
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNavigating the complexities of drug development for metallo-β-lactamase inhibitors
Nakita
Reddy
 ab,
Alessandra Moraes
Balieiro
c,
José Rogério A.
Silva
ac,
Christiaan A.
Gouws
a,
Awelani
Mutshembele
b,
Per I.
Arvidsson
ad,
Hendrik G.
Kruger
a,
Thavendran
Govender
*e and
Tricia
Naicker
*a
ab,
Alessandra Moraes
Balieiro
c,
José Rogério A.
Silva
ac,
Christiaan A.
Gouws
a,
Awelani
Mutshembele
b,
Per I.
Arvidsson
ad,
Hendrik G.
Kruger
a,
Thavendran
Govender
*e and
Tricia
Naicker
*a
aCatalysis and Peptide Research Unit, University of KwaZulu-Natal, Durban 4000, South Africa. E-mail: naickert1@ukzn.ac.za
bOffice of AIDS and TB, South African Medical Research Council, Pretoria 0084, South Africa
cLaboratory of Computer Modeling of Molecular Biosystems, Federal University of Pará, Belém 66075-110, Pará, Brazil
dScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, 17177, Sweden
eDepartment of Chemistry, University of Zululand, Private Bag X1001, KwaDlangezwa 3886, South Africa. E-mail: govenderthav@icloud.com
First published on 27th May 2025
Abstract
The rising antibiotic resistance rates, especially among carbapenem-resistant Enterobacterales with metallo-β-lactamases (MBLs), highlight the urgent need for effective MBL inhibitors (MBLIs). Navigating the complexities of drug development for MBLIs requires addressing the significant challenges that have hindered its progress. Despite numerous efforts in pre-clinical development, the lack of standardized approaches has led to disparities, stalling the translation of potential MBLIs from research into clinical use. Alarmingly, there is only one metallo-β-lactamase inhibitory candidate in the pre-registration phase of development. This review highlights the need for a global consensus on key aspects of MBLI development, including standardized in vitro testing, refined animal models, harmonized toxicity assessments, consistent pharmacokinetic data, and uniform in silico methods. It also proposes solutions to these challenges, aiming to bridge the gap between research and clinical application.
 Nakita Reddy | Dr. Nakita Reddy (UKZN/SAMRC) and Dr. Christiaan A. (Arno) Gouws (UP/NuMeRI) are postdoctoral researchers tackling critical healthcare challenges through innovative pharmaceutical research. Nakita's work targets antimicrobial resistance by evaluating novel drug candidates and exploring resistance mechanisms in WHO priority pathogens. Arno focuses on radiopharmaceutical development for PET imaging, designing tracers for infection and cancer diagnostics. Both obtained their PhDs from the Catalysis and Peptide Research Unit at University of KwaZulu-Natal in 2023 and 2024, respectively, and are passionate about translating cutting-edge science into impactful health solutions across Sub-Saharan Africa. |
 Alessandra Moraes Balieiro | Alessandra Moraes Balieiro holds a MSc in Chemistry and is currently a PhD candidate at the Federal University of Pará (UFPA, Brazil). Her research integrates molecular dynamics, docking, QM/MM and free energy calculations to investigate the structural and energetic mechanisms underlying antibiotic multiresistance, with a particular focus on metallo-β-lactamase enzymes. |
 José Rogério A. Silva | José Rogério A. Silva holds a PhD in Chemistry from the Federal University of Pará (UFPA, Brazil), with doctoral internships at the University of KwaZulu-Natal (UKZN, South Africa) and the University of Florida (USA). He is currently an Associate Professor at UFPA and an Honorary Research Fellow at UKZN. His research focuses on computational enzymology and drug design, applying molecular docking, molecular dynamics, QM/MM and free energy methods to investigate enzyme mechanisms, especially those linked to antimicrobial resistance. |
 Awelani Mutshembele | Dr Awelani Mutshembele is a Specialist Scientist at the South African Medical Research Council, in the Office of AIDS and TB. She leads the South African arm of the NIHR Global Health Research Programme and contributes to BRICS STI and AMR research initiatives. As a lead scientist in the East Africa AMR-STOP programme, she investigates antibiotic resistance and virulence genes in E. coli and K. pneumoniae across six African countries. She also supports One Health AMR surveillance in the Lake Victoria Basin. Awelani's expertise includes molecular surveillance, AI for AMR prediction, and strengthening health systems through capacity development and community engagement. |
 Per I. Arvidsson | Prof. Per I. Arvidsson is the founding Director of the national Swedish Drug Discovery & Development Platform at the Science for Life Laboratory (SciLifeLab). SciLifeLab DDD supports academic researchers with drug discovery infrastructure and expertise for small molecules, biologics, oligonucleotides, and new modalities. Since 2014, this academic infrastructure has evaluated >500 academic ideas and matured prioritized programs into 18 commercial exits, of which 5 programs have reached clinical trials. Before recruitment by SciLifeLab & Karolinska Institutet in 2013, Prof. Arvidsson held various roles at AstraZeneca as well as academic appointments at Uppsala University and the University of KwaZulu-Natal. |
 Tricia Naicker | Profs. Tricia Naicker, Thavendran Govender and Gert (H. G.) Kruger leads cutting-edge drug discovery efforts at the Catalysis and Peptide Research Unit. Combining strengths in organic synthesis, analytical, and computational chemistry, their interdisciplinary team pioneer's novel therapeutics targeting pressing global health challenges. With international training and decades of collective expertise, they have built a dynamic research environment that mentors emerging scientists while translating innovation into impactful solutions. Their collaborative efforts continue to shape the future of medicinal chemistry in Africa and beyond. |
1. Introduction
Antimicrobial resistance (AMR) has garnered significant attention in recent years, mainly due to the inefficacy of carbapenem antibiotics and the proliferation of multidrug-resistant pathogens.1 These pathogens are primarily of Gram-negative origin and carry genes responsible for expressing β-lactamases.2 These enzymes hydrolyze the amide bond in β-lactams, neutralizing the antibiotics and preventing them from interacting with bacterial penicillin-binding proteins (PBPs).3 Given the current global antimicrobial crisis and the World Health Organization's (WHO) call for more antibacterial interventions,4–6 currently, only about 60 antibacterial agents are in phase 1–3 clinical trials, with >50%7 targeting critical priority pathogens such as Carbapenem resistant – Enterobacterales, Acinetobacter baumanii, and Pseudomonas aeruginosa.8 Restoring the activity of existing antibiotics through the development of β-lactamase inhibitors is crucial in addressing the growing threat of AMR. This approach extends the lifespan of our current antibiotic arsenal, reduces the reliance on discovering new drugs, and helps curb the spread of resistant bacteria, ultimately safeguarding global public health, in the fight against AMR.9While many serine β-lactamase inhibitors (SBLIs) are effective in clinical practice, there is still a need for an approved metallo β-lactamase inhibitor (MBLI), to reach clinical dispensation (Fig. 1).10 Broad-spectrum cyclic boronate β-lactamase inhibitors (BLIs) taniborbactam (1)11–13 – previously known as VNRX-5133 and xeruborbactam (2)14–17 – previously known as QPX7728 (Fig. 2),18 are promising candidates that will provide patients with better treatment outcomes in a shorter timeframe.
 | ||
| Fig. 1 Schematic representation of the MBLI discovery workflow towards an investigational new drug. Created with https://Biorender.com. | ||
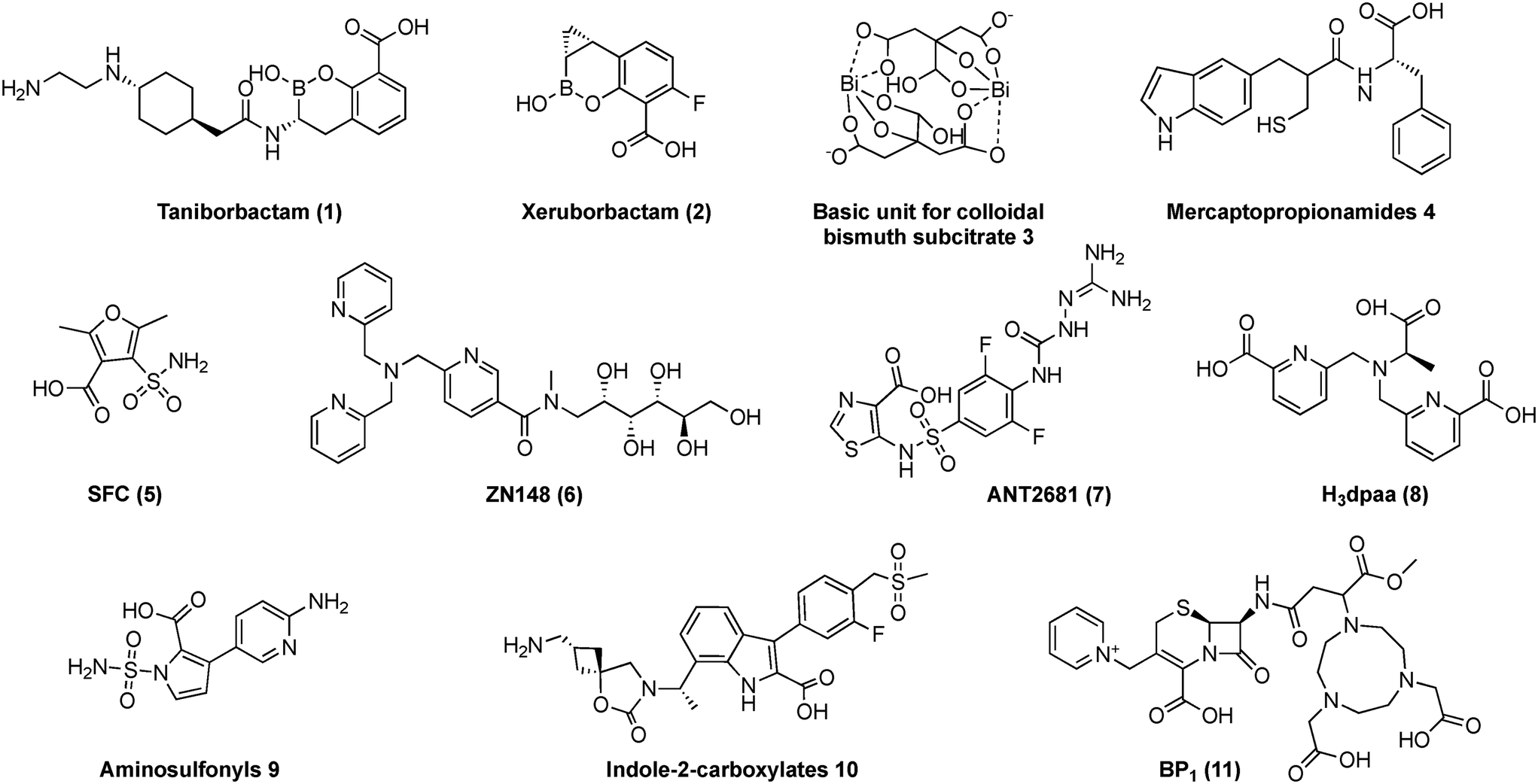 | ||
| Fig. 2 Representative MBLIs selected, that underwent comprehensive pre-clinical testing from 2018 to 2023 (1 and 2 have progressed to the clinical development stage, with 1 undergoing pre-registration). | ||
Cyclic boronates elicit their inhibitory activity by masking as analogues to the primary tetrahedral intermediate, which is shared by both serine β-lactamases (SBLs) and metallo β-lactamases (MBLs) during bicyclic β-lactam hydrolysis.19 They may also inhibit several PBPs upon interaction.20 Compound 1, in combination with cefepime (administered intravenously),11–13 has completed phase 3 clinical trials and is currently in the pre-registration phase of development.21 This compound inhibits Ambler class A, C, and D enzymes, as well as class B New-Delhi metallo-β-lactamase (NDM) and Verona integron-encoded metallo-β-lactamase (VIM) through reversible, competitive enzyme inhibition.13 Meanwhile, 2,14–17 in phase 1 clinical trials, is administered with meropenem intravenously or ceftibuten orally and targets a broader spectrum of β-lactamases, including the MBL imipenemase (IMP).14
In contrast to the rigorous regulatory testing and evaluation required for an Investigational New Drug (IND) proposal there is a noticeable lack of uniformity in the pre-clinical evaluation practices when reporting potential BLIs/MBLIs in the literature (Fig. 1). While some researchers perform in-depth evaluations with in vitro and in vivo assays, biochemical analyses, and computational methods, many challenges arise in comparing the results across the various research groups, underscoring the need for greater consistency in this field.
This review advocates for the implementation of standard protocols to help establish consensus guidance that can facilitate comparison, reproducibility and optimize the pre-clinical development of highly sought-after MBL inhibitors. The text is organized according to the hypothetical MBLI discovery workflow outlined in Fig. 1. It highlights shortcomings in these areas by utilizing the selected MBLIs depicted in Fig. 2 that have undergone thorough in vitro–in vivo–in silico examination using similar experiments with differing methodologies and outcomes. The authors acknowledge that many other representative MBLIs have been reported. However, this article does not aim to provide a comprehensive review but rather to offer a critical perspective with these examples.
2. Computational methods
2.1 High-throughput screening and in silico observations
High-throughput screening (HTS) is a popular drug discovery tool used to identify starting compounds with biological potential.22 HTS entails assaying and screening numerous biological effectors and modulators against a specific selected target when minimal information about the target exists.23 Screening assays are evaluated against a vast array of DNA, chemistry, and protein libraries, at a fast-paced rate, which can exceed thousands of compounds per day, thus accelerating the drug discovery process as “hit compounds” are identified.24 HTS comprises several steps including target recognition, compound management, reagent preparation, assay development, and screening.25 Assays used include; biochemical (enzyme-based), ligand-based or cell-based assays;22 with detection techniques comprising of; fluorescence resonance energy transfer (FRET), and colorimetric/photometric absorbance, among others.26HTS however, is disadvantaged by the practical limitations of not having the bioactive compound in existence, with synthesis-on-demand libraries, requiring a weeks/months that require significant material and experimental resources.27 Computational approaches have emerged as valuable tools that complement or, in some cases, serve as alternatives to traditional HTS, fragment-based screening (FBS), and newer platforms like DNA-encoded chemical libraries (DELs).27 Therefore, in the context of designing MBL inhibitors, in silico methods are an alternative to HTS. It is a crucial early step in drug discovery, enabling researchers to identify promising candidates before synthesis. Computational approaches such as molecular docking, virtual screening, molecular dynamics and artificial intelligence-driven drug discovery (discussed later in section 8) help predict how potential inhibitors interact with metallo-β-lactamases at the atomic level, guiding the optimization of binding affinity and specificity. Although computational methods offer a solution to improve HTS, it may not replace it,27 instead, it could be used in conjunction with HTS, to deliver a more robust outcome, as a drug discovery tool.
