 1–14, DOI:10.1038/s41419-024-06502-2.
- R. G. Khalifah, The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C, J. Biol. Chem., 1971, 246, 2561–2573 CrossRef CAS PubMed.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, Triple-negative breast cancer, N. Engl. J. Med., 2010, 363, 1938–1948, DOI:10.1056/NEJMra1001389.
- O. Gluz, C. Liedtke, N. Gottschalk, L. Pusztai, U. Nitz and N. Harbeck, Triple-negative breast cancer--current status and future directions, Ann. Oncol., 2009, 20, 1913–1927, DOI:10.1093/annonc/mdp492.
- S. M. Rodríguez-Pinilla, D. Sarrió, E. Honrado, G. Moreno-Bueno, D. Hardisson and F. Calero, et al., Vimentin and laminin expression is associated with basal-like phenotype in both sporadic and BRCA1-associated breast carcinomas, J. Clin. Pathol., 2007, 60, 1006–1012, DOI:10.1136/jcp.2006.042143.
- D. Sarrió, S. M. Rodriguez-Pinilla, D. Hardisson, A. Cano, G. Moreno-Bueno and J. Palacios, Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype, Cancer Res., 2008, 68, 989–997, DOI:10.1158/0008-5472.CAN-07-2017.
- Z. Liu, K. He, Q. Ma, Q. Yu, C. Liu and I. Ndege, et al., Autophagy inhibitor facilitates gefitinib sensitivity in vitro and in vivo by activating mitochondrial apoptosis in triple negative breast cancer, PLoS One, 2017, 12, e0177694, DOI:10.1371/journal.pone.0177694.
- F. Yamasaki, D. Zhang, C. Bartholomeusz, T. Sudo, G. N. Hortobagyi and K. Kurisu, et al., Sensitivity of breast cancer cells to erlotinib depends on cyclin-dependent kinase 2 activity, Mol. Cancer Ther., 2007, 6, 2168–2177, DOI:10.1158/1535-7163.MCT-06-0514.
- K. Fenn, M. Maurer, S. M. Lee, K. D. Crew, M. S. Trivedi and M. K. Accordino, et al., Phase 1 Study of Erlotinib and Metformin in Metastatic Triple-Negative Breast Cancer, Clin. Breast Cancer, 2020, 20, 80–86, DOI:10.1016/j.clbc.2019.08.004.
- N. T. Ueno and D. Zhang, Targeting EGFR in Triple Negative Breast Cancer, J. Cancer, 2011, 2, 324–328, DOI:10.7150/jca.2.324.
- C. Lefebvre, S. Pellizzari, V. Bhat, K. Jurcic, D. W. Litchfield and A. L. Allan, Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines, Biomedicines, 2023, 11, 2406, DOI:10.3390/biomedicines11092406.
- M. N. Dickler, H. S. Rugo, C. A. Eberle, E. Brogi, J. F. Caravelli and K. S. Panageas, et al., A Phase II Trial of Erlotinib in Combination with Bevacizumab in Patients with Metastatic Breast Cancer, Clin. Cancer Res., 2008, 14, 7878–7883, DOI:10.1158/1078-0432.CCR-08-0141.
- S. Ricciardi, S. Tomao and F. de Marinis, Efficacy and safety of erlotinib in the treatment of metastatic non-small-cell lung cancer, Lung Cancer: Targets Ther., 2010, 2, 1–9, DOI:10.2147/LCTT.S10167.
- S. M. Tolaney, H. Nechushtan, I.-G. Ron, P. Schöffski, A. Awada and C. A. Yasenchak, et al., Cabozantinib for metastatic breast carcinoma: results of a phase II placebo-controlled randomized discontinuation study, Breast Cancer Res. Treat., 2016, 160, 305–312, DOI:10.1007/s10549-016-4001-y.
- B. Bao, C. Mitrea, P. Wijesinghe, L. Marchetti, E. Girsch and R. L. Farr, et al., Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity, Sci. Rep., 2017, 7, 44125, DOI:10.1038/srep44125.
- E. Schiavon, S. Rezzola, E. Filippi, M. Turati, S. Parrasia and S. Bernardotto, et al., A novel mertansine conjugate for acid-reversible targeted drug delivery validated through the Avidin-Nucleic-Acid-NanoASsembly platform, Nanomedicine, 2024, 62, 102784, DOI:10.1016/j.nano.2024.102784.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00109a |
| This journal is © The Royal Society of Chemistry 2025 |