2.2 Computational methods used to identify MBL–MBLI interactions at a molecular level
The challenges in understanding MBL enzymes begin with their classification (Table 1). Particularly from a computational perspective, the difficulty in adequately describing the inhibition mode (Table 1) and the reaction mechanism induced by inhibitors stems from the challenging depiction of the MBL metal centers (Fig. 3). Although classical and quantum methods aim to describe the correct geometry and energetically coherent values relative to experimental data, this understanding still has several gaps to fill.| Compound | Chemical class28 | Inhibition mode |
|---|---|---|
| 1 | Boronic acid | Masks as analogues to the primary tetrahedral intermediate, common to SBLs and MBLs. 1 also formed bicyclic and tricyclic structures with NDM-1, during crystallography analyses11–13,28 |
| 2 | Boronic acid | Similar to 1, the cyclopropyl of 2 is positioned to occupy the hydrophobic pocket near the zinc ions of NDM-1 and VIM-2 (ref. 14–17 and 28) |
| 3 | Colloidal bismuth | NDM-1 was inhibited irreversibly by one Bi(III) displacing two zinc ions at the MBL active site29 |
| 4 | Thiols | Mimic substrate intermediates responsible for β-lactam ring opening. Through IMP-1 structural observations, thiol displaces the hydrolytic water molecule, responsible for bridging the two metal ions, carboxylate groups then form electrostatic hydrogen bonds with anchor residues, Lys224IMP-1 (ref. 28 and 30) |
| 5 | Sulfonamides | The carboxylate group of sulfamoyl heteroarylcarboxylic acid binds to the MBL active site by masking as a β-lactam substrate. The sulfamoyl group coordinates to the two zinc ions28,31 |
| 6 | Nitrogenous aromatic carboxylic acid | Tris-picolylamine meglumine derived acyclic chelator that mimics as a β-lactam substrate32 |
| 7 | Nitrogenous aromatic carboxylic acid | Masks as a β-lactam substrate. In VIM-2 the carboxylate and pyridine nitrogen bind to the second zinc ion, displacing two “non-bridging” water molecules present in the native structure28,33–35 |
| 8 | Nitrogenous aromatic carboxylic acid | Acylic dipicolinic acid pentadentate-chelating N–O ligands act by mimicking pharmacological substrates36 |
| 9 | Sulfonamides | The di-zinc bridging water molecule is displaced by the N-sulfamoyl group, whilst the carboxylate group facilitates ligation to the second zinc ion28,37 |
| 10 | Nitrogenous aromatic carboxylic acid | The C7 alkyl groups and the indole NH forms a stable retained di-zinc ion-bridging hydroxide complex. Whilst the carboxylate ligates to Zn2, to mimic the β-lactam substrate. Indole-2-carboxylates engage with various motifs during binding28,38 |
| 11 | Nitrogenous aromatic carboxylic acid | Masks as a β-lactam substrate, β-lactam derived cyclic amino chelator39 |
 | ||
| Fig. 3 Detailed 3D structure of NDM-1 in complex with hydrolyzed ampicillin (PDB code 5ZGE), showcasing the zinc-binding residues and hydrolyzed ampicillin in a ball-and-stick representation. The carbon atoms within the active residues are highlighted in orange, while those in the hydrolyzed ampicillin are depicted in green. Additionally, zinc ions are illustrated as grey spheres and hydroxide ions are represented by red spheres, providing a clear visualization of the system's complexity. | ||
As previously delineated in the scientific literature, it is widely accepted that MBL enzymes are reliant on Zn2+ ions for the execution of their biological activities.40,41 Therefore, excluding these ions from simulations is an impractical approach for faithfully representing the system from a computational perspective. There is no doubt that the most challenging aspect in the computational understanding of MBL enzymes lies in the accurate description of Zn2+ ions using the classical and quantum mechanical methods.42 Additionally, the nature of the inhibition reaction of MBL enzymes must be considered, as inhibitors can be classified as non-covalent and covalent,28 further contributing to the computational challenge. Various computational studies employing molecular docking, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations, density functional theory (DFT), and Molecular Dynamics (MD) are used to shed light on the intricate interactions and mechanisms involved in MBL inhibition.
Particularly, concerning the compounds 1–11 highlighted in this study, 733–35 and 1139 underwent computational analyses through molecular docking techniques,34,39 while 111–13 was assessed using classical molecular dynamics techniques, due to its theoretical and computational nature, only classical structural features were considered in the analyses.43 While thermodynamic parameters, such as free energy, play a crucial role and are derived from these classical simulations, it is critical to recognize that the energies associated with docked bindings offer only a basic approximation. To obtain more precise binding energies of the MBLI interactions, conducting QM/MM, Our Own N-layered Integrated molecular Orbital and molecular Mechanics (ONIOM) calculations or implementing QM/MM molecular dynamics simulations is essential.44,45
In the literature, it is possible to find some studies that provide computational insights for the description of MBLI systems: Olsen et al. (2004)46 conducted a detailed analysis of Bacillus cereus MBL interaction with benzylpenicillin, highlighting the influence of metal ions on the enzyme's mechanism and revealing distinct binding modes. Wang and Guo (2007)47 employed DFT and hybrid QM/MM simulations to elucidate the mode of action of inhibitors of the MBL IMP-1 from P. aeruginosa, emphasizing the crucial role of direct binding to the metal. Zhu et al. (2013)48 and Zheng and Xu (2013)49 investigated the hydrolysis mechanism of NDM-1 through MD and ONIOM methods, emphasizing the importance of ionized residues and water molecules in the catalytic process. Chen et al. (2014)50 explored the impact of mutations in NDM-1, while Levina et al. (2020)51 analyzed cephalosporin-L1 MBL complexes, employing potential energy surface (PES) calculations and QM/MM methods. More recently, Gervasoni et al. (2022)52 addressed the choice of QM methods, emphasizing the limitations of empirical models and advocating for higher-level calculations like DFT/MM. Medina and Jana (2022)53 investigated the catalytic mechanism of meropenem hydrolysis by VIM-1, identifying key residues and proposing mutants for inhibiting MBL activity. These studies emphasize the computational challenges of studying covalent and non-covalent inhibitors, stressing the importance of accurately describing the metal center in simulations.
3. Inhibition of enzyme activity
Another integral component of in vitro evaluation is the incorporation of enzyme analysis of an MBLI. The inhibitory candidate must be able to bind to the MBL at a suitable concentration, generally in the micro/nanomolar range, to be regarded as an efficient inhibitor. Other important factors include the mechanisms of inhibition expressed by the MBLI (Fig. 4). It is important to note that metal-binding agents function by binding to and extracting metal ions from enzymes, thereby disrupting enzyme activity without forming a permanent interaction. In contrast, covalent inhibitors establish a chemical bond with the enzyme, which can be either reversible (temporary) or irreversible (permanent), directly modifying the enzyme's function. Covalent irreversible inhibitors have the advantage of; prolonged action, target acquisition, and the ability to compete with high substrate concentrations.54 Successful covalent inhibition has been observed with many SBLIs.55 The MBLIs 3,296,32 and 1139 are classified as metal binders, which typically operate through an unclear irreversible binding mechanism, which may increase the enzyme's susceptibility to irreversible chemical modification. | ||
| Fig. 4 The different inhibition mechanisms of metallo-β-lactamase inhibitors. Abbreviations: MBL (metallo-β-lactamase), MBLI (metallo-β-lactamase inhibitor). Created with https://Biorender.com. | ||
A more comprehensive understanding of the mode of action of enzymatic inhibitors involves recognizing the presence of non-covalent and covalent states throughout the inhibition process. Consequently, relying solely on a straightforward examination of Ki (inhibition constant of the inhibitor) or IC50 (half the maximum inhibitory concentration) values is insufficient for determining the optimal MBLI potency, as the nature of inhibition should be considered.
Besides the covalent inhibitors' challenge, the traditional view in drug discovery is that irreversible inhibition is undesirable due to fears of off-target effects and toxicity. The reliance on equilibrium measures like IC50 values, which may underestimate the potency of inhibitors for slow-onset inhibitors, highlights the limitations of these metrics. Instead, kinetic parameters, particularly second-order parameters like kinact/Ki, offer a more accurate assessment by incorporating both inactivation rates and pseudo-affinity constants.56 Although it is the gold standard, this value is typically derived from in vitro assays, which can limit efficiency when studying enzyme families with multiple homologs, like PBPs. Moreover, enthalpy/entropy compensation warrants careful consideration for covalent inhibitors, as irreversible inhibitors often incur a more significant entropic penalty than their reversible counterparts.57 These points provide a clearer picture of the covalent inhibitors' effectiveness, sidestepping the pitfalls of equilibrium-based evaluations and affirming their therapeutic potential.
The IC50 of irreversible inhibitors is indeed time-dependent due to the formation of covalent bonds, unequivocally making Ki a more precise parameter for comparing binding affinities. In contrast, reversible inhibitors consistently exhibit time-independent IC50 values, with Ki directly reflecting their equilibrium binding affinity.58 Using an IC50 that approximates the Ki (Kiapp) will depend on the assay conditions (insufficient enzyme or weak substrate), which vary among research laboratories. Except for 10,38 all other compounds shown in Table 2 were evaluated for either the IC50 (1,11–133,299,371038) or Ki (2,14–174,305,316,327,33–35836) or both (11).39 In particular, the inhibition values for compound 10 (ref. 38) are in the nanomolar range. Then, its activity values are expressed as pIC50 (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50) and are reported to be a better indicator of enzyme inhibition for MBLs.38 MBLIs that function as metal chelators are known to be non-specific and may be limited in therapeutic use if they bind to other essential human enzymes that contain zinc.39
IC50) and are reported to be a better indicator of enzyme inhibition for MBLs.38 MBLIs that function as metal chelators are known to be non-specific and may be limited in therapeutic use if they bind to other essential human enzymes that contain zinc.39
| Compound | Inhibitory parameter assessed and binding mode | MBL: inhibition concentration (μM) | Mammalian metalloproteinase: inhibition concentration (μM) |
|---|---|---|---|
| ACE = angiotensin-converting enzyme, ECE = endothelin converting enzyme, Gly-2 = glyoxylase II, MMP = matrix metalloproteases.a <25% inhibition of important human MMPs indicating good selectivity towards bacterial MBLs.b IC50 measured in nanomolar units, pIC50 is unitless. | |||
| 1 (ref. 11–13) | IC50, covalent reversible | NDM-1: 0.19 | MMP – 1, 2, 3, 9: 100 |
| VIM-2: 0.026 | |||
| IMP-1: 39.8 | |||
| 2 (ref. 14–17) | K i, covalent reversible | NDM-1: 0.032 | ECE-1: 431 |
| VIM-1: 0.008 | ACE-2: >1000 | ||
| IMP-1: 0.22 | MMP-1: >1000 | ||
| MMP-9: >1000 | |||
| 3 (ref. 29) | IC50, metal ion binder and meta displacement mechanism | NDM-1: 2.81 | Not determined |
| VIM-2: 3.55 | |||
| IMP-4: 0.7 | |||
| 4 (ref. 30) | IC50, covalent irreversible | NDM-1: 0.12 | Not determined |
| 5 (ref. 31) | K i, uncharacterized | NDM-1: 9.82 | ACE: >1000 |
| VIM-2: 2.81 | |||
| IMP-1: 0.22 | |||
| 6 (ref. 32) | K i, metal ion binder and unclear irreversible inhibition | NDM-1: 310 | Gly-2: >500 |
| VIM-2: 2.24 | |||
| 7 (ref. 33–35) | K i, uncharacterized | NDM-1: 0.04 | ACE: 133 |
| VIM-1: 0.63 | Gly-2: >200 | ||
| IMP-1: 3.81 | MMP-2: 173 | ||
| MMP-9: >200 | |||
| 8 (ref. 36) | K i, uncharacterized | NDM-1: 1.70 | Not determined |
| VIM-2: 5.83 | |||
| IMP-1: 1.54 | |||
| 9 (ref. 37) | IC50, uncharacterized | NDM-1: 0.83 | MMP-1, 2, 3, 7, 8, 9, 12, 13, 14: % inhibition using 100 μM of 9: −0.1–22.2a |
| VIM-2: 32.4 | |||
| IMP-1: 0.43 | |||
| 10 (ref. 38) | IC50 (pIC50), non-covalent | NDM-1: <0.06 nM (>10.2) | Not determined |
| VIM-1: 0.4 nM (9.4) | |||
| VIM-2: 0.16 nM (9.8) | |||
| IMP-1: 10 nM (8.0) | |||
| 11 (ref. 39) | IC50 | 253.3 (NDM) and 45.8 (VIM-2) | Gly-2: >500 |
| K i, metal ion binder and unclear irreversible inhibition | 97.4 (NDM) and 24.8 (VIM-2) | ||
Metal binding inhibitors such as 3, 6 and 11 can overcome MBL resistance since they bind to the active site of MBLs, which is their defining feature and the most conserved part of the class B1 family.59 As highlighted previously, metal-binding inhibitors function by a metal ion stripping mechanism or ternary complex formation.54 In metal ion stripping, MBL active site metal ions are actively removed by inhibitors or sequestered when metal ions leave the active site.54 Ternary complex formation occurs when the inhibitor binds to the metal ions and surrounding protein residues, thereby blocking access to the antibiotics to be degraded by the MBL.54
Uncharacterized inhibitor mechanisms (compounds 5,317,33–358,36937) are solely identified through their potency, where the IC50 is determined and active site binding is ignored.60 This approach is unfavored since it does not consider the structure–activity relationship of MBL versus MBLI. In addition, a true IC50 cannot be determined for covalent inhibitors since it depends on time, the binding and kinetic components of inhibition.61 Although the IC50 is not an absolute constant, it remains a useful and ‘quick’ parameter for comparing compounds under consistent experimental conditions, regardless of the inhibition mechanism. We believe that determining the exact mode of inhibition and the corresponding kinetic constants (e.g., Ki, k2/K, or koff) is essential for a more mechanistic understanding of inhibitor behaviour. Regarding EC50, although not commonly reported for MBLIs, we consider this parameter relevant during the cellular validation phase of development. Once a candidate demonstrates potent enzyme inhibition in vitro, its efficacy can be evaluated in whole-cell assays where the EC50 reflects the concentration required to restore β-lactam activity in the presence of the inhibitor. Thus, for covalent (reversible and irreversible) MBLI, one must consider the contribution of non-covalent interactions that favour the removal of Zn, justifying the high values of Ki, since the entire inhibition process needs to be considered, advocating that higher Ki values do not equate to compound inferiority.
MBL concentrations are dependent on whether the enzyme is purchased (4,301139) or expressed (2,14–173,295,316,328,369,371038) and in what quantity. At the same time, activity is dependent on the sensitivity of each batch of enzyme, which can be influenced by temperature and freeze–thaw cycles.62 Freeze–thaw cycles of enzyme aliquots are not recommended. Still, they are practised in many labs due to the high cost of purchasing enzymes or the poor yield and technical difficulties in expressing and purifying MBLs of interest.63 Although these are standard biochemistry principles, they are often taken for granted by laboratory personnel in fast-paced environments or overlooked in labs that do not primarily focus on enzyme studies. The Assay Guidance Manual64 provides experimental information regarding enzyme assays and should be consulted when designing enzyme studies. For research labs lacking a dedicated enzymologist or the experience to troubleshoot assays, commercially available enzyme assay kits offer a promising solution to address this challenge and expedite the MBLI development process. These kits could include MBLs, MBLI controls, substrates, buffers, and optimized protocols, which aid in validating results. This standardization allows for results to be compared across compounds evaluated using the same assay kit. Furthermore, on a molecular level, heterologous MBL sequences (the reason why 111–13 inhibits NDM and VIM but not IMP B1 MBLs), shallow active sites, the presence of catalytic residues, and similarity to human metalloenzymes often impede the development of an MBLI.65
4. Inadequate predictive in vitro assays
Pre-clinical research necessitates robust in vitro assays to detect the presence of MBLs, evaluate the potency of BLIs and their capacity to restore the activity of β-lactam antibiotics.4.1 MBL detection using various diagnostic assays
IMP-1, the first MBL discovered in Japan in 1990 from P. aeruginosa and Serratia marcescens,66 currently comprises approximately 98 variants.67,68 A few years later, in 1997, VIM was reported in P. aeruginosa,69 with about 85 variants identified.67,68 NDM was subsequently reported in 2008,70 with 65 variants currently known.67,68 However, data collected by the SENTRY antimicrobial surveillance program in India during 2006–2007 revealed that 38.5% of CRE isolates harbored blaNDM-1,71 indicating its circulation before its discovery in 2008. NDM also shares a 32.4% amino acid similarity with VIM-1/VIM-2,72 underscoring the critical need for precise detection methods and the essential requirement for accurate diagnostic methods that can differentiate among MBLs.Another concern arising from antimicrobial susceptibility testing is the existence of two recognized guides for categorizing the generated minimal inhibitory concentration (MIC). These guidelines are from the Clinical and Laboratory Standards Institute (CLSI)73 and the European Committee for Antimicrobial Susceptibility Testing (EUCAST).74 Utilizing either of these guidelines in pre-clinical labs, will have an altering effect on the MIC categorization of the β-lactam that is used in combination with the MBLI, since most MBLIs work by restoring the efficacy of the β-lactam. Harmonizing these two guidelines will effectively reduce possible discrepancies in the MIC categorization.75 Speculation of an initiated agreement between the relevant stakeholders from EUCAST and CLSI has been mentioned, however, there is no conclusive evidence on this matter. Therefore, in the interim, the authors suggest providing an MIC interpretation using both guidelines.
Diagnostic tests vary in usage across countries due to factors such as accessibility, infrastructure, cost, and the expertise of laboratory personnel.72 Consequently, the specific protocols and techniques influence the uniformity of antimicrobial susceptibility testing (AST). The WHO does not support molecular methods, for the precise detection of carbapenemases.76 Instead, phenotypic detection is endorsed, through characterization of the MIC, which is reported by AST. This is currently regarded as the gold standard by WHO,76 with time-kill assays preferred over broth microdilution assays. Although there are flaws in the MIC and classification of antimicrobial susceptibility, it is still widely used and relied upon, particularly in many developing countries where access to specialized equipment is limited, and cost is a significant factor. Regrettably, it is in these countries where the burden of resistance is highest, and the need for surveillance and adequate reporting on prevalence and controlled antimicrobial usage is critical.72,77
4.2 Antimicrobial susceptibility testing and the associated shortcomings
Challenges in AST using the broth microdilution method have revealed significant discrepancies in the MIC of bacteria harboring MBLs. Adjusting the AST media to mimic in vivo conditions accurately should be explored when establishing predictive in situ resistance. For example, in contrast to in vitro resistance assessed by AST, many case studies have reported positive outcomes in patients infected with MBL-producing bacteria.78 This can occur from using a 2–3 antibiotic combination of β-lactams or non-β-lactams and a carbapenem, to target various PBPs. In Escherichia coli, meropenem targets PBP2,79 whilst cefepime/ceftazidime targets PBP3,80 using a combination of both antimicrobials, has shown clinical significance during treatment.78 Enhancers that bind to PBP2, such as Zidebactam, have also been used similarly. Zidebactam, in combination with PBP3-based β-lactams (cefepime), have successfully targeted and inhibited MBL-producing bacteria.81 However, bacteria are constantly evolving through resistance mechanisms (explained further in section 7), with NDM-1 and NDM-5 harbouring E. coli isolates, PBP-3 modifications have been observed, conferring resistance to the aztreonam-avibactam combination treatment.80MBL activity largely depends on the presence of the metal zinc, with in vivo homeostasis studies indicating that intracellular and plasma zinc exists in a bound state, with no observations of free zinc being present.82 However, the free zinc concentrations used in Mueller–Hinton broth (MHB) for microdilution AST are reportedly supraphysiological.83 A study by Asempa et al.84 found that all MBL-harboring pathogens producing NDM, IMP, and VIM MBLs were resistant to meropenem at concentrations of 16 to >64 mg L−1 when using cation-adjusted Mueller–Hinton broth (CAMHB). However, these MBL-producing pathogens were susceptible to meropenem (<0.06–0.5 mg L−1) when tested with chelex or EDTA-treated MHB. In mice infected with MBL-pathogens in the thigh and lung, respectively, a 2.24 and 3log10 decrease in the colony-forming units (CFU) was observed when utilizing meropenem monotherapy.84 Thus, the precise resistance level may be overestimated with in vitro tests compared to the resistance observed within in vivo models.
According to Bilinskaya et al.,85 another concerning factor in the AST of MBL-harboring pathogens is the complications of unspecified and variable zinc content among the different broth manufacturers and within lot batches of the same brand. This has led to an eight-fold difference in meropenem MIC among the commercial lots and has impacted the susceptibility categorization of MBL-producing bacteria.85 The utilization of zinc-limited media, by adding EDTA/chelex to the prepared broth, offers a more thorough approach to MIC classification for β-lactamase-producing bacteria in the absence of molecular testing. However, there are still some practical challenges in quantifying free zinc and EDTA-bound zinc before AST, as inductively coupled plasma mass spectrometry (ICP-MS) – the current method of choice, was unable to differentiate between these two types of zinc available to the pathogen.85–87 As a result, the efficacy of MBLIs under study will be compromised due to the extent of the EDTA/Chelex interference on susceptibility breakpoints. So far, there is no consensus on EDTA concentrations in such procedures, and the issues raised have not yet been settled.
A communication from Rennie88 states that manufacturers necessitate zinc measurement to attain low zinc ion concentrations for carbapenem susceptibility testing. However, this letter does not suggest an acceptable reference range for the zinc ion concentration used in the CAMHB. Instead, it stipulates that at low levels, the zinc ion concentration will indefinitely vary by more than 0.1 or 0.2 mg L−1 and criticizes the use of EDTA to deplete zinc ions, as other cations could be negatively impacted. In response to Rennie's letter, Asempa and Nicolau,89 stated unawareness of any such requirement and argued on what is regarded as “low zinc concentrations”. During the study, unsuccessful communication attempts with the respective MHB manufacturers prevented data validation, with each representative citing proprietary reasons. This observation implied potential zinc sequestration and/or supplementation in manufacturing. Asempa and Nicolau advocate for open access to these methods and the dissemination of zinc target requirements in CLSI standards and broth container labels. This is recommended to guide quality control efforts and investigational studies utilizing zinc ions to achieve performance standards.
Aligning to these concerns, the quantity of zinc in the broth directly affects MICs, which may be exaggerated for MBLIs such as 3,294,306,32 and 1139 that function by an unclear irreversible binding mechanism that will hamper the MBLI's full potency. Many laboratories do not have access to ICP-MS instrumentation used to quantify the zinc content pre- and post-addition of EDTA/Chelex in the broth. Therefore, it is unknown if 3,294,306,32 and 1139 partially bind to the zinc in the broth before encountering the MBL pathogen. Given the present challenges with the varying zinc content of CAMHB, as well as the mitigation of zinc-ion interference by the addition of EDTA, it is recommended that the effect on other cations, such as iron, be also considered. The inhibitor 6 exhibits irreversible inhibition, and it is essential to note that the mechanism behind this process is not yet fully understood.32 Nonetheless, multiple studies have suggested that chelating agents enhance the susceptibility of active-site amino acids to chemical modifications, such as the oxidation of Cys.90–92
Compounds 1–11 (Fig. 2) have all undergone AST using the broth microdilution technique in combination with meropenem against one or more MBLs. The meropenem MIC can vary depending on the amount of inhibitory compound used. The general reporting of MIC is done in mg L−1 units; however, 639 (Fig. 2) was reported in micromolar units, which considers the molar mass of the compound, which researchers do not always disclose. This presents an extra step before direct comparisons to other MBLIs can be made. It is also important to note that not all compounds were assessed for synergy with the partnering β-lactam. Compounds 3,295,318,36 and 1139 were the only MBLIs to incorporate the checkerboard assay with the FICI criteria for synergy assessment. The checkerboard assay involves studying a range of MBLI concentrations, and based on the produced activity, the researcher can select an MBLI-antibiotic combination, with the lowest concentration that produces effective inhibition. Utilizing a very high concentration of the MBLI may prove to be toxic, and detrimental to further biological evaluation. Increasing or decreasing the MBLI concentration directly impacts the antibiotic's MIC and the relationship shared between the two compounds. Hence this AST method is an excellent screening assay for MBLI/antibiotic compatibility.
The EUCAST interpretative criteria were used for compounds 1,11–133,294,306,32 and the CLSI guidelines were used for 2,14–175,318,369,37 whilst 7,33–3510,38 and 1139 utilized both guidelines (Table 3). Testing guidelines should list an approved number of isogenic MBL strains that are widely accessible and well-characterized for MIC testing. This will facilitate cross-comparisons of MBLIs, as MICs are species-specific and can be influenced by the presence of other β-lactamases (in clinical strains) when tested with the selected antibacterial agent.
| Compound | Clinical/reference/engineered strains | MBLI MIC(MBC): mg L−1 | Meropenem MIC(MBC): mg L−1 | Interpretive criteria and assay(s) used |
|---|---|---|---|---|
| Bacterial inhibition was achieved by co-administering meropenem with an MBLI (1–11). CLSI = Clinical Laboratory Standards Institute, EUCAST = European Committee on Antimicrobial Susceptibility Testing, FICI = fractional inhibitory concentration index, MBC = minimum bactericidal concentration, shown in brackets and obtained from the time-kill assay.a Cefepime is used instead of meropenem.b Biapenem is used instead of meropenem.Note compound 11 is a β-lactam-derived MBLI and possesses antimicrobial activity when used at concentrations >64 mg L−1. | ||||
| 1 (ref. 11–13) | Klebsiella pneumoniae VIM-4 | 4 | 0.5 | EUCAST broth microdilution method and time-kill assay |
| Pseudomonas aeruginosa VIM-2 | 4 | 0.06 | ||
| K. pneumoniae VIM-27 (CDC-0040) | 4 | 4 | ||
| Escherichia coli NDM (CDC-0452/CDC-0055) | 4 | 0.125–2 | ||
| P. aeruginosa VIM (CDC-0457) | 4 | 0.125 | ||
| K. pneumoniae NDM CDC-0049 | (4) | (4) | ||
| 2 (ref. 14–17) | K. pneumoniae IMP-26 (KP1160) | 2.5 | 1 | CSLI broth microdilution method |
| K. pneumoniae NDM-1 (KP1081) | 0.6 | 1 | ||
| K. pneumoniae VIM-1 (KP1054) | 0.16 | 1 | ||
| 3 (ref. 29) | E. coli NDM-1 | 32(64) | 2(24) | EUCAST broth microdilution method (checkerboard- with FICI calculation), and the time-kill assay |
| K. pneumoniae NDM-1 | 32 | 4 | ||
| Citrobacter freundii NDM-1 | 32 | 0.5 | ||
| E. coli BL21(DE3, pET-28a-VIM-2) | 32 | 0.5 | ||
| E. coli BL21(DE3, pET-28a-IMP-4) | 32 | 4 | ||
| 4 (ref. 30) | E. coli NDM-1 | 16 | 8 | EUCAST broth microdilution method with FICI calculation |
| K. pneumoniae NDM-1 | 32 | 32 | ||
| Sample ID 2470 | 32 | 64 | ||
| Sample ID2472 | ||||
| 5 (ref. 31) | E. coli IMP-1 NUBL-24 | (10) | (1) | CSLI broth microdilution method (checkerboard – with FICI calculation), and the time-kill assay |
| P. aeruginosa | ||||
| PAO1/pME-IMP-1 | 50 | 16 | ||
| PAO1/pME-NDM-1 | 10 | 4 | ||
| PAO1/pME-VIM-2 | 50 | 16 | ||
| E. coli | ||||
| DH5α/pBC-IMP-1 | 8 | 0.031 | ||
| DH5α/pBC-NDM-1 | 32 | ≤0.25 | ||
| BL21(DE3)/pET-SPM-1 | 8 | 0.063 | ||
| 6 (ref. 32) | K. pneumoniae NDM-1 (K66-45) | 50 μM(50 μM) | 0.125(4) | EUCAST broth microdilution method and the time-kill assay |
| P. aeruginosa VIM-2 (K34-7) | 50 μM | 0.125 | ||
| E. coli NDM-1/5 | 50 μM | 0.06–2 | ||
| E. coli IMP | 50 μM | ≤0.125 | ||
| 7 (ref. 33–35) | K. pneumoniae NDM | 8 | <0.06–32 | CLSI + EUCAST broth microdilution method |
| Enterobacter cloacae NDM | 8 | <0.06–8 | ||
| E. coli NDM | 8 | <0.06–4 | ||
| 8 (ref. 36) | E. coli-DH5a/pUC-NDM-1 | 16 | <0.03 | CLSI agar disc diffusion, broth microdilution method (checkerboard- with FICI calculation), and the time-kill assay |
| E. coli NDM-1/3/5 | 16 | <0.03–0.5 | ||
| E. coli IMP-4 | 16(16) | <0.03–0.25(0.25) | ||
| E. cloacae NDM-1/5 | 16 | <0.03–1 | ||
| K. pneumoniae IMP-4 | 16 | 0.125–2 | ||
| K. pneumoniae NDM-1 | 16(16) | <0.03–0.125(0.25) | ||
| P. aeruginosa VIM-1 | 16(16) | 0.25(1) | ||
| 9 (ref. 37) | K. pneumoniae NDM-1 | 4 | 0.25–4 | CLSI broth microdilution method and the time-kill assay |
| E. coli NDM-1: | ||||
| BAA-2452 | 4(4) | 0.03(0.12) | ||
| BAA-2469 | 4 | 0.06 | ||
| E. coli IMP | 4 | 0.03 | ||
| K. pneumoniae VIM-1 | 4 | 0.06 | ||
| 10 (ref. 38) | Acinetobacter baumanii NDM (76885-C) | 8 | 2 | CLSI + EUCAST broth microdilution method (checkerboard), and the agar dilution assay |
| A. baumanii NDM-1 (76030-E-G, CH3504, S7–29) | 8 | 2–8 | ||
| E. coli NDM (55 N/B53) | 8 | 0.25 | ||
| K. pneumoniae NDM (86259, 48F, I39, IR18, 76664-G) | 8 | 0.125–0.5 | ||
| C. freundii NDM-1 (84646-E-B, 85524-E-Pi, 85558-E-Pi, 85569-E-Pi) | 8 | 0.125–0.5 | ||
| 11 (ref. 39) | E. coli NDM-1/4 | 16 | 0.25 | CLSI + EUCAST broth microdilution method (checkerboard- with FICI calculation) and the time-kill assay |
| E. cloacae NDM-1 | 16 | 0.5 | ||
| K. pneumoniae NDM | 16(32) | 0.125 | ||
| E. coli IMP-1 | 8 | 0.5(2) | ||
| E. cloacae VIM-1 | 32 | 0.03 | ||
| 0.5 | ||||
5. Limited toxicity and pharmacokinetic data
5.1 Toxicity and drug safety
Many MBLIs have undergone toxicity testing and pharmacokinetic analysis in pre-clinical research. However, there is no consensus on how it is conducted, which cell line(s) are used for cytotoxicity, the protocol used to determine cytotoxicity, and the pharmacological parameters evaluated. Frequently, the choice of the cell line for cytotoxicity studies or the selection of software and equipment for pharmacokinetics assessment is dictated by the resources available within the laboratory. This underscores the critical importance of adequate infrastructure and accessibility in certain countries. For instance, some of the most commonly used cell lines for evaluating the cytotoxicity of a candidate MBLI (Table 4) are HeLa cells (the first human cancer cells used),11,31 human embryonic kidney cells,30 human hepatoma,32,34,37–39 and red blood cells.36 Human hepatoma cells (HepG2) were the most common cell line used among 6,327,33–359,3710,3811.39 However, 111–13 and 430 were assessed against three cell lines, which is important to identify possible discrepancies; in these cases, there were none. The frequently utilized laboratory tests include hemolysis, the 3(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric dye-reduction assay, the lactate dehydrogenase assay,39 and the Cell Titer-Blue assay31,37,93 to measure the effects on cell viability with and without the MBLI. This indicates that various assays and commercially available assay kits are available to the researcher; hence, the results generated will be validated for the specific assay used and cannot be extrapolated to other cell lines or assay procedures. Hence, it emphasizes the significance of a standardized test, or an assay kit designed to incorporate the commonly used assays and cell lines, that are accessible, cost-effective and allows for cross-comparisons among MBLIs in toxicity assessment.| Compound | Cell line/animal used | Assay conducted | Result |
|---|---|---|---|
| N/A = not applicable, HepG2 = hepatoblastoma, HeLa = cervical cancer, MRC-5 = human fetal lung fibroblast, 3T3 = mouse fibroblast, HUVEC = umbilical vein endothelial cells, LO2 = HeLa derivative, and HEK293 = human embryonic kidney. CCK-8 = cell counting kit assay, LDH = lactate dehydrogenase assay, MTT = cell metabolic activity, alamarBlue = nontoxic alternative to MTT assay. IC50 = half the maximum inhibitory concentration, LD50 = half the maximal lethal dose, HC50 = the concentration needed to cause 50% hemolysis of human red blood cells, CC50 = 50% cytotoxic concentration. | |||
| 1 (ref. 11–13) | HeLa, MRC-5, 3T3 | Cell viability – MTT | IC50 = 256 μM |
| Human primary renal proximal tubule cells | Toxicity assessments | Non-toxic up to 1000 μg mL−1 | |
| 2 (ref. 14–17) | Rodents | Acute toxicity | No change in tissue histology or clinical chemistry up to 300 mg kg−1 |
| 3 (ref. 29) | N/A | N/A | Low toxicity, repurposed drug that is in clinical use |
| 4 (ref. 30) | HUVEC, LO2, HEK293 cells | CCK-8 assay, LDH release assay | IC50 > 200 μM |
| ICR mice | Acute toxicity | LD50 = 350.1 mg kg−1 | |
| 5 (ref. 11–31) | HeLa cells | CellTiter 96 AQueous one solution cell proliferation assay | IC50 > 100 μM |
| CD1 mice | Acute toxicity | LD50 = 246 mg kg−1, i.v. | |
| LD50 > 1000 mg kg−1, i.p. | |||
| 6 (ref. 32) | HepG2 cells | AlamarBlue cell viability assay | IC50 > 100 μM |
| Balb/c mice | Acute toxicity | Up to 252 mg kg−1, s.c. | |
| 7 (ref. 33–35) | HepG2 cells | Cell viability | IC50 > 100 μM |
| 8 (ref. 36) | Red blood cells | Hemolysis | HC50 > 1024 μg mL−1, <7% hemolysis |
| Balb/c mice | Acute toxicity | Nontoxic up to 50 mg kg−1, i.p. | |
| 9 (ref. 37) | HepG2 cells | CellTiter-GloV viability assay | CC50 > 256 mg L−1 |
| 10 (ref. 38) | HepG2 cells | Cell viability | IC50 > 64 μM |
| NMRI mice | Acute toxicity | Up to 300 mg kg−1, s.c. | |
| 11 (ref. 39) | HepG2 cells | LDH and MTT assays | IC50 > 1000 μg mL−1 |
5.2 Pharmacokinetics and the absorption, distribution, metabolism, and excretion (ADME) parameters
The standard pharmacodynamic (PD) index for β-lactam antibiotics is T > MIC, which represents the time that the drug concentration exceeds the MIC.94 For β-lactamase inhibitors, maintaining a threshold concentration is often essential for efficacy. Pharmacokinetics (PK) plays a crucial role in drug discovery research, as compounds that may not be ideal therapeutics based on in vitro models can exhibit good absorption and distribution in vivo. This becomes even more important with the co-administration of a β-lactamase inhibitor and β-lactam antibiotic (combination therapy) since the inhibitor can restore the efficacy of the β-lactam. The efficacy of this combination therapy relies heavily on the PK and PD properties of both agents. PK parameters typically assessed are the peak drug concentration(Cmax), the time taken to reach peak concentration (Tmax), the terminal half-life (T1/2), the area under the curve (AUC), and clearance (CL) of the drug.95T max is a critical PK parameter indicating the time it takes for each drug to reach its highest concentration in the bloodstream. For combination therapy to be effective, the Tmax of the β-lactamase inhibitor should ideally align with that of the β-lactam antibiotic. This synchronization ensures that both agents are present at therapeutic levels simultaneously, maximizing their synergistic potential against bacteria. The MIC, a fundamental PD parameter, defines the lowest concentration of an antibiotic required to inhibit the visible growth of a microorganism. Achieving and maintaining drug concentrations above the MIC for a sufficient dosing/treatment interval (T > MIC) is vital to the antimicrobial efficacy of β-lactam antibiotics.
Optimizing therapeutic outcomes involves correlating Tmax and MIC values. This can be done by adjusting the dosing schedule to incorporate frequent doses of the β-lactamase inhibitor, or by using continuous infusions of the β-lactam/BLI (in a larger animal model), to maintain T > MIC. This alignment allows both agents to exert their maximal inhibitory effects simultaneously, thereby enhancing the overall success of the treatment.
The PK/PD profile of multiple β-lactams in clinical practice has been examined extensively; however, insufficient information exists for the PK/PD profile of MBLIs.96 For the past 20 years, dosing regimens have been optimized using time-dependent PK/PD,97 which has been enforced on MBLIs, although data are scarce.96 Thus far, PK data has only been made available for MBLIs (broad-spectrum BLIs) 111–13,98 and 2,14–17,99 which have reached clinical trials.
There are very few studies, such as those by Yan et al.,100 that conduct acute toxicity testing of MBLIs in animals. However, from the eleven compounds in Table 5; 2,14–174,305,316,398,36 and 1038 incorporated this test in the evaluation process. This test is important to determine if there are any side effects of the candidate MBLI from multiple doses or a single dose monitored over time. It can also be a cost-efficient way to prevent further development of the MBLI if found to be toxic in vivo.
| Compound | Animal model used | Bacteria used for infection | Administration route of drug | Dosage (mg kg−1) | log10 decrease in CFU |
|---|---|---|---|---|---|
| s.c. = subcutaneous, i.p. = intraperitoneal, i.v. = intravenously, NA = not applicable, ND = not determined.a Denotes that the survival rate of mice was assessed instead of a log10 decrease in the bacterial CFU. A survival rate of 50% was achieved for 3, 87.5% (i.p. 400 mg kg−1 and i.v. 300 mg kg−1) or 100% (i.p. 300 mg kg−1 and i.v. 200 mg kg−1) for 4, and 100% for 5.b Multiple in vivo studies done on 10, varying the dose, pathogen/strain and animal model infection site i.e. utilization of either peritonitis/septicemia infection model or thigh infection model. | |||||
| 1 (ref. 11–13) | Murine, lung | K. pneumoniae CTX-M-14 | s.c. | 16 | >4 |
| Murine, ascending urinary tract | E. coli CTX-M-15 | s.c. | 8 | >2 | |
| 2 (ref. 14–17) | Neutropenic murine, thigh | K. pneumoniae KP1244 | i.p. | 50 | >2 |
| 3 (ref. 29) | Murine, peritonitis | E. coli NDM-HK | i.p. | 10 | NDa |
| 4 (ref. 30) | Murine, acute toxicity | N/A | i.p. and i.v. | 50–400 | N/A |
| 5 (ref. 31) | Murine, systemic | E. coli IMP-1 | i.p. | 100 | NDa |
| 6 (ref. 32) | Neutropenic murine, peritonitis | K. pneumoniae NDM-1 | s.c. | 10 | >3 |
| 7 (ref. 33–35) | Neutropenic murine, thigh | Multiple NDM Enterobacterales | s.c. | 160 | 3 |
| 8 (ref. 36) | Murine, sepsis | K. pneumoniae NDM-1 | i.p. | 10 | ≥3 (liver and kidney), ≥2 (spleen) |
| 9 (ref. 37) | Murine, thigh | E. coli NDM-1 | s.c. | 100 | >3 (vehicle), >1 (meropenem monotherapy) |
| 10 (ref. 38)b | Murine, peritonitis | E. coli ATCC 25922 ISAba 125 blaNDM-7 | i.v. | 10 or 30 | >5 |
| Murine, thigh | i.v. | 10 or 30 | 2 | ||
| 11 (ref. 39) | Neutropenic murine, thigh | K. pneumoniae NDM | i.p. | 100 | >3 |
To guarantee the integrity of the generated bioanalytical data, the method validation providing the PK and ADME parameters of the MBLIs needs to conform to acceptance standards as part of the evaluation criteria. For example, in regulatory studies, the guidelines outlined by the European Medicines Agency (EMA) documents101 are utilized in facilities accredited for good laboratory practices (GLP). In contrast, non-regulatory studies do not adhere to such stringent practices as it is not a requirement to advance the research. However, implementing a single guideline for researchers conducting non-regulatory studies will help promote accurate results and eliminate discrepancies among candidate MBLIs.
6. Restricted animal models with variable pre-clinical study designs
Animal models are important tools used in pre-clinical research to bridge the gap between in vitro efficacy and clinical trials of MBLI candidates.102,103 They are utilized to assess the MIC generated by phenotypic tests, establish dosing regimens, and determine pharmacokinetic and pharmacodynamic (PK/PD) parameters.102,104Fig. 3 summarizes an evaluation report of in vivo efficacies published between the years 2000 and 2020.1056.1 Types of animal models used in MBLI efficacy studies
The thigh murine infection model was most frequently used in the evaluation report, with only a single study utilizing a rabbit model (Fig. 5). The neutropenic model (61%) was preferred over the immunocompetent model (39%)105 because it allowed for the infection to be established successfully without interference from the host's immune system and promoted the undisturbed evaluation of the antimicrobial agent against the target pathogen.106 The neutropenic murine thigh infection model was also a popular animal model utilized among compounds 2,14–177,33–359,371139 in Table 3. Although the remaining compounds experimented with using different models, all incorporated mice as the animal of choice to study. The use of mouse models as the standard choice for efficacy studies can provide researchers with a foundational basis for designing their MBLI studies. | ||
| Fig. 5 Animal model variabilities amongst 31 studies (from 27 publications) were used to study β-lactam and MBLI efficacy against MBL-harbouring bacterial genotypes between 2000–2020. Of these, 12 animal studies assessed the efficacy of novel experimental MBLIs. Image adapted from data provided by Asempa T. E. et al. (2021).105 Created with https://Biorender.com. | ||
6.2 Pathogen selection and inoculation
Apart from five studies107–111 in the evaluation report (Fig. 5), all animal trials established an Enterobacterale NDM-1 infection model with either clinical Klebsiella pneumoniae or Escherichia coli. However, such studies should not be limited to NDM-1 since the global prevalence of MBLs also includes VIM and IMP in significant numbers.112 Moreover, the evolution of MBL variants and their respective zinc sensitivities suggests that a wide collection of genotypes should be investigated when determining MBL efficacy in vivo.85,113,114An implementation that will improve the CFU data extracted from animal trials is the use of a standardized inoculum density, such as the 0.5 McFarland standard. Since a higher bacterial inoculum density will influence the treatment outcomes of the drug in neutropenic animal models, infecting with fewer bacteria may not lead to infection in immunocompetent hosts. Therefore, when incorporating the McFarland standard into the study design, researchers will need to plate an aliquot of the bacterial suspension. This will be done to determine the exact number of bacteria used to infect the animal.
Many of the reviewed studies in the evaluation report (Fig. 5) did not include CFU counts from 0 hours post-inoculation, such as an imipenem/ertapenem study using an engineered E. coli NDM-1 strain,115 and it was excluded, thereby raising concerns over the bacteriostatic effects produced and the effects of in vitro–in vivo inoculum density.116 Another concern with engineered MBL strains is the failure to establish infection in a mouse model, hindering the evaluation process of the MBLI.100 Therefore, genetically well-characterized bacterial strains harboring multiple MBLs should be deposited into commercial biobanks and made accessible to researchers from diverse ethnic and social backgrounds.
6.3 Antibiotic administration and dosage
Selecting the appropriate β-lactam antibiotic and dosage to co-administer with an MBLI is another concern in the design of animal models. Meropenem is the most frequently utilized β-lactam, often administered at a dose of 10 mg per kg body weight and provides stability when co-administered with a developing inhibitor (Fig. 5). This is further evidenced by all inhibitory compounds in Fig. 2 (1–11), utilizing meropenem as the partnering β-lactam antibiotic. However, the clearance rate requires administration every two hours, and this poses a technical challenge for its use in mouse models. While studies have compensated for this rapid clearance,37,39 injecting the animal repeatedly over the advised 24 hour study period is impractical.117Although monitoring an animal for >24 hours would provide in-depth efficacy data, such scrupulous studies are almost impossible to design. As they require larger research labs, more lab personnel, and are governed by animal welfare and ethical restrictions. Another concern reported by Moya et al.118 were failed murine infection models amongst four NDM-harboring isolates designed using a humanized meropenem regimen. These findings indicate that meropenem may not be the ideal β-lactam antibiotic for pre-clinical animal study assessment and necessitates the inclusion of a meropenem monotherapy treatment arm.
When designing an animal model, many administration routes can be used depending on the preference of the injector, the infection site, and the PK/PD properties of the drug. The most common administration routes include intraperitoneal (i.p.), sub-cutaneous (s.c.), and intravenous (i.v.).105 In Table 5, most of the compounds employed either the i.p. (2,14–173,295,318,361139) route or the s.c. route (1,11–136,327,33–35937), with only 1038 employing the i.v. route and 430 utilizing both i.v. and i.p. routes. The concern is that the results generated depend on the administration route used. Studies by Ooi et al.,37 and Everett et al.,119 demonstrated that utilizing i.v. and s.c. administration routes with identical meropenem dosing regimens produced varying degrees of efficacy in an E. coli NDM infection model.
These findings were in accordance with 4,30 as an identical dose led to one mouse dying using the i.p. route and seven mice dying with the i.v. route, thus highlighting inconsistencies based on the choice of administration route used. The choice between i.v. and i.p. routes carries significant implications for exposure, pharmacokinetics (including potential drug–drug interactions), and acute toxicity. It is acknowledged that the exposure profile and PK parameters can vary between these routes, with acute toxicity, particularly concerning effects on the heart, being a critical consideration.120,121 This suggests that acute toxicity, such as cardiac effects leading to fatalities in mice, may result from high maximum plasma concentration associated with i.v. administration. However, it is proposed that such toxicity could potentially be avoided with i.p. administration. Despite this, the recommendation is not an outright preference for i.p. administration, as i.v. administration is typically the chosen route in clinical settings. We would like to emphasize a nuanced approach: rather than deciding solely based on the route that appears to yield a more promising result, researchers of non-GLP studies should delve deeper into understanding the specific factors driving toxicity for each compound.
6.4 Determination of MBLI efficacy in animal studies
Another trend observed is that there is an in vitro–in vivo discordance among the generated efficacies (explained previously in 4.2, in greater detail). In an animal model, bacteria could also fail to express or lose MBL-harbored plasmids.122,123 Therefore, a β-lactam monotherapy treatment arm should be included in animal trials to account for this finding of pharmacodynamic susceptibility.124,125 While animal models provide a great amount of information regarding the therapeutic efficacy of the drug and guide approval applications by regulatory organizations,126,127 until the shortcomings mentioned above are addressed, it is advisable to exercise prudence in drawing in vivo-related conclusions when evaluating MBLI.7. Challenges in overcoming resistance mechanisms
Bacteria may possess intrinsic resistance to β-lactams expressed by chromosomal genes, or they may acquire resistance from mobile genetic elements such as plasmids.128 Acquired resistance is associated with producing multi-drug resistant bacterial strains, as other antibiotic resistance determinants are also carried on plasmids and may code for resistance to other classes of antimicrobials.129,130 For instance, the blaNDM gene also carries resistance to aminoglycosides since they are linked to ArmA and RmtB methyltransferase genes. While blaIMP and blaVIM genes are generally found on integrons that also have aac(6′) genes that code for acetyltransferase expression, which offers resistance to amikacin and tobramycin.131 Antimicrobial agents targeting the bacterial cell wall have a higher chance of reaching their target than antibiotics targeting the cytoplasm.1 Therefore, β-lactams are a popular treatment choice among clinicians.Nonetheless, there are many challenges to consider when examining β-lactam resistance, unlike other soluble MBLs in the periplasm, NDM is a lipoprotein in the outer membrane vesicles.132 Recent observations indicate that outer membrane vesicles protect the MBL and facilitate the transfer of the blaNDM-1 gene in carbapenem-resistant K. pneumoniae to other strains of K. pneumoniae.133 Other reports of NDM-1 expression convey that a fitness cost is not exerted on the bacteria in P. aeruginosa, E. coli, or A. baumannii. Expression of MBLs such as VIM-2 and Sãu Paulo metallo-β-lactamase 1 (SPM-1), are well tolerated in P. aeruginosa, but poorly tolerated when the same genes encoding for VIM-2 and SPM-1 are expressed in E. coli or A. baumannii. This indicates that MBL expression and compatibility may be species-specific.134
Since 2009, NDM-1 has undergone rapid evolution, as evidenced by the many existing variants.135,136 NDM-4, first detected in 2012,137 is one such variant that has gained importance. NDM-4 differs from NDM-1 by a single amino acid substitution (M154L), which increases the hydrolysis of carbapenems and cephalosporins,138 leading to increased pathogenicity in bacteria harboring the blaNDM-4 gene. Almost half of all NDM variants have the M154L amino acid substitution, among other amino acid substitutions.68 Consequently, the MBL inhibitor design process should consider targeting the M154L-containing gene in addition to other mechanisms of resistance.
Although similar activity is produced in vitro, NDM-1 is more prevalent than IMP-1.2 This is because a strongly conserved promoter (blaNDM-1 gene occurs downstream to the ISAba125 insertion sequence) facilitates higher NDM-1 expression.139 Additionally, the gene can frequently switch between the bacterial host genome and plasmid, as evidenced in many species.140 While IMP-1 is downstream-regulated, it has a variable promoter with differing strengths, leading to weaker resistance.139 For example, in E. coli, the binding of a protein upstream from the blaIMP-6 gene led to transcriptional repression of the gene, resulting in susceptibility to imipenem and meropenem,141 and therefore did not require the use of an MBLI. Upon inspection of the MICs of 1–11, clear observations can be noted of NDM-expressing bacterial strains undergoing more frequent assays, as compared to VIM or IMP harborants. This is often done because NDM variants possess higher MICs, contributing to the pathogens' virulence and, thus, treatment regimen. This further emphasizes the need for a better understanding of the various resistance mechanisms at play on a cellular level.
Gram-negative bacteria are intrinsically more resistant due to an outer membrane that functions as a permeability barrier to antimicrobial substances targeting the bacterial cell's periplasm and inner plasma membrane.142 Therefore, they mainly constitute the list of WHO's priority pathogens.143 The production of multidrug efflux pumps that can expel different antimicrobials out of the bacterial cell should not be ignored during the process of MBLI inhibitor synthesis. When efflux pumps are overexpressed in MBL-harboring pathogens, a significant amount of resistance is conferred to typically effective antibiotics.144 This can pose an enormous setback in MBLI development.
Other mechanisms of escaping the potent effects of antimicrobials include persistence and tolerance, which the scientific community has significantly underestimated.145 Bacterial cells that are “persisters” are dormant bacteria that can withstand the effects of antimicrobials without affecting the MIC and the host's immune response and are responsible for recurrent infections. Uropathogenic E. coli and some P. aeruginosa variants are persisters,145 and they also express MBLs. Consequently, when assessing MBLI potency with antimicrobial susceptibility assays, it is crucial to also incorporate diagnostic methods like molecular assays to confirm the results.
The sharp incline in MBL resistance rates, specifically with NDM, is primarily attributed to poor hygiene practices,146 subpar medical facilities, over-prescription of antimicrobials,147 underlying health conditions, bacterial resistance strategies,148 WHO priority pathogens,7,149 asymptomatic carriage,139 and environmental exposures and reservoirs.146 Although measures are in place to address these confounders, implementing strategies on a national scale with involved stakeholders may help alleviate resistance, persistence, and tolerance to antimicrobial chemotherapy, ultimately promoting the better development of MBL inhibitory compounds.
8. Artificial intelligence influence over drug discovery in AMR
Artificial intelligence (AI), is a field of computer science that mimics human intelligence,150 offers innovative approaches to accelerate scientific discoveries. This is common in medicinal chemistry, where AI has enabled the discovery of new drugs and expedited the overall process of drug development and clinical investigation. AI has become a central component of interdisciplinary efforts aimed at addressing the AMR crisis151–153 as the scale of biological data grows, various AI-driven methods have emerged to analyse it. AI technology enables computers to learn and improve automatically without explicit programming and to build and predict models using available data. The methodological domain of AI primarily involves reasoning, knowledge representation, solution search, and machine learning (ML).154Deep learning (DL), a subset of ML, involves neural networks that mimic the brain's structure to recognise and differentiate between patterns of language, imagery, and various biological data types.155,156 DL-related algorithms have advanced rapidly in recent years to include several typical algorithms; convolutional neural networks, recurrent neural networks, deep reinforcement learning, and generative adversarial networks (GANs).157 These are unsupervised learning algorithms that have been extensively applied in various fields of drug discovery.158–160
Recently, Ding et al. demonstrated that AI can enhance the accuracy and efficiency of BL detection.161 Traditional methods often struggle due to environmental factors such as variations in temperature and pH, which can lead to unreliable results. AI-assisted systems, including smartphone-based AI clouds, can automatically correct these errors, intelligently analyze data, and provide real-time outcomes. Integrating AI with fluorogenic probes and microfluidic devices facilitates the detection of rapid, low-cost, and highly sensitive BLs. This advancement is essential for the early identification of resistant bacteria, which aids in infection control and improves antibiotic treatment options. This method could also be applied to MBLs.
AI technology, therefore should not be ignored in drug discovery for combating AMR.162–166 For instance, data-driven methods can predict new antibiotic compounds, while image-based methods can aid in identifying resistant bacteria.167 AI-assisted compound library screening or the novel design of compound structures can help rapidly identify the most promising antimicrobial compounds. Additionally, AI can leverage known data, such as genomic information, to predict potential resistance sites and related enzymatic functions, laying the groundwork for the design of better antibiotics.168 Moreover, AI has facilitated target identification and dynamic modeling, the design and synthesis of peptides, the evaluation of structure–activity and structure-toxicity relationships, and drug repurposing.169 In this context, an ML model has recently identified a novel NDM-1 inhibitor,170 which significantly reduced the MIC of Meropenem against a panel of E. coli and K. pneumoniae clinical isolates expressing NDM-1, highlighting its potential in combating antibiotic resistance.
9. Outlook and conclusion
AMR poses a significant threat to global public health, with projections suggesting it could cause 10 million deaths annually by 2050, primarily due to infections affecting the respiratory and gastrointestinal tracts.171 The development of MBLIs to restore the activity of existing antibiotics is a critical advancement in the fight against AMR. As drug-resistant bacteria continue to render many antibiotics ineffective, these inhibitors target enzymes that degrade β-lactam antibiotics. By revitalizing the efficacy of these essential drugs, MBLIs can significantly reduce the need for new antibiotic development, mitigate the spread of resistant infections, and safeguard public health. This approach provides an immediate and cost-effective strategy for saving lives and preserving the effectiveness of our antibiotic arsenal. However, there are still relatively few compounds that have shown significant promise, highlighting the need for further development of MBLIs. Among recent advancements, compound 1 is progressing toward clinical registration as a treatment targeting MBL-expressing bacteria, though resistance to some NDM variants underscores the ongoing challenges in this field.172 Adjuvant 2 shows considerable promise as an ultra-broad-spectrum inhibitor of serine SBLs and MBLs, effectively evading resistance mechanisms such as efflux pump expression and porin structural modifications.28A critical aspect of advancing MBLI development lies in standardizing methodologies to facilitate comparison and reproducibility. Reliable assays for evaluating inhibitors' efficacy and specificity are necessary to expedite progress. A unified MIC breakpoint guideline could address discrepancies in assessing antibiotic efficacy. Moreover, zinc content quantification in testing media should become standard practice, with widely available protocols to ensure consistency. Incorporating EDTA/Chelex protocols and emphasizing enzyme inhibition potency through Ki values, IC50 during early enzymatic screening, and EC50 during cellular efficacy testing in the AST guidelines would enhance optimisation. For covalent (reversible and irreversible) MBL inhibitors, the full inhibition mechanism, including non-covalent interactions that promote zinc removal must be considered, supporting the view that higher Ki values do not necessarily indicate reduced compound efficacy. The development of low-cost MBLI enzyme inhibition kits, standardized protocols, and globally accessible materials would not only foster data comparability but also enable the integration of machine-learning models in inhibitor design.173
Pre-clinical research must also focus on addressing bacterial resistance mechanisms at multiple levels. Ethical and reproducible animal models are essential for assessing efficacy while mimicking β-lactamase-mediated resistance. Comprehensive pharmacokinetic data, toxicology studies, and optimized dosing strategies, including Cmax and T > MIC parameters, are critical to ensuring the clinical success of combination therapies. Cellular-level research and multifaceted approaches to bacterial phenotypes, β-lactamase activity, and B1 MBL sequence homology are also imperative to inform inhibitor development.
Emerging computational techniques, supported by artificial intelligence, offer significant potential in early-stage MBLI research. Methods such as ligand-based pharmacophore modeling, structure-based virtual screening, and molecular docking can streamline the initial stages of in silico assessment.174–176 Whether employing classical or quantum methods, researchers must carefully account for factors such as water molecule coordination in the Zn2+ ion active site to refine inhibition strategies.
To achieve meaningful progress in the fight against AMR, global collaboration is essential. Standardized approaches in MBLI development, supported by robust financial investments from organizations such as the Wellcome Trust, Gates Foundation, and CarbX, could enable a “Moonshot” initiative against superbugs. This vision would involve distributing standardized kits, establishing centralized data repositories, and implementing clear pre-clinical frameworks for decision-making. By aligning resources, uniform research methodologies, and stakeholder priorities, MBLIs can move closer to developing effective therapeutic strategies to combat resistant pathogens and help mitigate the AMR crisis.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
NR was responsible for data curation, and formal analysis, wrote the initial draft, and revised the manuscript accordingly. AMB and JRAS were responsible for some data curation, visualization aspects, funding acquisition and writing – review & editing. AM was responsible for writing – reviewing & editing and the funding acquisition. CAG was responsible for the visualization aspects and for writing – review & editing. HGK and PIA provided resources, funding acquisition and writing – review & editing. TN and TG conceptualized the idea, provided resources, funding acquisition, project administration, writing – review & editing and supervision of the manuscript.Conflicts of interest
There are none to declare.Acknowledgements
Respective authors thank the following funding agencies: JRAS (CNPQ 440053/2022-6, 402141/2023-7, 304610/2023-2 and 443533/2024-5), AMB (CAPES 88887.712684/2022-00), AM (SAMRC BRICS JAF 2021/095), for HGK, PIA, TN and TG (NRF 137961, 145774, 120419, 137979 and SAMRC BRICS JAF 2021/033). JRAS is also the recipient of a CNPq Research Productivity Fellowship.References
- D. Kim, S. Kim, Y. Kwon, Y. Kim, H. Park, K. Kwak, H. Lee, J. H. Lee, K.-M. Jang and D. Kim, Biomol. Ther., 2023, 31, 141–147, DOI:10.4062/biomolther.2023.008.
- G. Bahr, L. J. Gonzalez and A. J. Vila, Chem. Rev., 2021, 121, 7957–8094, DOI:10.1021/acs.chemrev.1c00138.
- K. M. Papp-Wallace, Expert Opin. Pharmacother., 2019, 20, 2169–2184, DOI:10.1080/14656566.2019.1660772.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans and Y. Carmeli, Lancet Infect. Dis., 2018, 18, 318–327, DOI:10.1016/S1473-3099(17)30753-3.
- S. R. Shrivastava, P. S. Shrivastava and J. Ramasamy, J. Med. Soc., 2018, 32, 76–77, DOI:10.4103/jms.jms_25_17.
- World Health Organization, Antibacterial products in clinical development for priority pathogens, https://www.who.int/observatories/global-observatory-on-health-research-and-development/monitoring/antibacterial-products-in-clinical-development-for-priority-pathogens, (accessed, 11 October, 2024).
- P. Beyer and S. Paulin, Bull. W. H. O., 2020, 98, 151, DOI:10.2471/BLT.20.251751.
- G. V. Asokan, T. Ramadhan, E. Ahmed and H. Sanad, Oman Med. J., 2019, 34, 184–193, DOI:10.5001/omj.2019.37.
- M. F. Mojica, M.-A. Rossi, A. J. Vila and R. A. Bonomo, Lancet Infect. Dis., 2022, 22, e28–e34, DOI:10.1016/S1473-3099(20)30868-9.
- N. Reddy, M. Shungube, P. I. Arvidsson, S. Baijnath, H. G. Kruger, T. Govender and T. Naicker, Expert Opin. Ther. Pat., 2020, 30, 541–555, DOI:10.1080/13543776.2020.1767070.
- B. Liu, R. E. L. Trout, G.-H. Chu, D. McGarry, R. W. Jackson, J. C. Hamrick, D. M. Daigle, S. M. Cusick, C. Pozzi and F. De Luca, J. Med. Chem., 2019, 63, 2789–2801, DOI:10.1021/acs.jmedchem.9b01518.
- J. C. Hamrick, J.-D. Docquier, T. Uehara, C. L. Myers, D. A. Six, C. L. Chatwin, K. J. John, S. F. Vernacchio, S. M. Cusick and R. E. Trout, Antimicrob. Agents Chemother., 2020, 64, e01963-19, DOI:10.1128/aac.01963-19.
- S. Mushtaq, A. Vickers, M. Doumith, M. J. Ellington, N. Woodford and D. M. Livermore, J. Antimicrob. Chemother., 2021, 76, 160–170, DOI:10.1093/jac/dkaa391.
- R. Tsivkovski, M. Totrov and O. Lomovskaya, Antimicrob. Agents Chemother., 2020, 64, e00130-20, DOI:10.1128/aac.00130-20.
- S. J. Hecker, K. R. Reddy, O. Lomovskaya, D. C. Griffith, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Sabet and Z. Tarazi, J. Med. Chem., 2020, 63, 7491–7507, DOI:10.1021/acs.jmedchem.9b01976.
- O. Lomovskaya, R. Tsivkovski, K. Nelson, D. Rubio-Aparicio, D. Sun, M. Totrov and M. N. Dudley, Antimicrob. Agents Chemother., 2020, 64, e00212–e00220, DOI:10.1128/aac.00212-20.
- O. Lomovskaya, M. Castanheira, J. Lindley, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Totrov, J. Loutit and M. Dudley, Antimicrob. Agents Chemother., 2023, 67, e00440-23, DOI:10.1128/aac.00440-23.
- S. E. Boyd, D. M. Livermore, D. C. Hooper and W. W. Hope, Antimicrob. Agents Chemother., 2020, 64, e00397-20, DOI:10.1128/aac.00397-20.
- S. T. Cahill, R. Cain, D. Y. Wang, C. T. Lohans, D. W. Wareham, H. P. Oswin, J. Mohammed, J. Spencer, C. W. Fishwick and M. A. McDonough, Antimicrob. Agents Chemother., 2017, 61, e02260-16, DOI:10.1128/aac.02260-16.
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J.-C. Jiménez-Castellanos, M. B. Avison, J. Spencer, C. W. Fishwick and C. J. Schofield, Nat. Commun., 2016, 7, 12406, DOI:10.1038/ncomms12406.
- B. Geibel, J. Dowell, D. Dickerson and T. Henkel, Open Forum Infect. Dis., 2018, 5, S431, DOI:10.1093/ofid/ofy210.1232.
- V. Blay, B. Tolani, S. P. Ho and M. R. Arkin, Drug Discovery Today, 2020, 25, 1807–1821, DOI:10.1016/j.drudis.2020.07.024.
- H. Aldewachi, R. N. Al-Zidan, M. T. Conner and M. M. Salman, Bioengineering, 2021, 8, 30, DOI:10.3390/bioengineering8020030.
- P. Szymański, M. Markowicz and E. Mikiciuk-Olasik, Int. J. Mol. Sci., 2012, 13, 427–452, DOI:10.3390/ijms13010427.
- E. Martis, R. Radhakrishnan and R. Badve, J. Appl. Pharm. Sci., 2011, 1, 02–10 Search PubMed , https://japsonline.com/abstract.php?article_id=2.
- M. J. Wildey, A. Haunso, M. Tudor, M. Webb and J. H. Connick, Annu. Rep. Med. Chem., 2017, 50, 149–195, DOI:10.1016/bs.armc.2017.08.004.
- I. Wallach, D. Bernard, K. Nguyen, G. Ho, A. Morrison, A. Stecula, A. Rosnik, A. M. O'Sullivan, A. Davtyan, B. Samudio, B. Thomas, B. Worley, B. Butler, C. Laggner, D. Thayer, E. Moharreri, G. Friedland, H. Truong, H. van den Bedem, H. L. Ng, K. Stafford, K. Sarangapani, K. Giesler, L. Ngo, M. Mysinger, M. Ahmed, N. J. Anthis, N. Henriksen, P. Gniewek, S. Eckert, S. de Oliveira, S. Suterwala, S. V. K. PrasadPrasad, S. Shek, S. Contreras, S. Hare, T. Palazzo, T. E. O'Brien, T. Van Grack, T. Williams, T.-R. Chern, V. Kenyon, A. H. Lee, A. B. Cann, B. Bergman, B. M. Anderson, B. D. Cox, J. M. Warrington, J. M. Sorenson, J. M. Goldenberg, M. A. Young, N. DeHaan, R. P. Pemberton, S. Schroedl, T. M. Abramyan, T. Gupta, V. Mysore, A. G. Presser, A. A. Ferrando, A. D. Andricopulo, A. Ghosh, A. G. Ayachi, A. Mushtaq, A. M. Shaqra, A. K. L. Toh, A. V. Smrcka, A. Ciccia, A. S. de Oliveira, A. Sverzhinsky, A. M. de Sousa, A. I. Agoulnik, A. Kushnir, A. N. Freiberg, A. V. Statsyuk, A. R. Gingras, A. Degterev, A. Tomilov, A. Vrielink, A. A. Garaeva, A. Bryant-Friedrich, A. Caflisch, A. K. Patel, A. V. Rangarajan, A. Matheeussen, A. Battistoni, A. Caporali, A. Chini, A. Ilari, A. Mattevi, A. T. Foote, A. Trabocchi, A. Stahl, A. B. Herr, A. Berti, A. Freywald, A. G. Reidenbach, A. Lam, A. R. Cuddihy, A. White, A. Taglialatela, A. K. Ojha, A. M. Cathcart, A. A. L. Motyl, A. Borowska, A. D'Antuono, A. K. H. Hirsch, A. M. Porcelli, A. Minakova, A. Montanaro, A. Müller, A. Fiorillo, A. Virtanen, A. J. O'Donoghue, A. Del Rio Flores, A. E. Garmendia, A. Pineda-Lucena, A. T. Panganiban, A. Samantha, A. K. Chatterjee, A. L. Haas, A. S. Paparella, A. L. S. John, A. Prince, A. ElSheikh, A. M. Apfel, A. Colomba, A. O'Dea, B. N. t. Diallo, B. M. R. M. Ribeiro, B. A. Bailey-Elkin, B. L. Edelman, B. Liou, B. Perry, B. S. K. Chua, B. Kováts, B. Englinger, B. Balakrishnan, B. Gong, B. Agianian, B. Pressly, B. P. M. Salas, B. M. Duggan, B. V. Geisbrecht, B. W. Dymock, B. C. Morten, B. D. Hammock, B. E. F. Mota, B. C. Dickinson, C. Fraser, C. Lempicki, C. D. Novina, C. Torner, C. Ballatore, C. Bon, C. J. Chapman, C. L. Partch, C. T. Chaton, C. Huang, C.-Y. Yang, C. M. Kahler, C. Karan, C. Keller, C. L. Dieck, C. Huimei, C. Liu, C. Peltier, C. K. Mantri, C. M. Kemet, C. E. Müller, C. Weber, C. M. Zeina, C. S. Muli, C. Morisseau, C. Alkan, C. Reglero, C. A. Loy, C. M. Wilson, C. Myhr, C. Arrigoni, C. Paulino, C. Santiago, D. Luo, D. J. Tumes, D. A. Keedy, D. A. Lawrence, D. Chen, D. Manor, D. J. Trader, D. A. Hildeman, D. H. Drewry, D. J. Dowling, D. J. Hosfield, D. M. Smith, D. Moreira, D. P. Siderovski, D. Shum, D. T. Krist, D. W. H. Riches, D. M. Ferraris, D. H. Anderson, D. R. Coombe, D. S. Welsbie, D. Hu, D. Ortiz, D. Alramadhani, D. Zhang, D. Chaudhuri, D. J. Slotboom, D. R. Ronning, D. Lee, D. Dirksen, D. A. Shoue, D. W. Zochodne, D. Krishnamurthy, D. Duncan, D. M. Glubb, E. L. M. Gelardi, E. C. Hsiao, E. G. Lynn, E. B. Silva, E. Aguilera, E. Lenci, E. T. Abraham, E. Lama, E. Mameli, E. Leung, E. Giles, E. M. Christensen, E. R. Mason, E. Petretto, E. F. Trakhtenberg, E. J. Rubin, E. Strauss, E. W. Thompson, E. Cione, E. M. Lisabeth, E. Fan, E. G. Kroon, E. Jo, E. M. García-Cuesta, E. Glukhov, E. Gavathiotis, F. Yu, F. Xiang, F. Leng, F. Wang, F. Ingoglia, F. van den Akker, F. Borriello, F. J. Vizeacoumar, F. Luh, F. S. Buckner, F. S. Vizeacoumar, F. B. Bdira, F. Svensson, G. M. Rodriguez, G. Bognár, G. Lembo, G. Zhang, G. Dempsey, G. Eitzen, G. Mayer, G. L. Greene, G. A. Garcia, G. L. Lukacs, G. Prikler, G. C. G. Parico, G. Colotti, G. De Keulenaer, G. Cortopassi, G. Roti, G. Girolimetti, G. Fiermonte, G. Gasparre, G. Leuzzi, G. Dahal, G. Michlewski, G. L. Conn, G. D. Stuchbury, G. R. Bowman, G. M. Popowicz, G. Veit, G. E. de Souza, G. Akk, G. Caljon, G. Alvarez, G. Rucinski, G. Lee, G. Cildir, H. Li, H. E. Breton, H. Jafar-Nejad, H. Zhou, H. P. Moore, H. Tilford, H. Yuan, H. Shim, H. Wulff, H. Hoppe and A. P. The Atomwise, Sci. Rep., 2024, 14, 7526, DOI:10.1038/s41598-024-54655-z.
- R. Li, X. Chen, C. Zhou, Q.-Q. Dai and L. Yang, Eur. J. Med. Chem., 2022, 242, 114677, DOI:10.1016/j.ejmech.2022.114677.
- R. Wang, T.-P. Lai, P. Gao, H. Zhang, P.-L. Ho, P. C.-Y. Woo, G. Ma, R. Y.-T. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439, DOI:10.1038/s41467-018-02828-6.
- Z. Meng, M.-L. Tang, L. Yu, Y. Liang, J. Han, C. Zhang, F. Hu, J.-M. Yu and X. Sun, ACS Infect. Dis., 2019, 5, 903–916, DOI:10.1021/acsinfecdis.8b00366.
- J.-I. Wachino, W. Jin, K. Kimura, H. Kurosaki, A. Sato and Y. Arakawa, mBio, 2020, 11, e03144-19, DOI:10.1128/mbio.03144-19.
- Ø. Samuelsen, O. A. H. Åstrand, C. Fröhlich, A. Heikal, S. Skagseth, T. J. O. Carlsen, H.-K. S. Leiros, A. Bayer, C. Schnaars and G. Kildahl-Andersen, Antimicrob. Agents Chemother., 2020, 64, e02415–e02419, DOI:10.1128/aac.02415-19.
- S. Das, A. Johnson, L. McEntee, N. Farrington, A. Kirby, J. Unsworth, A. Jimenez-Valverde, R. Kolamunnage-Dona, J. Bousquet and L. Alibaud, Antimicrob. Agents Chemother., 2020, 64, e01076-20, DOI:10.1128/aac.01076-20.
- D. T. Davies, S. Leiris, N. Sprynski, J. Castandet, C. Lozano, J. Bousquet, M. Zalacain, S. Vasa, P. K. Dasari and R. Pattipati, ACS Infect. Dis., 2020, 6, 2419–2430, DOI:10.1021/acsinfecdis.0c00207.
- M. Zalacain, C. Lozano, A. Llanos, N. Sprynski, T. Valmont, C. De Piano, D. Davies, S. Leiris, C. Sable and A. Ledoux, Antimicrob. Agents Chemother., 2021, 65, e00203–e00221, DOI:10.1128/aac.00203-21.
- F. Chen, M. Bai, W. Liu, H. Kong, T. Zhang, H. Yao, E. Zhang, J. Du and S. Qin, Eur. J. Med. Chem., 2021, 224, 113702, DOI:10.1016/j.ejmech.2021.113702.
- N. Ooi, V. E. Lee, N. Chalam-Judge, R. Newman, A. J. Wilkinson, I. R. Cooper, D. Orr, S. Lee and V. J. Savage, J. Antimicrob. Chemother., 2021, 76, 460–466, DOI:10.1093/jac/dkaa455.
- J. Brem, T. Panduwawala, J. U. Hansen, J. Hewitt, E. Liepins, P. Donets, L. Espina, A. J. Farley, K. Shubin and G. G. Campillos, Nat. Chem., 2022, 14, 15–24, DOI:10.1038/s41557-021-00831-x.
- B. K. Peters, N. Reddy, M. Shungube, L. Girdhari, S. Baijnath, S. Mdanda, L. Chetty, T. Ntombela, T. Arumugam and L. A. Bester, ACS Infect. Dis., 2023, 9, 486–496, DOI:10.1021/acsinfecdis.2c00485.
- Z. Yang, R. M. Twidale, S. Gervasoni, R. Suardíaz, C. K. Colenso, E. J. Lang, J. Spencer and A. J. Mulholland, J. Chem. Inf. Model., 2021, 61, 5658–5672, DOI:10.1021/acs.jcim.1c01109.
- O. Melse, I. Antes, V. R. Kaila and M. Zacharias, J. Comput. Chem., 2023, 44, 912–926, DOI:10.1002/jcc.27052.
- Q. Liao, A. Pabis, B. Strodel and S. C. L. Kamerlin, J. Phys. Chem. Lett., 2017, 8, 5408–5414, DOI:10.1021/acs.jpclett.7b02358.
- E. Lence and C. González-Bello, Front. Microbiol., 2021, 12, 721826, DOI:10.3389/fmicb.2021.721826.
- B. Honarparvar, T. Govender, G. E. Maguire, M. E. Soliman and H. G. Kruger, Chem. Rev., 2014, 114, 493–537, DOI:10.1021/cr300314q.
- V. T. Sabe, T. Ntombela, L. A. Jhamba, G. E. Maguire, T. Govender, T. Naicker and H. G. Kruger, Eur. J. Med. Chem., 2021, 224, 113705, DOI:10.1016/j.ejmech.2021.113705.
- L. Olsen, T. Rasmussen, L. Hemmingsen and U. Ryde, J. Phys. Chem. B, 2004, 108, 17639–17648, DOI:10.1021/jp0482215.
- C. Wang and H. Guo, J. Phys. Chem. B, 2007, 111, 9986–9992, DOI:10.1021/jp073864g.
- K. Zhu, J. Lu, Z. Liang, X. Kong, F. Ye, L. Jin, H. Geng, Y. Chen, M. Zheng and H. Jiang, J. Comput.-Aided Mol. Des., 2013, 27, 247–256, DOI:10.1007/s10822-012-9630-6.
- M. Zheng and D. Xu, J. Phys. Chem. B, 2013, 117, 11596–11607, DOI:10.1021/jp4065906.
- J. Chen, H. Chen, T. Zhu, D. Zhou, F. Zhang, X. Lao and H. Zheng, Phys. Chem. Chem. Phys., 2014, 16, 6709–6716, 10.1039/C3CP55069A.
- E. O. Levina, M. G. Khrenova, A. A. Astakhov and V. G. Tsirelson, RSC Adv., 2020, 10, 8664–8676, 10.1039/C9RA10649A.
- S. Gervasoni, J. Spencer, P. Hinchliffe, A. Pedretti, F. Vairoletti, G. Mahler and A. J. Mulholland, Proteins: Struct., Funct., Bioinf., 2022, 90, 372–384, DOI:10.1002/prot.26227.
- F. E. Medina and G. A. Jaña, ACS Catal., 2021, 12, 36–47, DOI:10.1021/acscatal.1c04786.
- L.-C. Ju, Z. Cheng, W. Fast, R. A. Bonomo and M. W. Crowder, Trends Pharmacol. Sci., 2018, 39, 635–647, DOI:10.1016/j.tips.2018.03.007.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201, DOI:10.1128/cmr.00037-09.
- B. Srinivasan, FEBS J., 2023, 290, 2292–2305, DOI:10.1111/febs.16404.
- E. Freire, Drug Discovery Today, 2008, 13, 869–874, DOI:10.1016/j.drudis.2008.07.005.
- B.-F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel and W. Huisinga, SLAS Discovery, 2009, 14, 913–923, DOI:10.1177/1087057109336751.
- M. M. González and A. J. Vila, An Elusive Task: A Clinically Useful Inhibitor of Metallo-β-Lactamases, in Zinc Enzyme Inhibitors, Topics in Medicinal Chemistry, ed. C. Supuran and C. Capasso, Springer, Cham, 2016, vol. 22, DOI:10.1007/7355_2016_6.
- Y. Xiang, Y.-N. Chang, Y. Ge, J. S. Kang, Y.-L. Zhang, X.-L. Liu, P. Oelschlaeger and K.-W. Yang, Bioorg. Med. Chem. Lett., 2017, 27, 5225–5229, DOI:10.1016/j.bmcl.2017.10.038.
- G. A. Holdgate, T. D. Meek and R. L. Grimley, Nat. Rev. Drug Discovery, 2018, 17, 115–132, DOI:10.1038/nrd.2017.219.
- S. Piszkiewicz and G. J. Pielak, Biochemistry, 2019, 58, 3825–3833, DOI:10.1021/acs.biochem.9b00675.
- H. Bisswanger, Perspect. Sci., 2014, 1, 41–55, DOI:10.1016/j.pisc.2014.02.005.
- S. Markossian, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, K. Brimacombe, T. D. Chung, N. P. Coussens and J. L. Dahlin, Assay guidance manual, Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD, 2004 Search PubMed.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105, DOI:10.1016/j.mib.2017.10.026.
- M. Watanabe, S. Iyobe, M. Inoue and S. Mitsuhashi, Antimicrob. Agents Chemother., 1991, 35, 147–151, DOI:10.1128/aac.35.1.147.
- T. Naas, S. Oueslati, R. A. Bonnin, M. L. Dabos, A. Zavala, L. Dortet, P. Retailleau and B. I. Iorga, J. Enzyme Inhib. Med. Chem., 2017, 32, 917–919, DOI:10.1080/14756366.2017.1344235.
- National Library of Medicine, https://www.ncbi.nlm.nih.gov/pathogens/refgene/# (accessed, October 5, 2023) Search PubMed.
- L. Lauretti, M. L. Riccio, A. Mazzariol, G. Cornaglia, G. Amicosante, R. Fontana and G. M. Rossolini, Antimicrob. Agents Chemother., 1999, 43, 1584–1590, DOI:10.1128/aac.43.7.1584.
- P. Nordmann, T. Naas and L. Poirel, Emerging Infect. Dis., 2011, 17, 1791–1798, DOI:10.3201/eid1710.110655.
- M. Castanheira, L. M. Deshpande, D. Mathai, J. M. Bell, R. N. Jones and R. E. Mendes, Antimicrob. Agents Chemother., 2011, 55, 1274–1278, DOI:10.1128/aac.01497-10.
- L. Dortet, L. Poirel and P. Nordmann, BioMed Res. Int., 2014, 2014, 249856, DOI:10.1155/2014/249856.
- M. P. Weinstein and J. S. Lewis, J. Clin. Microbiol., 2020, 58, e01864-19, DOI:10.1128/jcm.01864-19.
- EUCAST, 2015, https://www.eucast.org/clinical_breakpoints (accessed, October 5, 2024) Search PubMed.
- T. Cusack, E. Ashley, C. Ling, S. Rattanavong, T. Roberts, P. Turner, T. Wangrangsimakul and D. Dance, Clin. Microbiol. Infect., 2019, 25, 910–911, DOI:10.1016/j.cmi.2019.03.007.
- World Health Organization, Technical Report, 2019, https://www.who.int/publications/i/item/WHO-WSI-AMR-2019.1 (October 11, 2024) Search PubMed.
- E. Durante-Mangoni, R. Andini and R. Zampino, Clin. Microbiol. Infect., 2019, 25, 943–950, DOI:10.1016/j.cmi.2019.04.013.
- V. Chibabhai, T. Nana, N. Bosman, T. Thomas and W. Lowman, Infection, 2018, 46, 1–13, DOI:10.1007/s15010-017-1070-8.
- T. F. Durand-Reville, A. A. Miller, J. P. O'Donnell, X. Wu, M. A. Sylvester, S. Guler, R. Iyer, A. B. Shapiro, N. M. Carter and C. Velez-Vega, Nature, 2021, 597, 698–702, DOI:10.1038/s41586-021-03899-0.
- S. Freire, J. Findlay, E. Gruner, V. Bruderer, P. Nordmann and L. Poirel, J. Antimicrob. Chemother., 2024, 79, 930–932, DOI:10.1093/jac/dkae020.
- H. S. Sader, R. E. Mendes, L. R. Duncan, C. G. Carvalhaes and M. Castanheria, J. Antimicrob. Chemother., 2022, 77, 2642–2649, DOI:10.1093/jac/dkac233.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488–2492, DOI:10.1126/science.1060331.
- R. Girardello, P. J. Bispo, T. M. Yamanaka and A. C. Gales, J. Clin. Microbiol., 2012, 50, 2414–2418, DOI:10.1128/jcm.06686-11.
- T. E. Asempa, K. Abdelraouf and D. P. Nicolau, J. Antimicrob. Chemother., 2020, 75, 997–1005, DOI:10.1093/jac/dkz532.
- A. Bilinskaya, D. J. Buckheit, M. Gnoinski, T. E. Asempa and D. P. Nicolau, J. Clin. Microbiol., 2020, 58, e02019–e02020, DOI:10.1128/jcm.02019-20.
- D. Pröfrock and A. Prange, Appl. Spectrosc., 2012, 66, 843–868, DOI:10.1366/12-06681.
- M. D'Orazio, M. C. Mastropasqua, M. Cerasi, F. Pacello, A. Consalvo, B. Chirullo, B. Mortensen, E. P. Skaar, D. Ciavardelli and P. Pasquali, Metallomics, 2015, 7, 1023–1035, 10.1039/c5mt00017c.
- R. P. Rennie, J. Clin. Microbiol., 2021, 59, e01211-21, DOI:10.1128/jcm.00039-21.
- T. E. Asempa and D. P. Nicolau, J. Clin. Microbiol., 2021, 59, e01211–e01221, DOI:10.1128/jcm.01211-21.
- S. Siemann, D. Brewer, A. J. Clarke, G. I. Dmitrienko, G. Lajoie and T. Viswanatha, Biochim. Biophys. Acta, Gen. Subj., 2002, 1571, 190–200, DOI:10.1016/S0304-4165(02)00258-1.
- I. Garcia-Saez, J.-D. Docquier, G. Rossolini and O. Dideberg, J. Mol. Biol., 2008, 375, 604–611, DOI:10.1016/j.jmb.2007.11.012.
- D. Gardonio and S. Siemann, Biochem. Biophys. Res. Commun., 2009, 381, 107–111, DOI:10.1016/j.bbrc.2009.02.021.
- A. Proschak, G. Martinelli, D. Frank, M. J. Rotter, S. Brunst, L. Weizel, L. D. Burgers, R. Fürst, E. Proschak and I. Sosič, Eur. J. Med. Chem., 2022, 228, 113975, DOI:10.1016/j.ejmech.2021.113975.
- B. Kowalska-Krochmal and R. Dudek-Wicher, Pathogens, 2021, 10, 165, DOI:10.3390/pathogens10020165.
- R. Urso, P. Blardi and G. Giorgi, Riv. Eur. Sci. Med. Farmacol., 2002, 6, 33–44 CAS.
- R. L. Crass and M. P. Pai, Pharmacotherapy, 2019, 39, 182–195, DOI:10.1002/phar.2210.
- S. H. MacVane, J. L. Kuti and D. P. Nicolau, Int. J. Antimicrob. Agents, 2014, 43, 105–113, DOI:10.1016/j.ijantimicag.2013.10.021.
- J. A. Dowell, D. Dickerson and T. Henkel, Antimicrob. Agents Chemother., 2021, 65, e01053-21, DOI:10.1128/aac.01053-21.
- D. Griffith, J. Roberts, S. Wallis, M. P. Hernandez-Mitre, E. Morgan, S. Gehrke, M. Dudley and J. Loutit, Open Forum Infect. Dis., 2022, 9, ofac492.294, DOI:10.1093/ofid/ofac492.294.
- Y.-H. Yan, W. Li, W. Chen, C. Li, K.-R. Zhu, J. Deng, Q.-Q. Dai, L.-L. Yang, Z. Wang and G.-B. Li, Eur. J. Med. Chem., 2022, 228, 113965, DOI:10.1016/j.ejmech.2021.113965.
- European Medicines Agency, ICH Guideline M10 on Bioanalytical Method Validation - Step 5, 2023, pp. 1–45, https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline#current-version-9785 (accessed, October 5, 2024) Search PubMed.
- W. A. Craig, Clin. Infect. Dis., 1998, 26, 1–10, DOI:10.1086/516284.
- L. Santos Filho, J. L. Kuti and D. P. Nicolau, Braz. J. Microbiol., 2007, 38, 183–193, DOI:10.1590/S1517-83822007000200001.
- E. L. Gillespie, J. L. Kuti and D. P. Nicolau, Expert Opin. Drug Metab. Toxicol., 2005, 1, 351–361, DOI:10.1517/17425255.1.3.351.
- T. E. Asempa, K. Abdelraouf and D. P. Nicolau, Antimicrob. Agents Chemother., 2021, 65, 10–1128, DOI:10.1128/aac.02271-20.
- D. R. Andes and A. J. Lepak, Curr. Opin. Pharmacol., 2017, 36, 94–99, DOI:10.1016/j.coph.2017.09.004.
- K. Abdelraouf, S. Reyes and D. P. Nicolau, J. Antimicrob. Chemother., 2021, 76, 684–691, DOI:10.1093/jac/dkaa467.
- G. Daikos, A. Panagiotakopoulou, E. Tzelepi, A. Loli, L. Tzouvelekis and V. Miriagou, Clin. Microbiol. Infect., 2007, 13, 202–205, DOI:10.1111/j.1469-0691.2006.01590.x.
- M. Souli, E. Konstantinidou, I. Tzepi, T. Tsaganos, A. Pefanis, Z. Chryssouli, I. Galani, E. Giamarellos-Bourboulis and H. Giamarellou, J. Antimicrob. Chemother., 2011, 66, 611–617, DOI:10.1093/jac/dkq470.
- I. M. Ghazi, J. L. Crandon, E. P. Lesho, P. McGann and D. P. Nicolau, Heliyon, 2016, 2, e000121, DOI:10.1016/j.heliyon.2016.e00121.
- S. García-Fernández, M.-I. Morosini, D. Gijón, L. Beatobe, P. Ruiz-Garbajosa, L. Domínguez, R. Cantón and A. Valverde, J. Clin. Microbiol., 2016, 54, 464–466, DOI:10.1128/jcm.02580-15.
- K. Bush and P. A. Bradford, Clin. Microbiol. Rev., 2020, 33, 10–128, DOI:10.1128/cmr.00047-19.
- Z. Cheng, P. W. Thomas, L. Ju, A. Bergstrom, K. Mason, D. Clayton, C. Miller, C. R. Bethel, J. VanPelt and D. L. Tierney, J. Biol. Chem., 2018, 293, 12606–12618, DOI:10.1074/jbc.RA118.003835.
- N. Wade, K. H. Tehrani, N. C. Brüchle, M. J. van Haren, V. Mashayekhi and N. I. Martin, ChemMedChem, 2021, 16, 1651–1659, DOI:10.1002/cmdc.202100042.
- A. Roujansky, V. de Lastours, F. Guérin, F. Chau, G. Cheminet, L. Massias, V. Cattoir and B. Fantin, Antimicrob. Agents Chemother., 2020, 64, e00853-20, DOI:10.1128/aac.00853-20.
- J. R. Lenhard and Z. P. Bulman, J. Antimicrob. Chemother., 2019, 74, 2825–2843, DOI:10.1093/jac/dkz226.
- N. Reddy, L. Girdhari, M. Shungube, A. C. Gouws, B. K. Peters, K. K. Rajbongshi, S. Baijnath, S. Mdanda, T. Ntombela and T. Arumugam, Antibiotics, 2023, 12, 633, DOI:10.3390/antibiotics12040633.
- B. Moya, I. M. Barcelo, G. Cabot, G. Torrens, S. Palwe, P. Joshi, K. Umarkar, S. Takalkar, H. Periasamy and S. Bhagwat, Antimicrob. Agents Chemother., 2019, 63(5), e00128-19, DOI:10.1128/aac.00128-19.
- M. Everett, N. Sprynski, A. Coelho, J. Castandet, M. Bayet, J. Bougnon, C. Lozano, D. T. Davies, S. Leiris and M. Zalacain, Antimicrob. Agents Chemother., 2018, 62, 10–1128, DOI:10.1128/aac.00074-18.
- G. Kumar, V. Rao and N. Kumar, Nanomedicines for Breast Cancer Theranostics, Elsevier, 2020, pp. 299–329, DOI:10.1016/B978-0-12-820016-2.00013-6.
- S. Asati, V. Pandey, V. Gour, R. Tiwari, V. Soni, K. Rajpoot, M. Tekade, M. C. Sharma and R. K. Tekade, Pharmacokinetics and Toxicokinetic Considerations, Elsevier, 2022, pp. 401–424, DOI:10.1016/B978-0-323-98367-9.00001-9.
- D. E. Wiskirchen, P. Nordmann, J. L. Crandon and D. P. Nicolau, Antimicrob. Agents Chemother., 2014, 58, 1671–1677, DOI:10.1128/aac.01946-13.
- I. M. Ghazi, J. L. Crandon, E. P. Lesho, P. McGann and D. P. Nicolau, Antimicrob. Agents Chemother., 2015, 59, 7145–7147, DOI:10.1128/aac.00794-15.
- E. Growcott, T. Cariaga, L. Morris, X. Zang, S. Lopez, D. Ansaldi, J. Gold, L. Gamboa, T. Roth and R. Simmons, J. Antimicrob. Chemother., 2019, 74, 108–116, DOI:10.1093/jac/dky404.
- A. J. Lepak, M. Zhao and D. R. Andes, Antimicrob. Agents Chemother., 2019, 63, e01648-19, DOI:10.1128/aac.01648-19.
- J. B. Bulitta, W. W. Hope, A. E. Eakin, T. Guina, V. H. Tam, A. Louie, G. L. Drusano and J. L. Hoover, Antimicrob. Agents Chemother., 2019, 63, 10–1128, DOI:10.1128/aac.02307-18.
- U. Waack, E. A. Weinstein and J. J. Farley, Antimicrob. Agents Chemother., 2020, 64, e02242-19, DOI:10.1128/aac.02242-19.
- J.-D. Docquier and S. Mangani, Drug Resistance Updates, 2018, 36, 13–29, DOI:10.1016/j.drup.2017.11.002.
- P. Bose, A. Rangnekar and P. Desikan, Indian J. Med. Res., 2022, 155, 243–252, DOI:10.4103/ijmr.IJMR_685_19.
- K. A. Shenoy, E. Jyothi and R. Ravikumar, Indian J. Med. Res., 2014, 139, 625–631 Search PubMed.
- S. Huang, W. Dai, S. Sun, X. Zhang and L. Zhang, PLoS One, 2012, 7, e47636, DOI:10.1371/journal.pone.0047636.
- L. J. González, G. Bahr, T. G. Nakashige, E. M. Nolan, R. A. Bonomo and A. J. Vila, Nat. Chem. Biol., 2016, 12, 516–522, DOI:10.1038/nchembio.2083.
- J. Zakhour, L. E. W. El Ayoubi and S. S. Kanj, Expert Rev. Anti-infect. Ther., 2024, 22, 189–201, DOI:10.1080/14787210.2024.2311213.
- C. López, J. A. Ayala, R. A. Bonomo, L. J. González and A. J. Vila, Nat. Commun., 2019, 10, 3617, DOI:10.1038/s41467-019-11615-w.
- N. Farhat and A. U. Khan, Infect., Genet. Evol., 2020, 86, 104588, DOI:10.1016/j.meegid.2020.104588.
- T. Wang, Y. Zhou, C. Zou, Z. Zhu, J. Zhu, J. Lv, X. Xie, L. Chen, S. Niu and H. Du, mSphere, 2021, 6, e00776-21, DOI:10.1128/msphere.00776-21.
- P. Nordmann, A. E. Boulanger and L. Poirel, Antimicrob. Agents Chemother., 2012, 56, 2184–2186, DOI:10.1128/aac.05961-11.
- J. B. Thoden, B. M. Benin, A. Priebe, W. S. Shin, R. Muthyala, Y. Y. Sham and H. M. Holden, J. Biol. Chem., 2023, 299, 105135, DOI:10.1016/j.jbc.2023.105135.
- C. H. P. Cheung, M. Alorabi, F. Hamilton, Y. Takebayashi, O. Mounsey, K. J. Heesom, P. B. Williams, O. M. Williams, M. Albur and A. P. MacGowan, Antimicrob. Agents Chemother., 2021, 65, e02412–e02420, DOI:10.1128/aac.02412-20.
- P. Nordmann, L. Poirel, T. R. Walsh and D. M. Livermore, Trends Microbiol., 2011, 19, 588–595, DOI:10.1016/j.tim.2011.09.005.
- T. Segawa, T. Sekizuka, S. Suzuki, K. Shibayama, M. Matsui and M. Kuroda, PLoS One, 2018, 13, e0208976, DOI:10.1371/journal.pone.0208976.
- S. Kojima and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2629–E2634, DOI:10.1073/pnas.1310333110.
- World Health Organization, WHO publishes list of bacteria for which new antibiotics are urgently needed, https://www.who.int/news/item/27-02-2017-whopublishes-list-of-bacteria-for-which-new-antibiotics-are-urgentlyneeded, (accessed 29 June 2023) Search PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51, DOI:10.1038/nrmicro3380.
- O. Pacios, L. Blasco, I. Bleriot, L. Fernandez-Garcia, M. González Bardanca, A. Ambroa, M. López, G. Bou and M. Tomás, Antibiotics, 2020, 9, 65, DOI:10.3390/antibiotics9020065.
- A. Dixit, N. Kumar, S. Kumar and V. Trigun, Indian J. Community Med., 2019, 44, 4–8, DOI:10.4103/ijcm.IJCM_217_18.
- T. Nusrat, N. Akter, N. A. A. Rahman, B. Godman, D. T. D. Rozario and M. Haque, Hosp. Pract., 2020, 48, 128–136, DOI:10.1080/21548331.2020.1754687.
- P. Nori, W. Szymczak, Y. Puius, A. Sharma, K. Cowman, P. Gialanella, Z. Fleischner, M. Corpuz, J. Torres-Isasiga and R. Bartash, Int. J. Antimicrob. Agents, 2020, 56, 106179, DOI:10.1016/j.ijantimicag.2020.106179.
- V. V. Mogasale, P. Saldanha, V. Pai, P. Rekha and V. Mogasale, Sci. Rep., 2021, 11, 5116, DOI:10.1038/s41598-021-84293-8.
- J. G. Greener, S. M. Kandathil, L. Moffat and D. T. Jones, Nat. Rev. Mol. Cell Biol., 2022, 23, 40–55, DOI:10.1038/s41580-021-00407-0.
- E. J. Topol, Nat. Med., 2019, 25, 44–56, DOI:10.1038/s41591-018-0300-7.
- H. Wang, T. Fu, Y. Du, W. Gao, K. Huang, Z. Liu, P. Chandak, S. Liu, P. Van Katwyk and A. Deac, Nature, 2023, 620, 47–60, DOI:10.1038/s41586-023-06221-2.
- M. W. Mullowney, K. R. Duncan, S. S. Elsayed, N. Garg, J. J. van der Hooft, N. I. Martin, D. Meijer, B. R. Terlouw, F. Biermann and K. Blin, Nat. Rev. Drug Discovery, 2023, 22, 895–916, DOI:10.1038/s41573-023-00774-7.
- D. M. Camacho, K. M. Collins, R. K. Powers, J. C. Costello and J. J. Collins, Cell, 2018, 173, 1581–1592, DOI:10.1016/j.cell.2018.05.015.
- E. Gawehn, J. A. Hiss and G. Schneider, Mol. Inf., 2016, 35, 3–14, DOI:10.1002/minf.201501008.
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444, DOI:10.1038/nature14539.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Commun. ACM, 2020, 63, 139–144, DOI:10.1145/3422622.
- M. Lu, J. Yin, Q. Zhu, G. Lin, M. Mou, F. Liu, Z. Pan, N. You, X. Lian and F. Li, Engineering, 2023, 27, 37–69, DOI:10.1016/j.eng.2023.01.014.
- A. Lavecchia, Drug Discovery Today, 2019, 24, 2017–2032, DOI:10.1016/j.drudis.2019.07.006.
- D. Paul, G. Sanap, S. Shenoy, D. Kalyane, K. Kalia and R. K. Tekade, Drug Discovery Today, 2021, 26, 80–93, DOI:10.1016/j.drudis.2020.10.010.
- Y. Ding, J. Chen, Q. Wu, B. Fang, W. Ji, X. Li, C. Yu, X. Wang, X. Cheng and H. D. Yu, SmartMat, 2024, 5, e1214, DOI:10.1002/smm2.1214.
- K. Swanson, G. Liu, D. B. Catacutan, A. Arnold, J. Zou and J. M. Stokes, Nat. Mach. Intell., 2024, 6, 338–353, DOI:10.1038/s42256-024-00809-7.
- A. Cesaro and C. de la Fuente-Nunez, Nat. Chem. Biol., 2023, 19, 1296–1298, DOI:10.1038/s41589-023-01448-6.
- G. Liu and J. M. Stokes, Curr. Opin. Microbiol., 2022, 69, 102190, DOI:10.1016/j.mib.2022.102190.
- M. Miethke, M. Pieroni, T. Weber, M. Brönstrup, P. Hammann, L. Halby, P. B. Arimondo, P. Glaser, B. Aigle and H. B. Bode, Nat. Rev. Chem., 2021, 5, 726–749, DOI:10.1038/s41570-021-00313-1.
- T. Lluka and J. M. Stokes, Ann. N. Y. Acad. Sci., 2023, 1519, 74–93, DOI:10.1111/nyas.14930.
- D. Krentzel, S. L. Shorte and C. Zimmer, Trends Cell Biol., 2023, 33, 538–554, DOI:10.1016/j.tcb.2022.11.011.
- A. Talat and A. U. Khan, Drug Discovery Today, 2023, 28, 103491, DOI:10.1016/j.drudis.2023.103491.
- F. Urbina, A. C. Puhl and S. Ekins, Curr. Opin. Chem. Biol., 2021, 65, 74–84, DOI:10.1016/j.cbpa.2021.06.001.
- Z. Cheng, M. Aitha, C. A. Thomas, A. Sturgill, M. Fairweather, A. Hu, C. R. Bethel, D. D. Rivera, P. Dranchak and P. W. Thomas, J. Chem. Inf. Model., 2024, 64, 3977–3991, DOI:10.1021/acs.jcim.3c02015.
- J. O'Neill, CABI Databse, 2016, https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf, (accesed, October 5, 2024) Search PubMed.
- C. Le Terrier, V. Gruenig, C. Fournier, P. Nordmann and L. Poirel, Lancet Infect. Dis., 2023, 23, 401–402, DOI:10.1016/S1473-3099(23)00069-5.
- H. Achdout, A. Aimon, E. Bar-David and G. Morris, BioRxiv, 2020, preprint, DOI:10.1101/2020.10.29.339317.
- M. Kullappan, J. Mallavarapu Ambrose and K. M. Surapaneni, J. Mol. Recognit., 2021, 34, e2898, DOI:10.1002/jmr.2898.
- M. Ezati, A. Ahmadi, E. Behmard and A. Najafi, J. Biomol. Struct. Dyn., 2023, 10672–10687, DOI:10.1080/07391102.2023.2258406.
- T. Salih and P. G. Ali, Mol. Simul., 2023, 49, 1373–1387, DOI:10.1080/08927022.2023.2232468.
| This journal is © The Royal Society of Chemistry 2025 |